

Abstract
Evidence suggests that NMDA-type glutamate receptors contribute to degeneration of striatal medium-sized spiny neurons (MSNs) in Huntington's disease (HD). Previously, we demonstrated that NMDA receptor (NMDAR)-mediated current and/or toxicity is increased in MSNs from the yeast artificial chromosome (YAC) transgenic mouse model expressing polyglutamine (polyQ)-expanded (mutant) full-length human huntingtin (htt). Others have shown that membrane-associated guanylate kinases (MAGUKs), such as PSD-95 and SAP102, modulate NMDAR surface expression and excitotoxicity in hippocampal and cortical neurons and that htt interacts with PSD-95. Here, we tested the hypothesis that an altered association between MAGUKs and NMDARs in mutant huntingtin-expressing cells contributes to increased susceptibility to excitotoxicity. We show that htt coimmunoprecipitated with SAP102 in HEK293T cells and striatal tissue from wild-type and YAC transgenic mice; however, the association of SAP102 with htt or the NMDAR NR2B subunit was unaffected by htt polyQ length, whereas association of PSD-95 with NR2B in striatal tissue was enhanced by increased htt polyQ length. Treatment of cultured MSNs with Tat–NR2B9c peptide blocked binding of NR2B with SAP102 and PSD-95 and reduced NMDAR surface expression by 20% in both YAC transgenic and wild-type MSNs, and also restored susceptibility to NMDAR excitoxicity in YAC HD MSNs to levels observed in wild-type MSNs; a similar effect on excitotoxicity was observed after knockdown of PSD-95 by small interfering RNA. Unlike previous findings in cortical and hippocampal neurons, rescue of NMDA toxicity by Tat–NR2B9c occurred independently of any effect on neuronal nitric oxide synthase activity. Our results elucidate further the mechanisms underlying enhanced excitotoxicity in HD.
Introduction
NMDA-type glutamate receptors play a critical role in synaptic plasticity (Bliss and Collingridge, 1993) as well as mechanisms underlying neurodegeneration, including Huntington's disease (HD) (Waxman and Lynch, 2005; Cowan and Raymond, 2006). NMDA receptors (NMDARs) contain two NR1 subunits with two of NR2A, NR2B, NR2C, and/or NR2D (Dingledine et al., 1999). NR2 subunits determine receptor-channel properties and interactions with scaffolding proteins and signaling networks (Dingledine et al., 1999; Hardingham and Bading, 2003; Prybylowski and Wenthold, 2004), influencing the balance between neuronal survival and dysfunction or death.
Several lines of evidence support a role for altered NMDAR function in HD (DiFiglia, 1990; Fan and Raymond, 2007), which is caused by a polyglutamine (polyQ) repeat expansion >35 near the N terminus of the protein huntingtin (Huntington's Disease Collaborative Research Group, 1993). HD is characterized by selective neuronal degeneration, affecting striatal GABAergic medium-sized spiny neurons (MSNs) most severely (Vonsattel and DiFiglia, 1998). These neurons are enriched in NR2B-containing NMDARs compared with other NR2 subunits and other brain regions (Landwehrmeyer et al., 1995; Christie et al., 2000; Li et al., 2003). Previous work in mouse and cellular models of HD suggests NR2B-containing NMDARs are functionally altered in HD and contribute to neuronal dysfunction and susceptibility to apoptosis (Chen et al., 1999; Levine et al., 1999; Cepeda et al., 2001; Zeron et al., 2001, 2002; Song et al., 2003; Starling et al., 2005; Tang et al., 2005; Milnerwood et al., 2006; Shehadeh et al., 2006; Fan et al., 2007). However, the molecular mechanisms underlying mutant huntingtin's effect on NMDAR function have not been fully elucidated.
Membrane-associated guanylate kinases (MAGUKs), including PSD-93, PSD-95, SAP97, and SAP102, act as scaffolds to facilitate signaling by anchoring key enzymes close to glutamate receptors (Fujita and Kurachi, 2000). The second PSD-95/Discs large (Dlg)/ZO-1 (PDZ) domain in PSD-95 and SAP102 binds to the NR2A or NR2B C-terminal tSXV motif (Niethammer et al., 1996). This interaction regulates forward trafficking and stability of NMDARs at surface membranes and synapses (Roche et al., 2001; Lin et al., 2004; Prybylowski et al., 2005) and contributes to toxic signaling downstream of NMDAR activation via PSD-95-mediated coupling to neuronal nitric oxide synthase (nNOS) (Aarts et al., 2002). Notably, the PSD-95 Src homology 3 (SH3) domain interacts directly with the polyproline region of huntingtin in vitro, in a polyQ length-dependent manner, and these proteins coassociate in human brain tissue (Sun et al., 2001).
Here, we investigate the importance of the PSD-95 and SAP102 interaction with NR2B-containing NMDARs in mechanisms underlying enhanced NMDAR surface expression and excitotoxicity previously documented in mutant huntingtin-expressing MSNs (Zeron et al., 2002; Shehadeh et al., 2006; Fan et al., 2007). We used transfected HEK293T cells, as well as striatal MSNs and tissues from wild-type (WT) mice or the yeast artificial chromosome (YAC) transgenic HD mouse model expressing full-length human huntingtin with polyQ lengths of 18 (control), 72, and 128 (pathogenic) (Hodgson et al., 1999; Slow et al., 2003), to show that altered NR2B binding with PSD-95 contributes to enhanced NMDA toxicity independently of nNOS activation.
Materials and Methods
Cell culture and transient transfection.
HEK293T cells (human embryonic kidney cells expressing simian virus 40 large T antigen) were maintained in modified Eagle's medium (Invitrogen) with penicillin/streptomycin, α-glutamine, sodium pyruvate, and 10% fetal bovine serum (Invitrogen). Cells were transfected using the calcium–phosphate precipitation method (Chen and Okayama, 1987), with a 1:1 transfection solution of 250 mm CaCl2 and 2× N,N-bishydroxyethyl-2-aminoethane-sufonic acid (BES) (50 mm BES, 280 mm NaCl, and 1.5 mm Na2HPO4, pH 6.98). Ten to 12 μg of plasmids were mixed with 1 ml of transfection solution per 10 cm plate. Cells were transfected for 6–8 h in a 37°C, 3% CO2 incubator and then washed once with PBS to stop transfection. cDNA plasmids encoding NR1A, NR2B, htt15, and htt138 (htt15 and htt138 encode full-length human htt containing 15 and 138 polyQ, respectively) have been described previously (Chen et al., 1999). The cDNA construct for myc-tagged SAP102 (Müller et al., 1996) was a generous gift from Dr. R. L. Huganir (Johns Hopkins Medical Institutions, Baltimore, MD). The cDNA plasmid for PSD-95, described previously (Li et al., 2003), was a gift from Dr. A. El-Husseini (University of British Columbia, Vancouver, British Columbia, Canada).
Transgenic mice.
The following lines of YAC transgenic mice, YAC18 (line 212) (similar to line 29) (Leavitt et al., 2001), YAC72 (line 2511) (Hodgson et al., 1999), and YAC128 (line55) (Graham et al., 2006a), were used as models expressing full-length human htt containing 18 (control), 72, and 128 (pathogenic) polyQ repeats (18Q, 72Q, 128Q), respectively, and compared with FVB/N WT mice. Offspring of crosses between two homozygous YAC transgenic mice on a pure FVB/NJ strain background were used for experiments (Hodgson et al., 1999). All mice were housed and tested and tissue was harvested according to guidelines of the University of British Columbia and the Canadian Council for Animal Care.
Primary striatal neuronal culture.
Nitric-acid-treated 12 mm glass coverslips were placed in 24-well plates coated with poly-d-lysine (250 μg/ml). Striatal tissues were dissected from postnatal days 0–1 YAC transgenic mice or wild-type FVB/N mice in HBSS (Invitrogen) on ice, digested in papain, and further dissociated in trypsin inhibitor solution. MSNs were then transferred to serum-free plating medium [B27, penicillin/streptomycin, α-glutamine, and Neurobasal medium (Invitrogen)] and plated at a density of ∼2 × 105 cells per well as described previously (Zeron et al., 2002). Approximately 80–90% of the cells cultured in serum-free media have the characteristics of MSNs (Kovács et al., 2001; Zeron et al., 2004; Shehadeh et al., 2006).
Western blotting and immunoprecipitation.
Homogenized 4- or 8-week-old YAC transgenic mice striatal tissues, cultured MSNs at 9–10 d in vitro (DIV), or HEK293T cells (30–32 h after transfection) were harvested, lysed in 1% NP-40-containing lysis buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1 mm PMSF, 2 μg/ml aprotinin, 2 μg/ml leupeptin, 4 μg/ml pepstatin A, 30 mm NaF, 40 mm β-glycerophosphate, 20 mm sodium pyrophosphate, 1 mm sodium orthovanadate, and 10 μm ZVAD, in Milli-Q water), and solubilized by ultrasonication. The lysates were incubated overnight with equilibrated 50% Protein A/G beads and antibodies. Beads of each sample were then washed with Tris wash buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, and 1% Triton X-100 in Milli-Q water) and heated at 95–99°C in protein sample buffer (0.125 m Tris, pH 6.8, 2% SDS, 10% glycerol, and 72 mg/ml dithiothreitol, with Pyronin Y in Milli-Q water). Paired samples were run on 8% SDS-PAGE and then transferred from gels to polyvinylidene difluoride membranes (Bio-Rad) and subjected to immunoblotting. Densitometry of resulting bands was analyzed using NIH ImageJ or Scion Image software. The band densities for YAC72 and YAC128 were normalized to those of either WT or YAC18 (controls) that were run on the same gel because of variability in antibody sensitivity and exposure times between experiments.
Surface and internal NR1 and GluR1 staining.
Cultured MSNs were treated with 200 nm or 1 μm Tat–NR2B peptides for 1 h on DIV 9 or 10 and fixed with 4% paraformaldehyde (PFA) for 12 min. Fixed MSNs were numbered according to different conditions and coded before staining to ensure that the operator was blinded during the subsequent processing and analysis of immunofluorescence. MSNs were then blocked with 10% NGS and incubated with anti-NR1 or anti-GluR1 primary antibody for 1.5 h at room temperature (RT), washed with PBS, and then incubated with green secondary antibody (anti-mouse or anti-rabbit) for 45 min to 1 h at RT. MSNs were again washed with PBS and permeabilized with 0.5% Triton X-100 for 5 min. Then MSNs were blocked again with 10% NGS and incubated with anti-NR1 or anti-GluR1 primary antibody for 1.5 h at RT. MSNs were washed with PBS and incubated with red secondary antibody (anti-mouse or anti-rabbit) for 45 min to 1 h at RT. MSNs were washed with PBS and mounted on slides with Fluoromount-G (Southern Biotechnology Associates). Ten to 12 pictures of immunofluorescent single MSNs per each coverslip were taken using a 63× lens in the red and green fluorescent channels by Northern Eclipse software. Photomicrographs were analyzed by NIH ImageJ by measuring the mean fluorescence intensity of processes and soma of each MSN after background subtraction, as described previously (Fan et al., 2007). The surface/internal ratio of NR1 or GluR1 was calculated as the green/red fluorescent channel intensity for each MSN.
NMDA- and staurosporine-induced cytotoxicity.
Cultured MSNs were pretreated with 200 nm Tat–NR2B9c peptides for 1 h and/or 100 μm Nω-nitro-l-arginine (N-Arg) (nNOS inhibitor; from Sigma-Aldrich) for 40 min on DIV 9 and then incubated in warm balanced salt solution (BSS), pH 7.4, with or without 500 μm NMDA for 10 min. MSNs were washed twice with warm plating medium (PM) and then incubated in conditioned PM (without Tat peptides) for 24 h. In each experiment, apoptosis 24 h after 10 min treatment with BSS alone, without NMDA, was assessed and ranged from 4 to 25%; this value was subtracted from the total cell death induced by NMDA in each experiment to calculate the specific NMDAR-mediated apoptosis. In experiments assessing apoptosis induced by staurosporine, this drug was added to the medium to a final concentration of 10 μm after peptide treatment and maintained for 24 h. Then MSNs were washed with PBS once and fixed with 4% PFA for 1 h.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay and assessment of apoptosis.
Fixed MSNs were numbered according to different conditions and coded before staining to ensure that the operator was blinded during the subsequent processing and analysis of immunofluorescence. MSNs were further permeabilized with 0.25% Triton X-100 in PBS with 0.5% sodium citrate on ice for 2 min. MSNs were then washed once with PBS and incubated in PBS with 16 μg/ml RNase at RT for 30 min. MSNs were washed with PBS and incubated in terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) (Roche) reagent (at a ratio of label/enzyme of 9:1) per well in the dark for 1 h, followed by one wash with PBS and staining with 8 μm propidium iodide (PI) or 10 μm Hoechst 33342. MSNs were then washed with PBS and mounted on slides with Fluoromount-G. The second day after staining, the percentage of apoptotic cell death was assessed by counting the numbers of TUNEL-positive cells in the green fluorescent channel, which also showed condensed and blebbed nuclear morphology in the red (or blue) fluorescent channel, and then dividing by the total number of PI (or Hoechst)-positive cell nuclei in the red (or blue) fluorescent channel and multiplying by 100. A total of 1000 neurons were counted per condition. The percentage of apoptotic cell death of MSNs exposed to BSS alone was subtracted as a baseline from each of the other conditions.
Small interfering RNA knockdown.
The control [“scrambled” small interfering RNA (siRNA); sc-36869; or nontargeting siRNA, D-001950-01] and PSD-95 siRNA (sc-42012) and NE–Dlg (neuronal and endocrine DIg) (SAP102) siRNA (sc-42007) were purchased from Santa Cruz Biotechnology or Dharmacon (Thermo Fisher Scientific). At DIV 7 or 8, cultured MSNs were transfected with siRNA by DharmaFECT siRNA transfection reagent 3 (T-2003-01; Dharmacon/Thermo Fisher Scientific), in complete medium containing B-27 (Trushina et al., 2006). On DIV 9 or 10, MSNs were treated as described above (see NMDA- and staurosporine-induced cytotoxicity), fixed on DIV 10 or 11, and then forward to immunostaining. For detecting the knockdown efficiency by Western blotting, 35 mm dishes of MSNs with or without siRNA transfection were directly harvested 72 h later on DIV 10 or 11.
cGMP assay.
On DIV 9, cultured MSNs were pretreated 1 h with N-Arg and/or Tat–NR2B9c or ifenprodil (IFN), incubated in warm BSS with or without 500 μm NMDA for 1 min, and then immediately forwarded to Amersham cGMP Enzymeimmunoassay Biotrak (EIA) System kit (RPN226; GE Healthcare), as per the instructions of the manufacturer. The concentrations of cGMP in 96-well plates were determined by a microplate reader (Multiskan Ascent; Labsystems) at 630 nm.
Peptides.
The Tat–NR2B9c and Tat–NR2BAA peptides were originally a gift from Dr. M. Tymianski (University of Toronto, Toronto, Ontario, Canada) and later purchased from AnaSpec. The peptides were dissolved in sterile Milli-Q water to make 1 mm stock solution and stored at −80°C.
Antibodies.
The immunocytochemistry was done with a mouse monoclonal antibody against NR1A (2 μg/ml; catalog #MAB363; Millipore Bioscience Research Reagents), rabbit polyclonal anti-GluR1 antibody (1 μg/ml; catalog #06-306; Millipore Bioscience Research Reagents), anti-mouse Alexa Fluor 488 (1:2000; A-11029; Invitrogen), anti-rabbit Alexa Fluor 488 (1:2000; A-11008; Invitrogen), anti-rabbit Alexa Fluor 594 (1:2000; A-11012; Invitrogen), and anti-mouse cyanine 3 (1:200; 715-165-150; Jackson ImmunoResearch). Antibodies used for immunoprecipitation (IP) and/or to probe Western blots included the following: rabbit polyclonal anti-NR2B (1 μg/ml, catalog #06-600; Millipore Bioscience Research Reagents); mouse monoclonal anti-NR2B (1:250, catalog #610417; Transduction Laboratories); mouse monoclonal anti-NR2B (1 μg/ml; catalog #MA1-2014; Affinity BioReagents); rabbit polyclonal anti-NR2A (1 μg/ml, catalog #07-632; Millipore Bioscience Research Reagents); anti-huntingtin mouse monoclonal antibody (1 μg/ml; catalog #MAB2166; Millipore Bioscience Research Reagents); anti-huntingtin human-specific mouse monoclonal antibody (HD650, used at 1:500) (Warby et al., 2005); anti-SAP102 mouse monoclonal antibody (1 μg/ml; catalog #VAM PS006; Stressgene); anti-SAP102 rabbit polyclonal antibody (1:400; catalog #AB5170; Millipore Bioscience Research Reagents); anti-SAP102 mouse monoclonal antibody (1 μg/ml, catalog #75-058; NeuroMAB); anti-SAP102 rabbit polyclonal antibody (used at 8 μg/ml; a gift from Dr. R. Wenthold (National Institutes of Health, Bethesda, MD); anti-myc MAB-4A6 (1 μg/ml; catalog #05-724; Millipore Bioscience Research Reagents), anti-PSD-95 (1 μg/ml, catalog #75-028; NeuroMAB), anti-PSD-95 (1:200; MA1045; Affinity BioReagents), and anti-actin (1 μg/ml; sc-1616; Santa Cruz Biotechnology). Horseradish peroxidase-linked secondary antibodies for Western blotting are anti-mouse IgG antibody (NA931V) and anti-rabbit IgG antibody (NA934V), both from GE Healthcare and used at 1:5000.
Data analysis.
Figures, tables, and statistical analyses were generated using Microsoft Excel, Northern Eclipse, NIH ImageJ, Origin 6.0, Prism 4.0, or Adobe Photoshop 7.0 software. Data or bars are presented as the mean ± SEM. Significant differences were determined using the unpaired or paired, two-tailed Student's t test, one-way ANOVA, or two-way ANOVA, as appropriate.
Results
Association of htt with PSD-95 and SAP102
Previous studies in transfected heterologous cells and human brain tissue have demonstrated that htt associates with PSD-95 via the polyproline and SH3 domains, respectively, and that polyQ expansion weakens that interaction (Sun et al., 2001). We tested these observations in the YAC transgenic mouse model of HD. First, we found no significant difference in expression levels of PSD-95 across YAC genotypes (18, 72, and 128) and WT mouse striatal tissue (for representative blot, see Fig. 1A) (quantitative data not shown). As predicted from the previous studies in heterologous cells and human brain tissue, we also found that coassociation of htt and PSD-95 was decreased significantly in striatal tissue from YAC128 transgenic mice compared with WT (Fig. 1A). In the example blot, there is a clear decrease in co-IP of PSD-95 with htt for YAC128 but not for YAC72; this reflects the group data (Fig. 1A, shown in the bar graph below the blot) in that the YAC72 results were quite variable and not significantly different from WT.
Figure 1.
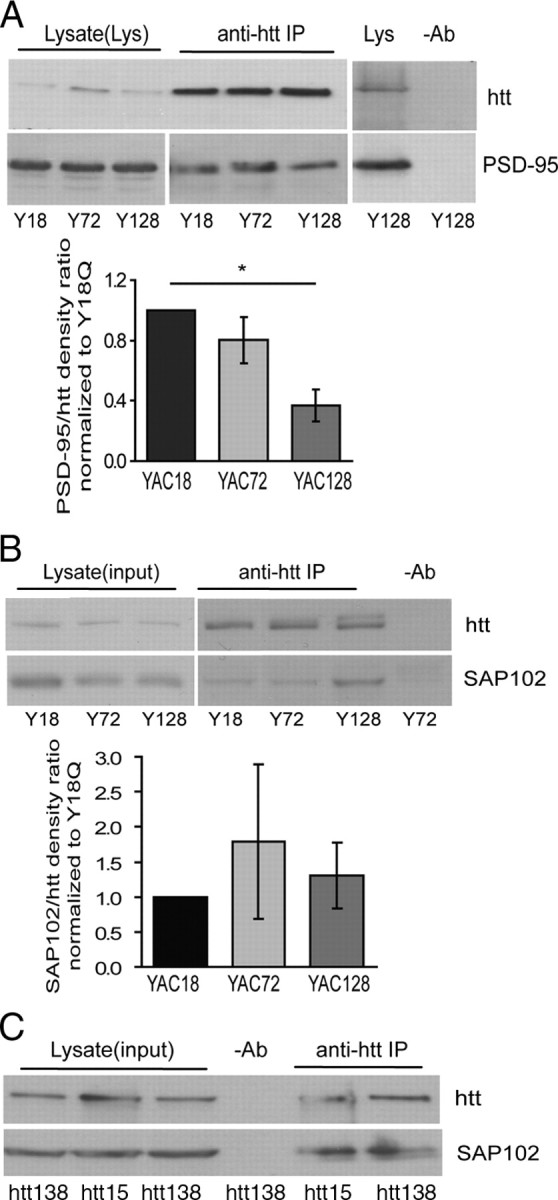
Association of huntingtin with PSD-95 and SAP102. A, PSD-95 associates with htt in YAC transgenic mouse striatal tissues. Lysates from 4- to 8-week-old mouse striatal tissue were immunoprecipitated with MAB2166 or HD650 (an anti-human htt-specific antibody). The blot was cut into two parts along the 160 kDa marker and probed with anti-htt and anti-PSD-95 antibodies, respectively. The mean band density ratio of PSD-95 to huntingtin for pooled data is shown below (in each experiment, YAC72 and/or YAC128 were run on same gel as YAC18, and band density ratios were normalized to those in YAC18 lane). Significant by one-way ANOVA and Bonferroni's post hoc tests, *p < 0.05 (n = 3). B, SAP102 associates with huntingtin in YAC transgenic mouse striatal tissue. Lysates from 4-week-old YAC transgenic mouse striatal tissue were immunoprecipitated with MAB2166 or HD650. The blot was cut into two parts along the 160 kDa marker and probed with anti-htt and anti-SAP102 antibodies, respectively. Pooled data for SAP102/htt density ratios are shown below representative blot. Ratios are similar across all three genotypes (in each experiment, YAC72 and/or YAC128 were run on same gel as YAC18, and band density ratios were normalized to those in YAC18 lane). p > 0.05 by one-way ANOVA; n = 7 for YAC18 and YAC72, n = 5 for YAC128. C, SAP102 associates with huntingtin in HEK293T cells. Lysates from HEK239T cells transfected with full-length htt15 or htt138, and SAP102 were immunoprecipitated and probed as described in B. Note that SAP102/htt density ratios are similar for htt15 and htt138.
SAP102 is a member of the PSD-95 family of MAGUK proteins and shows high homology with PSD-95 in the SH3 as well as PDZ domains (Kim and Sheng, 2004). To determine whether SAP102 could also associate in a complex with htt, we repeated the previous experiments probing for SAP102. Similar to results for PSD-95, expression levels of SAP102 were not significantly different in YAC18, YAC72, and YAC128 striatal tissues (as illustrated in Fig. 1B) (quantitative data not shown). Moreover, SAP102 coimmunoprecipitated with htt in striatal tissue from all genotypes of YAC transgenic mice (Fig. 1B), but no significant differences in coassociation of SAP102 and htt were found between genotypes. Notably, SAP102 also coimmunoprecipitated with htt (both full-length human htt containing 15 and 138 polyQ: htt15 and htt138, respectively) in lysates from transfected HEK293T cells (Fig. 1C). SAP102/htt band density ratio was 0.72 ± 0.14 for htt15-transfected cells and 0.50 ± 0.06 for htt138-transfected cells (p = 0.23 by Student's paired t test for nine experiments). This result suggests that the association between these proteins may be direct as it is for htt and PSD-95 (Sun et al., 2001), although there is no significant difference in the htt-SAP102 association with htt15 or htt138 expression.
Effect of mutant htt expression on interaction of NR2B with PSD-95 and SAP102
Both SAP102 and PSD-95 interact with NR2A and NR2B subunits (Niethammer et al., 1996; Sans et al., 2000), and interaction of NR2B with these MAGUKs has been shown to regulate NMDAR surface expression and synaptic localization (Roche et al., 2001; Chung et al., 2004; Prybylowski et al., 2005), as well as excitotoxicity (Aarts et al., 2002). Previously, we have shown that, in cultured MSNs from YAC transgenic mice, NMDAR-mediated apoptosis increases with htt polyQ length (Zeron et al., 2002; Shehadeh et al., 2006) and that NR2B-containing NMDAR surface expression is significantly increased in YAC72 compared with WT MSNs (Fan et al., 2007).
To determine whether mutant htt expression alters interaction between NR2B-containing NMDARs and SAP102 or PSD-95, we immunoprecipitated NR2B from striatal tissue of YAC transgenic mice and probed for these MAGUK proteins. As illustrated in Figure 2A, we found that association between NR2B and PSD-95 significantly increased with increasing polyQ length (PSD-95/NR2B band density ratio was 0.69 ± 0.13 for WT, 1.14 ± 0.23 for YAC72, and 1.47 ± 0.27 for YAC128 MSNs). In contrast, the extent of NR2B binding with SAP102 was similar for all YAC genotypes (Fig. 2B). Although we quantified results from experiments in which WT tissue served as a control for the PSD-95/NR2B co-IP whereas YAC18 tissue was the control for the SAP102/NR2B co-IP, when we did compare YAC18 and WT results with the other YAC genotypes in one or more experiments, these two controls showed highly similar results for co-IP of either PSD-95 or SAP102 with NR2B. Notably, the enhanced interaction of PSD-95 with the NMDAR is specific for the NR2B type, because the binding of NR2A and PSD-95 was similar for WT and all YAC genotypes (Fig. 2C).
Figure 2.
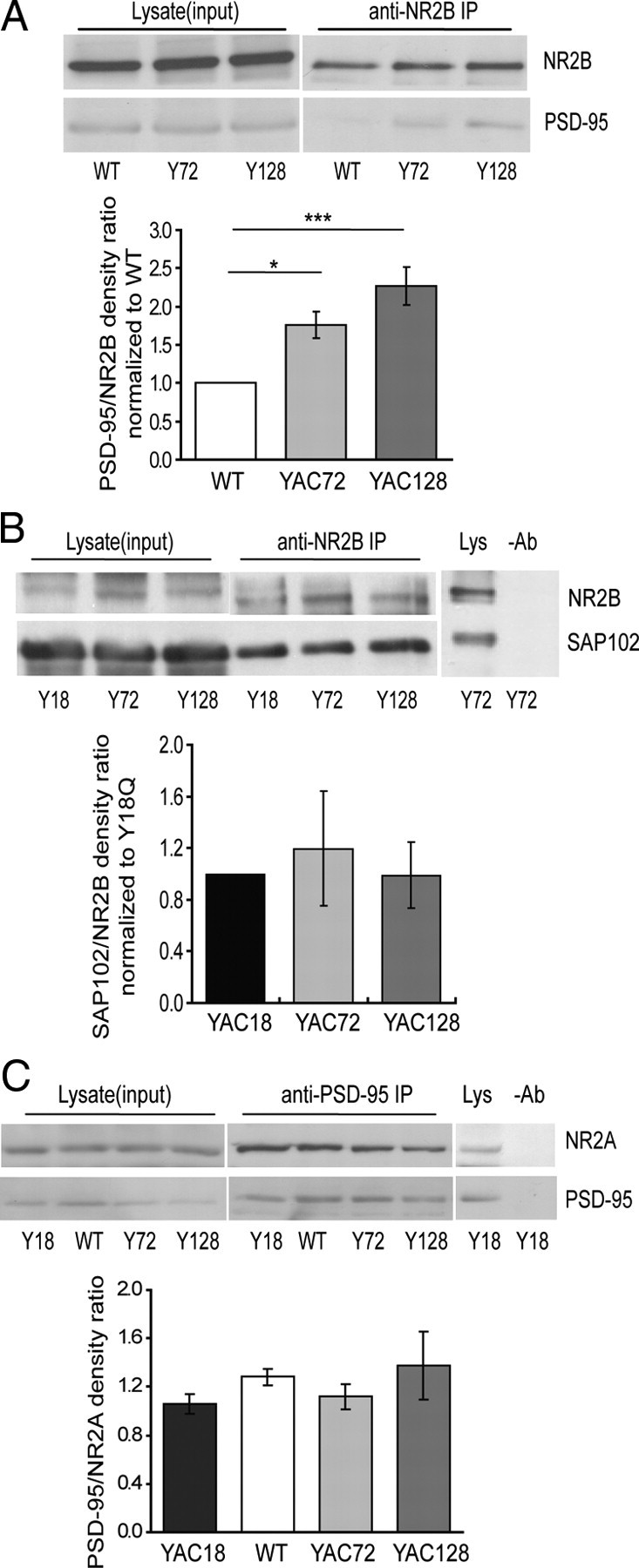
Effect of mutant htt expression on interaction of NR2B with PSD-95 and SAP102. A, Mutant htt expression alters the association of PSD-95 with NR2B in vivo. NR2B was immunoprecipitated from lysates of 8-week-old WT and YAC transgenic mouse striatal tissue using an anti-NR2B antibody (Affinity BioReagents), and proteins were subjected to Western blot analysis. The blot was cut into two parts along the 160 kDa marker and probed with anti-NR2B and anti-PSD-95 antibodies, respectively. The mean band density ratio of PSD-95 to NR2B for pooled data is shown below (in each experiment, YAC72 and/or YAC128 were run on same gel as WT, and band density ratios were normalized to those in WT lane). Significant by one-way ANOVA and Bonferroni's post hoc tests, *p < 0.05, ***p < 0.001 (n = 8 for WT; n = 7 for YAC72 and YAC128). B, Representative blot showing that coimmunoprecipitation of SAP102 with NR2B in 4-week-old YAC mouse striatal tissue is not altered by htt repeat length. Pooled data for the mean band density ratio of SAP102 to NR2B is shown (in each experiment, YAC72 and/or YAC128 were run on same gel as YAC18, and band density ratios were normalized to those in YAC18 lane). There is no significant difference by one-way ANOVA, p > 0.05 (n = 6 for YAC18 and YAC128; n = 5 for YAC72). C, Representative blot showing that coimmunoprecipitation of PSD-95 with NR2A in 8-week-old YAC mouse striatal tissue is not altered by htt polyQ repeat length. Pooled data for the mean band density ratio of PSD-95 to NR2A is shown. There is no significant difference by one-way ANOVA, p > 0.05 (n = 3 for YAC18; n = 5 for WT and YAC72; n = 4 for YAC128).
Tat–NR2B9c peptide perturbs NR2B interaction with PSD-95 and SAP102 in cultured MSNs
We hypothesized that polyQ-expanded htt in a complex with MAGUKs and NMDARs functionally alters the interaction between NR2B and PSD-95 and/or SAP102, thereby contributing to the enhanced NMDAR surface expression and/or toxicity described previously in YAC mutant transgenic mouse MSNs (Zeron et al., 2002; Shehadeh et al., 2006; Fan et al., 2007). To test this hypothesis, we used a Tat-fused peptide identical to the C-terminal 9 aa of NR2B (Tat–NR2B9c) to disrupt NR2B binding to PDZ-containing proteins, as described previously (Aarts et al., 2002). In that study, 100 μm Tat-NR2B9c was required to disrupt PSD-95 binding to NR2B in rat forebrain lysates ex vivo.
To determine the peptide concentration range required to uncouple NR2B from PSD-95 and SAP102 in live cells, we treated transfected HEK293T cells for 1 h with various peptide concentrations. In experiments in HEK293 cells, we found that, compared with treatment with the control peptide Tat–NR2BAA, Tat–NR2B9c significantly reduced the amount of PSD-95 (36 ± 9% by 1 μm Tat-NR2B9c, *p < 0.05, n = 5; and 42 ± 9% by 200 nm Tat-NR2B9c, **p < 0.01, n = 6; normalized to Tat–NR2BAA and tested by one-sample t test; data not shown), as well as SAP102 (49 ± 11% by 1 μm Tat–NR2B9c, *p < 0.05 by paired Student's t test, n = 5; data not shown) that coimmunoprecipitated with NR2B. We also tested the efficacy of 1 μm and 200 nm Tat–NR2B9c in uncoupling NR2B and PSD-95 or SAP102 in live, cultured YAC128 MSNs. Similar to results in transfected HEK293T cells, we found a reduction (by ∼50% for both 1 μm and 200 nm peptide concentrations) in the amount of PSD-95 (Fig. 3A,C), as well as SAP102 (by ∼50% for 1 μm and ∼65% for 200 nm peptide concentrations) (Fig. 3B,C), that coimmunoprecipitated with NR2B compared with neurons treated with Tat–NR2BAA. The pooled data of the reduction in PSD-95 or SAP102 to NR2B band intensity ratios (normalized to paired Tat–NR2BAA at same concentration) by both 200 nm and 1 μm Tat–NR2B9c concentrations are shown in Figure 3D.
Figure 3.

Tat–NR2B9c (9c) disrupts coimmunoprecipitation of NR2B with PSD-95 and SAP102 in cultured MSNs. A, Representative blot showing that 1 μm Tat–NR2B9c disrupts co-IP of NR2B with PSD-95 in YAC128 cultured MSNs. B, Representative blot showing that 1 μm Tat–NR2B9c disrupts co-IP of NR2B with SAP102 in YAC128 cultured MSNs. C, Representative blot showing that 200 nm Tat–NR2B9c disrupts co-IP of NR2B with both PSD-95 and SAP102 in YAC128 cultured MSNs. A–C, The concentration of Tat–NR2BAA (AA) used in each experiment matched that of the Tat–NR2B9c (9c). D, The pooled data of the reduction of either PSD-95 or SAP102 (MAGUK) to NR2B band intensity ratio by both 200 nm and 1 μm Tat–NR2B9c concentrations (normalized to its paired Tat–NR2BAA at the same concentration as Tat–NR2B9c). For the four bars from left to right, *p < 0.05, n = 3; *p < 0.05, n = 8; *p < 0.05, n = 4; **p < 0.01, n = 3, compared with its paired Tat–NR2BAA at the same concentration (data not shown, all equal to 1), by paired one-sample t test, respectively.
Tat–NR2B9c or knockdown of PSD-95 reduces NMDA-induced apoptosis in YAC72 and YAC128 MSNs but not WT MSNs
Interaction between NMDARs and PDZ-containing proteins, especially PSD-95, has been postulated to contribute to NMDAR-mediated excitotoxicity by anchoring signaling proteins such as nNOS in close proximity with NMDAR-mediated calcium influx (Aarts et al., 2002; Sattler and Tymianski, 2000). Furthermore, the number of surface NMDARs would be expected to regulate calcium influx in response to excitotoxic insults and therefore affect the level of NMDA-induced apoptosis.
We therefore treated cultured MSNs with Tat peptides to determine whether Tat–NR2B9c could protect these neurons from NMDA-induced toxicity. We chose to use a concentration of 200 nm rather than 1 μm because we found 200 nm to be the lowest effective concentration for reducing surface NMDAR expression (see Fig. 6C) (also, in pilot experiments, 50 nm Tat–NR2B9c showed no effect on NMDA-induced surface expression or toxicity; data not shown). Plates of WT, YAC72, and YAC128 MSNs were processed in parallel to assess the proportion of apoptotic cells, using the TUNEL stain to identify apoptotic neurons and the PI stain to identify all nuclei (representative photomicrograph shown in Fig. 4A).
Figure 6.
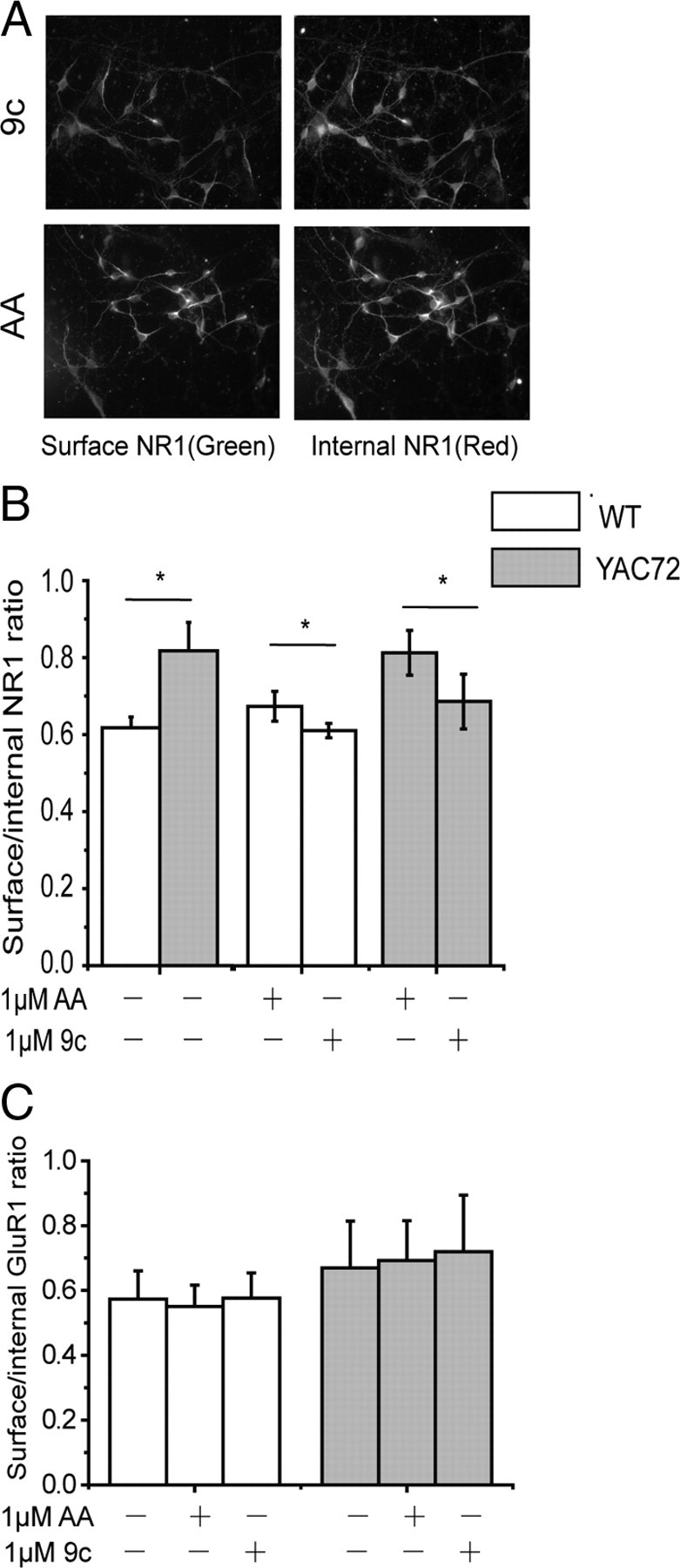
Tat–NR2B9c peptide reduces surface NMDAR levels in cultured MSNs. A, Representative photomicrographs showing 1 h pretreatment with 1 μm Tat–NR2B9c (9c) peptide reduces surface NR1 expression compared with treatment with 1 μm Tat–NR2BAA (AA) peptide, in YAC72 MSNs. B, Pretreatment with Tat–NR2B9c peptide at 1 μm reduces surface/internal NR1 ratio by 15.3 ± 4.6% in YAC72 and 8.9 ± 3.4% in WT MSNs relative to 1 μm Tat–NR2BAA-treated MSNs. n = 4 for both genotypes. F(1,18) = 10.35, p < 0.005 for genotype; F(2,18) = 1.80, p = 0.19 for treatment; F(2,18) = 0.71 for interaction, p = .0.50. After normalizing each peptide-treated group to the untreated group, F(1,12) = 0.50 for genotype, p = 0.49; F(1,12) = 32.80, p < 0.0001 for treatment; F(1,12) = 8.37 for interaction, p = 0.01, by two-way ANOVA, *p < 0.05 by Bonferroni's post hoc tests. C, Pretreatment with 1 μm Tat-NR2B9c peptide has no effect on surface/internal GluR1 ratio of WT and YAC72 MSNs. Three batches of WT and four batches of YAC72 MSNs; not significant for genotype and treatment, by two-way ANOVA. F(1,15) = 1.39 for genotype, p > 0.05; F(2,15) = 0.03, p > 0.05 for treatment; F(2,15) = 0.02 for interaction, p > 0.05.
Figure 4.
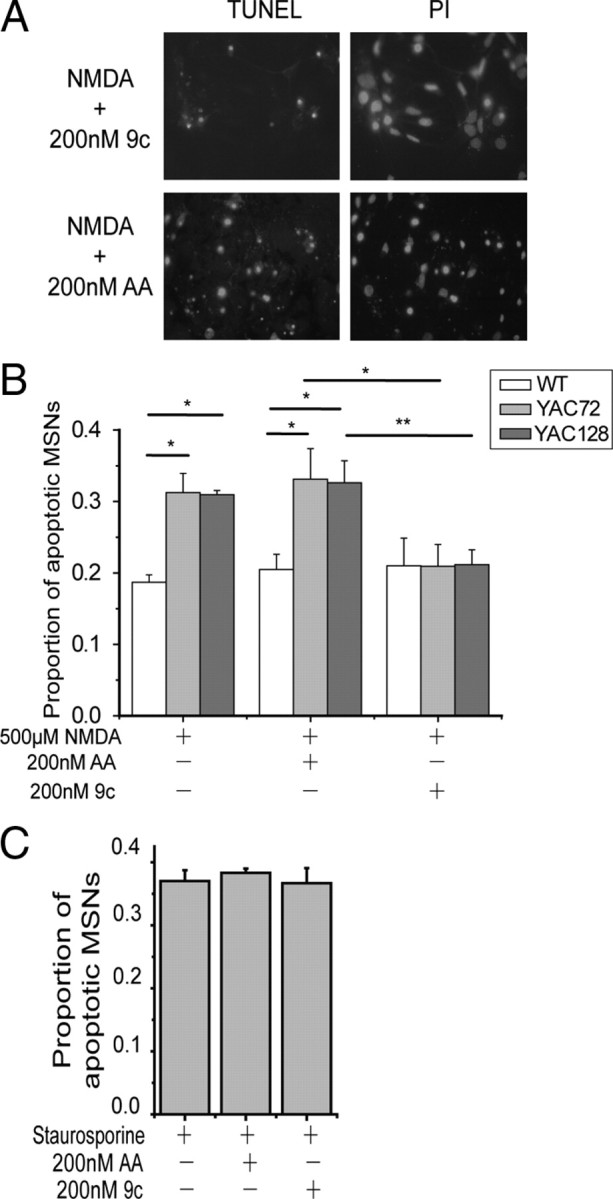
Tat–NR2B9c reduces NMDA-induced toxicity in YAC72 and YAC128 but not WT cultured MSNs. A, Representative photomicrographs showing TUNEL- and PI-stained YAC128 MSNs pretreated with 200 nm Tat–NR2B9c (top, 9c) or Tat–NR2BAA (bottom, AA) for 1 h before 10 min exposure to NMDA. B, One hour pretreatment with 200 nm Tat–NR2B9c peptide reduces NMDA-induced toxicity in YAC72 and YAC128 but not WT cultured MSNs. Average proportion of apoptotic MSNs after pretreatment with 200 nm Tat–NR2B9c and exposure to 500 μm NMDA showed a 37.4 ± 2.7% reduction for YAC72 and 33.6 ± 6.6% reduction for YAC128 compared with 200 nm Tat–NR2BAA pretreatment and 500 μm NMDA exposure (mean values calculated after subtraction of percentage apoptosis in NMDA-untreated condition for each experiment). Tested by two-way ANOVA, significant for both genotype and treatment, n = 4; F(2,50) = 6.34, p < 0.005 for genotype; F(5,50) = 63.9, p < 0.0001 for treatment (*p < 0.05, **p < 0.01, by Bonferroni's post hoc tests). C, One hour pretreatment of Tat–NR2B peptide at 200 nm has no effect on 10 μm staurosporine-induced cell death of cultured YAC72 MSNs. Mean values calculated after subtraction of percentage apoptosis in staurosporine-untreated condition for each experiment (n = 3; no significant difference between three groups treated with staurosporine by one-way ANOVA and post hoc tests).
Remarkably, we found that 200 nm Tat–NR2B9c significantly reduced NMDA-induced cell death in YAC72 and YAC128 MSNs (by ∼35% compared with treatment with NMDA alone or in combination with 200 nm Tat–NR2BAA, as shown in Fig. 4B). Moreover, the proportion of NMDA-induced apoptotic neurons in Tat–NR2B9c-treated YAC72 and YAC128 MSNs was equivalent to that observed for WT MSNs under all three experimental conditions (Tat–NR2B9c had no apparent effect on NMDA-induced apoptosis in WT MSNs). Similar to previous findings from our laboratory (Shehadeh et al., 2006), we found that the proportion of apoptotic MSNs in groups treated with NMDA alone (without peptides) was significantly higher for YAC72 and YAC128 MSNs compared with WT MSNs (∼19% for WT, 31% for YAC72, and 31% for YAC128 MSNs) (Fig. 4B). Importantly, treatment with Tat–NR2BAA had no significant effect on NMDA-treated MSNs. Furthermore, 200 nm Tat–NR2B9c or Tat–NR2BAA alone did not cause significant apoptosis (data not shown). As a control for specificity of the neuroprotective effect of Tat–NR2B9c, we found that 1 h pretreatment with 200 nm Tat–NR2B9c did not alter the extent of apoptosis induced by 10 μm staurosporine in cultured YAC72 MSNs (Fig. 4C).
To test whether PSD-95 or SAP102 is necessary for elevated NMDA-induced toxicity in YAC HD MSNs, we used siRNA to knockdown expression of PSD-95 (Fig. 5A) or SAP102 (for representative blots, see Fig. 5D) (knockdown of SAP102 expression levels, measured by SAP102/actin ratio, ranged from ∼30 to 80% and was significant for both WT and YAC128 in comparing lysates from control siRNA versus SAP102 siRNA-treated cultured MSNs, n = 3, p < 0.05 by paired t test for each genotype). After 72 h treatment with siRNA, MSNs were exposed to 500 μm NMDA with or without 200 nm Tat–NR2B9c. We found that the NMDA-induced toxicity in YAC128 MSNs was reduced to WT levels by PSD-95 siRNA (Fig. 5B,C) and that the effect of Tat–NR2B9c peptide was not additive to that of PSD-95 knockdown. As a control, we found that the nontargeting siRNA did not alter NMDA-induced toxicity in either WT or YAC128 MSNs. In contrast, SAP102 siRNA had no effect on NMDA-induced toxicity in both WT and YAC128 MSNs (Fig. 5E), whereas Tat–NR2B9c peptide still reduced NMDA-induced toxicity of YAC128 to that of WT in both the presence and absence of SAP102 knockdown.
Figure 5.

PSD-95 but not SAP102 knockdown by siRNA reduces NMDA toxicity in YAC128 MSNs but not WT MSNs. A, Representative Western blots and pooled data showing the knockdown of PSD-95 expression in WT and YAC128 MSNs 72 h after addition of PSD-95 siRNA but not control siRNA to culture medium. **p < 0.01, ***p < 0.001, by one-way ANOVA and Bonferroni's post hoc tests (n = 3 for WT and YAC128). B, Representative photomicrographs showing TUNEL- and Hoechst-stained WT and YAC128 MSNs pretreated with control siRNA (top) or PSD-95 siRNA (bottom) 72 h before 10 min challenge of NMDA. C, The effect of PSD-95 knockdown by siRNA on NMDA-induced toxicity in WT and YAC128 MSNs. Average proportion of apoptotic MSNs are shown (mean values calculated after subtraction of percentage apoptosis in NMDA-untreated condition for each experiment). Two-way ANOVA revealed significant difference for genotype, treatment, and interaction, n = 3; F(1,20) = 22, p < 0.001 for genotype; F(4,20) = 25, p < 0.001 for treatment; F(4,20) = 29, p < 0.001 for interaction (***p < 0.001 by Bonferroni's post hoc tests). D, Representative Western blots showing the knockdown of SAP102 expression in WT and YAC128 MSNs 72 h after addition of SAP102 siRNA but not control siRNA to culture medium (n = 3 for WT and YAC128). E, The effect of SAP102 knockdown on NMDA-induced toxicity in WT and YAC128 MSNs. Average proportion of apoptotic MSNs are shown (mean values calculated after subtraction of percentage apoptosis in NMDA-untreated condition for each experiment). Two-way ANOVA revealed significant difference for genotype, treatment, and interaction; n = 3 for WT and n = 4 for YAC128; F(1,25) = 52, p < 0.001 for genotype; F(4,25) = 11, p < 0.001 for treatment; F(4,25) = 14, p < 0.001 for interaction (***p < 0.001 by Bonferroni's post hoc tests). 9c, NR2B9c, N, NMDA.
Tat-NR2B9c peptide reduces surface NMDAR levels in cultured MSNs
After establishing the peptide concentrations required for dissociating NR2B from PSD-95 and SAP102 in live cells, we assessed the effect of 1 μm Tat–NR2B9c on the proportion of NR1 expressed at the plasma membrane of cultured MSNs (Fig. 6A,B). This peptide concentration significantly decreased NR1 surface expression in WT and YAC72 MSNs for treatment with Tat–NR2B9c compared with Tat–NR2BAA, although the effect was relatively small. We obtained similar results for treatment with 200 nm Tat–NR2B9c versus Tat–NR2BAA (the surface to internal NR1 ratio after treatment with Tat–NR2B9C normalized to that after Tat–NR2BAA treatment was 0.73 ± 0.04 for WT and 0.82 ± 0.03 for YAC72, n = 4, p < 0.05 by paired t test for each genotype); therefore, at this concentration of Tat peptides, which was also used in the toxicity assays, NMDAR surface expression remained enhanced for YAC72 compared with WT MSNs. As a control, GluR1 surface expression was also assessed and showed no difference between MSNs treated with Tat–NR2B9c or the control peptide (Fig. 6C). Our results demonstrate that peptide concentrations shown to disrupt the binding of NR2B with PSD-95 or SAP102 also reduce NMDAR surface expression equally well in WT and YAC72 MSNs. This finding indicates that the decreased surface NMDAR level cannot fully explain the specific effect of Tat–NR2B9c peptide on NMDA-induced toxicity in YAC HD MSNs.
nNOS inhibitor reduces NMDA-induced apoptosis in YAC72 and YAC128 MSNs but not WT MSNs
PSD-95 has been postulated to contribute to NMDAR-mediated excitotoxicity by anchoring signaling proteins, such as nNOS, in close proximity with NMDAR-mediated calcium influx (Sattler and Tymianski, 2000; Aarts et al., 2002). Furthermore, a previous study showed that nitric oxide released from nNOS-interneurons could promote release of intracellular calcium from mitochondria in MSNs (Horn et al., 2002) and thus contribute to NMDA-induced cell death. Therefore, we treated cultured MSNs with nNOS inhibitor (N-Arg) and/or Tat peptides to determine whether the effect of Tat–NR2B9c is equivalent to that of an nNOS inhibitor on NMDA-induced toxicity. Interestingly, we found that 200 nm Tat–NR2B9c has a similar effect as an nNOS inhibitor on NMDA-induced cell death in both WT and YAC HD MSNs, and the combination of these two treatments did not show any significant additive effect (as shown in Fig. 7A,B). This finding suggested that inhibition of nNOS signaling could contribute to the protection afforded by Tat–NR2B9c against NMDAR-dependent toxicity in YAC HD MSNs.
Figure 7.
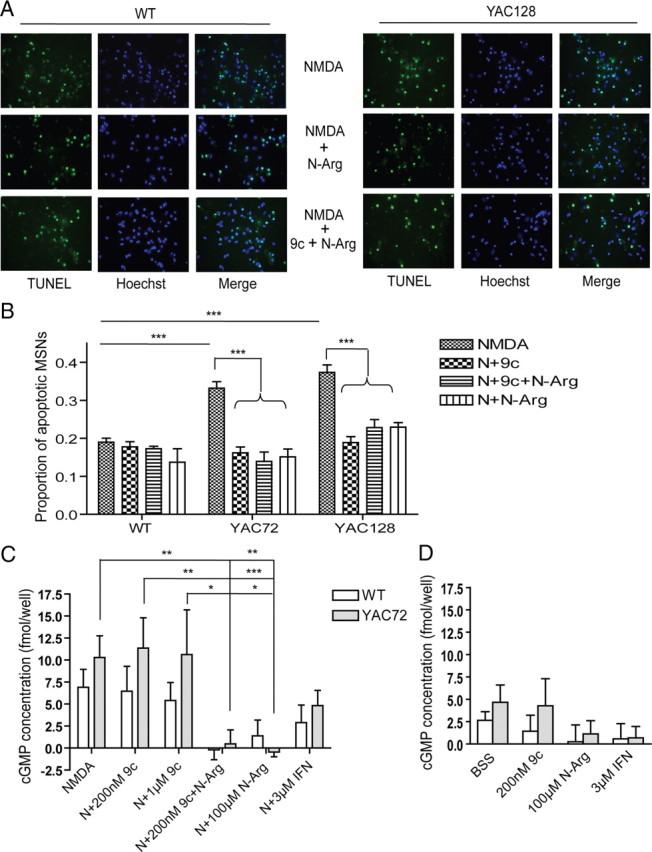
nNOS inhibitor (N-Arg) reduces NMDA-induced toxicity and occludes effect of Tat–NR2B9c in YAC72 and YAC128 but not WT cultured MSNs. A, Representative photomicrographs showing TUNEL- and Hoechst-stained WT and YAC128 MSNs pretreated with 100 μm N-Arg (middle) and 200 nm Tat–NR2B9c plus 100 μm N-Arg (bottom) for 1 h before 10 min exposure to NMDA. B, Average proportion of apoptotic MSNs after pretreatment with 100 μm N-Arg and/or 200 nm Tat–NR2B9c (9c) and exposure to 500 μm NMDA (N) showed a similar reduction in YAC72 and YAC128 MSNs and no significant reduction in WT MSNs (mean values calculated after subtraction of percentage apoptosis in NMDA-untreated condition for each experiment). Tested by two-way ANOVA, significant for both genotype and treatment, n = 6; F(1,42) = 48, p < 0.0001 for genotype; F(5,42) = 70, p < 0.0001 for treatment F(5,42) = 9, p < 0.0001 for interaction (***p < 0.001, by Bonferroni's post hoc tests). C, The 500 μm NMDA-induced cGMP production of WT and YAC72 MSNs after pretreatment with 100 μm N-Arg and/or 200 nm/1 μm Tat–NR2B9c or 3 μm IFN. Significance was tested by two-way ANOVA; n = 5 for WT and n = 6 for YAC72, significant for treatment. F(1,46) = 3, p > 0.05 for genotype; F(5,46) = 6, p < 0.0001 for treatment; F(5,46) = 0.7, p > 0.05 for interaction (*p < 0.05, **p < 0.01, ***p < 0.001, by Bonferroni's post hoc tests). D, Basal cGMP production of WT and YAC72 MSNs after pretreatment with 100 μm N-Arg and/or 200 nm/1 μm Tat–NR2B9c, BSS, or 3 μm IFN. Significance tested by two-way ANOVA, n = 5 for WT and n = 6 for YAC72. F(1,33) = 1, p > 0.05 for genotype; F(3,33) = 1, p > 0.05 for treatment; F(3,33) = 0.2, p > 0.05 for interaction.
To further assess a role for nNOS signaling in the effect of Tat–NR2B9c on NMDA toxicity, we repeated these treatments and measured the cGMP production as a readout for nNOS activity. We found that NMDA-stimulated cGMP production was abolished by 100 μm N-Arg, partially reduced by ifenprodil, and not significantly affected by either 200 nm or 1 μm Tat–NR2B9c (as shown in Fig. 7C,D). Thus, interference with the nNOS pathway cannot explain the protective effect of Tat–NR2B9c on NMDA toxicity in YAC HD MSNs.
Discussion
Altered NMDA receptor function and increased neuronal sensitivity to excitotoxicity have been proposed as candidate mechanisms for selective neuronal dysfunction and degeneration in HD based on studies in human autopsy brain tissue, as well as cellular and animal models of HD (Young et al., 1988; Albin et al., 1990; Ferrante et al., 1993; Chen et al., 1999; Zeron et al., 2002; Fan and Raymond, 2007). Attention has focused on NR2B-containing NMDARs, which are enriched in striatal tissue (Landwehrmeyer et al., 1995; Christie et al., 2000; Li et al., 2003) and show specificity for regulation by mutant huntingtin in heterologous cell lines and cultured MSNs (Chen et al., 1999; Zeron et al., 2001, 2002; Song et al., 2003; Tang et al., 2005; Fan et al., 2007). Here, we demonstrate that huntingtin associates with SAP102 and polyQ expansion in huntingtin increases interaction between NR2B-containing NMDARs and PSD-95. Moreover, a peptide that interferes with NR2B binding to SAP102 and PSD-95 rescues the increased NMDA-induced toxicity in MSNs expressing polyQ-expanded htt, restoring excitotoxic susceptibility to WT levels. Together, our results provide evidence that the NR2B interaction with MAGUKs plays an important role in enhanced excitotoxicity in YAC HD MSNs. Notably, the peptide used in our studies to uncouple NMDARs from SAP102/PSD-95 has been shown previously to protect against ischemic neuronal damage in a rodent model of stroke (Aarts et al., 2002).
Modulation of NMDAR surface expression by PDZ domain interactions in striatal MSNs
PSD-95 family members interact with NR2A- and NR2B-containing NMDARs via binding of the PDZ1,2 domains to the C-terminal 4 aa of NR2 subunits (Kornau et al., 1995; Kim et al., 1996; Müller et al., 1996; Niethammer et al., 1996). Previous studies demonstrate a critical role for this interaction in NMDAR: trafficking to the neuronal surface, synaptic localization, and signaling to cell death pathways (Sattler et al., 1999; Roche et al., 2001; Prybylowski et al., 2005; Sans et al., 2005).
PSD-95 binding to the NR2B C-terminal tSXV motif reduces endocytosis from the surface (Roche et al., 2001) and stabilizes NR2B-containing NMDARs in the synapse (Prybylowski et al., 2005). We previously published that mutant htt expression alters steady-state NMDAR distribution by enhancing forward trafficking to the surface rather than reducing removal and degradation of receptors from the surface (Fan et al., 2007), making it unlikely that PSD-95 plays a role.
The interaction of SAP102 with NR2B regulates delivery of NMDARs to the plasma membrane (Sans et al., 2003), raising the possibility that the SAP102/htt interaction is involved in accelerating NMDAR surface delivery in YAC72 MSNs. We speculate that a reduction in the SAP102/NR2B interaction with Tat–NR2B9c treatment contributes to the decrease in NMDAR surface expression; however, the similar reduction in WT and YAC72 MSNs indicates that htt polyQ length does not influence the MAGUK/NMDAR interaction that modulates receptor surface expression.
A previous report suggested that Tat–NR2B9c had no effect on NMDAR surface expression in cultured hippocampal neurons; that study tested only a 50 nm concentration (Aarts et al., 2002), which also did not alter NMDAR surface expression in cultured MSNs (data not shown). However, at higher concentrations (1 μm), which we have shown are effective in uncoupling PSD-95/SAP102 from NR2B in live cultured MSNs, NMDAR surface expression was significantly reduced.
Interaction of PSD-95 with NR2B modulates mutant htt enhancement of NMDAR apoptosis
PSD-95 binds directly to htt, and binding is reduced with increasing htt polyQ length (Sun et al., 2001), leading to the hypothesis that expression of polyQ-expanded htt could augment the NR2B/PSD-95 interaction. Here, we show increased amounts of PSD-95 coimmunoprecipitated with NR2B from YAC HD mouse striatal tissue, providing direct evidence for an altered NR2B/PSD-95 interaction. Previous work showed that glutamate-induced excitotoxicity in an NMDAR-transfected neuronal cell line is enhanced by mutant htt expression (Sun et al., 2001) and that this toxicity is influenced by PSD-95-dependent src family kinase-mediated NR2B phosphorylation (Song et al., 2003). However, these studies did not directly demonstrate that interaction between NR2B and PSD-95 contributed to mutant htt-enhanced excitotoxicity in striatal neurons.
We have shown that pretreatment of live, cultured MSNs with a peptide that significantly reduces (by ∼50%) the interaction between NR2B and PSD-95/SAP102 also decreases NMDAR-mediated apoptosis in YAC72 and YAC128 MSNs to WT levels. This effect was specific for NMDA excitotoxicity, because Tat–NR2B9c had no effect on staurosporine-induced apoptosis. Notably, the peptide effect on NMDAR surface expression was found equally for WT and YAC72 MSNs, whereas the effect on toxicity was exclusive to YAC HD MSNs and not observed in WT MSNs. Therefore, we propose that the Tat–NR2B-mediated reduction in NMDA-induced apoptosis is predominantly a consequence of uncoupling NMDAR activation from downstream, cell death-signaling proteins that would normally be anchored near the receptors by PSD-95 or other MAGUKs. Based on the known association between PSD-95 and the cell death-signaling protein nNOS and our data showing increasing NR2B/PSD-95 coassociation with htt polyQ expansion, we hypothesized that the NR2B/PSD-95/htt complex (rather than NR2B/SAP102/htt) is critical for enhanced excitotoxicity in the YAC transgenic HD mouse model. Indeed, siRNA knockdown of PSD-95, but not of SAP102, reduced NMDAR-mediated toxicity in YAC72 MSNs, with no effect in WT, a result similar (and not additive) to the effect of Tat–NR2B9c. These data are consistent with a critical role of the NR2B/PSD-95/htt complex in enhanced excitotoxicity in mutant htt-expressing MSNs.
The fact that inhibition of nNOS also mimicked the protective effect of Tat–NR2B9c and that its protection was not additive to that of the Tat–NR2B9c peptide suggests a role for nNOS in enhanced NMDA excitotoxicity in YAC72 and YAC128 MSNs. However, Tat–NR2B9c failed to influence NMDA-stimulated nNOS activity (reflected by cGMP production) in either WT or YAC72 MSNs. We conclude that nNOS activation by NMDA occurs independently of PSD-95/NR2B interaction and cannot explain the protective effect of Tat–NR2B9c on excitotoxicity in YAC HD MSNs. Conversely, NMDAR-activated signaling pathways that depend on NMDAR/PSD-95 association may converge with those that involve NMDAR-mediated nNOS activation in the same final pathway that mediates enhanced excitotoxic vulnerability of YAC HD MSNs; if so, inhibition of just one of these upstream pathways may be sufficient to abolish enhanced toxicity.
One possibility for nNOS-independent, PSD-95-dependent NMDAR cell death signaling is through the phosphatase calcineurin, which is involved in induction of the mitochondrial permeability transition and release of apoptotic factors (Springer et al., 2000). Calcineurin and PSD-95 are linked through binding with A-kinase anchoring protein (AKAP79/150) (Colledge et al., 2000; Bhattacharyya et al., 2009). Moreover, NMDAR-induced calcineurin activity is enhanced in mutant htt-expressing striatal cells, and inhibition of calcineurin protects against excitotoxicity better in mutant htt-expressing than WT cells (Xifró et al., 2008). Another apoptotic pathway downstream of PSD-95 is through binding to synaptic Ras-GTPase activating protein, known to regulate synaptic plasticity and neuronal apoptosis through p38–MAPK signaling (Kim et al., 1998; Rumbaugh et al., 2006). Notably, NMDAR-dependent p38 activation relies on neuronal context and is disrupted by Tat–NR2B9c (Soriano et al., 2008). Additional experiments are required to fully elucidate signaling pathways mediating enhanced excitotoxicity in YAC HD MSNs.
The highly homologous MAGUK proteins SAP102 and PSD-95 both bind NR2 subunits and associate in a complex with htt yet show differential interactions with polyQ-expanded htt and play distinct roles in mhtt-induced alterations of NMDAR trafficking and signaling. Although PSD-95 and SAP102 bind NR2 and htt through PDZ and SH3 domains, respectively, other regions of these MAGUKs likely affect the strength of this binding. Both PDZ1 and PDZ2 contribute to MAGUK interactions with NR2 subunits (Kornau et al., 1995), whereas regions upstream of the NR2 C-terminal PDZ ligand, which differ between NR2A and NR2B, contribute to interaction of these subunits with PSD-95 (Cousins et al., 2008) and may explain differences in strength of binding to various MAGUKs (Cui et al., 2007). Similarly, sequence differences around the SH3 domain of PSD-95 and SAP102 may contribute to differential interaction of these two MAGUKs with the polyproline of htt.
Role of NR2B interactions with PSD-95 family members in HD
In the YAC transgenic mouse models expressing human htt with expanded polyQ, the middle-age onset of motor and cognitive deficits, along with neuronal degeneration that is relatively striatal MSN specific (Hodgson et al., 1999; Slow et al., 2003; Van Raamsdonk et al., 2005), are tightly linked with enhanced susceptibility to NMDAR-mediated excitotoxicity; mice expressing htt with 128 polyQ truncated after exon 2—(“short-stop”) (Slow et al., 2005) or modified to eliminate cleavage by caspase-6 (“C6R”) (Graham et al., 2006b)—lack the HD motor and cognitive phenotype, show no striatal degeneration, and are resistant to NMDAR-mediated toxicity. However, because of the adverse side effects associated with direct NMDAR inhibition, therapy targeted to proteins or molecular mechanisms that mediate the enhanced NMDAR death signaling in HD may be more specific, effective, and have fewer side effects. The fact that the Tat–NR2B9c peptide eliminated the excess NMDAR toxicity in YAC72 and YAC128 MSNs without affecting cell death in WT MSNs suggests that uncoupling NMDARs from PSD-95 (and/or other MAGUKs) may be a target for therapy in HD.
Footnotes
The research was funded by Canadian Institutes of Health Research (CIHR) Grants MOP 12699 and 129029 (L.A.R.) and MOP 84438 (M.R.H.), the Cure Huntington's Disease Initiative (L.A.R., M.R.H.), Huntington Disease Society of America (HDSA) (M.R.H.), and a Michael Smith Foundation for Health Research (MSFHR) infrastructure grant. C.M.C. was supported by a fellowship award from the HDSA. L.A.R. was a CIHR Investigator and MSFHR Senior Scholar. M.R.H. holds a Canada Research Chair in Human Genetics and is a University Killam Professor. We are grateful to the late Dr. A. El-Husseini for many helpful discussions, and we dedicate this report to his loving memory. We thank T. Luo and E. Yu for technical assistance.
References
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science. 2002;298:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB, Handelin B, Balfour R, Anderson KD, Markel DS, Tourtellotte WW, Reiner A. Abnormalities of striatal projection neurons and N-methyl-d-aspartate receptors in presymptomatic Huntington's disease. N Engl J Med. 1990;322:1293–1298. doi: 10.1056/NEJM199005033221807. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Biou V, Xu W, Schlüter O, Malenka RC. A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat Neurosci. 2009;12:172–181. doi: 10.1038/nn.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Ariano MA, Calvert CR, Flores-Hernández J, Chandler SH, Leavitt BR, Hayden MR, Levine MS. NMDA receptor function in mouse models of Huntington disease. J Neurosci Res. 2001;66:525–539. doi: 10.1002/jnr.1244. [DOI] [PubMed] [Google Scholar]
- Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Luo T, Wellington C, Metzler M, McCutcheon K, Hayden MR, Raymond LA. Subtype-specific enhancement of NMDA receptor currents by mutant huntingtin. J Neurochem. 1999;72:1890–1898. doi: 10.1046/j.1471-4159.1999.0721890.x. [DOI] [PubMed] [Google Scholar]
- Christie JM, Jane DE, Monaghan DT. Native N-methyl-D-aspartate receptors containing NR2A and NR2B subunits have pharmacologically distinct competitive antagonist binding sites. J Pharmacol Exp Ther. 2000;292:1169–1174. [PubMed] [Google Scholar]
- Chung HJ, Huang YH, Lau LF, Huganir RL. Regulation of the NMDA receptor complex and trafficking by activity-dependent phosphorylation of the NR2B subunit PDZ ligand. J Neurosci. 2004;24:10248–10259. doi: 10.1523/JNEUROSCI.0546-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Cousins SL, Papadakis M, Rutter AR, Stephenson FA. Differential interaction of NMDA receptor subtypes with the post-synaptic density-95 family of membrane associated guanylate kinase proteins. J Neurochem. 2008;104:903–913. doi: 10.1111/j.1471-4159.2007.05067.x. [DOI] [PubMed] [Google Scholar]
- Cowan CM, Raymond LA. Selective neuronal degeneration in Huntington's disease. Curr Top Dev Biol. 2006;75:25–71. doi: 10.1016/S0070-2153(06)75002-5. [DOI] [PubMed] [Google Scholar]
- Cui H, Hayashi A, Sun HS, Belmares MP, Cobey C, Phan T, Schweizer J, Salter MW, Wang YT, Tasker RA, Garman D, Rabinowitz J, Lu PS, Tymianski M. PDZ protein interactions underlying NMDA receptor-mediated excitotoxicity and neuroprotection by PSD-95 inhibitors. J Neurosci. 2007;27:9901–9915. doi: 10.1523/JNEUROSCI.1464-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M. Excitotoxic injury of the neostriatum: a model for Huntington's disease. Trends Neurosci. 1990;13:286–289. doi: 10.1016/0166-2236(90)90111-m. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Fan MM, Raymond LA. N-methyl-d-aspartate (NMDA) receptor function and excitotoxicity in Huntington's disease. Prog Neurobiol. 2007;81:272–293. doi: 10.1016/j.pneurobio.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Fan MM, Fernandes HB, Zhang LY, Hayden MR, Raymond LA. Altered NMDA receptor trafficking in a yeast artificial chromosome transgenic mouse model of Huntington's disease. J Neurosci. 2007;27:3768–3779. doi: 10.1523/JNEUROSCI.4356-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Cipolloni PB, Storey E, Beal MF. Excitotoxin lesions in primates as a model for Huntington's disease: histopathologic and neurochemical characterization. Exp Neurol. 1993;119:46–71. doi: 10.1006/exnr.1993.1006. [DOI] [PubMed] [Google Scholar]
- Fujita A, Kurachi Y. SAP family proteins. Biochem Biophys Res Commun. 2000;269:1–6. doi: 10.1006/bbrc.1999.1893. [DOI] [PubMed] [Google Scholar]
- Graham RK, Slow EJ, Deng Y, Bissada N, Lu G, Pearson J, Shehadeh J, Leavitt BR, Raymond LA, Hayden MR. Levels of mutant huntingtin influence the phenotypic severity of Huntington disease in YAC128 mouse models. Neurobiol Dis. 2006a;21:444–455. doi: 10.1016/j.nbd.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006b;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003;26:81–89. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- Horn TF, Wolf G, Duffy S, Weiss S, Keilhoff G, MacVicar BA. Nitric oxide promotes intracellular calcium release from mitochondria in striatal neurons. FASEB J. 2002;16:1611–1622. doi: 10.1096/fj.02-0126com. [DOI] [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable in Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Kim E, Cho KO, Rothschild A, Sheng M. Heteromultimerization and NMDA receptor-clustering activity of Chapsyn-110, a member of the PSD-95 family of proteins. Neuron. 1996;17:103–113. doi: 10.1016/s0896-6273(00)80284-6. [DOI] [PubMed] [Google Scholar]
- Kim JH, Liao D, Lau LF, Huganir RL. SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron. 1998;20:683–691. doi: 10.1016/s0896-6273(00)81008-9. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Kovács AD, Cebers G, Cebere A, Moreira T, Liljequist S. Cortical and striatal neuronal cultures of the same embryonic origin show intrinsic differences in glutamate receptor expression and vulnerability to excitotoxicity. Exp Neurol. 2001;168:47–62. doi: 10.1006/exnr.2000.7576. [DOI] [PubMed] [Google Scholar]
- Landwehrmeyer GB, Standaert DG, Testa CM, Penney JB, Jr, Young AB. NMDA receptor subunit mRNA expression by projection neurons and interneurons in rat striatum. J Neurosci. 1995;15:5297–5307. doi: 10.1523/JNEUROSCI.15-07-05297.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavitt BR, Guttman JA, Hodgson JG, Kimel GH, Singaraja R, Vogl AW, Hayden MR. Wild-type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am J Hum Genet. 2001;68:313–324. doi: 10.1086/318207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MS, Klapstein GJ, Koppel A, Gruen E, Cepeda C, Vargas ME, Jokel ES, Carpenter EM, Zanjani H, Hurst RS, Efstratiadis A, Zeitlin S, Chesselet MF. Enhanced sensitivity to N-methyl-d-aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J Neurosci Res. 1999;58:515–532. [PubMed] [Google Scholar]
- Li L, Fan M, Icton CD, Chen N, Leavitt BR, Hayden MR, Murphy TH, Raymond LA. Role of NR2B-type NMDA receptors in selective neurodegeneration in Huntington disease. Neurobiol Aging. 2003;24:1113–1121. doi: 10.1016/j.neurobiolaging.2003.04.003. [DOI] [PubMed] [Google Scholar]
- Lin Y, Skeberdis VA, Francesconi A, Bennett MV, Zukin RS. Postsynaptic density protein-95 regulates NMDA channel gating and surface expression. J Neurosci. 2004;24:10138–10148. doi: 10.1523/JNEUROSCI.3159-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood AJ, Cummings DM, Dallérac GM, Brown JY, Vatsavayai SC, Hirst MC, Rezaie P, Murphy KP. Early development of aberrant synaptic plasticity in a mouse model of Huntington's disease. Hum Mol Genet. 2006;15:1690–1703. doi: 10.1093/hmg/ddl092. [DOI] [PubMed] [Google Scholar]
- Müller BM, Kistner U, Kindler S, Chung WJ, Kuhlendahl S, Fenster SD, Lau LF, Veh RW, Huganir RL, Gundelfinger ED, Garner CC. SAP102, a novel postsynaptic protein that interacts with NMDA receptor complexes in vivo. Neuron. 1996;17:255–265. doi: 10.1016/s0896-6273(00)80157-9. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Kim E, Sheng M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci. 1996;16:2157–2163. doi: 10.1523/JNEUROSCI.16-07-02157.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski K, Wenthold RJ. N-Methyl-D-aspartate receptors: subunit assembly and trafficking to the synapse. J Biol Chem. 2004;279:9673–9676. doi: 10.1074/jbc.R300029200. [DOI] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- Rumbaugh G, Adams JP, Kim JH, Huganir RL. SynGAP regulates synaptic strength and mitogen-activated protein kinases in cultured neurons. Proc Natl Acad Sci U S A. 2006;103:4344–4351. doi: 10.1073/pnas.0600084103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos J, 2nd, Hell JW, Wenthold RJ. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20:1260–1271. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, Prybylowski K, Petralia RS, Chang K, Wang YX, Racca C, Vicini S, Wenthold RJ. NMDA receptor trafficking through an interaction between PDZ proteins and the exocyst complex. Nat Cell Biol. 2003;5:520–530. doi: 10.1038/ncb990. [DOI] [PubMed] [Google Scholar]
- Sans N, Wang PY, Du Q, Petralia RS, Wang YX, Nakka S, Blumer JB, Macara IG, Wenthold RJ. mPins modulates PSD-95 and SAP102 trafficking and influences NMDA receptor surface expression. Nat Cell Biol. 2005;7:1179–1190. doi: 10.1038/ncb1325. [DOI] [PubMed] [Google Scholar]
- Sattler R, Tymianski M. Molecular mechanisms of calcium-dependent excitotoxicity. J Mol Med. 2000;78:3–13. doi: 10.1007/s001090000077. [DOI] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- Shehadeh J, Fernandes HB, Zeron Mullins MM, Graham RK, Leavitt BR, Hayden MR, Raymond LA. Striatal neuronal apoptosis is preferentially enhanced by NMDA receptor activation in YAC transgenic mouse model of Huntington disease. Neurobiol Dis. 2006;21:392–403. doi: 10.1016/j.nbd.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Slow EJ, Graham RK, Osmand AP, Devon RS, Lu G, Deng Y, Pearson J, Vaid K, Bissada N, Wetzel R, Leavitt BR, Hayden MR. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc Natl Acad Sci U S A. 2005;102:11402–11407. doi: 10.1073/pnas.0503634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Zhang Y, Parsons CG, Liu YF. Expression of polyglutamine-expanded huntingtin induces tyrosine phosphorylation of N-methyl-d-aspartate receptors. J Biol Chem. 2003;278:33364–33369. doi: 10.1074/jbc.M304240200. [DOI] [PubMed] [Google Scholar]
- Soriano FX, Martel MA, Papadia S, Vaslin A, Baxter P, Rickman C, Forder J, Tymianski M, Duncan R, Aarts M, Clarke P, Wyllie DJ, Hardingham GE. Specific targeting of pro-death NMDA receptor signals with differing reliance on the NR2B PDZ ligand. J Neurosci. 2008;28:10696–10710. doi: 10.1523/JNEUROSCI.1207-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer JE, Azbill RD, Nottingham SA, Kennedy SE. Calcineurin-mediated BAD dephosphorylation activates the caspase-3 apoptotic cascade in traumatic spinal cord injury. J Neurosci. 2000;20:7246–7251. doi: 10.1523/JNEUROSCI.20-19-07246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starling AJ, André VM, Cepeda C, de Lima M, Chandler SH, Levine MS. Alterations in N-methyl-d-aspartate receptor sensitivity and magnesium blockade occur early in development in the R6/2 mouse model of Huntington's disease. J Neurosci Res. 2005;82:377–386. doi: 10.1002/jnr.20651. [DOI] [PubMed] [Google Scholar]
- Sun Y, Savanenin A, Reddy PH, Liu YF. Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-d-aspartate receptors via post-synaptic density 95. J Biol Chem. 2001;276:24713–24718. doi: 10.1074/jbc.M103501200. [DOI] [PubMed] [Google Scholar]
- Tang TS, Slow E, Lupu V, Stavrovskaya IG, Sugimori M, Llinás R, Kristal BS, Hayden MR, Bezprozvanny I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proc Natl Acad Sci U S A. 2005;102:2602–2607. doi: 10.1073/pnas.0409402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushina E, Singh RD, Dyer RB, Cao S, Shah VH, Parton RG, Pagano RE, McMurray CT. Mutant huntingtin inhibits clathrin-independent endocytosis and causes accumulation of cholesterol in vitro and in vivo. Hum Mol Genet. 2006;15:3578–3591. doi: 10.1093/hmg/ddl434. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Pearson J, Slow EJ, Hossain SM, Leavitt BR, Hayden MR. Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington's disease. J Neurosci. 2005;25:4169–4180. doi: 10.1523/JNEUROSCI.0590-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Warby SC, Chan EY, Metzler M, Gan L, Singaraja RR, Crocker SF, Robertson HA, Hayden MR. Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum Mol Genet. 2005;14:1569–1577. doi: 10.1093/hmg/ddi165. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Lynch DR. N-methyl-d-aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist. 2005;11:37–49. doi: 10.1177/1073858404269012. [DOI] [PubMed] [Google Scholar]
- Xifró X, García-Martínez JM, Del Toro D, Alberch J, Pérez-Navarro E. Calcineurin is involved in the early activation of NMDA-mediated cell death in mutant huntingtin knock-in striatal cells. J Neurochem. 2008;105:1596–1612. doi: 10.1111/j.1471-4159.2008.05252.x. [DOI] [PubMed] [Google Scholar]
- Young AB, Greenamyre JT, Hollingsworth Z, Albin R, D'Amato C, Shoulson I, Penney JB. NMDA receptor losses in putamen from patients with Huntington's disease. Science. 1988;241:981–983. doi: 10.1126/science.2841762. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Chen N, Moshaver A, Lee AT, Wellington CL, Hayden MR, Raymond LA. Mutant huntingtin enhances excitotoxic cell death. Mol Cell Neurosci. 2001;17:41–53. doi: 10.1006/mcne.2000.0909. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Fernandes HB, Krebs C, Shehadeh J, Wellington CL, Leavitt BR, Baimbridge KG, Hayden MR, Raymond LA. Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington's disease. Mol Cell Neurosci. 2004;25:469–479. doi: 10.1016/j.mcn.2003.11.014. [DOI] [PubMed] [Google Scholar]