

Abstract
Paraplegin and AFG3L2 are ubiquitous nuclear-encoded mitochondrial proteins that form hetero-oligomeric paraplegin-AFG3L2 and homo-oligomeric AFG3L2 complexes in the inner mitochondrial membrane, named m-AAA proteases. These complexes ensure protein quality control in the inner membrane, jointly with a chaperone-like activity on the respiratory chain complexes. Despite coassembling in the same complex, mutations of either paraplegin or AFG3L2 cause two different neurodegenerative disorders. Indeed, mutations of paraplegin are responsible for a recessive form of hereditary spastic paraplegia, whereas mutations of AFG3L2 have been recently associated to a dominant form of spinocerebellar ataxia (SCA28). In this work, we report that the mouse model haploinsufficient for Afg3l2 recapitulates important pathophysiological features of the human disease, thus representing the first SCA28 model. Furthermore, we propose a pathogenetic mechanism in which respiratory chain dysfunction and increased reactive oxygen species production caused by Afg3l2 haploinsufficiency lead to dark degeneration of Purkinje cells and cerebellar dysfunction.
Introduction
Mitochondria are the primary energy-generating system in most eukaryotic cells. Additionally, they play a crucial role in regulating calcium homeostasis and are integrated in a number of signaling pathways, including cell death cascades (McBride et al., 2006). Neuronal health relies heavily on mitochondrial functionality and integrity (Chang and Reynolds, 2006). It is therefore not surprising that alteration of mitochondrial physiology is linked to several neurodegenerative disorders and aging (Kwong et al., 2006). Many of these diseases are caused by mutations of nuclear genes encoding proteins residing in mitochondria (Schapira, 2006). Among them, loss-of-function mutations of the paraplegin-coding gene lead to a recessive form of hereditary spastic paraplegia (HSP), characterized by degeneration of the longest motor and sensory axons of the CNS (Casari et al., 1998).
Paraplegin belongs to the AAA-protease superfamily (ATPases associated with a variety of cellular activities). By assembling with the homologous protein AFG3L2, paraplegin forms the m-AAA complex in the inner membrane, which takes part of an evolutionarily conserved proteolytic system mediating the complete degradation of organellar proteins (Leonhard et al., 1999; Koppen and Langer, 2007). The m-AAA protease can also exert chaperone-like activity, being required for the assembly of the respiratory chain complexes (Arlt et al., 1998; Atorino et al., 2003; Maltecca et al., 2008), and mediates the proteolytic activation of substrates (Nolden et al., 2005). Differently from human yeast and Caenorhabditis elegans, a third homolog, AFG3L1, is present in the mouse. Paraplegin, AFG3L2, and AFG3L1 can form mixed-composition oligomers, but only AFG3L1 and AFG3L2 have the ability to form functional high-molecular-weight homo-complexes (Koppen et al., 2007). Thus, a variety of m-AAA protease isoforms can be formed in mammalian and especially in murine mitochondria. Moreover, considering that paraplegin, AFG3L2, and AFG3L1 are differentially expressed in different tissues, mammalian m-AAA protease shows a versatile assembly, as it varies its subunit composition in different tissues according to subunit availability (Koppen et al., 2007).
We previously demonstrated that paraplegin and Afg3l2 loss-of-function mutations result in strikingly different outcomes in the mouse. Indeed, the paraplegin null model displays a late-onset axonal degeneration, whereas Afg3l2 homozygous mutants show impaired axonal development and delayed myelination, together with widespread neuropathological alterations in both the CNS and PNS. Moreover, loss of Afg3l2 strongly affects cerebellum, which is spared in the paraplegin mouse model (Ferreirinha et al., 2004; Maltecca et al., 2008). These observations indicate that different neuronal populations have a different threshold of sensitivity to the amount and composition of m-AAA complexes. To address this possibility, we investigated the gene-dosage effect of the m-AAA complex by characterizing a mouse model haploinsufficient for Afg3l2 (Afg3l2+/Emv66 heterozygous mouse) (Maltecca et al., 2008). In this work, we demonstrate that cerebellum is the neuronal tissue with the highest susceptibility to reduced Afg3l2 dosage. Indeed, Afg3l2+/Emv66 mice show a progressive deficit in motor coordination and balance associated with mitochondrial dysfunction and Purkinje cell (PC) dark degeneration. Interestingly, AFG3L2 mutations have been recently associated with spinocerebellar ataxia type 28 (SCA28) (Cagnoli et al., 2006, 2008; DiBella et al., 2008). We thus propose the Afg3l2 heterozygous mouse as the excellent model to unravel the pathological cascade leading to this form of SCA.
Materials and Methods
Murine strains.
Procedures involving animals and their care were conducted in conformity with guidelines of the Institutional Animal Care and Use Committee at San Raffaele Hospital (Milan, Italy) in compliance with national (D.L. No. 116, G.U. Suppl. 40, Feb. 18, 1992, Circolare No. 8, G.U., 14 Lug. 1994) and international (EEC Council Directive 86/609, OJ L 358, 1 DEC.12, 1987; National Institutes of Health Guide for the Care and Use of Laboratory Animals, U.S. National Research Council, 1996) laws and policies.
Antibodies.
Rabbit polyclonal antibodies directed against AFG3L2 were generated previously (Atorino et al., 2003). For Western blot analysis, commercially available monoclonal antibodies were used for the detection of HSP60 (Nventa Biopharmaceuticals), respiratory chain complex I 39 kDa subunit, and respiratory complex III 25.6 kDa subunit (Invitrogen). A rabbit polyclonal antibody was used for the detection of dinitrophenyl hydrazine (DNPH; Invitrogen). ECL anti-mouse and anti-rabbit IgG and horseradish peroxidase (HRP)-linked species-specific whole antibodies were purchased from GE Healthcare. Polyclonal rabbit anti-goat Igs/HRP were obtained from Dako Denmark A/S. For immunofluorescence experiments, the following antibodies were used: goat anti-GFAP (Santa Cruz Biotechnology) and mouse anti-Calbindin D-28K (Sigma). Secondary antibodies were conjugated with Alexa 488 and Alexa 596 (Invitrogen). For the immunoperoxidase procedure, a secondary biotinylated antiserum (goat anti-rabbit for the polyclonal antibody, goat anti-mouse for the monoclonal antibody; Vector Laboratories) was used.
Behavioral analysis.
We performed a modified version of the SHIRPA protocol primary screening developed by Irwin (Irwin et al., 1968). A score was assigned to each behavioral test (supplemental material, available at www.jneurosci.org). To test clasping response, mice were suspended by the tail above an open cage for 30 s for 10 trials. Each mouse was classified as tending to clasp if, during any of its 10 trials, the mouse stopped struggling and held its front paws together near its torso. Usually once a mutant mouse clasped, it would continue for the duration of the trial (Garden et al., 2002). Motor ability was assessed by rotarod analysis (instrument; Ugo Basile). Briefly, Afg3l2+/Emv66 and control mice were tested in two sessions of three trials each per day (6 h rest between the two daily sessions) for 3 consecutive days. The first day of the test was considered training. Data are reported for the second and third days. During the test, the rod accelerated from 4 to 40 rotations per minute, and the time that the animal remained on the rod (maximum, 600 s) was measured. In the beam-walking analysis, mice were trained to walk from a start platform along a balance beam (1 cm wide × 90 cm long) suspended 30 cm above bedding. Two trials were performed for each mouse, and the number of hindfoot missteps was counted while the mouse walked along the length of the beam. The time required to cross the beam from start to end (latency) was also evaluated. The better of two trials was scored. Mice were trained to walk the length of the beam for 3 d before testing (Carter et al., 1999).
Morphological analysis.
Mice were anesthetized with Avertin (2–2-2 tribromoethanol; 0.75 mg/g, i.p.; Fluka) and perfused transcardially with 20 ml of saline and subsequently with 50 ml of sodium phosphate-buffered 4% paraformaldehyde solution. The cerebellum and spinal cord were removed rapidly, postfixed for 2 h with sodium phosphate-buffered 4% paraformaldehyde solution, transferred to 20% sucrose solution in PBS overnight and then to 30% sucrose solution until they sank, and finally frozen in 2-methylbutane at −45°C. Sections (20 μm thick) were cryostat cut (Leica) at −20°C. To avoid structural variability, all the experiments described in this work were conducted on the primary fissure of cerebellum. Quantification of molecular layer (ML) thickness was performed analyzing an average of three nonoverlapping fields for each mouse (n = 3). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive nuclei were quantified as the percentage of TUNEL-positive nuclei relative to 4′,6-diamidino-2-phenylindole (DAPI)-stained nuclei total count per field. An average of five nonoverlapping fields for each mouse (n = 4) were considered. Lumbar spinal cord was cut in the transverse plane at the L2–L3 level. For immunohistochemical detection of protein carbonyls, cerebella were removed rapidly, fixed overnight in methacarn (methanol:chloroform:acetic acid, 60:30:10), and mounted in paraffin (Smerjac and Bizzozero, 2008). Tissues were sectioned in the sagittal plane (10 μm thick). Immunofluorescence on tissue sections was performed as described previously (Veglianese et al., 2006). They were blocked with 10% donkey serum and 0.4% Triton X-100 in 1× PBS, incubated overnight at 4°C with the primary antibody, washed in 1× PBS, and incubated with the appropriate Alexa488/Alexa596-conjugated donkey secondary antibodies.
For the immunoperoxidase procedure, tissue sections were treated with 1% H2O2 in PBS to inhibit endogenous peroxidase, blocked in 10% goat serum in PBS containing 0.1% Triton X-100 for 60 min, and incubated overnight with the primary antibodies diluted in PBS containing 3% goat serum. Immune reactions were revealed by a 60 min incubation in the appropriate secondary biotinylated antiserum (1:200), followed by a 60 min incubation in the avidin–biotin–peroxidase complex (Vector Laboratories) using diaminobenzidine as the chromogen. Control sections processed with omission of the primary antiserum and developed under the same conditions gave no immunostaining.
Semithin and ultrathin sections were conducted as described previously (Quattrini et al., 1996; Previtali et al., 2000). Tissues were removed, postfixed with 2% glutaraldehyde in 0.12 m phosphate buffer, sectioned into 2 mm blocks, postfixed with 1% osmium tetroxide, and embedded in Epon (Fluka). Semithin sections (0.5–1 μm thick) were stained with toluidine blue and examined by light microscopy (Olympus BX51). Counting of PCs was performed in a blinded and nonbiased manner on an average of 12 nonoverlapping microscopic fields per mouse (n = 3). Ultrathin sections (100–120 nm thick) were stained with uranile acetate and lead citrate and examined by electron microscopy (EM) (magnification, 3000×; Leo 912 Omega).
TUNEL assay.
TUNEL assay for detection of apoptotic nuclei was performed on cryostat tissue sections using the In situ cell death detection kit, TMR red kit (Roche), following manufacturer's instructions.
Mitochondria isolation.
Mitochondria were isolated by differential centrifugation from different tissue homogenates (Robinson, 1996). In brief, tissues were homogenized in an appropriate isotonic buffer (0.25 m sucrose, 20 mm 3-(N-morpholino) propanesulfonic acid (MOPS), pH 7.2, 1 mm EDTA, 0.1% BSA fatty acid free) using a glass-Teflon homogenizer. Cell debris and nuclei were pelleted by centrifugation at 2500 × g for 5 min at 4°C. Supernatants were centrifuged at 12,000 × g for 25 min at 4°C, and the mitochondrial pellet was resuspended in an isotonic buffer (0.5 m sucrose, 20 mm MOPS, pH 7.2, 1 mm EDTA).
Protein concentration was measured using the Bio-Rad Protein Assay according to the manufacturer's instructions.
Respiratory chain activity and luminometric assay of ATP.
Tests for respiratory chain defects were performed as described previously (Maltecca et al., 2008). Results are expressed as nanomoles of ATP per milligram of protein. Data, in triplicates, are indicated as the mean of four independent experiments ± SD. Three mice per genotype were analyzed at 4, 6, and 12 months. The two-tailed t test was applied for significance calculation.
Assembly of respiratory chain complexes.
The experiment was conducted as described previously (Maltecca et al., 2008). Briefly, isolated mitochondria were solubilized by dodecyl maltoside (2% final concentration) and centrifuged at 12,000 × g for 25 min at 4°C. Supernatants were loaded on a linear 5–13% gradient polyacrylamide gel. Gels were transblotted onto a nitrocellulose membrane and immunodecorated with anti-39 kDa and anti-core2 (25.6 kDa subunit) antibodies (Schagger et al., 1994). Band quantifications relative to Western blots were performed by densitometric analysis. Three mice per genotype were analyzed. The two-tailed t test was used for significance calculation.
Biochemical and histochemical detection of oxidized proteins.
Protein carbonyls were detected as described previously (Levine et al., 1990) with slight modifications. Tissues were homogenized in 0.44 m sucrose, 20 mm MOPS, pH 7.2, 1 mm EDTA, 1 mm PMSF, and 0.5% Triton X-100 using a glass-Teflon homogenizer. Cell debris was discarded by centrifugation at 8000 × g for 10 min at 4°C. Mitochondria were isolated by differential centrifugation as described above. Total lysate (50 μg) and isolated mitochondria (25 μg) were derivatized in an equal volume of DNPH (Fluka) dissolved in 10% trifluoroacetic acid (TFA). After neutralization with an equal volume of 2 m Tris and 30% glycerol, dinitrophenyl (DNP)-derivatized protein samples were mixed with an equal volume of 2× sample buffer (4% SDS, 20% glycerol, 4% 2-mercaptoethanol, 0.04% bromophenol blue, 120 mm Tris-HCl, pH 6.8) and resolved by SDS-PAGE on 10% gradient gels (15 μg protein/well). Carbonylated proteins were detected using an antibody against DNP moiety. For immunohistochemical detection of protein carbonyls, cerebellum sections were treated with 10 mm 2,4-dinitrophenyl-hydrazine prepared in 2N HCl for 60 min to convert the carbonyls group into DNP hydrazones. Sections were blocked with 10% normal goat serum (v/v) and incubated overnight with rabbit anti-DNPH (1:100) antibody. The reaction was revealed by the immunoperoxidase method. Quantitative analysis was performed counting the number of DNP-positive and -negative PCs in three nonoverlapping fields at 5× magnification to cover the entire cerebellar sections.
Statistical analyses.
For SHIRPA protocol primary screening, comparisons were performed with the Mann–Whitney nonparametric test. Repeated-measures ANOVA was performed for behavioral tests requiring repeated tests (clasping response, rotarod, and beam walking). A critical p < 0.05 was used for statistical significance in all analyses.
The two-tailed t test was used for significance calculation in morphometric analyses, respiratory chain activity assay, and densitometric analysis.
Results
We previously generated the Afg3l2 null model (Afg3l2Emv66/Emv66) by an ecotropic murine leukemia proviral reinsertion, which completely eliminates full-length expression of the Afg3l2 gene. Homozygous Afg3l2Emv66/Emv66 mice display an extremely severe neuromuscular syndrome that broadly affects central and peripheral neurons, leading to death at postnatal day 14 (Maltecca et al., 2008). Heterozygous Afg3l2+/Emv66 mice with a halved amount of AFG3L2 protein are phenotypically indistinguishable from wild type at this age. To investigate whether Afg3l2 haploinsufficiency could cause a late-onset neurodegeneration and to identify the neuronal tissues mainly sensitive to reduced AFG3L2 dosage, we performed a phenotypic characterization of adult Afg3l2 heterozygous mice.
Afg3l2 heterozygous mice display defects in motor coordination
Afg3l2+/Emv66 mice develop normally and show normal appearance and fertility. To have an overview of motor, sensory, autonomic, and neuropsychiatric functions, we performed SHIRPA protocol primary screening (Irwin et al., 1968; Rogers et al., 1997) on Afg3l2+/Emv66 mice at 4, 6, and 12 months as described in Materials and Methods. A full list of the scored parameters is summarized (supplemental Table 1, available at www.jneurosci.org). By comparing Afg3l2+/Emv66 mice and their wild-type littermates (Table 1), we detected alterations in gait, pelvic elevation, and negative-geotaxis reflex. The latter is a widely used test for the evaluation of cerebellar and labyrinth function (Hermans et al., 1993; Coluccia et al., 2004). These defects appear at 4 months and worsen with age, and mutants older than 12 months present abnormal gait characterized by uncoordinated hindlimb movement. Another sign of abnormality is clasping on tail suspension, beginning at 8–10 months of age (Fig. 1b, picture). By 12 months, almost all mutant animals show an abnormal clasping response (84.7% vs 17.8%; n = 20).
Table 1.
SHIRPA primary tests; SHIRPA primary screening is used to assess sensory–motor function of Afg3l2+/Emv66 and syngenic wild-type mice (n = 10) at 4, 6, and 12 months
| Behaviors | 4 months |
6 months |
12 months |
|||
|---|---|---|---|---|---|---|
| +/+ | +/Emv66 | +/+ | +/Emv66 | +/+ | +/Emv66 | |
| Negative geotaxis | 0.0 ± 0.0 | 0.9 ± 1.0** | 0.0 ± 0.0 | 1.1 ± 0.67** | 0.0 ± 0.0 | 1.67 ± 1.03* |
| Gait | 0.0 ± 0.0 | 0.4 + 0.5 | 0.0 ± 0.0 | 0.3 ± 0.5* | 0.0 ± 0.0 | 2 ± 0* |
| Transfer arousal | 5 ± 0.0 | 5 ± 0.0 | 5 ± 0.0 | 5 ± 0.0 | 5 ± 0.0 | 3.67 ± 1.03* |
| Pelvic elevation | 2 ± 0.0 | 2 ± 0.0 | 2 ± 0.0 | 2 ± 0.0 | 2 ± 0.0 | 1 ± 0.0* |
Data are expressed as mean ± SD (Mann–Whitney nonparametric test; *p < 0.05; **p < 0.001).
Figure 1.

Afg3l2+/Emv66 mice display motor impairment. a, Western blot using an antibody against AFG3L2 showing that AFG3L2 protein amount is halved in Afg3l2+/Emv66 mice compared with wild-type controls. HSP60 was used to verify equal loading. b, Afg3l2+/Emv66 mice display clasping phenotype after tail suspension. The wild-type mouse shows normal splaying after tail suspension, whereas the Afg3l2+/Emv66 mouse clasps. c, Performance of Afg3l2+/Emv66 and wild-type mice on an accelerating rotating rod apparatus at 2, 4, and 6 months. Afg3l2+/Emv66 animals showed impaired performance compared with wild-type, age-matched animals beginning at 4 months of age. Repeated-measures ANOVA confirmed a significant genotype effect (heterozygous vs wild type) at 4 and 6 months of age (**p < 0.001). n = 10. D, Day; T, trial. d, The beam-walking test was used to better assess the fine motor coordination and balance capabilities. Afg3l2+/Emv66 mice made significantly more footslips compared with wild type and showed increased latency to cross the beam. Repeated-measures ANOVA, **p < 0.001; n = 10. Error bars represent ±SD.
Although negative geotaxis and clasping are sensitive indicators of neurological disease, they are observed in many pyramidal and extrapyramidal phenotypes. To better assess the motor phenotype of heterozygous mice, Afg3l2+/Emv66 and wild-type littermates were subjected to rotarod analysis at different ages. Afg3l2+/Emv66 mice failed to maintain balance on the rotarod at 4 months and showed a progressive decline of performance over the ensuing months (Fig. 1c). Mutants fell earlier compared with controls and performed even worse in the following trials (i.e., latency to fall decreased), indicating that they have a reduced resistance to motor exercise and an increased difficulty of walking in synchrony with the rotating beam.
The rotarod is quite effective and sensitive to assess motor coordination and balance (Dunham and Miya, 1957; Crawley, 1999), for detection of neuromuscular defects, but also to cerebellar lesions in mouse models (Lalonde et al., 2002). We therefore performed a beam-walking test, which is more sensitive in revealing fine coordination defects and motor ataxia attributable to cerebellar dysfunction (Carter et al., 1999; Herson et al., 2003; Sausbier et al., 2004). At 4 months, Afg3l2+/Emv66 mice could barely walk along the beam and made significantly more foot slips compared with wild-type controls, the frequency of which increases with age (Fig. 1d, left graph). Moreover, whereas controls adopted a stable upright posture in traversing the beam, mutant mice flattened their abdomen and thorax against the upper surface of the beam and showed increased latency to cross (Fig. 1d, right graph).
We monitored mutant mice up to 18 months and observed that none developed other signs of neurological disease such as tremor, rigidity, or kyphosis. They instead seem to have strictly an ataxic phenotype of cerebellar origin, thus recapitulating clinical features of SCA28 patients. This evidence, together with the severe cerebellar phenotype previously observed in Afg3l2 homozygous mutants (Maltecca et al., 2008), prompted us to evaluate cerebellar degeneration in Afg3l2 heterozygous mice.
Motor coordination deficit correlates with alteration of cerebellar morphology
We started evaluating the overall morphology of the cerebellum on cryostat sagittal sections from Afg3l2+/Emv66 stained with hematoxylin and eosin. The size of the cerebellum was comparable between mutants and controls with normal development of folia and formation of cortical structure up to 4 months of age. Two months later, mutants show thickness reduction of the ML, which consists mostly of PC dendrites (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Since atrophy of the ML is generally correlated with degeneration of PC dendrites (He et al., 2006), we performed immunohistochemistry using calbindin (a specific marker of PCs) on the cerebellar section of Afg3l2+/Emv66 mice at 6 and 12 months. At 6 months, mutant PCs retain normal flask-shaped cell body morphology but appear smaller compared with wild-type controls. Moreover, dendritic stalks are abbreviated and hypertrophic with poorly arborized dendritic branches, leading to evident thickness reduction of the ML (139.679 μm ± 12.7915 SD in mutants vs 212.026 μm ± 22.26 SD in controls; t test, p < 0.001) (Fig. 2, top). At 12 months, mutant PCs appear atrophic and are reduced in number, leaving empty spaces in the PC monolayer compared with wild type (Fig. 2, bottom). At this age, ML thickness is 111.764 μm ± 17.893 SD in mutants versus 190.756 μm ± 10.377 in controls; t test, p < 0.001).
Figure 2.
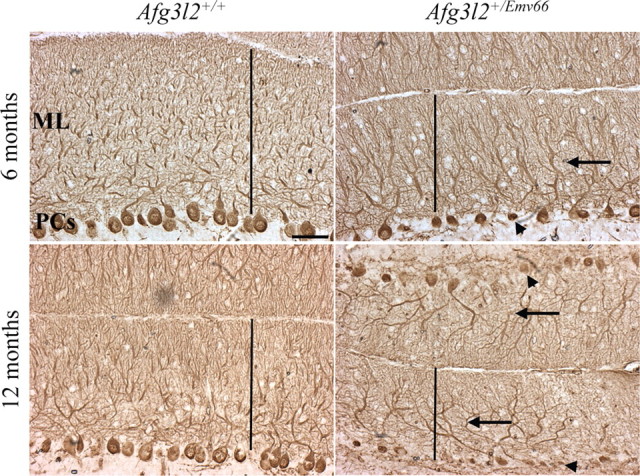
Cerebella from Afg3l2+/Emv66 mice show alteration of the PC dendritic tree and reduced thickness of the ML. Calbindin immunohistochemistry on cryostat sections of the midsagittal portion of the cerebellum of Afg3l2+/Emv66 and control mice at 6 months (top) and 12 months (bottom) is shown. In the mutant, several PCs disappear, leaving empty spaces in the PC monolayer (arrowheads). PC dendrites appear hypertrophic and less arborized (arrows), which leads to the ML's reduced thickness compared with controls in Afg3l2+/Emv66. Scale bar, 50 μm.
To better evaluate the extent of PC alterations and to make a possible correlation with the onset of motor coordination defects, we performed semithin sections on Afg3l2+/Emv66 mice at 4, 6, and at 12 months. We indeed observed progressive degeneration and loss of PCs in Afg3l2+/Emv66 cerebella. At 4 months, PCs start to display degenerating features, such as altered morphology and dark cytoplasm (Fig. 3b). As shown in Figure 3d, these alterations become more severe at 6 months, when mutant PCs display evident shrinkage and darkening of the cytoplasm accompanied by condensation of the nucleus. We also observed cases in which these cells exhibit defects in spatial alignment and reside outside of the defined layer. Quantitative analysis revealed a significant loss of PCs in Afg3l2+/Emv66 mice, which is already detectable at 6 months and become more extensive at 12 months. As shown in the graphs, most of the remaining cells are degenerating PCs.
Figure 3.
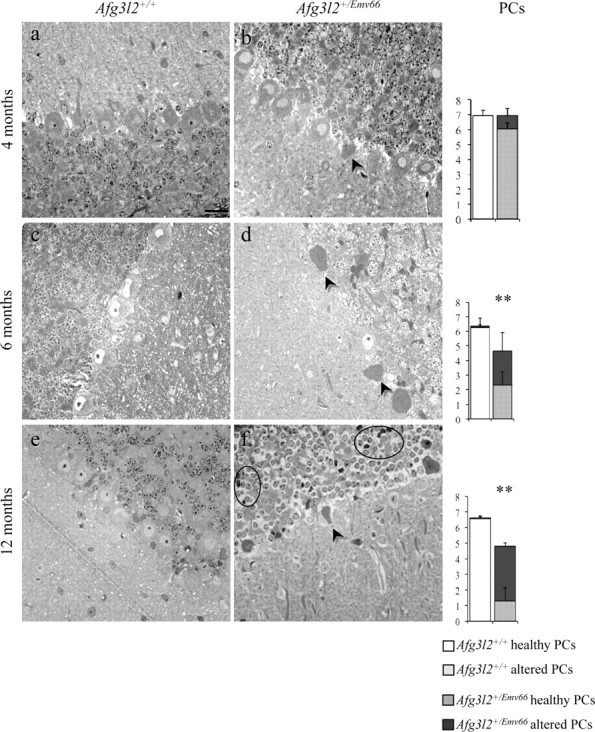
Afg3l2+/Emv66 mice show degeneration and loss of PCs. Semithin sections of cerebellum from Afg3l2+/Emv66 and syngenic control mice at 4, 6, and 12 months are shown. Afg3l2+/Emv66 mice show PC dark degeneration (arrowheads; b, d, f). At 12 months, GCs of Afg3l2+/Emv66 mice show empty cytoplasm and condensed nuclei (ovals; f). Scale bar, 20 μm. The graphs show quantitative evaluation of healthy and degenerated PCs in mutants versus controls. Error bars represent ±SD; Student's t test, **p < 0.001; n = 3.
Morphological changes were also detected in the granule cell (GC) layer, mostly at 12 months, where the vast majority of GCs show the presence of a swollen, empty cytoplasm and condensed nucleus (Fig. 3f). At this age, quantitative analysis also revealed a significant loss of GCs in the mutants (51.65 ± 1 SD/area in the mutants vs 70.95 ± 2.89 SD in controls; t test, p < 0.05).
PCs undergo dark degeneration in Afg3l2 heterozygous mice
To go deep inside the characterization of PC and GC degeneration, we performed ultrathin sections of Afg3l2+/Emv66 and controls, which revealed several morphological abnormalities in both cell types. Notably, dramatic defects in mitochondrial morphology, distribution, and cristae organization were detected already at 4 months, contemporary to the onset of motor coordination defects (Fig. 4a,b).
Figure 4.
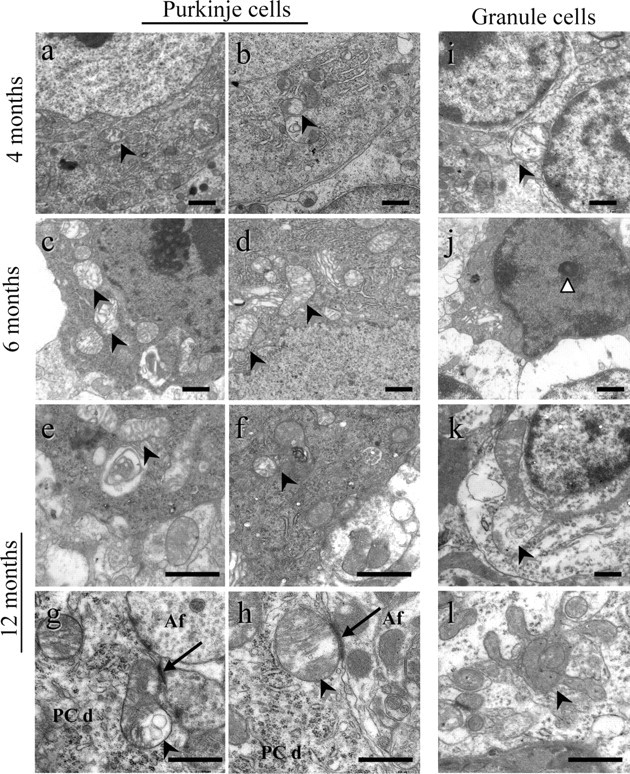
Aberrant mitochondria in PCs and GCs. Ultrathin sections of cerebellum from Afg3l2+/Emv66 and syngenic control mice at 4, 6, and 12 months are shown. a–h, Mutant PCs show aberrant mitochondria (arrowheads) in the cellular bodies and also in the dendritic synapses. i–l, GCs display apoptotic nuclei (white arrowhead; j), clusters of mitochondria surrounded by membrane (arrowhead; l), and giant mitochondria with altered cristae (arrowhead; k). PCd, PC dendrite; Af, afferent excitatory fiber. Arrows in g and h show synapses. Scale bars, 1 μm.
Mutant mitochondria appear larger and lose the classic elongated shape found in wild-type mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). They also show vesciculation of the inner membrane and abnormal cristae structure. Organellar alterations worsen with disease progression in mutant PCs, as shown at 6 months by the presence of clusters of aberrant mitochondria close to the plasma membrane. At this age, mutant PCs also start displaying increased electron density and condensed cytoplasm and nucleus but no signs of nuclear blebbing or apoptotic bodies (Fig. 4c). These features are peculiar of the type of neuronal cell death known as dark cell degeneration. This is characteristic of excitotoxic injury, in which excessive glutamate stimulation or excessive sensitivity to synaptic glutamate causes cellular toxicity (Barenberg et al., 2001; Strahlendorf et al., 2003). Intriguingly, this specific type of cell death has been recently described in mouse models of SCA7 and SCA5. In both cases, it has been linked to glutamate signaling (Custer et al., 2006; Ikeda et al., 2006), indicating that enhanced sensitivity of PCs to glutamate-induced excitotoxicity could be central for SCA pathogenesis. It is worth noting that we detected enlarged, vacuolated mitochondria in PC dendrites (Fig. 4g,h), exactly where PC cells receive small synapses mostly from parallel fibers (the branches of ascending axons of GCs) and from climbing fibers (afferents arising from the inferior olivary nucleus). Since both parallel and climbing fibers are glutamatergic and use AMPA and metabotropic glutamate receptor for transmission, these synapses are exposed to high concentrations of calcium (Ito, 2002).
In the GC layer, as well as in the PCs, EM revealed the appearance of aberrant mitochondria at 4 months, the number of which increases over time. At 12 months, in addition to mitochondria with destroyed cristae, organelles with unusual shapes and spatial organization were detected. Indeed, clusters of polylobulated mitochondria contained within a double membrane structure of undefined origin are reported in Figure 4l. An example in which an abnormally enlarged mitochondrion occupies the entire cytoplasmic space is shown in Figure 4k.
GCs progressively deteriorate, and at 6 months, many cells display empty cytoplasm and nucleus with totally condensed chromatin (Fig. 4i,j). These EM features correspond with those of late-stage apoptosis, namely apoptotic body (Ihara et al., 1998). Indeed, GCs clearly undergo apoptosis, as demonstrated by the TUNEL assay, which revealed a signal throughout the granular layer at 6 months (Fig. 5b) and a much stronger signal at 12 months (Fig. 5f) in Afg3l2+/Emv66 mice, in contrast to the almost complete absence of reactivity observed in age-matched controls (Fig. 5a, e, respectively). The percentage of TUNEL-positive nuclei relative to the total count of DAPI-stained nuclei was 6.803 ± 1.421 SD in mutants versus 0.633 ± 0.33 SD in controls at 6 months (t test, p < 0.001) and 12.967 ± 1.755 SD in mutants versus 1.424 ± 0.595 at 12 months (t test, p < 0.001).
Figure 5.
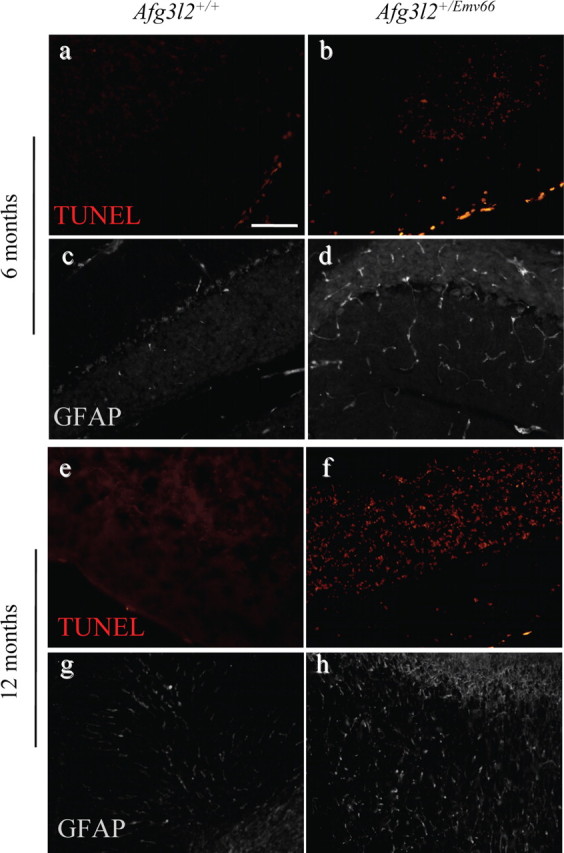
Apoptotic cell death of cerebellar GCs and massive gliosis in the granule layer in Afg3l2+/Emv66 mice. a, b, e, f, TUNEL-labeled cells (red) in cerebellar sections of Afg3l2+/Emv66 mice and syngenic controls at 6 and 12 months. At both ages, TUNEL-positive cells appear located in the GC layer (b, f). No TUNEL-positive staining was detected in PCs. c, d, g, h, Immunofluorescence on cerebellar sections of Afg3l2+/Emv66 mice and syngenic controls at 6 and 12 months showed a strong GFAP immunoreactivity in the granule layer. Scale bar, 100 μm.
Remarkably, no TUNEL-positive PCs were observed, thus confirming that these cells are not dying by apoptosis.
Consistently with the findings reported above, we detected an extensive reactive gliosis, especially prominent in the granule layer at 12 months. As shown in Figure 5h, Afg3l2+/Emv66 cerebella exhibit a marked staining for GFAP, which highlights activated astrocytes.
Spinal cord is unaffected in Afg3l2 heterozygous mice
We previously showed that the loss of Afg3l2 causes dramatic and widespread neuropathological alterations in both the CNS and PNS. Indeed, Afg3l2 homozygous mutant mice exhibit delayed myelination and poor radial growth of both primary and secondary motoneuron axons and the presence of enlarged mitochondria, clustering in the neuronal cell body of lumbar motoneurons, dorsal root ganglia (DRGs), and PCs (Maltecca et al., 2008). Based on these observations, we expected that the spinal cord was affected to the same extent of the cerebellum by Afg3l2 depletion in Afg3l2+/Emv66 mice. Interestingly, semithin sections of spinal cord revealed no signs of degeneration in lumbar motoneurons (Fig. 6, top) and the appearance of very rare vacuolated DRGs only at 12 months (Fig. 6, bottom), indicating that haploinsufficiency of AFG3L2 does not compromise mitochondrial functionality. Additionally, paraplegin and AFG3L1 could compensate for the reduced amount of AFG3L2 in this tissue. In accordance, no TUNEL-positive cells and no reactive gliosis were detected in heterozygous mice at the same age (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). These data suggest that different neuronal populations have different sensitivity to m-AAA defects and, more importantly, that the cerebellum is the neuronal tissue with the highest susceptibility to a reduced Afg3l2 dosage.
Figure 6.
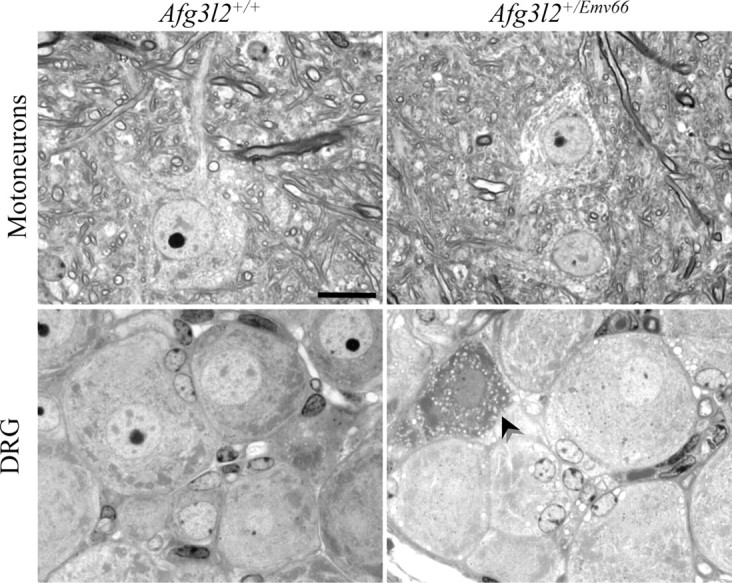
Afg3l2+/Emv66 mice do not display motoneurons and DRGs alteration. Semithin sections of spinal cord from Afg3l2+/Emv66 and syngenic control mice at 12 months are shown. Motoneurons in Afg3l2+/Emv66 show no signs of degeneration, whereas only sparse vacuolated DRG neurons (arrowhead) were detected.
Mutant PCs show impaired mitochondrial bioenergetics and increased reactive oxygen species production
The combination of enlarged spheroidal mitochondria and abnormal cristae structure is often associated with compromised mitochondrial bioenergetics and alterations of electron transport activity (Bereiter-Hahn and Voth, 1994). Moreover, impaired respiration due to m-AAA protease dysfunction has been well documented both in yeast and in mammals. Yeast cells lacking the m-AAA protease are indeed respiratory deficient, and this phenotype is rescued after exogenous expression of both paraplegin and AFG3L2 (Arlt et al., 1996; Atorino et al., 2003). We previously demonstrated that loss of Afg3l2 causes a dramatic reduction of ATP synthesis in brain and spinal cord of mutant mice because of insufficient assembly of respiratory complexes I and III (Maltecca et al., 2008). We thus investigated whether mitochondrial morphological alterations detected in the cerebellum of Afg3l2+/Emv66 mice correlate with defects of mitochondrial metabolism.
We tested ATP production in brain, spinal cord, and cerebellum of Afg3l2+/Emv66 mutant mice and controls in the presence of different substrates and inhibitors of the respiratory chain. The basal activity of the respiratory chain was similar in controls and mutants in the three analyzed tissues. To dissect the individual contribution of each complex to the total ATP production, we provided pyruvate and glutamate (which stimulate the activity of the overall respiratory chain complexes), succinate and rotenone (to assay the activity of complexes II–V), and ascorbate–tetramethyl-p-phenylenediamine (TMPD; to assay the activity of complex IV). At 6 and 12 months, the assay revealed no significant difference in ATP synthesis both in mutant brain and spinal cord compared with wild type (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). On the contrary, we detected a substantial reduction of ATP synthesis in mutant mitochondria from cerebellum at 6 months with pyruvate, glutamate, and succinate/rotenone. These defects become more severe at 12 months. The ATP level measured in the presence of ascorbate was the same in mutants and in controls, suggesting a comparable activity of complexes IV and V even at 12 months (Fig. 7a). Consistent with what we observed in Afg3l2 homozygous mutants, Blue Native-PAGE (BN-PAGE) followed by immunoblot revealed a decreased amount of assembled complexes I and III (Fig. 7b). Considering that the chaperone-like activity of the m-AAA complex is required in yeast for the assembly of the ATP synthase (Arlt et al., 1996), these data suggest an active role of Afg3l2 in assembling respiratory chain complexes and demonstrate the conservation of this function in mammals.
Figure 7.

ATP production deficiency in Afg3l2+/Emv66 mice. a, ATP synthesis rates in mitochondria isolated from cerebellum at 4, 6, and 12 months. Basal, No substrates (basal activity); PM, pyruvate and l-malate (complexes I–V); GM, glutamate and l-malate (complexes I–V); SR, rotenone and succinate (complexes II–V); AT, ascorbate and TMPD (complexes IV and V). Afg3l2+/Emv66 mice show impairment of ATP production starting at 6 months of age. Student's t test, *p < 0.05, **p < 0.001; n = 3. b, BN-PAGE–immunoblot analysis of complex I revealed by an anti-39 kDa antibody and immunoblot analysis of complex III revealed by anti-core2 (25.6 kDa subunit) antibody at 6 and 12 months; immunoblotting with anti-HSP60 antibody was used to verify equal loading. The graph shows protein amount of complexes I and III were quantified by densitometric analysis. Student's t test, *p < 0.05; n = 3. Error bars represent ±SD.
Defects in the mitochondrial respiratory chain complexes can increase reactive oxygen species (ROS) production as well as reduce ROS removal. When ROS generation exceeds protective mechanisms, multiple destructive redox reactions may ensue, such as lipid peroxidation, protein oxidation, and oxidative damage to DNA. In Afg3l2+/Emv66 mutants, we detected deficiency in respiratory chain complexes I and III, major producers of ROS in physiological and especially in pathological conditions (Murphy, 2009). We thus evaluated whether these respiratory defects cause oxidative damage in Afg3l2+/Emv66 mutants by measuring carbonyl formation, which is an easy detectable marker of protein oxidation (Requena et al., 2003). Carbonyl levels after reaction with DNPH (see Materials and Methods) were measured by Western blot in cerebellar total lysate and in cerebellar mitochondrial enrichments from Afg3l2+/Emv66 mutant mice and controls. We found that levels of protein carbonylation are strikingly increased in Afg3l2+/Emv66 mutants compared with wild type in both total lysate and mitochondria, despite the protein content that remains unchanged (Fig. 8a). This evidence demonstrates that half dosage of AFG3L2 impairs mitochondrial respiration and increases mitochondrial ROS production, leading to oxidative modification of mitochondrial proteins. Moreover, as a consequence of increased hydrogen peroxide efflux from the organelle, oxidative damage also affects cytosolic proteins.
Figure 8.
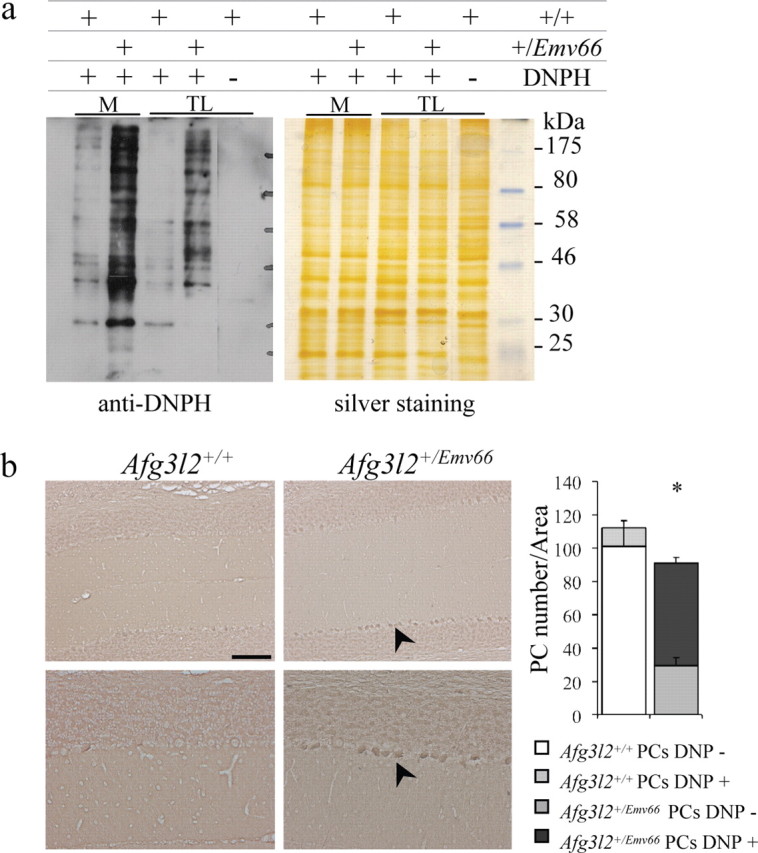
Increased level of oxidized proteins in mutant PCs. a, Total lysates and mitochondrial enrichments from cerebellum tissue from Afg3l2+/Emv66 and syngenic control mice were treated with DNPH. Derivatized proteins were analyzed by SDS-PAGE using an antibody against DNP moiety. Silver staining was used to verify equal loading. The untreated sample (treated with TFA only) was used as the negative control. M, Mitochondrial enrichment; TL, total lysate. b, Immunohistochemical localization of protein carbonyls in the Afg3l2+/Emv66 mouse cerebellum at 12 months of age. PC bodies are heavily stained in the Afg3l2+/Emv66 cerebellum (arrows). Scale bar: top, 100 μm; bottom, 50 μm. The graphs represent the count of DNP+ cells over the total cell number in mutants versus controls. Error bars represent ±SD; Student's t test, *p < 0.05; n = 2.
To verify that the increased ROS production was effectively a consequence of respiratory chain defects and not a secondary effect as a result of apoptotic process of GCs, we next evaluated the levels of protein carbonyls in situ at 12 months. Immunohistochemistry on cerebellar sagittal sections using an anti-DNPH antibody revealed that the vast majority of mutant PCs display a strong DNP-positive signal and only scattered GCs are positively stained (Fig. 8b). Interestingly, the percentage of DNP-positive PCs (Fig. 8b, graph) approximately correlates with the relative percentage of dark-degenerated PCs at the same age (Fig. 3f, graph). Probably because of technique sensitivity, we failed to detect differences at younger stages (data not shown).
Discussion
Despite the ubiquitous expression of m-AAA subnits, neurons seem selectively affected by alterations of paraplegin and AFG3L2. Indeed, loss-of-function mutations of paraplegin cause HSP, and mutations of AFG3L2 recently have been associated with SCA28 (Cagnoli et al., 2008; DiBella et al., 2008). Absence of paraplegin or AFG3L2 in the mouse causes strikingly different neurological defects: whereas paraplegin knock-out mice show late-onset degeneration mainly involving primary motoneurons, Afg3l2 null mutants display a severe defect of axonal development and widespread morphological alterations in both the CNS and PNS. Moreover, loss of Afg3l2 affects tissues that are spared in paraplegin knock-out mice (e.g., cerebellum) (Ferreirinha et al., 2004; Maltecca et al., 2008). It is thus conceivable that tissue-, cell-, or even subcellular-specific threshold effects exist, also considering the versatile assembly of m-AAA complexes (Koppen et al., 2007). In this work, we investigated whether different subsets of neurons are selectively susceptible to m-AAA gene dosage effect by characterizing a mouse model haploinsufficient for Afg3l2.
Selective susceptibility of cerebellum to reduced AFG3L2 dosage
Both behavioral and morphological analyses conducted on Afg3l2+/Emv66 mice indicate that the cerebellum is mainly affected by AFG3L2 haploinsufficiency. Indeed, Afg3l2+/Emv66 mice show progressive decline in motor coordination and balance, as assessed by rotarod and beam-walking tests. These findings demonstrate that the Afg3l2 haploinsufficient model recapitulates features of SCA28 patients, such as unbalance standing and slowly progressive gait incoordination (Cagnoli et al., 2006).
In agreement with behavioral signs, heterozygous mice present severe alteration of cerebellar morphology. They indeed show progressive degeneration and loss of PCs, dendritic atrophy, and significant thinning of the ML starting from 5–6 months of age. Remarkably, these signs are typical neuropathological hallmarks of SCAs. Cerebellar pathology is also evident by apoptotic death of GCs associated with massive substitutive gliosis in the granule layer.
The mitochondrial origin of the defect is demonstrated at EM examination by the appearance of morphologically altered organelles in both PCs and GCs at 4 months, which correlates with the onset of motor impairment. While disease progresses, an increasing number of swollen mitochondria with damaged cristae and vacuoles is detected in degenerating PCs, close to the plasma membrane, and also in the dendritic tract in proximity of synapses. This observation is notable, since mitochondria are required at synapses to maintain ion homeostasis, especially in dendritic spines of excitatory glutamatergic synapses that experience a large amount of Ca2+ influx (Mattson et al., 2008).
Interestingly, in the spinal cord of heterozygous mice at 12 months, the number of motoneurons and DRGs appears conserved, and no evident signs of degeneration were detected, supporting cerebellar sensitivity to AFG3L2 dysfunction. Many factors may be responsible for this specific susceptibility: for instance, the higher expression of Afg3l2 in the cerebellum compared with paraplegin and Afg3l1 (supplemental Fig. 5, available at www.jneurosci.org as supplemental material) and the ability of AFG3L2 to form both homo- and the hetero-oligomeric m-AAA complexes (Koppen et al., 2007). Since mammalian m-AAA protease shows a versatile assembly, as it varies its subunit composition in different tissues according to subunit availability, it is conceivable that AFG3L2-containing complexes are indeed the predominant m-AAA protease present in cerebellar mitochondria. This hypothesis is also supported by the ineffective compensation of AFG3L2 haploinsufficiency by paraplegin and AFG3L1 in the cerebellum. Conversely, in the spinal cord, a half amount of AFG3L2 may be sufficient to preserve mitochondrial functionality.
In agreement with these observations, we recently reported that the phenotype of Afg3l2+/Emv66 markedly worsens when placed on a paraplegin-null background (Spg7−/−/Afg3l2+/Emv66). In fact, these mice display anticipation and increased severity of motoneuron degeneration found in Spg7−/− mice, suggesting that Afg3l2 is able to partially compensate the lack of paraplegin in the spinal cord. Moreover, as expected from the reduced amount of AFG3L2-containing m-AAA complexes, these animals undergo anticipated degeneration of PCs with respect to Afg3l2+/Emv66 mice (Martinelli et al., 2009).
Dark degeneration of PCs in Afg3l2 heterozygous mice
PCs are crucial to cerebellar function, because they constitute the sole efferent projections from the cerebellum. Glutamatergic synapses of GC parallel fibers to PC dendrites contribute to >90% of all PC synaptic connections, providing important reciprocal interactions for the survival of both cell types. As an obvious consequence, PC loss results in a functional lesion in the cerebellum (Sotelo, 2004). PC death is usually ascribed to apoptosis, even if after acute insults (e.g., hypoxia or ischemia) necrotic death can occur (Sarna and Hawkes, 2003).
In Afg3l2+/Emv66 mice, the number of PCs decreases progressively between 4 and 12 months, but no TUNEL-positive PCs were observed, suggesting that they are not dying by apoptosis. Moreover, degenerating PCs present peculiar features, such as irregular shape, atrophy, condensed chromatin, and dark cytoplasm. These findings are consistent with an ongoing process of dark cell degeneration that has been documented in PCs exposed to excessive glutamate stimulation. The appearance of marked cytoskeletal changes in response to abusive AMPA receptor stimulation, coupled with increased intracellular Ca2+ concentration, suggests activation of Ca2+-dependent cysteine proteases as the mechanism leading to darkening of the cytoplasm in degenerating PCs (Barenberg et al., 2001; Strahlendorf et al., 2003). Intriguingly, PC dark degeneration has been recently described in mouse models for SCA7 and SCA5. In both cases, increased levels of glutamate available in the synaptic cleft for neuronal uptake has been demonstrated as the basis of degeneration, but with striking different mechanisms. Increased glutamate concentrations are elicited in SCA7 by reduced expression of the Bergmann glia-specific glutamate transporter GLAST, whereas they are elicited in SCA5 by impaired trafficking of neuron-specific glutamate transporter EEAT4 to the plasma membrane (Custer et al., 2006; Ikeda et al., 2006). These mechanisms of cerebellar pathogenesis underline the high susceptibility of PCs to glutamate dysregulation. We report that in Afg3l2+/Emv66 GCs undergo apoptosis starting from 6 months of age. At this time point, PCs are already extensively lost, and most of the remaining cells present degenerating features. One possible explanation could be that GC loss is secondary to insufficient trophic support from PCs or to cerebellar afferent alterations. Indeed, apoptosis of GCs is generally reported as a consequence of PC loss (Sarna and Hawkes, 2003). The alternative scenario of GC loss as a direct consequence of AFG3L2 haploinsufficiency in these cells is equally possible. PCs and GCs could die independently by activating different mechanisms of cell death, as supported by the concomitant appearance of morphologically altered mitochondria in both cell types at the same age. In this case, reduced Afg3l2 dosage in GCs could trigger an intrinsic pathway of apoptosis, which is not secondary to PC degeneration.
Mitochondrial dysfunction and oxidative stress mediate PC dark degeneration
Although PC dark degeneration has been already documented in SCA (Custer et al., 2006; Ikeda et al., 2006), this is the first report in which this type of degeneration is mediated by mitochondrial dysfunction rather than by prolonged glutamate stimulation at the synaptic cleft.
We indeed demonstrated that in Afg3l2+/Emv66 mutant PCs, the appearance of morphologically altered mitochondria precedes cell shrinkage, cytoplasm darkening, and nucleus condensation, which are the typical ultrastructural signs of excitotoxicity-mediated dark cell degeneration. Swollen mitochondria with damaged cristae are also metabolically dysfunctional in Afg3l2+/Emv66 mutants. We indeed detected marked reduction of ATP synthesis associated with a reduced activity of respiratory complexes I and III. Consistent with what we previously observed in Afg3l2 homozygous mutants, faulty assembly caused by reduced AFG3L2 chaperone-like activity rather than decreased availability of complex subunits is at the basis of this deficit of respiratory activities (Maltecca et al., 2008).
Inhibition of complexes I and III is known to diminish ATP production, to increase leakage of electrons and production of ROS, which attack intracellular biomolecules (Murphy, 2009). Proteins are one of the major targets of oxygen-free radicals and other reactive species. In line with this scenario, in Afg3l2+/Emv66 mutants we detected strikingly increased levels of protein carbonyls in cerebellar mitochondrial enrichments as well as in total lysates. Carbonylation has irreversible and unrepairable consequences modulating biochemical characteristics of proteins such as enzymatic activity susceptibility to proteolytic degradation (Nystrom, 2005). This evidence indicates that dysfunction of the respiratory chain caused by the lack of AFG3L2 cannot be counterbalanced by the mitochondrial antioxidant system, leading to increased ROS production and oxidative damage.
It is worth noting that we detected protein carbonylation mainly in PCs, whereas only sparse GCs are positively stained. Moreover, the percentage of dark degenerating PCs correlates with the fraction of PCs undergoing oxidative stress, indicating that the two phenomena are strictly interconnected.
One important consequence of respiratory chain defects and increased ROS production is alteration of ΔΨm, an electric steep gradient that is essential for Ca2+ internalization in mitochondria (Brookes et al., 2004). Consequences of insufficient mitochondrial Ca2+ sequestration include not only disruption of normal [Ca2+]i cycling needed to maintain appropriate Ca2+-dependent signaling pathways and enzyme activities but also pathological accumulation of [Ca2+]i. Excessive Ca2+ influx into cells is a key event in glutamate excitotoxicity and in dark degeneration (Barenberg et al., 2001; Strahlendorf et al., 2003). Many groups indeed demonstrated the importance of mitochondrial Ca2+uptake rather than just elevated [Ca2+]i in mediating excitotoxic cell death of neurons after glutamate treatment (Stout et al., 1998; Nicholls et al., 2003).
Since PC dendrites exclusively receive excitatory synapses, it is conceivable that inefficient Ca2+ sequestration by mitochondria causes pathological accumulation of [Ca2+]i in mutant PCs, thus mimicking excitotoxic-mediated dark degeneration in Afg3l2+/Emv66 mutants.
Taking all our data together, we propose the Afg3l2+/Emv66 mouse as the first model of SCA28. We also hypothesize a pathogenetic mechanism in which respiratory chain dysfunction and increased ROS production caused by AFG3L2 haploinsufficiency alters Ca2+ buffering capacity of mitochondria, thus leading to PC dark degeneration.
Footnotes
This work was supported by the Italian Telethon Foundation (Grant GGP08138), Istituto Superiore di Sanità, and Fondo per gli investimenti della ricerca di base TissueMet. We greatly acknowledge Giorgia Dina for neuropathology technical assistance.
The authors declare no competing financial interests.
References
- Arlt H, Tauer R, Feldmann H, Neupert W, Langer T. The YTA10-12 complex, an AAA protease with chaperone-like activity in the inner membrane of mitochondria. Cell. 1996;85:875–885. doi: 10.1016/s0092-8674(00)81271-4. [DOI] [PubMed] [Google Scholar]
- Arlt H, Steglich G, Perryman R, Guiard B, Neupert W, Langer T. The formation of respiratory chain complexes in mitochondria is under the proteolytic control of the m-AAA protease. EMBO J. 1998;17:4837–4847. doi: 10.1093/emboj/17.16.4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atorino L, Silvestri L, Koppen M, Cassina L, Ballabio A, Marconi R, Langer T, Casari G. Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J Cell Biol. 2003;163:777–787. doi: 10.1083/jcb.200304112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barenberg P, Strahlendorf H, Strahlendorf J. Hypoxia induces an excitotoxic-type of dark cell degeneration in cerebellar Purkinje neurons. Neurosci Res. 2001;40:245–254. doi: 10.1016/s0168-0102(01)00234-6. [DOI] [PubMed] [Google Scholar]
- Bereiter-Hahn J, Voth M. Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsc Res Tech. 1994;27:198–219. doi: 10.1002/jemt.1070270303. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Cagnoli C, Mariotti C, Taroni F, Seri M, Brussino A, Michielotto C, Grisoli M, Di Bella D, Migone N, Gellera C, Di Donato S, Brusco A. SCA28, a novel form of autosomal dominant cerebellar ataxia on chromosome 18p11.22-q11.2. Brain. 2006;129:235–242. doi: 10.1093/brain/awh651. [DOI] [PubMed] [Google Scholar]
- Cagnoli C, Stevanin G, Durr A, Ribai P, Forlani S, Brussino A, Pappi P, Pugliese L, Barberis M, Margolis R, Holmes S, Padovan S, Migone N, Di Bella D, Taroni F, Brice A, Brusco A. Mutations of AFG3L2 gene (SCA28) in autosomal dominant cerebellar ataxias. Paper presented at Annual Meeting of The American Society of Human Genetics; November; Philadelphia, PA. 2008. [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, De Michele G, Filla A, Cocozza S, Marconi R, Dürr A, Fontaine B, Ballabio A. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–983. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- Chang DT, Reynolds IJ. Mitochondrial trafficking and morphology in healthy and injured neurons. Prog Neurobiol. 2006;80:241–268. doi: 10.1016/j.pneurobio.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Coluccia A, Tattoli M, Bizzoca A, Arbia S, Lorusso L, De Benedictis L, Buttiglione M, Cuomo V, Furley A, Gennarini G, Cagiano R. Transgenic mice expressing F3/contactin from the transient axonal glycoprotein promoter undergo developmentally regulated deficits of the cerebellar function. Neuroscience. 2004;123:155–166. doi: 10.1016/j.neuroscience.2003.08.025. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Behavioral phenotyping of transgenic and knockout mice: experimental design and evaluation of general health, sensory functions, motor abilities, and specific behavioral tests. Brain Res. 1999;835:18–26. doi: 10.1016/s0006-8993(98)01258-x. [DOI] [PubMed] [Google Scholar]
- Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, Guyenet SJ, Deller T, Westrum LE, Sopher BL, La Spada AR. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci. 2006;9:1302–1311. doi: 10.1038/nn1750. [DOI] [PubMed] [Google Scholar]
- DiBella D, Lazzaro F, Brusco A, Battaglia G, Pastore A, Finardi A, Fracasso V, Plumari M, Cagnoli C, Tempia F, Brussino A, Gellera C, Mariotti C, Plevani P, DiDonato S, Langer T, Muzi-Falconi M, Taroni F. AFG3L2 mutations cause autosomal dominant ataxia SCA28 and reveal an essential role of the m-AAA AFG3L2 homocomplex in the cerebellum. Paper presented at Annual Meeting of The American Society of Human Genetics; November; Philadelphia, PA. 2008. [Google Scholar]
- Dunham NW, Miya TS. A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharm Assoc Am Pharm Assoc (Baltim) 1957;46:208–209. doi: 10.1002/jps.3030460322. [DOI] [PubMed] [Google Scholar]
- Ferreirinha F, Quattrini A, Pirozzi M, Valsecchi V, Dina G, Broccoli V, Auricchio A, Piemonte F, Tozzi G, Gaeta L, Casari G, Ballabio A, Rugarli EI. Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J Clin Invest. 2004;113:231–242. doi: 10.1172/JCI20138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden GA, Libby RT, Fu YH, Kinoshita Y, Huang J, Possin DE, Smith AC, Martinez RA, Fine GC, Grote SK, Ware CB, Einum DD, Morrison RS, Ptacek LJ, Sopher BL, La Spada AR. Polyglutamine-expanded ataxin-7 promotes non-cell-autonomous purkinje cell degeneration and displays proteolytic cleavage in ataxic transgenic mice. J Neurosci. 2002;22:4897–4905. doi: 10.1523/JNEUROSCI.22-12-04897.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zu T, Benzow KA, Orr HT, Clark HB, Koob MD. Targeted deletion of a single Sca8 ataxia locus allele in mice causes abnormal gait, progressive loss of motor coordination, and Purkinje cell dendritic deficits. J Neurosci. 2006;26:9975–9982. doi: 10.1523/JNEUROSCI.2595-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans RH, McGivern RF, Chen W, Longo LD. Altered adult sexual behavior in the male rat following chronic prenatal hypoxia. Neurotoxicol Teratol. 1993;15:353–363. doi: 10.1016/0892-0362(93)90051-o. [DOI] [PubMed] [Google Scholar]
- Herson PS, Virk M, Rustay NR, Bond CT, Crabbe JC, Adelman JP, Maylie J. A mouse model of episodic ataxia type-1. Nat Neurosci. 2003;6:378–383. doi: 10.1038/nn1025. [DOI] [PubMed] [Google Scholar]
- Ihara T, Yamamoto T, Sugamata M, Okumura H, Ueno Y. The process of ultrastructural changes from nuclei to apoptotic body. Virchows Arch. 1998;433:443–447. doi: 10.1007/s004280050272. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Dick KA, Weatherspoon MR, Gincel D, Armbrust KR, Dalton JC, Stevanin G, Durr A, Zuhlke C, Burk K, Clark HB, Brice A, Rothstein JD, Schut LJ, Day JW, Ranum LP. Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet. 2006;38:184–190. doi: 10.1038/ng1728. [DOI] [PubMed] [Google Scholar]
- Irwin S, Banuazizi A, Kalsner S, Curtis A. One trial learning in the mouse. I. Its characteristics and modification by experimental-seasonal variables. Psychopharmacologia. 1968;12:286–302. doi: 10.1007/BF00401408. [DOI] [PubMed] [Google Scholar]
- Ito M. The molecular organization of cerebellar long-term depression. Nat Rev Neurosci. 2002;3:896–902. doi: 10.1038/nrn962. [DOI] [PubMed] [Google Scholar]
- Koppen M, Langer T. Protein degradation within mitochondria: versatile activities of AAA proteases and other peptidases. Crit Rev Biochem Mol Biol. 2007;42:221–242. doi: 10.1080/10409230701380452. [DOI] [PubMed] [Google Scholar]
- Koppen M, Metodiev MD, Casari G, Rugarli EI, Langer T. Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia. Mol Cell Biol. 2007;27:758–767. doi: 10.1128/MCB.01470-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ, Beal MF, Manfredi G. The role of mitochondria in inherited neurodegenerative diseases. J Neurochem. 2006;97:1659–1675. doi: 10.1111/j.1471-4159.2006.03990.x. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Dumont M, Staufenbiel M, Sturchler-Pierrat C, Strazielle C. Spatial learning, exploration, anxiety, and motor coordination in female APP23 transgenic mice with the Swedish mutation. Brain Res. 2002;956:36–44. doi: 10.1016/s0006-8993(02)03476-5. [DOI] [PubMed] [Google Scholar]
- Leonhard K, Stiegler A, Neupert W, Langer T. Chaperone-like activity of the AAA domain of the yeast Yme1 AAA protease. Nature. 1999;398:348–351. doi: 10.1038/18704. [DOI] [PubMed] [Google Scholar]
- Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz AG, Ahn BW, Shaltiel S, Stadtman ER. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990;186:464–478. doi: 10.1016/0076-6879(90)86141-h. [DOI] [PubMed] [Google Scholar]
- Maltecca F, Aghaie A, Schroeder DG, Cassina L, Taylor BA, Phillips SJ, Malaguti M, Previtali S, Guenet JL, Quattrini A, Cox GA, Casari G. The mitochondrial protease AFG3L2 is essential for axonal development. J Neurosci. 2008;28:2827–2836. doi: 10.1523/JNEUROSCI.4677-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli P, La Mattina V, Bernacchia A, Magnoni R, Cerri F, Cox G, Quattrini A, Casari G, Rugarli EI. Genetic interaction between the m-AAA protease isoenzymes reveals novel roles in cerebellar degeneration. Hum Mol Genet. 2009;18:2001–2013. doi: 10.1093/hmg/ddp124. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008;60:748–766. doi: 10.1016/j.neuron.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16:R551–560. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Vesce S, Kirk L, Chalmers S. Interactions between mitochondrial bioenergetics and cytoplasmic calcium in cultured cerebellar granule cells. Cell Calcium. 2003;34:407–424. doi: 10.1016/s0143-4160(03)00144-1. [DOI] [PubMed] [Google Scholar]
- Nolden M, Ehses S, Koppen M, Bernacchia A, Rugarli EI, Langer T. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell. 2005;123:277–289. doi: 10.1016/j.cell.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Nystrom T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 2005;24:1311–1317. doi: 10.1038/sj.emboj.7600599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Previtali SC, Quattrini A, Fasolini M, Panzeri MC, Villa A, Filbin MT, Li W, Chiu SY, Messing A, Wrabetz L, Feltri ML. Epitope-tagged P(0) glycoprotein causes Charcot-Marie-Tooth-like neuropathy in transgenic mice. J Cell Biol. 2000;151:1035–1046. doi: 10.1083/jcb.151.5.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quattrini A, Previtali S, Feltri ML, Canal N, Nemni R, Wrabetz L. Beta 4 integrin and other Schwann cell markers in axonal neuropathy. Glia. 1996;17:294–306. doi: 10.1002/(SICI)1098-1136(199608)17:4<294::AID-GLIA4>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Requena JR, Levine RL, Stadtman ER. Recent advances in the analysis of oxidized proteins. Amino Acids. 2003;25:221–226. doi: 10.1007/s00726-003-0012-1. [DOI] [PubMed] [Google Scholar]
- Robinson BH. Use of fibroblast and lymphoblast cultures for detection of respiratory chain defects. Methods Enzymol. 1996;264:454–464. doi: 10.1016/s0076-6879(96)64041-5. [DOI] [PubMed] [Google Scholar]
- Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome. 1997;8:711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- Sarna JR, Hawkes R. Patterned Purkinje cell death in the cerebellum. Prog Neurobiol. 2003;70:473–507. doi: 10.1016/s0301-0082(03)00114-x. [DOI] [PubMed] [Google Scholar]
- Sausbier M, Hu H, Arntz C, Feil S, Kamm S, Adelsberger H, Sausbier U, Sailer CA, Feil R, Hofmann F, Korth M, Shipston MJ, Knaus HG, Wolfer DP, Pedroarena CM, Storm JF, Ruth P. Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci U S A. 2004;101:9474–9478. doi: 10.1073/pnas.0401702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagger H, Cramer WA, von Jagow G. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal Biochem. 1994;217:220–230. doi: 10.1006/abio.1994.1112. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Mitochondrial disease. Lancet. 2006;368:70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- Smerjac SM, Bizzozero OA. Cytoskeletal protein carbonylation and degradation in experimental autoimmune encephalomyelitis. J Neurochem. 2008;105:763–772. doi: 10.1111/j.1471-4159.2007.05178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotelo C. Cellular and genetic regulation of the development of the cerebellar system. Prog Neurobiol. 2004;72:295–339. doi: 10.1016/j.pneurobio.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- Strahlendorf J, Box C, Attridge J, Diertien J, Finckbone V, Henne WM, Medina MS, Miles R, Oomman S, Schneider M, Singh H, Veliyaparambil M, Strahlendorf H. AMPA-induced dark cell degeneration of cerebellar Purkinje neurons involves activation of caspases and apparent mitochondrial dysfunction. Brain Res. 2003;994:146–159. doi: 10.1016/j.brainres.2003.09.048. [DOI] [PubMed] [Google Scholar]
- Veglianese P, Lo Coco D, Bao Cutrona M, Magnoni R, Pennacchini D, Pozzi B, Gowing G, Julien JP, Tortarolo M, Bendotti C. Activation of the p38MAPK cascade is associated with upregulation of TNF alpha receptors in the spinal motor neurons of mouse models of familial ALS. Mol Cell Neurosci. 2006;31:218–231. doi: 10.1016/j.mcn.2005.09.009. [DOI] [PubMed] [Google Scholar]