

Abstract
GluN2B (GluRε2/NR2B) subunit is involved in synapse development, synaptic plasticity, and cognitive function. However, its roles in synaptic expression and function of NMDA receptors (NMDARs) in the brain remain mostly unknown because of the neonatal lethality of global knock-out mice. To address this, we generated conditional knock-out mice, in which GluN2B was ablated exclusively in hippocampal CA3 pyramidal cells. By immunohistochemistry, GluN2B disappeared and GluN1 (GluRζ1/NR1) was moderately reduced, whereas GluN2A (GluRε1/NR2A) and postsynaptic density-95 (PSD-95) were unaltered in the mutant CA3. This was consistent with protein contents in the CA3 crude fraction: 9.6% of control level for GluN2B, 47.7% for GluN1, 90.6% for GluN2A, and 98.0% for PSD-95. Despite the remaining NMDARs, NMDAR-mediated currents and long-term potentiation were virtually lost at various CA3 synapses. Then, we compared synaptic NMDARs by postembedding immunogold electron microscopy and immunoblot using the PSD fraction. In the mutant CA3, GluN1 was severely reduced in both immunogold (20.6-23.6%) and immunoblot (24.6%), whereas GluN2A and PSD-95 were unchanged in immunogold but markedly reduced in the PSD fraction (51.4 and 36.5%, respectively), indicating increased detergent solubility of PSD molecules. No such increased solubility was observed for GluN2B in the CA3 of GluN2A-knock-out mice. Furthermore, significant decreases were found in the ratio of filamentous to globular actin (49.5%) and in the density of dendritic spines (76.2%). These findings suggest that GluN2B is critically involved in NMDAR channel function, organization of postsynaptic macromolecular complexes, formation or maintenance of dendritic spines, and regulation of the actin cytoskeleton.
Introduction
NMDA-type glutamate receptors (NMDARs) are characterized by high Ca2+ permeability and unique gating governed by both ligand and voltage (Mayer et al., 1984; Nowak et al., 1984; MacDermott et al., 1986). Ca2+ influx through NMDARs induces long-term potentiation (LTP) and depression (LTD) of synaptic transmission (Bliss and Collingridge, 1993; Bear and Malenka, 1994; Yang et al., 1999), which are thought to underlie activity-dependent synapse development and cognitive function (Li et al., 1994; Tsien et al., 1996; Iwasato et al., 2000).
Major NMDARs consist of the GluN1 (formerly GluRζ or NR1) and GluN2 (GluRε or NR2) subfamilies. GluN1 subunit, the sole member of the GluN1 subfamily, is essential for functional NMDAR channels and their exit from the endoplasmic reticulum, whereas four members of the GluN2 subfamily (GluRε1-4 or NR2A–D) are the major determinants of functional diversity and synaptic localization (Seeburg, 1993; Nakanishi et al., 1994; Mori and Mishina, 1995; Cull-Candy et al., 2001; Fukaya et al., 2003; Abe et al., 2004). GluN2A (GluRε1 or NR2A) and GluN2B (GluRε2 or NR2B) subunits were thought to play important roles in synaptic plasticity and cognitive function, based on their high Mg2+ sensitivity and abundant expression in the adult forebrain (Meguro et al., 1992; Mori et al., 1992, 1995; Watanabe et al., 1993). Indeed, these roles have been evidenced using gene-engineered mice of these subunits (Sakimura et al., 1995; Kutsuwada et al., 1996; Kiyama et al., 1998; Tang et al., 1999; Zhao et al., 2005). During development, GluN2B expression starts from early embryonic stages, whereas GluN2A subunit appears postnatally (Watanabe et al., 1992; Komuro and Rakic, 1993; Monyer et al., 1994; X. B. Liu et al., 2004). In accordance with the distinct temporal profiles, GluN2A-knock-out (KO) mice are viable and show reduced hippocampal LTP and spatial learning in adulthood (Sakimura et al., 1995; Lu et al., 2001), whereas GluN2B-KO mice exhibit neonatal lethality, impaired somatosensory synapse refinement, and impaired LTD (Kutsuwada et al., 1996). Although the two subunits confer distinct properties in pharmacological profiles, synaptic plasticity, receptor trafficking, and left–right asymmetrical synaptic delivery (Ito et al., 1997; Quinlan et al., 1999, 2004; Barria and Malinow, 2002; Kawakami et al., 2003; L. Liu et al., 2004; Wu et al., 2005; Shinohara et al., 2008), it remains unknown how they play roles in synaptic expression and function of NMDARs in the brain, mainly because of the lack of viable GluN2B-KO animal models.
Here, we generated CA3-targeted GluN2B-KO (CA3-GluN2B-KO) mice, in which GluN2B subunit is preferentially ablated from CA3 pyramidal cells. We show that, despite remaining NMDAR subunits, GluN2B subunit deletion leads to almost complete loss of NMDAR-mediated synaptic currents and LTP, increased detergent solubility of GluN2A and postsynaptic density-95 (PSD-95), decreased filamentous actin, and reduced spine density in the mutant CA3. Considering that NMDAR-mediated synaptic currents and plasticity remain in the CA3 of GluN2A-KO mice (Sakimura et al., 1995; Ito et al., 1997), the GluN2B subunit thus plays indispensable roles in NMDAR channel function, organization of postsynaptic macromolecular complex and dendritic spines, and regulation of the actin cytoskeleton in CA3 pyramidal cells.
Materials and Methods
Generation of CA3-GluN2B-KO mice.
We designed a targeting vector in which Cre gene was placed just behind the translational initiation site of the GluK4 gene (Grik4) in frame. Therefore, two genomic DNA clones carrying the Grik4 translational start site were isolated from a C57BL/6 genomic library (Stratagene). A 272 bp PstI–AgeI fragment was generated by two-step PCR using Grik4 genomic DNA and pNCre (Tsujita et al., 1999) as templates. A knock-in vector pγ1TVCre contained a 3 kb fragment at the 5′ side, a Cre gene placed behind the Grik4 translational start, a Pgk-neo-p(A) cassette, a 7.5 kb fragment at the 3′ side, and a MC1 promoter-driven diphtheria toxin gene.
To generate Grin2b-flox mice, a genomic clone containing the transmembrane segment M4 of the GluN2B gene (Grin2b) was isolated from a C57BL/6 genomic library. The 0.3 kb BamHI fragment carrying a Pgk-1 polyadenylation signal was inserted into the BamHI site of pLFNeo (Takeuchi et al., 2002), which was digested with SmaI and NotI (blunted), to yield pLFNeopA. The 2.2 kb HindIII–SacI (blunted) fragment from pLFNeopA was inserted to the HindIII site in the intron followed by the M4 containing exon. Another loxP sequence was inserted into the Bpu1102I site in the intron after the M4-containing exon to obtain a vector pSTVfloxε2. It contained a 2.6 kb Grin2b at the 5′ side and 7.5 kb Grin2b at the 3′ side, followed by a MC1 promoter-driven diphtheria toxin gene.
Culture of ES cells and generation of chimeric mice were performed as described previously (Kitayama et al., 2001). Briefly, linearized pγ1CreTV or pSTVfloxε2 was introduced into C57BL/6 mouse ES cells, and then G418-resistant clones were picked up. Each homologous recombined ES clone was identified by Southern blotting. To produce germ line chimera, they were microinjected into eight cell-stage embryos of CD-1 mouse strain. We crossed Grin2b-flox and Grik4-Cre mice to obtain CA3-GluN2B-KO (Grin2bflox/flox; Grik4Cre/+) mice. Control animals in all experiments were littermate homozygous Grin2b-flox or wild-type mice. Breeding and maintenance of mice were performed under our institutional guidelines.
Genotypes were identified by Southern blot hybridization or PCR. Tail genomic DNA was digested with NheI (Grin2b locus) or EcoT22-I (Grik4 locus) and hybridized with probe C (Grin2b locus) or 3′ probe (Grik4 locus), respectively. PCR was performed with specific three primers for each line. The sequence of each primer and the approximate length of the amplified DNA fragments are described as follows: Grik4Cre, γ1-1-5 (5′-AACTGCAGTCTTGCATGCTCTCTGGAGCC-3′), γ1Met2 (5′-GGAGCGGAGAACACGGGGCAT-3′), and CreR1 (5′-TTGCCCCTGTTTCACTATCC-3′); wild type, 170 bp; mutant, 1 kbp; Grin2bflox, M4S4 (5′-GTCAGAGAGTTTAACCTCAG-3′), Pgk-pr2 (5′-GCTGCTAAAGCGCATGCTCC-3′), and M4A3 (5′-ACAAGCACCTTCTTGGTCTC-3′); wild type, 940 bp; mutant, 200 bp; null, 100 bp.
β-Galactosidase assay.
CAG-CAT-Z11, a Cre-inducible lacZ mouse line (Araki et al., 1995), was crossed with the Grik4-Cre mouse line (Grik4Cre/+) for detecting the Cre recombinase activity. β-Galactosidase expression was examined by X-gal staining (Tsujita et al., 1999) in parasagittal brain sections of mice heterozygous for each transgene (Grik4Cre/+; CAG-CAT-Z11/+).
Immunohistochemistry and postembedding immunogold electron microscopy.
To identify the cell type expressing Cre recombinase in the brain of mouse line (Grik4Cre/+; CAG-CAT-Z11/+), parasagittal sections (1 mm thick) subjected to the X-gal staining were embedded in paraffin wax for immunohistochemical analysis. Paraffin sections (5 μm thick) were successively incubated with 10% normal goat serum for 30 min, primary antibodies against calbindin (1:10,000) (Nakagawa et al., 1998), parvalbumin (1 μg/ml) (Nakamura et al., 2004), or glutamine synthase (1 μg/ml; antigen; amino acid residues 346-375 of mouse glutamine synthase; accession number M60803) overnight, biotinylated goat anti-guinea pig or anti-rabbit IgG for 1 h, and streptavidin–peroxidase (Histofine SAB-PO kit; Nichirei) for 30 min. Immunoreactions were visualized with DAB (3,3-diaminobenzidine). Nuclear fast red (Nakarai) was used for counterstaining.
In immunohistochemistry for NMDA and AMPA receptor subunits and PSD-95/synapse-associated protein 90 (SAP90), paraffin sections (5 μm) of control and mutant mice were subjected to pepsin pretreatment, as reported previously (Watanabe et al., 1998; Fukaya and Watanabe, 2000; Yamada et al., 2001; Oshima et al., 2002). The sections were immunoreacted with rabbit or guinea pig primary antibodies (all at 1 μg/ml) against GluN2AC (formerly termed GluRε1C), GluN2BN (GluRε2N), GluN2BC (GluRε2C) (Watanabe et al., 1998), GluN1-C2 (GluRζ1-C2) subunits of NMDARs (Abe et al., 2004), GluA1 (GluR1, GluR-A, or GluRα1) subunit of AMPA receptors (AMPARs) (Watanabe et al., 1998), PSD-95 (Fukaya and Watanabe, 2000), β-galactosidase (ab616; Abcam), and Cre recombinase (69050-3; Novagen). We also used Cre recombinase antibody, which was raised in the guinea pig against full-length bacteriophage P1 Cre recombinase (GenBank accession number X03453).
Postembedding immunogold electron microscopy was performed similarly as described previously (Fukaya and Watanabe, 2000; Abe et al., 2004). Briefly, ultrathin sections obtained from two control and two mutant mice at 2 months of age were immunoreacted overnight with GluN2AC, GluN2BN, GluN1-C2, GluN1-C2′ (Abe et al., 2004), or PSD-95 antibody (10 μg/ml for each), and then with colloidal gold (10 nm)-conjugated anti-rabbit or anti-guinea pig IgG (British Biocell International) for 2 h. Grids were stained with 2% uranyl acetate for 20 min. The number of gold particles associated with the postsynaptic membrane, which were defined as those apart <30 nm from the postsynaptic membrane, was counted in the stratum radiatum of hippocampal CA3 region.
Golgi staining and quantification of dendritic spines.
For Golgi-Cox staining, brains from postnatal day 60 (P60) mice were processed using the FD Rapid GolgiStain kit (FD Neurotechnologies). In brief, brains were rapidly removed from deeply anesthetized mice, washed in distilled water, and immersed in impregnation solution for 2 weeks at room temperature. After incubation with cryoprotectant solution, transverse sections through left hippocampi (60 μm thick) were cut on a cryostat (Leica) and mounted on gelatinized glass slides. Sections were counterstained with cresyl violet, rinsed in distilled water, dehydrated, and mounted with Permount (Thermo Fisher Scientific). Apical dendrites of CA3 pyramidal cells, which were isolated from those of neighboring neurons, were selected and reconstructed using 100× objective lens and Neurolucida software (MBF Biosciences). Spine numbers on primary and secondary dendrites in the stratum radiatum were counted, and spine density along dendrites was calculated by Neuroexplorer (MBF Biosciences). Statistical significance was assessed by the Mann–Whitney U test.
Electrophysiology.
Transverse hippocampal slices (400 μm thick) were prepared from 6- to 12-week-old mice and kept for at least 1 h before recording. All experiments were performed at room temperature (24–28°C) in accordance with the animal welfare guidelines of the Physiological Society of Japan. For LTP experiments, field EPSPs (fEPSPs) were recorded with a glass microelectrode of ∼10 μm tip diameter filled with extracellular solution. The composition of the standard artificial CSF (ACSF) was as follows (in mm): 127 NaCl, 1.5 KCl, 1.2 KH2PO4, 1.3 MgSO4, 2.4 CaCl2, 26 NaHCO3, and 10 glucose. The solution was equilibrated with 95% O2 and 5% CO2. Stimuli (0.1 ms duration) were delivered at 0.1 Hz through a concentric bipolar tungsten electrode. Commissural (Com) inputs to CA3 neurons were stimulated at the fimbria, and fEPSPs were recorded from the stratum oriens. For commissural/associational (C/A) fEPSPs, stimulating and recording electrodes were placed in the stratum radiatum of the CA3 region. Mossy fibers (MFs) were stimulated at the stratum granulosum in the dentate gyrus and recorded from the stratum lucidum of the CA3 region. Schaffer collateral/commissural (SC) fibers were stimulated and recorded from the stratum radiatum of the CA1 region. A train of high-frequency stimulation (100 Hz; 1 s) was used to elicit LTP at these synaptic inputs. For LTP experiment at MF synapses, 25 μm d-AP5 was applied 10 min before the high-frequency stimulation to prevent contamination of NMDAR-dependent potentiation at the CA3 recurrent collaterals. In separate set of experiments, whole-cell recordings were made with pipettes filled with an internal solution containing the following (in mm): 150 Cs gluconate, 8 NaCl, 0.2 EGTA, 10 HEPES, and 2 Mg-ATP, pH 7.2. To quantify functional NMDAR channels at postsynaptic sites, we compared the ratio of synaptic currents mediated by NMDARs (NMDA EPSCs) to those mediated by non-NMDARs (non-NMDA EPSCs) (Sakimura et al., 1995). Non-NMDA EPSCs were first recorded at −80 mV, and then NMDA EPSCs were recorded at +50 mV in the presence of 6-cyano- 7-nitroquinoxaline-2,3-dione (CNQX) (10 μm), a blocker of non-NMDARs. In these experiments, picrotoxin (100 μm) was added to ACSF to block GABAA receptor-mediated synaptic responses, and concentrations of both Ca2+ and Mg2+ were raised to 4 mm to prevent generation of seizure discharges. In addition, non-NMDAR- and NMDAR-mediated currents were elicited by direct application of glutamate (0.1 m), pH 8.0, to CA3 neurons by iontophoresis. These currents were expected to reflect mainly activation of extrasynaptic receptors. Current pulses (100 ms duration; ∼80 nA intensity) were used for ejecting glutamate, whereas smaller retaining currents of reverse polarity were used to prevent leakage of glutamate and steady-state desensitization of the receptors. Non-NMDAR-mediated currents were recorded at −80 mV, and then NMDAR-mediated currents were recorded at +50 mV in the presence of 10 μm CNQX.
Production of chimeric protein for titration of GluN2A and GluN2B antibodies.
Original cDNAs of GluN2A and GluN2B were obtained from pBKSAε1 (Meguro et al., 1992) and pBKSAε2 (Kutsuwada et al., 1992). Chimeric cDNA of GluN2B-2A (1-853 of GluN2B, 857-1442 of GluN2A) and GluN2A-2B (1-856 of GluN2A, 854-1456 of GluN2B) were obtained by two-step PCR using pBKSAε1 and pBKSAε2 as templates. All four of these cDNAs were cloned into pEF-BOS vector (Mizushima and Nagata, 1990), which were introduced into COS-7 cells by the Lipofectamine Plus Reagent (Invitrogen). Twenty-six hours after transfection, cells were harvested in ice-cold PBS, and lysed in 200 μl of 1× sample buffer (62.5 mm Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 1% 2-mercaptoethanol, 0.002% BPB), and lysate of COS-7 cells was sonicated and centrifuged at 10,000 × g for 10 min at 2°C to obtain supernatant as chimeric protein samples. To evaluate relative titers of GluN2AC and GluN2BN antibodies, graded amounts of the lysate were subjected to Western blotting. By comparison of band intensity, the titer of GluN2AC antibody relative to GluN2BN antibody was obtained to be 1.3. This ratio was used to estimate relative amounts of GluN2A and GluN2B subunits in the hippocampal CA3 region.
Subcellular fractionation and Western blot analysis.
Mouse hippocampi were excised from 8- to 12-week-old mice, and the CA3 area was dissected out in ice-cold ACSF under dissecting microscope, and then homogenized in homogenization buffer [320 mm sucrose and 5 mm EDTA, pH 7.5, containing 0.5 mm phenylmethylsulfonyl fluoride (PMSF) (Sigma-Aldrich), 2 μm leupeptin (Peptide Institute), 1 μm pepstatin (Peptide Institute), and phosphatase inhibitor (PhosSTOP; Roche)]. The homogenate was centrifuged at 1000 × g for 10 min to obtain the supernatant (S1) as the crude fraction. The cell fractionation was performed as described previously (Carlin et al., 1980). To obtain the PSD fraction, the synaptosomal fraction of the hippocampal CA3 region from five mice was treated with ice-cold Triton solution (containing 0.5% Triton X-100, 0.16 m sucrose, and 6 mm Tris-HCl, pH 8.0) for 15 min on ice, and then centrifuged at 32,000 × g for 25 min at 2°C. The resultant pellet was resuspended in 40 mm Tris-HCl, pH 8.0, and further treated with ice-cold Triton solution for 15 min on ice and then centrifuged at 201,800 × g for 65 min at 2°C. The resultant pellet (PSD fraction) obtained was dissolved in 1% SDS, 40 mm Tris-HCl, pH 8.0. F-actin and G-actin fractions were separated as described previously (Aoki et al., 2005). Briefly, the synaptosomal fraction of the hippocampal CA3 region from five mice was centrifuged at 228,000 × g for 60 min at 2°C. The pellet suspended in 5 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.5 mm DTT, 1% Nonidet P-40 (NP-40), and protease inhibitors (0.5 mm PMSF, 2 μm leupeptin, and 1 μm pepstatin) was centrifuged at 200,000 × g for 60 min at 2°C. The resultant supernatant and pellet were used as the G-actin and the F-actin fractions, respectively. Protein concentration of the fractions was determined by the method of Lowry et al. (1951) or using BCA Protein Assay Reagent (Pierce). SDS-PAGE and immunoblotting were performed as described previously (Abe et al., 2004). The following antibodies were used: GluN2BN, GluN2BC, GluN2AC, GluN1N (1 μg/ml) (Watanabe et al., 1998), GluK4C (1 μg/ml), rabbit PSD-95 (1 μg/ml) (Fukaya and Watanabe, 2000), rabbit N-cadherin (1 μg/ml; H-63; sc-7939; Santa Cruz Biotechnology), rabbit NSE (1 μg/ml) (Sakimura et al., 1980), mouse monoclonal α-actinin (1 μg/ml; A5044; Sigma-Aldrich), mouse monoclonal actin (1 μg/ml; MAB1501R; Millipore Bioscience Research Reagents), rabbit cofilin (1:500; C8736; Sigma-Aldrich), mouse monoclonal CaMKIIα (1 μg/ml; MAB8699; Millipore Bioscience Research Reagents), rabbit CaMKIIβ (1 μg/ml), rabbit active CaMKII (1:500; V1111; Promega), rabbit GluRα1C (1 μg/ml) (Shimuta et al., 2001), and mouse monoclonal SAP97 (1 μg/ml; RPI197.4; Assay Designs) antibodies. Rabbit GluK4C and rabbit CaMKIIβ antibodies were raised against C-terminal 91 aa residues of mouse GluK4 (GenBank accession number AK032029) and 353–388 aa residues of mouse CaMKIIβ (NM_007595), respectively. For semiquantification of the protein level, images on x-ray film (Kodak) were scanned to determine the relative gray intensities by using NIH Image software. To prevent saturated signals, densitometric quantification was performed in the range of good linearity between signal intensity and exposure time.
Results
Generation of Grik4-Cre and Grin2b-flox mouse lines
To establish the Cre expression system targeting hippocampal CA3 pyramidal cells, we used the promoter of the glutamate receptor GluK4 subunit (GluRγ1 or KA1) gene (Grik4), which is predominantly expressed in the CA3 pyramidal cell (Werner et al., 1991). We constructed a targeting vector, in which Cre gene was inserted into the translational initiation site of the Grik4 in frame. The vector was introduced into the C57BL/6 ES cells by electroporation (Fig. 1A). Chimeric mice were generated from a recombinant ES clone (Fig. 1B) and crossed with C57BL/6 mice to produce the Grik4-Cre mutant line. The heterozygous (Grik4Cre/+) mice were viable, fertile, and did not display any overt behavioral and anatomical phenotypes (data not shown). To detect Cre recombinase activity, we intercrossed the Grik4-Cre line with the Cre-inducible lacZ reporter mouse line (CAG-CAT-Z11) (Araki et al., 1995) and produced reporter mice heterozygous for each transgene (Grik4Cre/+; CAG-CAT-Z11/+). Blue staining representing Cre recombinase activity first appeared in the hippocampal CA3 region at P0. Cre recombinase expression increased progressively in the CA3 until P8 (supplemental Figs. S1, S2A, available at www.jneurosci.org as supplemental material). Thereafter, high recombinase activity was maintained and virtually restricted to this region with gradual appearance of low activities in the layer VI of the cerebral cortex and in several brainstem regions (Fig. 1Ca; supplemental S1, available at www.jneurosci.org as supplemental material).
Figure 1.
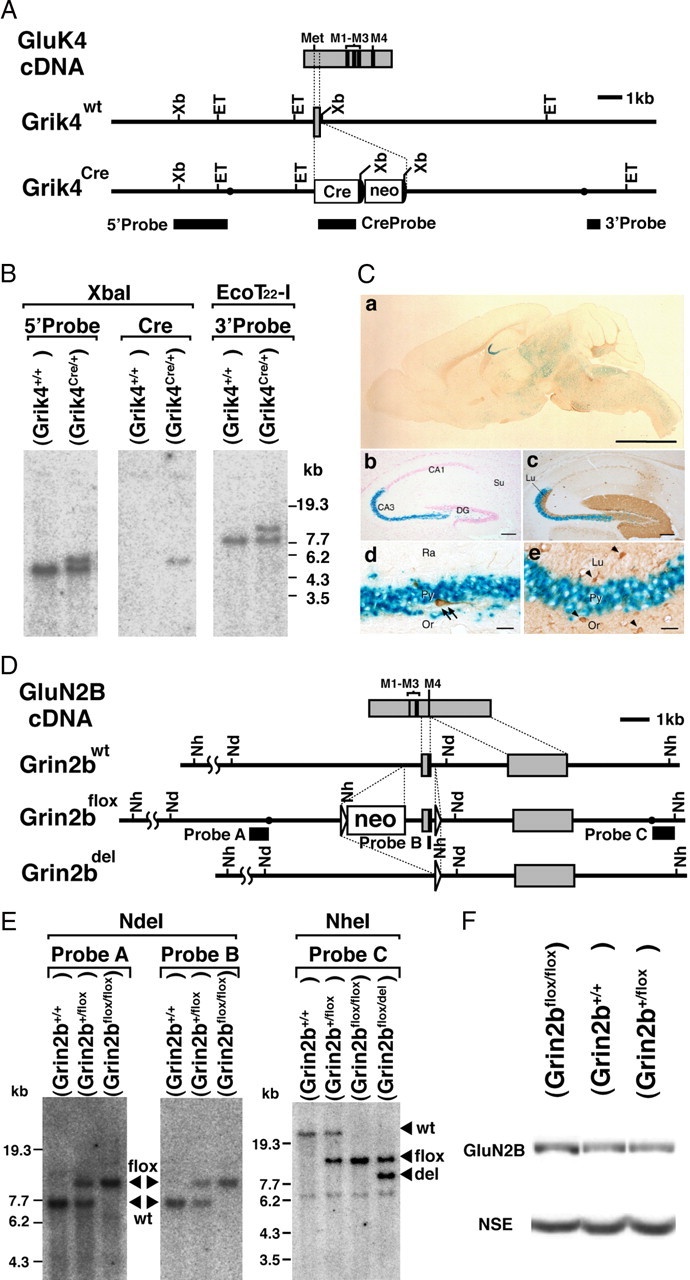
Generation of Grin2b-flox and Grik4-Cre mice. A, Schematic representations of GluK4 cDNA, genomic DNA (Grik4wt), and targeted genome (Grik4Cre). The gray boxes indicate exons. The black boxes indicate the probes for Southern blot analysis. The filled circles in the Grik4Cre allele delineate the 5′ and 3′ termini of the targeting vector. The semicircles indicate FRT sequences. Met, Initial methionine; Cre, Cre recombinase gene; neo, neomycin-resistant gene expression cassette; Xb, XbaI; ET, EcoT22-I. B, Southern blot analysis of XbaI- or EcoT22-I-digested genomic DNA prepared from the wild-type (Grik4+/+) and heterozygous Cre (Grik4Cre/+) mice. C, β-Galactosidase activity induced by Grik4-Cre in 4-week-old reporter mice. a, β-Galactosidase staining of a whole-brain slice (50 μm thick). b, c, Hippocampus, counterstained with nuclear fast red (NFR) (b) or reacted with anti-calbindin antibody (c). d, e, CA3 region. Parvalbumin (d) and glutamine synthase (e) were stained with respective antibodies as markers for interneurons and glial cells, respectively. Scale bars: a, 3 mm; b, c, 100 μm; d, e, 10 μm. D, Schematic representations of GluN2B cDNA, genomic DNA (Grin2bwt), targeted genome (Grin2bflox), and Cre-mediated deleted genome (Grin2bdel). The gray boxes indicate exons. The black boxes indicate the probes for Southern blot analysis. The triangles indicate loxP sequences. The filled circles in the Grin2bflox allele delineate the 5′ and 3′ termini of the targeting vector. Nh, NheI; Nd, NdeI. E, Southern blot analysis of NdeI- or NheI-digested genomic DNA prepared from the wild-type (Grin2b+/+), heterozygous floxed (Grin2b+/flox), homozygous floxed (Grin2bflox/flox), and heterozygous floxed and deleted (Grin2bflox/del) mice. F, Immunoblot analysis of whole-brain crude fraction prepared from the wild-type, heterozygous (Grin2b+/flox), and homozygous floxed (Grin2bflox/flox) mice at P0.5 with anti-GluN2BC and anti-NSE antibodies.
Within the hippocampus, Cre-mediated recombination was almost selective to the CA3 region and its continuous hilar region (Fig. 1Ca–c; supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). Recombination in the dentate gyrus, which is known to express GluK4 subunit at low levels (Werner et al., 1991), occurred at apparently low frequencies. By counting the number of blue cells, all (100%) of the CA3 pyramidal neurons were positive as early as P8, whereas positive cells in dentate gyrus granule cells were 0.4% at P8 and 6.9% at postnatal 10 weeks (P10W). β-Gal-positive cells in the hilus were polymorphic cells (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). No positive cells were found in CA1 pyramidal cells even at P10W. In contrast, Cre-mediated recombination was negative in glutamine synthase-positive astrocytes and parvalbumin-positive interneurons in the CA3 region (Fig. 1Cd,e). In the reporter mice, the distribution of blue cells was consistent with that of β-galactosidase (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material) or Cre recombinase immunoreactivity (supplemental Fig. S2D, available at www.jneurosci.org as supplemental material). These results indicate that the Grik4-Cre mouse line can drive Cre-mediated recombination mainly in CA3 pyramidal cells.
In our preliminary experiments using the Xenopus oocyte expression system, we confirmed that mutant GluN2B subunit lacking the transmembrane segment M4 constituted no functional NMDAR channels (data not shown). We thus constructed a targeting vector in which the exon encoding the transmembrane segment M4 was flanked by two loxP sequences in the same orientation. The vector was introduced into the C57BL/6 ES cells by electroporation, and chimeric mice were generated from a recombinant ES clone. The chimeric mice were further crossed with C57BL/6 mice to establish the neomycin resistant gene (neo)-containing Grin2b-flox mouse line (Fig. 1D,E). Because expression level of GluN2B subunit in neo-containing Grin2b-flox mice was comparable with that in wild-type mice (Fig. 1F; supplemental Fig. S4A, available at www.jneurosci.org as supplemental material), we did not remove the neo cassette and hereafter used them as control mice (Fig. 1F). Homozygous Grin2b-flox mice (Grin2bflox/flox) were viable, fertile, and did not display any overt behavioral and anatomical phenotypes (data not shown).
Preferential GluN2B deletion in CA3 pyramidal cells within mutant hippocampus
By intercrossing Grik4-Cre mice with Grin2b-flox mice, we obtained CA3-GluN2B-KO (Grin2bflox/flox; Grik4Cre/+) mice, which are termed mutant mice hereafter. They were viable, fertile, and displayed no overt behavioral phenotypes. In Grik4-Cre mice, a low level of Cre recombinase activity appeared in germ line (data not shown), and we obtained heterozygous Grin2b-flox and deleted mice (Grin2bflox/del) at the frequency of <1% (Fig. 1E). The size, proportion, and histology of the brain, including the hippocampus, were apparently normal in mutant mice at 2 months of age (Fig. 2A–D). Immunohistochemical changes of GluN2B and other postsynaptic molecules were evaluated by pepsin-pretreated immunoperoxidase with paraffin sections (Figs. 2E–J, 3A), because postsynaptic antigens were detected very weakly without pepsin pretreatment (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material), and paraffin sections yielded immunohistochemical staining with higher signal-to-noise ratios than frozen sections (supplemental Fig. S3B, available at www.jneurosci.org as supplemental material). We observed that GluN2B subunit was lowered in the CA3 region of mutant mice to around the background level using both GluN2BN (Fig. 2E,F) and GluN2BC (data not shown) antibodies. No apparent loss of GluN2B subunit was discerned in the CA1 region, whereas mild reduction was found in the molecular layer of the dentate gyrus and the hilus. By using calbindin as a marker for subfields of the hippocampal formation (Fig. 2I,J), this pattern of GluN2B ablation was confirmed in both the dorsal and ventral hippocampi (Fig. 2G,H). At 2 months of age, mild to moderate reduction of GluN2B immunoreactivities was also noted in deep cortical layers and some brainstem regions (Fig. 2A,B), as expected from β-galactosidase assay (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). In the crude fraction prepared from the microdissected CA3 region, immunoblot with GluN2BN antibody showed that GluN2B bands, which was comparable in size with that in control mice, was greatly reduced in mutant mice (9.59 ± 5.15% of control level; n = 3 independent preparations; t test, p < 0.004) (mean ± SEM) (Fig. 3B). The lack of truncated GluN2B band as well as immunohistochemical invisibility with use of GluN2BN antibody (supplemental Fig. S4B, available at www.jneurosci.org as supplemental material) suggests accelerated premature degradation of GluN2B subunit lacking the M4. Therefore, it is conceivable that a trace level of residual GluN2B subunit might reflect intact expression in nonpyramidal cells and/or contamination of adjacent dentate gyrus or CA1 region.
Figure 2.
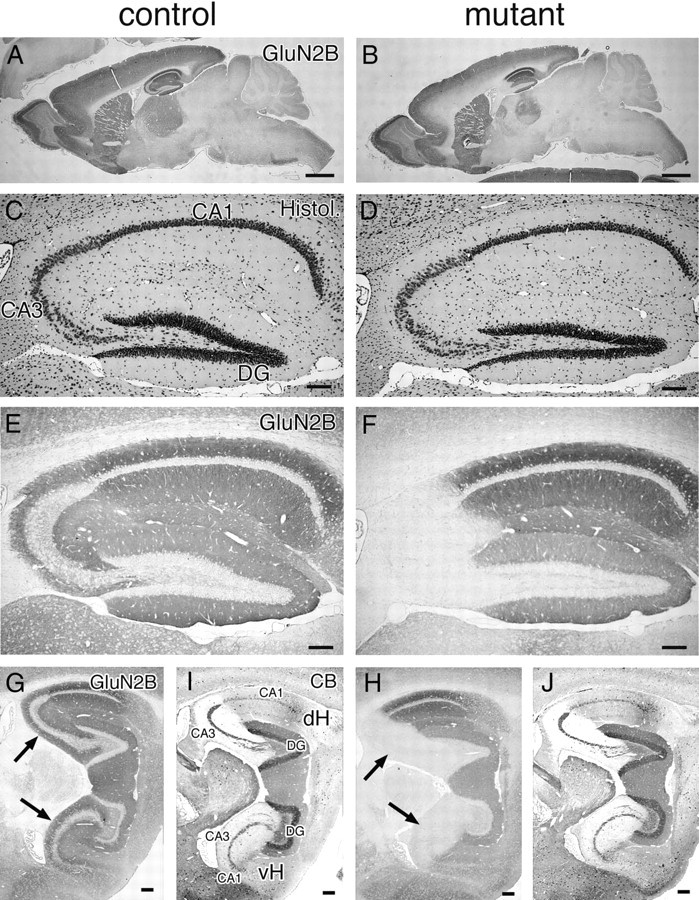
Selective deletion of GluN2B subunit in CA3 pyramidal cells of CA3-GluN2B-KO mice. Immunohistochemistry for GluN2B in mutant and control mice at 2 months of age. Immunoreactivities for GluN2B in the whole brain (A, B), hippocampus (E, F), and the dorsal and ventral hippocampi (G, H). Normal histology of the hippocampus in mutant mice (C, D). Immunoreactivities for calbindin used as a marker for subfields of the hippocampal formation (I, J): dense somal and dendritic labeling in the dentate gyrus (DG), dense mossy fiber labeling in the CA3 (CA3), and low diffuse labeling in the CA1. The arrows in G–J indicate the CA3 subfield. CB, Calbindin; dH, dorsal hippocampus; vH, ventral hippocampus. Scale bars: A, B, 1 mm; C–J, 0.1 mm.
Figure 3.

Effects of CA3 region-selective GluN2B deletion on PSD-associated proteins. A, Immunohistochemistry for GluN2A, GluN1-C2, PSD-95, and GluA1 in the mutant and control hippocampus. Scale bar, 0.1 mm. B, Immunoblot analysis of crude fraction from CA1 and CA3 of control and mutant mice with respective antibodies (GluN2B, GluN2A, GluN1, PSD-95, GluK4, and N-cadherin). Each lane was loaded with 20 μg of protein.
Moreover, a substantial reduction was noted in immunohistochemical signals for GluN1 subunit in the mutant CA3 region (Fig. 3A). This was consistent with immunoblot analysis using crude fraction (Fig. 3B); the amount of GluN1 subunit was significantly reduced in the mutant CA3 region to one-half level of the control one (47.7 ± 12.3%; n = 5; p < 0.02). In contrast, almost comparable levels by immunohistochemistry and immunoblot were observed for GluN2A subunit (90.6 ± 16.3% of control; n = 4; p = 0.61), GluA1 subunit (111 ± 13.2% of control; n = 5; p = 0.45), and PSD-95, a scaffolding protein for ionotropic glutamate receptors (98.0 ± 6.85% of control; n = 3; p = 0.79) (Fig. 3A,B; supplemental Fig. S4B, available at www.jneurosci.org as supplemental material). N-cadherin, a homophilic adhesion molecule comprising the adhesive junction between presynaptic and postsynaptic membranes, was unchanged in the mutant CA3 region (77.6 ± 1.80% of control; n = 4; p > 0.05). In the mutant CA1 region, however, no significant changes were observed for GluN2A subunit (110 ± 21.9% of control; n = 3; p = 0.69), GluN2B subunit (67.2 ± 20.6% of control; n = 3; p = 0.25), and GluN1 subunit (90.1 ± 30.2% of control; n = 3; p = 0.77).
GluK4 subunit was reduced to one-half level in the mutant CA3 (Fig. 3B), because Cre recombinase cDNA was inserted into the translational initiation site of the Grik4 gene in frame in Grik4-Cre mice (see Materials and Methods). As no reduction of immunohistochemical signals for GluN1 subunit was discerned in the CA3 of Grik4-Cre mice compared with wild-type mice (data not shown), the reduced GluN1 subunit in the mutant CA3 region is unlikely to be caused by the ablation of single Grik4. Therefore, GluN2B subunit is preferentially ablated in CA3 pyramidal cells within mutant hippocampus, which accompanies reduction of GluN1 subunit to one-half level without affecting other synaptic molecules, including GluN2A subunit and PSD-95.
Almost complete loss of NMDAR-mediated synaptic currents at mutant CA3 synapses
We next examined the effects of conditional GluN2B ablation on NMDAR-mediated synaptic currents at three excitatory CA3 synapses: the C/A–CA3 synapse, Com–CA3 synapse, and MF–CA3 synapse (Fig. 4A). The ratio of NMDAR to non-NMDAR components of EPSCs decreased drastically at all three synapses in the mutant mice: C/A–CA3 synapse (30.5 ± 4.5% in control, n = 12; 3.9 ± 0.8% in mutant, n = 13; p < 0.001), Com–CA3 synapse (17.9 ± 5.2% in control, n = 3; 0.3 ± 0.3% in mutant, n = 4; p < 0.001), and MF–CA3 synapse (17.1 ± 2.2% in control, n = 7; 2.0 ± 1.5% in mutant, n = 5; p < 0.001). In contrast, there was no significant difference in NMDAR components at the SC–CA1 synapse between control and mutant mice (38.9 ± 8.0% in control, n = 9; 31.8 ± 4.0% in mutant, n = 8; p = 0.46).
Figure 4.

Effects of GluN2B deficiency on NMDAR-mediated EPSCs and LTPs in CA3 area. A, Effects on NMDAR-mediated EPSCs at the synapses in hippocampal CA3 (C/A–CA3, Com–CA3, and MF–CA3) and CA1 (SC–CA1). The upward and downward traces show NMDAR-mediated EPSCs in the presence of CNQX and non-NMDAR-mediated EPSCs in the control solution, respectively. B, Effects on extracellular field potentials after tetanic stimulation (100 Hz, 1 s). The averaged time course of the field EPSP slope. The open and filled circles represent control and mutant slices, respectively.
We also measured LTP (Fig. 4B). In mutant slices, LTP was abolished at the Com–CA3 synapse (212.1 ± 32.2% in control, n = 5; 107.7 ± 19.8% in mutant, n = 6; p < 0.02) and C/A–CA3 synapse (139.6 ± 8.4% in control, n = 5; 96.6 ± 9.0% in mutant, n = 6; p < 0.01). In contrast, robust LTP was induced at the MF–CA3 synapse (131.1 ± 9.9% in control, n = 6; 141.6 ± 23.2% in mutant, n = 4; p = 0.65), in which LTP does not depend on NMDAR activation (Harris and Cotman, 1986; Zalutsky and Nicoll, 1990). As to LTP at the SC–CA1 synapse, there were no significant differences between control and mutant slices (161.8 ± 11.7% in control, n = 6; 149.0 ± 8.0% in mutant, n = 4; p = 0.45). Therefore, NMDAR-mediated synaptic currents and plasticity are abolished by the ablation of GluN2B subunit.
The 1:2 ratio of GluN2A and GluN2B amounts in CA3 region
Our results show that synaptic NMDAR-mediated currents are almost lost despite the presence of remaining GluN2A and GluN1 subunits. To explain the apparent discrepancy, we tested several possibilities. The first possibility was that the amount of GluN2B subunit is overwhelming to that of GluN2A subunit in the CA3 region, and the ablation of the former thereby leads to almost complete loss of NMDAR function. To estimate relative subunit contents in the CA3, we first determined the titer ratio of GluN2AC and GluN2BN antibodies using COS cell lysates expressing chimeric GluN2A/GluN2B protein (supplemental Fig. S5, available at www.jneurosci.org as supplemental material).
Chimeric 2A–2B protein was constructed from the N-terminal side of GluN2A subunit and the C-terminal side of GluN2B, and this is opposite for chimeric 2B–2A protein (supplemental Fig. S5A, available at www.jneurosci.org as supplemental material). In immunoblot analysis of COS-7 cell extracts, GluN2AC antibody recognized specific band of GluN2A subunit and chimeric 2B–2A protein, whereas GluN2BN antibody did GluN2B subunit and chimeric 2B–2A protein (supplemental Fig. S5B, available at www.jneurosci.org as supplemental material). By graded dilutions of these chimeric proteins, the titration of GluN2AC and GluN2BN antibodies was performed. The signal intensity of protein bands produced by GluN2AC antibody was found to be ∼1.3-fold higher than that by GluN2BN antibody (supplemental Fig. S5C, left; D, left, available at www.jneurosci.org as supplemental material). Because the intensity of protein bands in the CA3 crude fraction was ∼1.8-fold higher for GluN2B subunit than for GluN2A subunit (supplemental Fig. S5C, right; D, right, available at www.jneurosci.org as supplemental material), the relative amount of GluN2B subunit was estimated to be approximately twice (1.8 × 1.3 = 2.3) as much as that of GluN2A subunit in the hippocampal CA3 region. Therefore, the ∼1:2 ratio in amounts of GluN2A and GluN2B subunits in the CA3 is apparently insufficient to account for almost complete loss of NMDAR-mediated synaptic currents by GluN2B ablation.
Normal synaptic expression of GluN2A and PSD-95 and severe reduction of GluN1 in mutant CA3
The second possibility was attributable to the inability of NMDARs to localize at the postsynapse when GluN2B subunit is absent, and this was tested by quantitative postembedding immunogold electron microscopy (Fig. 5). The number of gold particles on the postsynaptic membrane and the length of PSD were measured to calculate the density of synaptic immunogold labeling. No significant difference was found for GluN2A subunit (10.8 particles/μm in control, n = 22 synapses; 11.4 in mutant, n = 20; p = 0.81) (106% of control level) (Fig. 5A,B). Synaptic labeling density was also comparable for PSD-95 (103% of control level; n = 20 for each; p = 0.52) (Fig. 5I,J).
Figure 5.
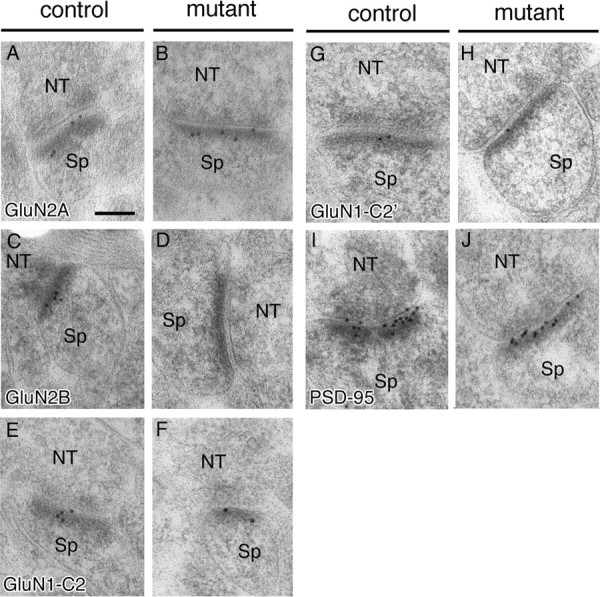
Synaptic localization of NMDA receptor subunits in the stratum radiatum of hippocampal CA3 region. Postembedding immunogold labeling for GluN2A (A, B), GluN2B (C, D), GluN1-C2 (E, F), GluN1-C2′ (G, H), and PSD-95 (I, J) in the stratum radiatum of control and mutant CA3 region. NT, Nerve terminal; Sp, spine. Scale bar, 100 nm.
GluN1 subunit ends with four different C termini, being generated by optional usage of the C1 cassette and by alternative usage of the C2 and C2′ cassettes (Sugihara et al., 1992; Hollmann et al., 1993). We examined GluN1 subunit by using GluN1-C2 and GluN1-C2′ antibodies. Synaptic labeling density was reduced severely in the mutant: GluN1-C2 subunit was 23.6% of the control level (25.0 in control, n = 23; 5.9 in mutant, n = 19; p < 0.001) (Fig. 5E,F), and GluN1-C2′ subunit was 20.6% of the control level (7.98 in control, n = 27; 1.64 in mutant, n = 16; p < 0.003) (Fig. 5G,H). Thus, GluN2A subunit and PSD-95 are normally localized at mutant CA3 synapses, whereas GluN1 subunit is severely reduced to one-fifth or one-fourth levels. Considering that functional NMDARs require GluN1 subunit and any of four GluN2 subunits (Seeburg, 1993; Nakanishi et al., 1994; Mori and Mishina, 1995), mutant CA3 synapses should possess, at least, one-fifth to one-fourth levels of functional NMDARs. Again here, this is insufficient to account, by itself, for almost complete loss of NMDAR-mediated synaptic currents by GluN2B ablation.
Reduced but detectable extrasynaptic NMDAR-mediated currents in mutant CA3 pyramidal cells
Then, we pursued the third possibility that NMDARs lacking GluN2B subunit are nonfunctional in the brain, although coexpression of GluN2A and GluN1 subunits in Xenopus oocytes and cell lines yields functional NMDARs (Meguro et al., 1992; Stern et al., 1994; Wyllie et al., 1998; Popescu and Auerbach, 2003). To test this possibility, we measured NMDAR- and non-NMDAR-mediated currents evoked by iontophoretic glutamate application onto the soma and dendrites of CA3 pyramidal cells (Fig. 6A,B). These currents were supposed to mainly reflect extrasynaptic receptors, although possible activation of synaptic receptors could not be excluded. The ratio of glutamate-evoked NMDAR to non-NMDAR components in mutant slices was significantly decreased: 31.3% of control level by glutamate application on the soma (58.1 ± 6.5% in control, n = 14; 18.2 ± 5.9% in mutant, n = 12; p < 0.01) and 44.1% on dendrites (55.5 ± 10.3% in control, n = 6; 24.5 ± 5.7% in mutant, n = 6; p < 0.05) with the average being 35.4% (57.3 ± 5.3% in control, n = 20; 20.3 ± 4.3% in mutant, n = 18; p < 0.01). Interestingly, the extents of reduced NMDAR responses in the mutant CA3 region appear to match with estimated ratio (1:2) of GluN2A and GluN2B subunits in the control one. Considering almost complete loss of synaptic NMDAR-mediated currents, NMDARs lacking GluN2B subunit, which may be composed of GluN1 and GluN2A subunits and mainly expressed at extrasynaptic site, are functional. Thus, the ablation of GluN2B subunit has produced more severe effects on synaptic NMDARs than extrasynaptic ones.
Figure 6.

NMDAR-mediated currents evoked by glutamate application in CA3-GluN2B-KO mice. A, Currents were evoked by iontophoretic glutamate (Glu) application to the soma or dendrites of CA3 pyramidal cells. The downward and upward traces show non-NMDAR-mediated EPSCs at −80 mV (a) and NMDAR- and/or non-NMDAR-mediated EPSCs at +50 mV (b–d), respectively. B, AMPAR-mediated (b − c) and NMDAR-mediated (c − d) responses.
Increased detergent solubility of GluN2A subunit and PSD-95 in mutant CA3
The above results collectively indicate that the ablation of GluN2B subunit did cause considerable reduction of GluN1 subunit, but this cannot fully explain almost complete loss of synaptic NMDAR function. Some other biochemical changes should occur to dampen the function of synaptic NMDARs. We then examined synaptic NMDARs and other synaptic molecules by immunoblot using the PSD fraction prepared from microdissected CA3 region (Fig. 7).
Figure 7.

Effects of GluN2B deletion on the proteins in PSD fraction from CA3 region. Immunoblots with respective antibodies (GluN2A, GluN2B, GluN1, PSD-95, N-cadherin, GluA1, and SAP97) in PSD fraction from CA3 region of control and mutant. Each lane was loaded with 2 μg of protein.
Severe reduction of GluN1 subunit was confirmed in mutants; the relative amount was significantly reduced to 24.6 ± 8.70% of control level (n = 6 independent preparations for control and n = 4 for mutant; p < 0.005), consistent with the result from postembedding immunogold (Fig. 5). To our surprise, significant reduction further occurred for GluN2A subunit [51.1 ± 10.1% of control level; n = 6 (control) and n = 4 (mutant); p < 0.02] and PSD-95 [36.5 ± 13.4%; n = 5 (control) and n = 3 (mutant); p < 0.05]. However, no significant decrease or increase was noted in the amounts of N-cadherin (77.6 ± 1.80%; n = 4 for each; p > 0.05), SAP97 (102 ± 7.56%; n = 3 for each; p = 0.79), a scaffolding protein for shaker K channel, and GluA1 subunit (80.8 ± 6.09% of control level; n = 4 for each; p = 0.051) (Fig. 7). Thus, despite comparable synaptic localization and tissue contents in the mutant CA3 region, GluN2A subunit and PSD-95 became more sensitive to solubilization by detergent treatment. Importantly, no such change was noted for GluN2B subunit (95.2 ± 42.7% of control; n = 5 for each; p = 0.91) and PSD-95 (64.4 ± 17.7% of control; n = 5 for each; p = 0.11) in the PSD fraction prepared from the CA3 of GluN2A-KO mice, in which GluN1 subunit was reduced to one-half level of control one (47.1 ± 11.6% of control; n = 5 for each; p < 0.02). Together, the loss of GluN2B subunit has profound effects on the organization of macromolecular complexes in the PSD.
Marked decrease of F-actin/G-actin ratio in mutant CA3 synaptosomes
The increased detergent sensitivity of PSD molecules could result from changes in the interaction with cytoskeletal proteins. We focused on the actin cytoskeleton, because it is the main cytoskeletal component in dendritic spines, in which NMDARs and related PSD scaffolding proteins accumulate and interact with the actin cytoskeleton (Wyszynski et al., 1997; Böckers et al., 2001; Hering and Sheng, 2003). We prepared NP-40-soluble and -insoluble fractions from the CA3 synaptosomal fraction to distinguish globular monomeric actin (G-actin) and filamentous polymerized actin (F-actin), respectively (Fig. 8A). In the synaptosomal fraction, no significant differences were found in the total actin level (92.7 ± 12.0% of control; n = 5 for each; p = 0.58) (n indicates the number of independent preparations) (Fig. 8B). However, in mutant, G-actin content tended to increase (168 ± 28.7% of control; n = 4 for each; p = 0.09), whereas F-actin content was significantly decreased (84.5 ± 4.48% of control; n = 4 for each; p < 0.05) (Fig. 8B). The ratio of F-actin to G-actin was 1.03 ± 0.19 in control (n = 6) and 0.51 ± 0.05 in mutant (n = 6; 49.5% of control), showing significant reduction (p < 0.03) (Fig. 8B).
Figure 8.
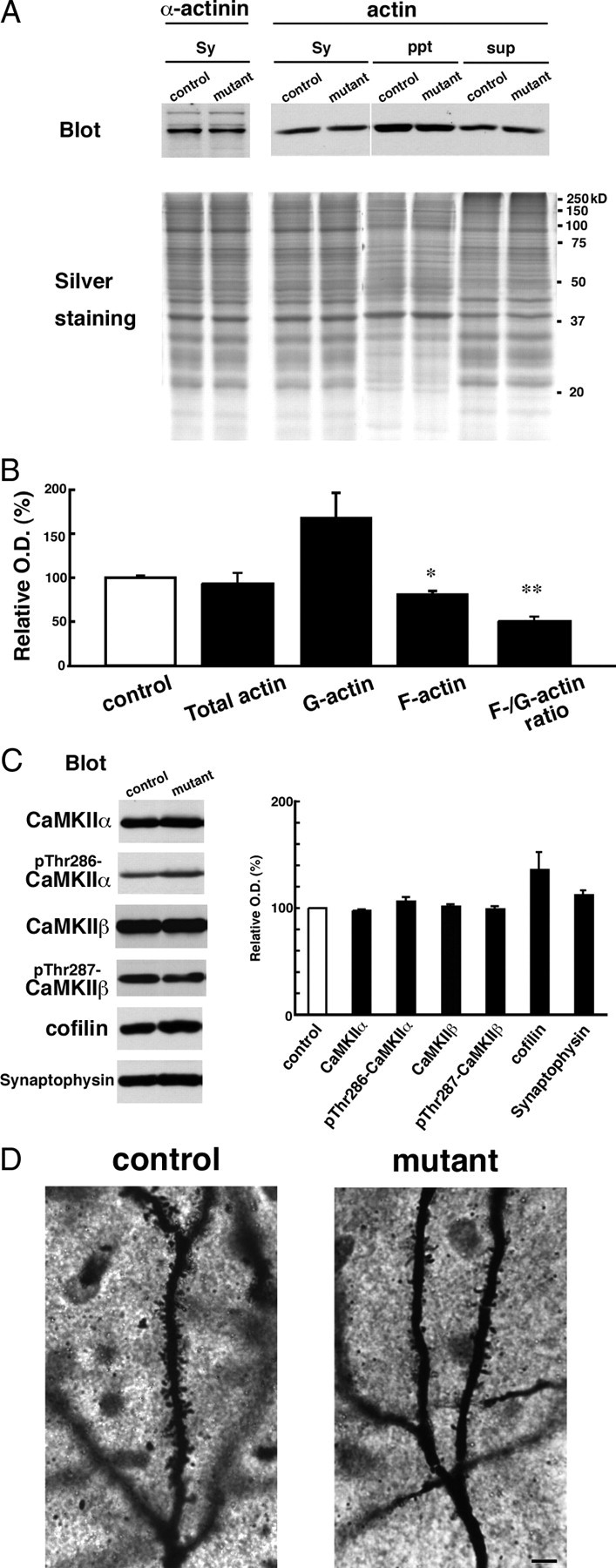
GluN2B ablation decreases F-actin content in CA3 synaptosomes. A, Synaptosomal fraction (Sy) was prepared from hippocampal CA3 region. G-actin and F-actin fractions were obtained from the synaptosomal fraction as NP-40 soluble (sup) and insoluble (ppt) fractions, respectively. In each lane, 10 μg of protein was loaded. Each fraction was separated by SDS-PAGE, and silver-stained (bottom panel) or immunoblotted with an anti-α-actinin or anti-actin antibody. B, Summary graphs indicate relative content of total actin, G-actin, F-actin, and relative ratio of F-actin to G-actin in the mutant to control. **p < 0.03; *p < 0.05. Error bars indicate SEM. C, Immunoblots with antibodies to CaMKIIα, phospho-Thr286-CaMKIIα, CaMKIIβ, phospho-Thr287-CaMKIIβ, cofilin, and synaptophysin (left panel). In each lane, 20 μg of protein of CA3 crude fraction was loaded. Summary graphs (right panel) indicate density of immunoreactivity in mutant normalized to that in control for each molecule. D, Photomicrographs showing Golgi-impregnated apical dendrites of CA3 pyramidal cells from control (left panel) and mutant (right panel) mice. Note apparent reduction of dendritic spines in CA3 pyramidal cells of CA3-GluN2B-KO mice. Scale bar, 5 μm.
Next, we compared expressions of actin-binding proteins and regulators of actin dynamics. In the CA3 synaptosomal fraction, no significant change was seen for α-actinin (82.7 ± 10.0% of control; n = 5 for each; p = 0.16) (Fig. 8A), which binds to both actin filaments and the C-terminal domain of GluN2B subunit (Wyszynski et al., 1997). Using the CA3 crude fraction, we examined actin-related proteins enriched in the cytosol (Fig. 8C). It has been reported that phospho-Thr268-CaMKIIα can simultaneously interact with α-actinin and GluN2B subunit (Robison et al., 2005). No significant differences were found for CaMKIIα (97.6 ± 1.15% of control; n = 4; p = 0.057) and phospho-Thr286-CaMKIIα (106 ± 4.62% of control; n = 3; p = 0.34). The relative amount of the actin-depolymerizing factor cofilin tended to increase (137 ± 15.9% of control; n = 6; p = 0.070). Phospho-Ser3-cofilin, which inhibits the F-actin-severing activity of cofilin, was scarcely detected (data not shown). CaMKIIβ interacts with F-actin and promotes the bundling of actin fibers (Okamoto et al., 2007); no significant differences were observed for CaMKIIβ (103 ± 1.18% of control; n = 4; p = 0.087) and phospho-Thr287-CaMKIIβ (99.9 ± 1.79% of control; n = 4; p = 0.95). Synaptic terminal marker synaptophysin was not changed (113 ± 3.46% of control; n = 3; p = 0.058) (Fig. 8C). These observations suggest that the loss of GluN2B subunit rather selectively affects the dynamics and/or equilibrium of the actin cytoskeleton, without any overt changes in expression levels and phosphorylation state of actin-binding and -regulating molecules.
Finally, we examined whether the absence of GluN2B affected dendritic spine density. By reconstructing Golgi-impregnated apical dendrites, the spine density in primary and secondary dendrites of CA3 pyramidal cells were quantitatively compared in the stratum radiatum. We reconstructed 34 dendrites from three control mice and 33 dendrites from three mutant mice and found that spine density was significantly reduced by 24% in mutant mice (0.84 ± 0.17 μm−1 for control, 0.64 ± 0.17 μm−1 for KO; p < 0.001, Mann–Whitney U test) (mean ± SD) (Fig. 8D).
Discussion
GluN2B subunit is expressed from early embryonic stages in brain development (Watanabe et al., 1992; Monyer et al., 1994), and GluN2B-containing NMDARs confer high Mg2+ sensitivity, large channel conductance, and potentiation by protein kinase C (Kutsuwada et al., 1992; Mori et al., 1992, 1993). These properties have led to the proposal that this subunit is critically involved in activity-dependent brain functions, such as synapse refinement, synaptic plasticity, and cognitive function (Mori and Mishina, 1995). Some of these postulated roles have been experimentally evidenced using animal models, as exemplified by impaired hippocampal LTD, whisker-related neuronal patterning, and suckling reflex in GluN2B−/− newborn mice (Kutsuwada et al., 1996), reduced hippocampal LTP in GluN2B+/− adult mice (Ito et al., 1997), and enhanced hippocampal LTP and superior learning ability in GluN2B-overexpressing mice (Tang et al., 1999).
In the present study, we developed a viable animal model, CA3-GluN2B-KO mice, in which GluN2B subunit is preferentially ablated in CA3 pyramidal cells and also in hilar polymorphic cells within the hippocampus. In the mutant CA3, GluN1 subunit was reduced from the postsynaptic site (20.6–23.6% of control levels by immunogold and 24.6% by immunoblot), whose reduction is apparently severer than that in the crude fraction (47.7%). The severe reduction of synaptic GluN1 subunit is attributable mainly to the loss of GluN2B subunit, because GluN2 subunits are essential for both postsynaptic localization and protein stability of GluN1 subunit (Abe et al., 2004). Nevertheless, considering normal postsynaptic localization of GluN2A subunit and the presence of remaining GluN1 subunit, virtual loss of NMDAR-mediated synaptic currents and plasticity in CA3-GluN2B-KO mice was unexpected.
GluN2A and GluN2B subunits are two major GluN2 subunits in the hippocampus (Kutsuwada et al., 1992; Meguro et al., 1992; Monyer et al., 1992). In the CA3, they are distributed at much higher levels in the stratum radiatum and oriens, in which C/A–CA3 and Com–CA3 synapses are formed, than in the stratum lucidum, in which MF–CA3 synapses reside (Watanabe et al., 1998). In GluN2A−/− and GluN2B+/− mice, NMDAR-mediated synaptic currents are preferentially impaired at C/A–CA3 or Com–CA3 synapses, respectively (Ito et al., 1997). From these findings, we predicted that GluN2B knock-out would differentially disrupt NMDAR-mediated responses at CA3 synapses in an input-dependent manner. However, our observation that NMDAR-mediated currents were almost lost at all these CA3 synapses was also unexpected. To account for these unexpected observations, we pursued three possibilities: whether the amount of GluN2B subunit is overwhelming against that of GluN2A subunit within the CA3, whether synaptic localization of NMDARs is aborted in the absence of GluN2B subunit, and whether NMDARs lacking GluN2B subunit are nonfunctional. Our data show that the first two possibilities cannot sufficiently explain almost total loss of NMDAR function, and that NMDARs lacking GluN2B subunit are functional at extrasynaptic sites of GluN2B-lacking CA3 pyramidal cells. We finally addressed biochemical changes in CA3 synapses (i.e., increased detergent solubility of NMDAR subunits and PSD-95) and significant decrease of F-actin in the synaptosomal fraction.
F-actin is most concentrated in dendritic spines (Matus et al., 1982; Allison et al., 1998; Capani et al., 2001), and recent studies indicate a variety of interactions between the actin cytoskeleton and PSD molecules (Wyszynski et al., 1997; Böckers et al., 2001; Hering and Sheng, 2003). Depolymerization of F-actin with latrunclin-A leads to drastic reductions in synaptic clusters of NMDARs, AMPARs, CaMKIIα, and PSD scaffolds, such as GKAP (guanylate kinase-associated protein), Shank, and Homer (Allison et al., 1998, 2000; Kuriu et al., 2006). Notably, the C termini of GluN1 and GluN2B subunits bind directly to α-actinin (Wyszynski et al., 1997), an actin-binding protein that is concentrated in the PSD and regulates dendritic spine morphology (Wyszynski et al., 1998; Nakagawa et al., 2004; Racz and Weinberg, 2004). NMDARs are also anchored to the PSD through interaction of the PSD-95/SAP90 protein family with the C termini of GluN2A and GluN2B subunits (Kornau et al., 1995; Kim et al., 1996; Niethammer et al., 1996; Bassand et al., 1999). Thus, the C terminus of GluN2B is capable of interacting with both the actin cytoskeleton and PSD-95/SAP90 protein family. When the C-terminal region of GluN2B is truncated, postsynaptic clustering and localization of NMDARs are severely impaired in hippocampal neurons (Mori et al., 1998), suggesting its importance in the organization of postsynaptic molecules. Our finding that increased detergent solubility in the PSD fraction was seen for GluN2A subunit in CA3-GluN2B-KO mice, but not for GluN2B subunit in GluN2A-KO mice, further indicates that GluN2A subunit cannot compensate for this role by GluN2B subunit. Furthermore, dendritic spine density showed a mild but significant reduction (∼24%) in CA3-GluN2B-KO mice. These observations are similar to those obtained from RNA interference (RNAi) knockdown experiments using cultured hippocampal neurons (Kim et al., 2005); GluN2B-RNAi, but not GluN2A-RNAi, leads to great reductions of synaptic NMDARs and spine density. Therefore, GluN2B subunit likely plays an indispensable role in the formation or maintenance of postsynaptic macromolecular complex and dendritic spines.
Ca2+-induced depolymerization of F-actin also leads to rundown of NMDAR function (Rosenmund and Westbrook, 1993). Conversely, increased stability of F-actin, which occurs in mice lacking Eps8, a regulator of actin dynamics, enhances NMDAR-mediated currents (Offenhäuser et al., 2006). Furthermore, LTP induction is associated with long-lasting increase of F-actin contents in the activated spines, and this increase of F-actin is dependent on the activation of NMDARs and the inactivation of actin depolymerizing factor cofilin (Fukazawa et al., 2003). In the present study, we noted reduced F-actin/G-actin ratio and tendency of cofilin to increase in the CA3 of CA3-GluN2B-KO mice, without showing any overt changes in expression levels and phosphorylation state of other actin-binding and -regulating molecules. The mechanism of how the actin cytoskeleton regulates NMDAR activity and vice versa is currently unknown. In this regard, a mechanosensitive nature of NMDARs seems interesting; NMDAR-mediated currents alter by changing the osmolarity of external solution and the pressure of patch pipettes (Paoletti and Ascher, 1994). From these findings, it is conceivable that GluN2B-containing NMDARs not only regulate structural and molecular organization in the postsynapse and may also potentiate NMDAR function, presumably, through modulating the integrity of F-actin in dendritic spines.
Some of our present findings need to be discussed in relation to previous notions and findings. First, previous electrophysiological studies using cultured neurons and cortical slices reported that GluN2B-containing NMDARs are mostly distributed on the extrasynaptic site, whereas GluN2A-containing NMDARs are localized on the synaptic site (Stocca and Vicini, 1998; Tovar and Westbrook, 1999). However, we observed by postembedding immunogold microscopy that both GluN2A and GluN2B subunits are accumulated at synaptic sites (Fig. 5). Our observation is rather consistent with recent studies in the hippocampus using electrophysiological recording (Thomas et al., 2006; Harris and Pettit, 2007; Miwa et al., 2008) and GluN2 antibody-mediated live imaging (Groc et al., 2006). Second, using organotypic hippocampal slices transfected with electrophysiologically tagged GluN1, Barria and Malinow (2002) have shown that GluN2A-containing receptors replace synaptic GluN2B-containing NMDARs in a ligand binding-dependent manner. This model of use-dependent subunit switching is consistent with developmental and use-dependent increases of GluN2A-containing NMDARs (Quinlan et al., 1999, 2004), developmental reduction and shortening of synaptic NMDAR currents (Carmignoto and Vicini, 1992; Hestrin, 1992), and reduced plasticity as animals age (Crair and Malenka, 1995; Feldman et al., 1998). In the present study, we found that GluN2A and GluN1 subunits are present at synapses of CA3-GluN2B-KO mice (Fig. 5), suggesting that GluN2A-containing NMDARs can be recruited to synapses in the absence of preexisting GluN2B-containing ones. Third, in contrast to almost complete functional loss of NMDARs at CA3 synapses in CA3-GluN2B-KO mice, NMDARs lacking GluN2B subunit are known to be functional at autapses of cultured hippocampal neurons prepared from GluN2B-null mutant mice (Tovar et al., 2000). GluN2A/GluN1 receptors are also active at cerebellar mossy fiber–granule cell synapses of GluN2C-null mutant mice (Kadotani et al., 1996) (our unpublished observations). Furthermore, we found different effects of GluN2B ablation between synaptic and extrasynaptic NMDARs (Figs. 4, 6). Fourth, against mild reduction in the present study, the spine density in CA3 pyramidal cells has been reported to be comparable in GluN1-deficient mice (Fukushima et al., 2009). Although the reason for this discrepancy is uncertain, the two studies differ in that Fukushima et al. (2009) analyzed basal dendrites, whereas we analyzed apical dendrites. Considering that GluN2B subunit is differentially allocated between apical and basal dendrites of single pyramidal cells (Kawakami et al., 2003; Wu et al., 2005; Shinohara et al., 2008), the role of GluN2B subunit for dendritic spine formation or maintenance could be different according to its asymmetrical allocation.
In conclusion, the present study has shed light on functional importance of GluN2B subunit in NMDAR channel function, organization of postsynaptic macromolecular complexes, dendritic spine formation or maintenance, and regulation of the actin cytoskeleton. Mechanisms of these novel roles need to be clarified in future studies using pertinent animal and cellular models.
Footnotes
This study was supported in part by Grants-in-Aid for Scientific Research (M.W., K.S.) and for Scientific Research on Priority Areas (M.W., K.S.) from the Ministry of Education, Culture, Sports and Technology, Japan, and by Grant for Promotion of Niigata University Research Projects (K.A., M.A.). This study was also supported in part by the Uehara Foundation and Akiyama Foundation (M.W.). We thank Estuko Kushiya and Yuriko Ishikawa for technical assistance.
References
- Abe M, Fukaya M, Yagi T, Mishina M, Watanabe M, Sakimura K. NMDA receptor GluRε/NR2 subunits are essential for postsynaptic localization and protein stability of GluRζ1/NR1 subunit. J Neurosci. 2004;24:7292–7304. doi: 10.1523/JNEUROSCI.1261-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison DW, Gelfand VI, Spector I, Craig AM. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci. 1998;18:2423–2436. doi: 10.1523/JNEUROSCI.18-07-02423.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison DW, Chervin AS, Gelfand VI, Craig AM. Postsynaptic scaffolds of excitatory and inhibitory synapses in hippocampal neurons: maintenance of core components independent of actin filaments and microtubules. J Neurosci. 2000;20:4545–4554. doi: 10.1523/JNEUROSCI.20-12-04545.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki C, Sekino Y, Hanamura K, Fujisawa S, Mahadomrongkul V, Ren Y, Shirao T. Drebrin A is a postsynaptic protein that localizes in vivo to the submembranous surface of dendritic sites forming excitatory synapses. J Comp Neurol. 2005;486:384–402. doi: 10.1002/cne.20449. [DOI] [PubMed] [Google Scholar]
- Araki K, Araki M, Miyazaki J, Vassalli P. Site-specific recombination of a transgene in fertilized eggs by transient expression of Cre recombinase. Proc Natl Acad Sci U S A. 1995;92:160–164. doi: 10.1073/pnas.92.1.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–353. doi: 10.1016/s0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- Bassand P, Bernard A, Rafiki A, Gayet D, Khrestchatisky M. Differential interaction of the tSXV motifs of the NR1 and NR2A NMDA receptor subunits with PSD-95 and SAP97. Eur J Neurosci. 1999;11:2031–2043. doi: 10.1046/j.1460-9568.1999.00611.x. [DOI] [PubMed] [Google Scholar]
- Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Böckers TM, Mameza MG, Kreutz MR, Bockmann J, Weise C, Buck F, Richter D, Gundelfinger ED, Kreienkamp HJ. Synaptic scaffolding protein in rat brain. Ankyrin repeats of the multidomain Shank protein family interact with the cytoskeletal protein alpha-fodrin. J Biol Chem. 2001;276:40104–40112. doi: 10.1074/jbc.M102454200. [DOI] [PubMed] [Google Scholar]
- Capani F, Martone ME, Deerinck TJ, Ellisman MH. Selective localization of high concentrations of F-actin in subpopulations of dendritic spines in rat central nervous system: a three-dimensional electron microscopic study. J Comp Neurol. 2001;435:156–170. doi: 10.1002/cne.1199. [DOI] [PubMed] [Google Scholar]
- Carlin RK, Grab DJ, Cohen RS, Siekevitz P. Isolation and characterization of postsynaptic densities from various brain regions: enrichment of different types of postsynaptic densities. J Cell Biol. 1980;86:831–845. doi: 10.1083/jcb.86.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmignoto G, Vicini S. Activity-dependent decrease in NMDA receptor responses during development of the visual cortex. Science. 1992;258:1007–1011. doi: 10.1126/science.1279803. [DOI] [PubMed] [Google Scholar]
- Crair MC, Malenka RC. A critical period for long-term potentiation at thalamocortical synapses. Nature. 1995;375:325–328. doi: 10.1038/375325a0. [DOI] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–335. doi: 10.1016/s0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- Feldman DE, Nicoll RA, Malenka RC, Isaac JT. Long-term depression at thalamocortical synapses in developing rat somatosensory cortex. Neuron. 1998;21:347–357. doi: 10.1016/s0896-6273(00)80544-9. [DOI] [PubMed] [Google Scholar]
- Fukaya M, Watanabe M. Improved immunohistochemical detection of postsynaptically located PSD-95/SAP 90 protein family by protease section pretreatment: a study in the adult mouse brain. J Comp Neurol. 2000;426:572–586. [PubMed] [Google Scholar]
- Fukaya M, Kato A, Lovett C, Tonegawa S, Watanabe M. Retention of NMDA receptor NR2 subunits in the lumen of endoplasmic reticulum in targeted NR1 knockout mice. Proc Natl Acad Sci U S A. 2003;100:4855–4860. doi: 10.1073/pnas.0830996100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- Fukushima F, Nakao K, Shinoe T, Fukaya M, Muramatsu S, Sakimura K, Kataoka H, Mori H, Watanabe M, Manabe T, Mishina M. Ablation of NMDA receptors enhances the excitability of hippocampal CA3 neurons. PLoS One. 2009;4:e3993. doi: 10.1371/journal.pone.0003993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Heine M, Cousins SL, Stephenson FA, Lounis B, Cognet L, Choquet D. NMDA receptor surface mobility depends on NR2A–2B subunits. Proc Natl Acad Sci U S A. 2006;103:18769–18774. doi: 10.1073/pnas.0605238103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AZ, Pettit DL. Extrasynaptic and synaptic NMDA receptors form stable and uniform pools in rat hippocampal slices. J Physiol. 2007;584:509–519. doi: 10.1113/jphysiol.2007.137679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris EW, Cotman CW. Long-term potentiation of guinea pig mossy fiber responses is not blocked by N-methyl-d-aspartate antagonists. Neurosci Lett. 1986;70:132–137. doi: 10.1016/0304-3940(86)90451-9. [DOI] [PubMed] [Google Scholar]
- Hering H, Sheng M. Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci. 2003;23:11759–11769. doi: 10.1523/JNEUROSCI.23-37-11759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hestrin S. Developmental regulation of NMDA receptor-mediated synaptic currents at a central synapse. Nature. 1992;357:686–689. doi: 10.1038/357686a0. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Boulter J, Maron C, Beasley L, Sullivan J, Pecht G, Heinemann S. Zinc potentiates agonist-induced currents at certain splice variants of the NMDA receptor. Neuron. 1993;10:943–954. doi: 10.1016/0896-6273(93)90209-a. [DOI] [PubMed] [Google Scholar]
- Ito I, Futai K, Katagiri H, Watanabe M, Sakimura K, Mishina M, Sugiyama H. Synapse-selective impairment of NMDA receptor functions in mice lacking NMDA receptor ε1 or ε2 subunit. J Physiol. 1997;500:401–408. doi: 10.1113/jphysiol.1997.sp022030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knöpfel T, Erzurumlu RS, Itohara S. Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature. 2000;406:726–731. doi: 10.1038/35021059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadotani H, Hirano T, Masugi M, Nakamura K, Nakao K, Katsuki M, Nakanishi S. Motor discoordination results from combined gene disruption of the NMDA receptor NR2A and NR2C subunits, but not from single disruption of the NR2A or NR2C subunit. J Neurosci. 1996;16:7859–7867. doi: 10.1523/JNEUROSCI.16-24-07859.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami R, Shinohara Y, Kato Y, Sugiyama H, Shigemoto R, Ito I. Asymmetrical allocation of NMDA receptor ε2 subunits in hippocampal circuitry. Science. 2003;300:990–994. doi: 10.1126/science.1082609. [DOI] [PubMed] [Google Scholar]
- Kim E, Cho KO, Rothschild A, Sheng M. Heteromultimerization and NMDA receptor-clustering activity of Chapsyn-110, a member of the PSD-95 family of proteins. Neuron. 1996;17:103–113. doi: 10.1016/s0896-6273(00)80284-6. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Kitayama K, Abe M, Kakizaki T, Honma D, Natsume R, Fukaya M, Watanabe M, Miyazaki J, Mishina M, Sakimura K. Purkinje cell-specific and inducible gene recombination system generated from C57BL/6 mouse ES cells. Biochem Biophys Res Commun. 2001;281:1134–1140. doi: 10.1006/bbrc.2001.4492. [DOI] [PubMed] [Google Scholar]
- Kiyama Y, Manabe T, Sakimura K, Kawakami F, Mori H, Mishina M. Increased thresholds for long-term potentiation and contextual learning in mice lacking the NMDA-type glutamate receptor ε1 subunit. J Neurosci. 1998;18:6704–6712. doi: 10.1523/JNEUROSCI.18-17-06704.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro H, Rakic P. Modulation of neuronal migration by NMDA receptors. Science. 1993;260:95–97. doi: 10.1126/science.8096653. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Kuriu T, Inoue A, Bito H, Sobue K, Okabe S. Differential control of postsynaptic density scaffolds via actin-dependent and -independent mechanisms. J Neurosci. 2006;26:7693–7706. doi: 10.1523/JNEUROSCI.0522-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsuwada T, Kashiwabuchi N, Mori H, Sakimura K, Kushiya E, Araki K, Meguro H, Masaki H, Kumanishi T, Arakawa M, Mishina M. Molecular diversity of the NMDA receptor channel. Nature. 1992;358:36–41. doi: 10.1038/358036a0. [DOI] [PubMed] [Google Scholar]
- Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E, Natsume R, Watanabe M, Inoue Y, Yagi T, Aizawa S, Arakawa M, Takahashi T, Nakamura Y, Mori H, Mishina M. Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor ε2 subunit mutant mice. Neuron. 1996;16:333–344. doi: 10.1016/s0896-6273(00)80051-3. [DOI] [PubMed] [Google Scholar]
- Li Y, Erzurumlu RS, Chen C, Jhaveri S, Tonegawa S. Whisker-related neuronal patterns fail to develop in the trigeminal brainstem nuclei of NMDAR1 knockout mice. Cell. 1994;76:427–437. doi: 10.1016/0092-8674(94)90108-2. [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Liu XB, Murray KD, Jones EG. Switching of NMDA receptor 2A and 2B subunits at thalamic and cortical synapses during early postnatal development. J Neurosci. 2004;24:8885–8895. doi: 10.1523/JNEUROSCI.2476-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Lu HC, Gonzalez E, Crair MC. Barrel cortex critical period plasticity is independent of changes in NMDA receptor subunit composition. Neuron. 2001;32:619–634. doi: 10.1016/s0896-6273(01)00501-3. [DOI] [PubMed] [Google Scholar]
- MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- Matus A, Ackermann M, Pehling G, Byers HR, Fujiwara K. High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sci U S A. 1982;79:7590–7594. doi: 10.1073/pnas.79.23.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- Meguro H, Mori H, Araki K, Kushiya E, Kutsuwada T, Yamazaki M, Kumanishi T, Arakawa M, Sakimura K, Mishina M. Functional characterization of a heteromeric NMDA receptor channel expressed from cloned cDNAs. Nature. 1992;357:70–74. doi: 10.1038/357070a0. [DOI] [PubMed] [Google Scholar]
- Miwa H, Fukaya M, Watabe AM, Watanabe M, Manabe T. Functional contributions of synaptically localized NR2B subunits of the NMDA receptor to synaptic transmission and long-term potentiation in the adult mouse CNS. J Physiol. 2008;586:2539–2550. doi: 10.1113/jphysiol.2007.147652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima S, Nagata S. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Mori H, Mishina M. Structure and function of the NMDA receptor channel. Neuropharmacology. 1995;34:1219–1237. doi: 10.1016/0028-3908(95)00109-j. [DOI] [PubMed] [Google Scholar]
- Mori H, Masaki H, Yamakura T, Mishina M. Identification by mutagenesis of a Mg2+-block site of the NMDA receptor channel. Nature. 1992;358:673–675. doi: 10.1038/358673a0. [DOI] [PubMed] [Google Scholar]
- Mori H, Yamakura T, Masaki H, Mishina M. Involvement of the carboxyl-terminal region in modulation by TPA of the NMDA receptor channel. Neuroreport. 1993;4:519–522. doi: 10.1097/00001756-199305000-00014. [DOI] [PubMed] [Google Scholar]
- Mori H, Manabe T, Watanabe M, Satoh Y, Suzuki N, Toki S, Nakamura K, Yagi T, Kushiya E, Takahashi T, Inoue Y, Sakimura K, Mishina M. Role of the carboxy-terminal region of the GluRε2 subunit in synaptic localization of the NMDA receptor channel. Neuron. 1998;21:571–580. doi: 10.1016/s0896-6273(00)80567-x. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Watanabe M, Isobe T, Kondo H, Inoue Y. Cytological compartmentalization in the staggerer cerebellum, as revealed by calbindin immunohistochemistry for Purkinje cells. J Comp Neurol. 1998;395:112–120. doi: 10.1002/(sici)1096-9861(19980525)395:1<112::aid-cne8>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Engler JA, Sheng M. The dynamic turnover and functional roles of α-actinin in dendritic spines. Neuropharmacology. 2004;47:734–745. doi: 10.1016/j.neuropharm.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Sato K, Fukaya M, Araishi K, Aiba A, Kano M, Watanabe M. Signaling complex formation of phospholipase Cβ4 with metabotropic glutamate receptor type 1α and 1,4,5-trisphosphate receptor at the perisynapse and endoplasmic reticulum in the mouse brain. Eur J Neurosci. 2004;20:2929–2944. doi: 10.1111/j.1460-9568.2004.03768.x. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, Masu M, Bessho Y, Nakajima Y, Hayashi Y, Shigemoto R. Molecular diversity of glutamate receptors and their physiological functions. Experientia Suppl. 1994;71:71–80. doi: 10.1007/978-3-0348-7330-7_8. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Kim E, Sheng M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci. 1996;16:2157–2163. doi: 10.1523/JNEUROSCI.16-07-02157.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Offenhäuser N, Castelletti D, Mapelli L, Soppo BE, Regondi MC, Rossi P, D'Angelo E, Frassoni C, Amadeo A, Tocchetti A, Pozzi B, Disanza A, Guarnieri D, Betsholtz C, Scita G, Heberlein U, Di Fiore PP. Increased ethanol resistance and consumption in Eps8 knockout mice correlates with altered actin dynamics. Cell. 2006;127:213–226. doi: 10.1016/j.cell.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Narayanan R, Lee SH, Murata K, Hayashi Y. The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc Natl Acad Sci U S A. 2007;104:6418–6423. doi: 10.1073/pnas.0701656104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima S, Fukaya M, Masabumi N, Shirakawa T, Oguchi H, Watanabe M. Early onset of NMDA receptor GluRε1 (NR2A) expression and its abundant postsynaptic lacalization in developing motoneurons of the mouse hypoglossal neucleus. Neurosci Res. 2002;43:239–250. doi: 10.1016/s0168-0102(02)00035-4. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Ascher P. Mechanosensitivity of NMDA receptors in cultured mouse central neurons. Neuron. 1994;13:645–655. doi: 10.1016/0896-6273(94)90032-9. [DOI] [PubMed] [Google Scholar]
- Popescu G, Auerbach A. Modal gating of NMDA receptors and the shape of their synaptic response. Nat Neurosci. 2003;6:476–483. doi: 10.1038/nn1044. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci. 1999;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Lebel D, Brosh I, Barkai E. A molecular mechanism for stabilization of learning-induced synaptic modifications. Neuron. 2004;41:185–192. doi: 10.1016/s0896-6273(03)00874-2. [DOI] [PubMed] [Google Scholar]
- Racz B, Weinberg RJ. The subcellular organization of Cortactin in hippocampus. J Neurosci. 2004;24:10310–10317. doi: 10.1523/JNEUROSCI.2080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ, Bartlett RK, Bass MA, Colbran RJ. Differential modulation of Ca2+/calmodulin-dependent protein kinase II activity by regulated interactions with N-methyl-d-aspartate receptor NR2B subunits and α-actinin. J Biol Chem. 2005;280:39316–39323. doi: 10.1074/jbc.M508189200. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Westbrook GL. Calcium-induced actin depolymerization reduces NMDA channel activity. Neuron. 1993;10:805–814. doi: 10.1016/0896-6273(93)90197-y. [DOI] [PubMed] [Google Scholar]
- Sakimura K, Yoshida Y, Nabeshima Y, Takahashi Y. Biosynthesis of the brain-specific 14-3-2 protein in a cell-free system from wheat germ extract directed with poly(A)-containing RNA from rat brain. J Neurochem. 1980;34:687–693. doi: 10.1111/j.1471-4159.1980.tb11198.x. [DOI] [PubMed] [Google Scholar]
- Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S, Inoue Y, Sugiyama H, Mishina M. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor ε1 subunit. Nature. 1995;373:151–155. doi: 10.1038/373151a0. [DOI] [PubMed] [Google Scholar]
- Seeburg PH. The molecular biology of mammalian glutamate receptor channels. Trends Neurosci. 1993;16:359–365. doi: 10.1016/0166-2236(93)90093-2. [DOI] [PubMed] [Google Scholar]
- Shimuta M, Yoshikawa M, Fukaya M, Watanabe M, Takeshima H, Manabe T. Postsynaptic modulation of AMPA receptor-mediated synaptic responses and LTP by the type 3 ryanodine receptor. Mol Cell Neurosci. 2001;17:921–930. doi: 10.1006/mcne.2001.0981. [DOI] [PubMed] [Google Scholar]
- Shinohara Y, Hirase H, Watanabe M, Itakura M, Takahashi M, Shigemoto R. Left-right asymmetry of the hippocampal synapses with different subunit allocation of glutamate receptors. Proc Natl Acad Sci U S A. 2008;105:19498–19503. doi: 10.1073/pnas.0807461105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern P, Cik M, Colquhoun D, Stephenson FA. Single channel properties of cloned NMDA receptors in a human cell line: comparison with results from Xenopus oocytes. J Physiol. 1994;476:391–397. doi: 10.1113/jphysiol.1994.sp020140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol. 1998;507:13–24. doi: 10.1111/j.1469-7793.1998.013bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugihara H, Moriyoshi K, Ishii T, Masu M, Nakanishi S. Structures and properties of seven isoforms of the NMDA receptor generated by alternative splicing. Biochem Biophys Res Commun. 1992;185:826–832. doi: 10.1016/0006-291x(92)91701-q. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Nomura T, Tsujita M, Suzuki M, Fuse T, Mori H, Mishina M. Flp recombinase transgenic mice of C57BL/6 strain for conditional gene targeting. Biochem Biophys Res Commun. 2002;293:953–957. doi: 10.1016/S0006-291X(02)00321-2. [DOI] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Thomas CG, Miller AJ, Westbrook GL. Synaptic and extrasynaptic NMDA receptor NR2 subunits in cultured hippocampal neurons. J Neurophysiol. 2006;98:1727–1734. doi: 10.1152/jn.00771.2005. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Sprouffske K, Westbrook GL. Fast NMDA receptor-mediated synaptic currents in neurons from mice lacking the ε2 (NR2B) subunit. J Neurophysiol. 2000;83:616–620. doi: 10.1152/jn.2000.83.1.616. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Tsujita M, Mori H, Watanabe M, Suzuki M, Miyazaki J, Mishina M. Cerebellar granule cell-specific and inducible expression of Cre recombinase in the mouse. J Neurosci. 1999;19:10318–10323. doi: 10.1523/JNEUROSCI.19-23-10318.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Inoue Y, Sakimura K, Mishina M. Developmental changes in distribution of NMDA receptor channel subunit mRNAs. Neuroreport. 1992;3:1138–1140. doi: 10.1097/00001756-199212000-00027. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Inoue Y, Sakimura K, Mishina M. Distinct distributions of five N-methyl-d-aspartate receptor channel subunit mRNAs in the forebrain. J Comp Neurol. 1993;338:377–390. doi: 10.1002/cne.903380305. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Fukaya M, Sakimura K, Manabe T, Mishina M, Inoue Y. Selective scarcity of NMDA receptor channel subunits in the stratum lucidum (mossy fibre-recipient layer) of the mouse hippocampal CA3 subfield. Eur J Neurosci. 1998;10:478–487. doi: 10.1046/j.1460-9568.1998.00063.x. [DOI] [PubMed] [Google Scholar]
- Werner P, Voigt M, Keinänen K, Wisden W, Seeburg PH. Cloning of a putative high-affinity kainate receptor expressed predominantly in hippocampal CA3 cells. Nature. 1991;351:742–744. doi: 10.1038/351742a0. [DOI] [PubMed] [Google Scholar]
- Wu Y, Kawakami R, Shinohara Y, Fukaya M, Sakimura K, Mishina M, Watanabe M, Ito I, Shigemoto R. Target-cell-specific left-right asymmetry of NMDA receptor content in Schaffer collateral synapses in ε1/NR2A knock-out mice. J Neurosci. 2005;25:9213–9226. doi: 10.1523/JNEUROSCI.2134-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie DJ, Béhé P, Colquhoun D. Single-channel activations and concentration jumps: comparison of recombinant NR1a/NR2A and NR1a/NR2D NMDA receptors. J Physiol. 1998;510:1–18. doi: 10.1111/j.1469-7793.1998.001bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M. Competitive binding of α-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Kharazia V, Shanghvi R, Rao A, Beggs AH, Craig AM, Weinberg R, Sheng M. Differential regional expression and ultrastructural localization of α-Actinin-2, a putative NMDA receptor-anchoring protein, in rat brain. J Neurosci. 1998;18:1383–1392. doi: 10.1523/JNEUROSCI.18-04-01383.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Fukaya M, Shimizu H, Sakimura K, Watanabe M. NMDA receptor subunits GluRε1, GluRε3 and GluRζ1 are enriched at the mossy fibre-granule cell synapse in the adult mouse cerebellum. Eur J Neurosci. 2001;13:2025–2036. doi: 10.1046/j.0953-816x.2001.01580.x. [DOI] [PubMed] [Google Scholar]
- Yang SN, Tang YG, Zucker RS. Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. J Neurophysiol. 1999;81:781–787. doi: 10.1152/jn.1999.81.2.781. [DOI] [PubMed] [Google Scholar]
- Zalutsky RA, Nicoll RA. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science. 1990;248:1619–1624. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Lee YS, Wu LJ, Ko SW, Zhang XH, Jia Y, Shum F, Xu H, Li BM, Kaang BK, Zhuo M. Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron. 2005;47:859–872. doi: 10.1016/j.neuron.2005.08.014. [DOI] [PubMed] [Google Scholar]