

Abstract
Although nitric oxide (NO) has been implicated as a messenger molecule in hippocampal long-term potentiation (LTP) for almost 20 years, its precise function has not been elucidated because presynaptic and/or postsynaptic actions of NO have been reported. Most of the effects of NO as a signaling molecule are mediated by the NO receptor guanylyl cyclases (NO-GCs), two heme-containing enzymes with pronounced homology in which cGMP-forming activity is stimulated on NO binding. Here we report on knock-out (KO) mice in which either one of the NO-GC receptors has been genetically deleted. By measuring NO-induced cGMP levels, similar quantities of both NO-GC receptors were determined in the hippocampus. Surprisingly, hippocampal LTP was abolished in either one of the KO strains, demonstrating that both NO-GC receptors are required in the course of LTP. Expression of LTP was restored with a cGMP analog in one of the KO strains but did not recover in the other one. Moreover, single-cell recordings of paired pulse facilitation revealed a presynaptic role of one of the NO-GC isoforms in neurotransmitter release, confirming different roles of the NO-GC receptors in LTP. Because neither one of the NO/cGMP-induced responses by itself is sufficient for LTP, two divergent, possibly presynaptically and postsynaptically localized NO-stimulated cGMP pathways are apparently required for the expression of LTP. The unexpected role of cGMP at two sites of the synaptic cleft explains many of the controversial results in former NO research in LTP and demonstrates the necessity of presynaptic and postsynaptic changes for LTP expression.
Introduction
Long-term potentiation (LTP), the use-dependent increase in synaptic transmission, in hippocampal CA1 synapses is a prototype of synaptic mechanisms involved in memory formation and learning. Already in the early 1990s, the nitric oxide (NO)/cGMP signaling cascade had been suggested to participate in LTP, and a role for NO as a retrograde messenger had been postulated (Böhme et al., 1991; O'Dell et al., 1991; Schuman and Madison, 1991). In this concept, activation of NMDA receptors required for LTP also increases NO production by stimulating Ca2+/calmodulin-dependent neuronal NO synthase (nNOS) (Christopherson et al., 1999). The diffusible NO can cause changes in the presynaptic neurotransmitter release machinery; yet, reports about the postsynaptic NO action also exist (Ko and Kelly, 1999). Accordingly, LTP was reduced in mice lacking both NO-generating enzymes (Son et al., 1996). Although the general contribution of NO/cGMP signaling in LTP appears to be beyond doubt, the precise contribution and the molecular mechanism of how NO participates in certain forms of LTP are unclear.
Apparently, most of the NO effects are mediated by the NO receptor guanylyl cyclases (NO-GCs), NO-sensitive enzymes that form cGMP, thereby causing activation of cGMP effector molecules [protein kinases, ion channels, phosphodiesterases (Garthwaite, 2008)]. In addition to NO, CO has been suggested as a GC activator, and colocalization of the CO forming heme oxygenase-2 and NO-GC has been reported (Verma et al., 1993). Nevertheless, stimulation of NO-GC by CO is not pronounced and a physiological role of CO as GC activator is only conceivable in the case of a CO-sensitizing factor (Friebe et al., 1996).
The NO receptor GC consists of two different subunits (α, β) and occurs in two isoforms. The isoforms share a common β subunit but differ in their α subunit, and are designated NO-GC1 and NO-GC2, respectively. No functional differences were detected between the isoforms (Russwurm et al., 1998), but the ability of NO-GC2 to interact with PDZ (PSD-95/Dlg/ZO-1) domains, for example, of PSD-95, suggests a certain subcellular localization in brain (Russwurm et al., 2001). Whereas in the vascular system, NO-GC1 is the dominant NO receptor, both NO-GC receptors are distributed equally in brain (Mergia et al., 2003). Mice deficient in either one of the NO-GC receptors are powerful tools to evaluate their physiological function (Mergia et al., 2006). In previous experiments, we studied LTP in the visual cortex of the knock-out (KO) mice (Haghikia et al., 2007). Surprisingly, both NO-GC receptors were required for LTP in this brain region since LTP was abolished in both KO mouse lines but was restorable with a cGMP analog.
Here, we studied LTP in the hippocampal CA1 area of both NO-GC isoform-deficient mice. NO-stimulated cGMP levels revealed equal distribution of the NO receptors in this brain region. Similar to the visual cortex, hippocampal LTP was abolished in either KO line. Moreover, single-cell recordings of paired pulse facilitation revealed a presynaptic role of NO-GC1 in neurotransmitter release pointing to distinct roles of each NO-GC receptor in hippocampal LTP.
Materials and Methods
Generation and genotyping of the NO-GC1 or NO-GC2 KO mice have been described previously (Mergia et al., 2006). Briefly, exon 4 of the α1- or α2-subunits floxed by loxP-sites (α1+/flox and α2+/flox) were removed by crossing with the general deleter mouse EIIa-Cre, respectively. The resulting heterozygous KO mice were crossed with wild-type (WT) mice (C57BL/6) to remove the Cre recombinase gene. Finally, the generated heterozygotes were intercrossed to yield homozygous mice (NO-GC1 KO and NO-GC2 KO). Most experiments were performed with 6- to 10-week-old mice of either sex with a hybrid background (129/SvJ × C57BL/6; F6–F9 generation). For whole-cell patch-clamp recordings, mice at the age of 21–26 d were used. In WT experiments, the respective littermates of NO-GC1 or NO-GC2 KOs were used when available; otherwise, WTs of the same background strain were measured. Because no differences were detected between WTs and the respective WTs of either KO strain, all are designated WTs.
Electrophysiological recordings.
The present study was performed in full accordance with the guidelines of the local animal ethic commission and German laws. Mice were deeply anesthetized with ether and decapitated. The brains were quickly removed and transferred into ice-cold artificial CSF (ACSF) containing (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 d-glucose, 2 CaCl2, 1.5 MgCl2, bubbled with 95% O2 and 5% CO2 to pH 7.4. A vibratome (VT 1000 S, Leica) was used to cut transversal hippocampal slices of 350 μm thickness. The slices were allowed to recover in a submerged holding chamber with oxygenated ACSF at room temperature for at least 1 h. Subsequently, the slices were transferred into a standard interface-type chamber superfused with oxygenated ACSF at 32 ± 2°C. A concentric bipolar stimulation electrode was placed in the Schaffer collateral-commissural pathway to electrically stimulate afferent fibers projecting to the CA1 area. The resulting field EPSPs (fEPSPs) were recorded in the stratum radiatum of CA1 area using glass electrodes from borosilicate glass capillaries (GB 150-8P, Science Products) filled with ACSF to reach resistances of 1–3 MΩ. If not otherwise mentioned, the bipolar stimulation electrode was set to deliver monophasic currents of 100 μs duration and intensities of 80–150 μA. The fEPSPs were low-pass filtered at 3 kHz and amplified with a differential amplifier (EPMS07, NPI Electronic). Traces were sampled off-line using Clampex 9.2 (Molecular Devices). Measurements of the fEPSP slopes were made during their initial phase (20 and 50% of the peak). The input–output relation of fEPSPs was calculated by first determining the stimulus intensity that evoked a saturated maximal signal at a stimulus duration of 200 μs. Second, this intensity, as determined above, was kept constant while the stimulus duration was gradually increased from 40 to 200 μs in steps of 20 μs. For LTP experiments, the stimulus intensity was adjusted to elicit 30% of the maximum response signal and kept constant throughout the experiment. Baseline synaptic responses were stimulated and recorded at a frequency of 0.033 Hz for at least 30 min before tetanic stimulation (Bon and Garthwaite, 2003). The recording was discarded if the baseline fEPSP slope was not stable for 30 min and/or if the reproducible fEPSP amplitude was <1 mV. LTP was induced by one train of high-frequency synaptic stimulation (HFS) at 100 Hz (1 s duration) and with an intensity of the normal baseline stimulation (Hopper and Garthwaite, 2006). Changes in the fEPSP slope were calculated in relation to the baseline fEPSP responses during the last 10 min before HFS (100%). The time course of the fEPSP response was then normalized to this baseline and averaged across experiments. Long-term synaptic changes were evaluated by the averaged 20 responses at 51–60 min after HFS compared with the averaged 20 baseline signals before HFS. Positive control LTP measurements were run alternately with tests (e.g., WT with and without treatment as well as WT and KOs).
Paired-pulse ratios (PPRs) were derived from two consecutive synaptic stimulations at an interstimulus interval (ISI) of 50 ms before HFS and 2 min after. The PPR was calculated as the ratio of the slope of the second fEPSP (fEPSP2) in relation to the slope of the first fEPSP (fEPSP1).
The NOS inhibitors [NG-nitro-l-arginine (l-NNA) (Tocris Bioscience) and N5-(1-imino-3-butenyl)-l-ornithine (l-VNIO) (Alexis)] were added 30 min before, and 1H-[1,2,4]oxadiazolo[4,3-a]guinoxalin-1-one (ODQ) (Sigma) and cGMP (BIOLOG Life Science Institute) 10 min before HFS, and stayed in the samples for the entire recording period. None of the compounds led to alterations of the baseline field potential (FP) signals (data not shown).
Whole-cell patch-clamp recordings.
Paired-pulse ratio experiments from single pyramidal neurons in the stratum pyramidale of the CA1 region were performed by whole-cell patch-clamp recordings under visual control using differential interference contrast optics. Patch pipettes (4–6 MΩ) were pulled from borosilicate glass capillaries (GB 150F-8P; Science Products). Individual slices were transferred to a submerged-type recording chamber mounted on the stage of an upright microscope (Olympus BX50-WI) equipped with a 40× submerged objective. EPSCs were acquired with an Axoclamp 200B amplifier (Molecular Devices) in voltage clamp mode. The synaptic stimulation conditions were similar to those of the FP recordings. By bath application of picrotoxin (0.5 μm) to block GABAergic inhibition and by d-AP5 (25 μm) to prevent NMDA receptor activity, AMPA receptor-mediated EPSCs were pharmacologically isolated. PPRs were derived from two consecutively evoked EPSCs at a holding potential of −80 mV. The ISI ranged from 20 to 200 ms. PPR was calculated from the mean slopes of six to eight paired stimulations.
Determination of cGMP content in hippocampal slices.
Hippocampal transversal slices (350 μm) were cut and equilibrated as described above. Slices were incubated with the phosphodiesterase inhibitor isobutyl-3-methylxanthine (IBMX) (300 μm; Sigma) for 10 min at 37°C. Subsequently, the slices were incubated either with 2-(N,N-diethylamino)-diazenolate-2-oxide (DEA-NO) (10 μm) (Alexis) or NMDA (300 μm) (Sigma-Aldrich) for an additional 2 min. To extract cGMP, slices were snap frozen in liquid nitrogen, homogenized in 70% (v/v) ice-cold ethanol using a glass/glass homogenizer, and then centrifuged (14,000 × g, 15 min, 4°C). Supernatants were dried at 95°C and the cGMP content was measured by RIA. To standardize the different samples, protein pellets were dissolved in 0.1 m NaOH/0.1% SDS, and protein content was determined using the bicinchoninic acid method (Uptima).
Cyclic GMP levels determined in duplicates were measured in hippocampal slices of WT mice (23 slices from 11 mice, 2 male and 9 female), and NO-GC1 (16–18 slices from 9 mice, 2 male and 7 female)- and NO-GC2 (16–18 slices from 8 mice, 5 male and 3 female)-deficient mice.
Statistical analysis.
All data are presented as means ± SEM. Student's t and Mann–Whitney U tests were performed for statistical analysis of the electrophysiological data. The p-values represent significance of the data compared with baseline unless stated otherwise. Cyclic GMP levels were analyzed using two-tailed Student's t test.
Results
KO mice in which either one of the NO-GC receptors, NO-GC1 or NO-GC2, has been deleted do not reveal any obvious pathophysiological deviations (Mergia et al., 2006). In these mice, the remaining NO-stimulated cGMP-forming activity can be attributed to the nondeleted NO-GC receptor. To determine the NO-GC1 and NO-GC2 content in hippocampus, NO-stimulated cGMP levels (10 μm DEA-NO, 2 min) were measured in hippocampal slices of WT and KO mice in the presence of the phosphodiesterase inhibitor IBMX (300 μm). Cyclic GMP levels in the NO-GC1 (22 ± 3 pmol/mg) and NO-GC2 KO (24 ± 3 pmol/mg) amounted to ∼50% of the cGMP levels measured in WT hippocampal slices (43 ± 5 pmol/mg), suggesting equal distribution of the NO-GC receptor isoforms. Under nonstimulated conditions, cGMP levels of both KOs and WTs were comparable (WT, 9 ± 2 pmol/mg; NO-GC1 KO, 8 ± 2 pmol/mg; NO-GC2 KO, 7 ± 2 pmol/mg).
To examine the properties of basal synaptic transmission, we first examined the input–output relationship of extracellular recorded fEPSPs in hippocampal slices as described in Materials and Methods. Although no difference was observed between WT and NO-GC2 KO mice, the NO-GC1 KO mice showed significantly reduced slopes of evoked fEPSPs during maximal stimulus conditions (Fig. 1). However, when using a stimulation strength that evoked fEPSPs with 30% of the maximum signal, the fEPSPs did not differ significantly, and this relatively weak stimulation was exclusively used in the following LTP measurements.
Figure 1.

Input–output curves of KO and WT slices. Shown are slopes of fEPSPs recorded in CA1 region of hippocampal slices from NO-GC1 KOs (8 slices from 4 mice, 4 female), NO-GC2 KOs (12 slices from 4 mice, 1 male and 3 female), and WT (11 slices from 6 mice, 2 male and 4 female), as described in Materials and Methods, in response to stimulation of afferent fibers projecting to the CA1 area. *p < 0.05, differences considered significant.
To study LTP in the NO-GC1- and NO-GC2-deficient mice, we first characterized hippocampal CA1 LTP induced with HFS (100 Hz for 1 s). As can be seen in Figure 2a, HFS resulted in an approximately 220% increased fEPSP slope in WT slices. To determine the cGMP-dependent fraction, LTP induction was performed in the presence of the NO-GC-specific inhibitor ODQ (10 μm). As can be seen in Figure 2b, the major fraction of LTP was indeed cGMP dependent although, especially in the induction phase of LTP, ODQ did not completely inhibit the fEPSP increase. Recently, NO formed by NOS, nNOS, and eNOS has been suggested to play a role in LTP (Hopper and Garthwaite, 2006). Accordingly, we used the specific nNOS inhibitor l-VNIO (0.1 μm) and the inhibitor of eNOS and nNOS, l-NNA (100 μm), respectively (Fig. 2c,d). Because both inhibitors caused an almost complete impairment of LTP, we conclude that nNOS-derived NO is involved under the conditions tested, although a possible contribution of eNOS-derived NO could not be ruled out.
Figure 2.
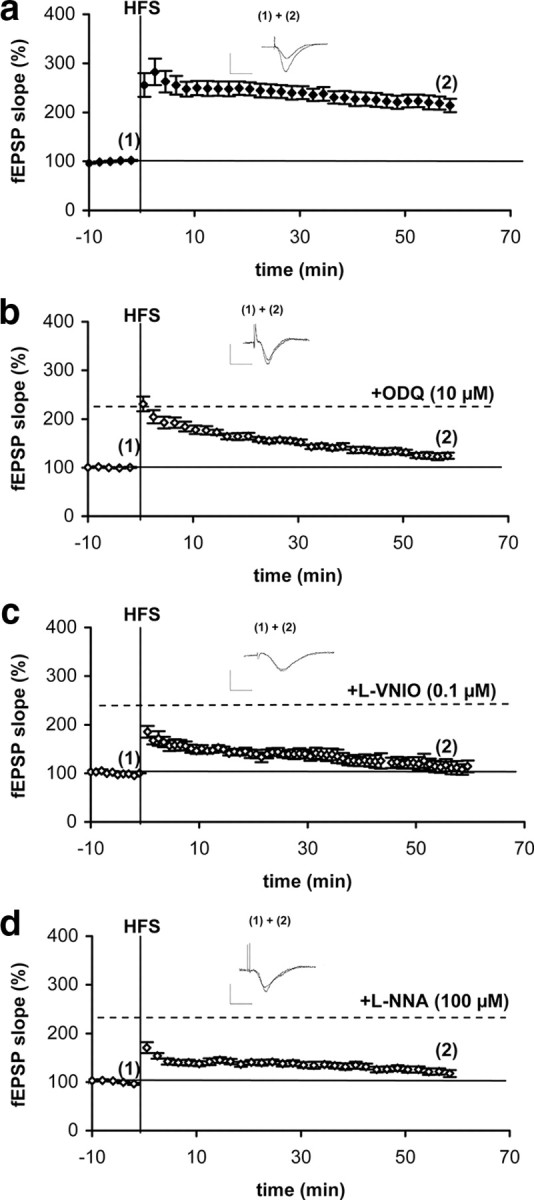
Hippocampal CA1 LTP is cGMP dependent. a, HFS-induced enhancement of fEPSP slope in WT slices (9 slices from 6 mice, 2 male and 4 female). b–d, HFS fails to induce LTP in the presence of the NO-GC receptor inhibitor, ODQ (b) (8 slices from 5 mice, 2 male and 3 female), the specific nNOS inhibitor, l-VNIO (c) (7 slices from 6 mice, 3 male and 3 female), and the inhibitor of NOS, l-NNA (d) (7 slices from 5 mice, 3 male and 2 female). Slices were treated 10 min with ODQ and 30 min with the respective NOS inhibitor before HFS and the inhibitors stayed in the samples for the entire recording period. Representative fEPSPs before (1) and after HFS (2) are shown. Calibration: 1 mV, 5 ms for all traces shown.
Subsequently, LTP was studied in the KO mice. As can be seen in Figure 3, neither in hippocampi of the NO-GC1 nor in the ones of the NO-GC2 KO mice, HFS led to an fEPSP increase similar to that in WT. These results indicate that both NO-GC receptors, NO-GC1 and NO-GC2, are required for LTP expression but serve different functions.
Figure 3.

LTP is absent in hippocampal CA1 area of NO-GC1 and NO-GC2 KO mice. a, b, HFS-induced enhancement of fEPSP slope of NO-GC1 (a) (7 slices from 4 mice, 1 male and 3 female)- and NO-GC2 (b) (9 slices from 4 mice, 2 male and 2 female)-deficient hippocampal slices. Representative fEPSPs before (1) and after HFS (2) are shown above the respective diagram. Calibration: 1 mV, 5 ms.
To substitute for the deleted NO-GC receptor, we used a cGMP analog [8-bromo-β-phenyl-1,N2-ethenoguanosine-3′, 5′-cycle monophosphate (8-Br-PET-cGMP), 50 μm] to reconstitute LTP. The analog by itself did not alter synaptic transmission in WT (data not shown). WT slices in which LTP was pharmacologically impaired by ODQ were used to substantiate our reconstitution conditions. Application of HFS to the ODQ-treated slices alone did not cause any increase in fEPSP (Fig. 4b), whereas in the presence of the cGMP analog, HFS was able to elicit normal LTP expression in the ODQ-treated slices, reinsuring the cGMP dependency of LTP (Fig. 4a). The successful reconstitution of LTP in WT shows that the cGMP analog reaches the relevant locations in sufficient concentrations. Next, we tried to reconstitute LTP in KO slices. In the NO-GC2 KO mice, application of HFS in the presence of the cGMP analog resulted in expression of LTP comparable to that in WT mice (Fig. 4d). Unexpectedly, LTP was not restorable in the NO-GC1 KO mice under the same conditions (Fig. 4c).
Figure 4.

LTP can be restored with a cGMP analog. a, Pharmacologically impaired LTP by ODQ is reconstituted with the cGMP analog, 8-Br-PET-cGMP (7 slices from 7 mice, 2 male and 5 female). b, A second application of HFS alone is not sufficient to induce LTP in the ODQ-treated slices (6 slices from 5 mice, 2 male and 3 female). c, The cGMP analog does not restore LTP in NO-GC1 KO mice (7 slices from 4 mice, 2 male and 2 female). d, Recovery of LTP in NO-GC2-deficient slices with the cGMP analog (7 slices from 4 mice, 3 male and 1 female). Representative fEPSPs before (1), and after the first HFS (2) and second HFS (3) are shown. Calibration: 1 mV, 5 ms.
The impaired LTP in both KO strains indicates that the cGMP formed by one of the NO-GC receptors does not substitute for the cGMP formed by the other isoform, suggesting a possible presynaptic or postsynaptic localization or, at least, compartmentalization of the NO-GC receptor isoforms. We measured NMDA-induced cGMP increases in the hippocampal slices of the KO mice to identify the receptor for NO produced in response to NMDA receptor activation. NMDA led to a cGMP increase in the NO-GC2 KO hippocampus comparable to the ∼2-fold cGMP increase in WT hippocampus, whereas no cGMP increase was observed in the NO-GC1 KO mice (Fig. 5). These results indicate that the NO-GC1 isoform still present in the NO-GC2-deficient mice is activated in response to NMDA receptor activation and implicate a different localization for NO-GC2.
Figure 5.

NMDA does not induce cGMP increase in NO-GC1 KO mice. Cyclic GMP levels were determined in hippocampal slices of WT (23 slices from 11 mice, 2 male and 9 female), and NO-GC1 (16–18 slices from 9 mice, 2 male and 7 female)- and NO-GC2 (16–18 slices from 8 mice, 5 male and 3 female)-deficient mice preincubated with IBMX (300 μm, 10 min) and stimulated with NMDA (300 μm, 2 min). *,§p < 0.001, differences considered significant.
To identify possible functions of the NO-GC isoforms in LTP, we determined the PPR before and after HFS stimulation. PPR was induced as described in Materials and Methods, with a decreased PPR being considered to result from an increase in the probability of presynaptic neurotransmitter release (Zucker, 1989). In hippocampal WT and NO-GC2-deficient slices, PPR was significantly decreased after HFS stimulation, indicating an enhanced neurotransmitter release within LTP (Fig. 6). In contrast, PPR in NO-GC1-deficient slices did not significantly differ before and after HFS, suggesting that the presynaptic neurotransmitter release was not augmented by HFS. Together, these results suggest a HFS-induced presynaptic enhancement of neurotransmitter release in WT and NO-GC2 KO mice that is lost in the NO-GC1 KOs.
Figure 6.

HFS-induced reduction in PPR is missing in the NO-GC1 KO mice. Summary of paired-pulse fEPSP slope ratios (left) and example paired-pulse fEPSP traces (right), showing a decreased paired-pulse ratio after HFS in WT (9 slices from 8 mice, 4 male and 4 female) and NO-GC2-deficient slices (9 slices from 4 mice, 2 male and 2 female), which is not observed in the NO-GC1-deficient slices (7 slices from 4 mice, 1 male and 3 female). Calibration: 1 mV, 5 ms. *p < 0.001, differences between pre- and post-HFS within a group considered significant.
To substantiate a possible reduced neurotransmitter release probability in the NO-GC1 KO mice, we performed further PPR experiments in single-cell recording. Picrotoxin and AP-5 were added to exclude inhibitory GABAergic and NMDA receptor-mediated effects. As shown in Figure 7, PPRs were similar in WT and NO-GC2 KO over the whole range of ISIs, whereas in the NO-GC1 KO, PPR was significantly increased at short ISIs (20–50 ms), indicative of a reduced neurotransmitter release probability. We conclude that NO-GC1 appears as the likely candidate to stimulate the presynaptic neurotransmitter release.
Figure 7.

Single-cell recorded PPR is increased in the NO-GC1 KO mice. a, Summary of paired-pulse EPSC slope ratios of WT (13 cells from 4 mice, 3 male and 1 female) slices, and NO-GC1 (15 cells from 3 mice, 1 male and 2 female)- and NO-GC2 (10 cells from 3 mice, 1 male and 2 female)-deficient slices. b, Representative voltage traces at ISIs between 20 and 200 ms obtained in the presence of picrotoxin (100 μm) and AP-5 (25 μm). *p < 0.05, differences considered significant.
Discussion
In the early days of the NO research, a multitude of results was published about the role of NO as a retrograde messenger in LTP (Garthwaite and Boulton, 1995). In most of these experiments, the contribution of NO was demonstrated by the use of NOS inhibitors causing a reduction of LTP (Haley et al., 1993; Williams et al., 1993). In further approaches, NO donors or cGMP analogs paired with a weak stimulus were used to induce LTP, often with controversial results (Zhuo et al., 1994; Selig et al., 1996; Bon and Garthwaite, 2003). However, up to now, no unifying concept has been proposed, although a role of NO in LTP appears to be widely accepted, and consensus that NO is produced by the postsynaptically localized nNOS exists. Activation of nNOS occurs in the course of NMDA receptor-induced Ca2+ influx; the produced NO acts as a retrograde messenger and increases neurotransmitter release in the presynaptic nerve terminal (Garthwaite and Boulton, 1995). Yet, intact LTP reported in the nNOS KO mice (O'Dell et al., 1994) is not in support of that notion but is likely attributable to the presence of a catalytically active splice variant of nNOS in the KO mouse strain. However, NO produced by eNOS may play a role in LTP as well (Hopper and Garthwaite, 2006).
The role of NO acting via stimulation of NO-GC in LTP as shown by others (Boulton et al., 1995) is also supported in our study with the NO-GC inhibitor ODQ, which caused a reduction in LTP similar to the ones observed with the NO synthase inhibitors. These results underline that within LTP expression, the NO effects are solely mediated by the NO receptor GC. Although the NO-GC is the only NO effector molecule identified so far, not much attention has been paid to the fact that two NO receptors exist, NO-GC1 and NO-GC2. Because of the indistinguishable properties of the receptor isoforms, the functional impact of two similarly regulated NO receptors remained unclear. The similar expression levels of the NO receptor isoforms in brain, however, indicate possible functions of both isoforms in the neuronal system. Moreover, a possible neuronal role of NO-GC2 is supported by its reported interaction with the PDZ domains of PSD-95 (Russwurm et al., 2001). With the KO mice lacking either one of the NO-GC receptors, the question of the respective contribution of the two NO-GC receptors in LTP could be addressed for the first time and, in addition, the general involvement of NO in LTP could be reconsidered in clear-cut experiments. In our recent studies in the visual cortex of the KOs, crucial roles of NO and of both NO-GC receptors in LTP emerged as LTP was completely abolished in either one of the KO mouse strains, but was reconstituted with a cGMP analog (Haghikia et al., 2007).
Here, we studied whether the two NO-GC receptors also participate in hippocampal CA1 LTP, the best studied LTP form. Indeed, WT-like LTP was absent in either one of the NO-GC receptor-deficient mice. The impairment of LTP in either KO strain demonstrates that the cGMP formed by one NO-GC receptor cannot make up for the cGMP produced by the other one, a finding possibly explained by a presynaptic and postsynaptic localization of the NO receptors. In addition, our finding that NMDA-induced NO generation increases cGMP levels only in the presence of the NO-GC1 receptor underlines once more the postulate of different localizations of the NO-GC receptors. Consistent with our hypothesis, different synaptic localizations of the NO-GC receptors have also been demonstrated by others using immunochemistry and in situ hybridization methods (Szabadits et al., 2007).
Generally it is believed that presynaptic and postsynaptic alterations are required for the induction and maintenance of LTP (Lisman et al., 2007). An increase in postsynaptic AMPA receptor number or efficacy (or both) causes an enhanced postsynaptic response, whereas presynaptically, the neurotransmitter release is increased. An increased PPR in the NO-GC1 KO in single-cell recordings indicates a possible presynaptic localization of the NO-GC1. Accordingly, we propose that NO formed by nNOS as a retrograde messenger stimulates NO-GC1 in the presynaptic nerve terminal to increase neurotransmitter release, which also has been shown by others (Arancio et al., 1996). However, activation of NO-GC1, only, does not suffice for the induction of LTP by itself, as indicated by the complete lack of LTP in the NO-GC2-deficient animals. Accordingly, we have to postulate a role of NO-GC2 in the increase of the postsynaptic response. In this context, a recent publication showing a role of the cGMP-dependent protein kinase II in the regulation of AMPA receptor trafficking is of major importance (Serulle et al., 2007). Hence, neither the presynaptic nor the postsynaptic cGMP increase alone is sufficient for LTP induction, but the combined presynaptic and postsynaptic action on transmitter release and postsynaptic responsiveness appears necessary. Of course, these assumptions are speculative but provide a model for further experimental approaches.
In reconstitution experiments, we were able to substitute the function of NO-GC2 in LTP with a cGMP analog. In contrast, the function of the deleted NO-GC1, which we assume to play a role in presynaptic neurotransmitter release, was not restorable with the cGMP analog. Several reasons for this have to be considered. (1) A general impairment of LTP caused by the lack of the isoform during brain developmental cannot completely be ruled out, although the recovery of LTP with a cGMP analog in the visual cortex argues against this assumption (Haghikia et al., 2007). (2) Longer cGMP action might be required. In WT, cGMP is constantly being produced by the NO receptors. If, for example, cGMP is responsible not only for the transmitter release but also for the recycling of the vesicles, there may not be enough vesicles to supply a pronounced neurotransmitter release in the absence of constant cGMP levels (Micheva et al., 2003). (3) Finally, instead of just flooding the slices with cGMP, precise temporally and spatially regulated cGMP increases may be required for the LTP induction, as has been reported by others (Son et al., 1998).
In sum, our data indicate that two possibly presynaptic and postsynaptic localized NO receptors are required for hippocampal CA1 LTP. The unexpected finding that cGMP has to act on two sites, perhaps on both sides of the synaptic cleft, explains many of the former controversies about the role of NO in LTP. With the help of the NO receptor KO mice, we hope that in the near future we will be able to identify the cGMP targets in the synaptic nerve terminals to elucidate the functional consequences of NO/cGMP signaling in LTP and in neuronal transmission and networks in general.
Footnotes
This work is supported by the Deutsche Forschungsgemeinschaft. We thank Medah Özcan and Petra Küsener for excellent technical assistance.
References
- Arancio O, Kiebler M, Lee CJ, Lev-Ram V, Tsien RY, Kandel ER, Hawkins RD. Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell. 1996;87:1025–1035. doi: 10.1016/s0092-8674(00)81797-3. [DOI] [PubMed] [Google Scholar]
- Böhme GA, Bon C, Stutzmann JM, Doble A, Blanchard JC. Possible involvement of nitric oxide in long-term potentiation. Eur J Pharmacol. 1991;199:379–381. doi: 10.1016/0014-2999(91)90505-k. [DOI] [PubMed] [Google Scholar]
- Bon CL, Garthwaite J. On the role of nitric oxide in hippocampal long-term potentiation. J Neurosci. 2003;23:1941–1948. doi: 10.1523/JNEUROSCI.23-05-01941.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton CL, Southam E, Garthwaite J. Nitric oxide-dependent long-term potentiation is blocked by a specific inhibitor of soluble guanylyl cyclase. Neuroscience. 1995;69:699–703. doi: 10.1016/0306-4522(95)00349-n. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Hillier BJ, Lim WA, Bredt DS. PSD-95 assembles a ternary complex with the N-methyl-d-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J Biol Chem. 1999;274:27467–27473. doi: 10.1074/jbc.274.39.27467. [DOI] [PubMed] [Google Scholar]
- Friebe A, Schultz G, Koesling D. Sensitizing soluble guanylyl cyclase to become a highly CO-sensitive enzyme. EMBO J. 1996;15:6863–6868. [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27:2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- Haghikia A, Mergia E, Friebe A, Eysel UT, Koesling D, Mittmann T. Long-term potentiation in the visual cortex requires both nitric oxide receptor guanylyl cyclases. J Neurosci. 2007;27:818–823. doi: 10.1523/JNEUROSCI.4706-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley JE, Malen PL, Chapman PF. Nitric oxide synthase inhibitors block long-term potentiation induced by weak but not strong tetanic stimulation at physiological brain temperatures in rat hippocampal slices. Neurosci Lett. 1993;160:85–88. doi: 10.1016/0304-3940(93)90919-c. [DOI] [PubMed] [Google Scholar]
- Hopper RA, Garthwaite J. Tonic and phasic nitric oxide signals in hippocampal long-term potentiation. J Neurosci. 2006;26:11513–11521. doi: 10.1523/JNEUROSCI.2259-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko GY, Kelly PT. Nitric oxide acts as a postsynaptic signaling molecule in calcium/calmodulin-induced synaptic potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 1999;19:6784–6794. doi: 10.1523/JNEUROSCI.19-16-06784.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Raghavachari S, Tsien RW. The sequence of events that underlie quantal transmission at central glutamatergic synapses. Nat Rev Neurosci. 2007;8:597–609. doi: 10.1038/nrn2191. [DOI] [PubMed] [Google Scholar]
- Mergia E, Russwurm M, Zoidl G, Koesling D. Major occurrence of the new alpha2beta1 isoform of NO-sensitive guanylyl cyclase in brain. Cell Signal. 2003;15:189–195. doi: 10.1016/s0898-6568(02)00078-5. [DOI] [PubMed] [Google Scholar]
- Mergia E, Friebe A, Dangel O, Russwurm M, Koesling D. Spare guanylyl cyclase NO receptors ensure high NO sensitivity in the vascular system. J Clin Invest. 2006;116:1731–1737. doi: 10.1172/JCI27657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Buchanan J, Holz RW, Smith SJ. Retrograde regulation of synaptic vesicle endocytosis and recycling. Nat Neurosci. 2003;6:925–932. doi: 10.1038/nn1114. [DOI] [PubMed] [Google Scholar]
- O'Dell TJ, Hawkins RD, Kandel ER, Arancio O. Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proc Natl Acad Sci U S A. 1991;88:11285–11289. doi: 10.1073/pnas.88.24.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dell TJ, Huang PL, Dawson TM, Dinerman JL, Snyder SH, Kandel ER, Fishman MC. Endothelial NOS and the blockade of LTP by NOS inhibitors in mice lacking neuronal NOS. Science. 1994;265:542–546. doi: 10.1126/science.7518615. [DOI] [PubMed] [Google Scholar]
- Russwurm M, Behrends S, Harteneck C, Koesling D. Functional properties of a naturally occurring isoform of soluble guanylyl cyclase. Biochem J. 1998;335:125–130. doi: 10.1042/bj3350125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russwurm M, Wittau N, Koesling D. Guanylyl cyclase/PSD-95 interaction: targeting of the nitric oxide-sensitive alpha2beta1 guanylyl cyclase to synaptic membranes. J Biol Chem. 2001;276:44647–44652. doi: 10.1074/jbc.M105587200. [DOI] [PubMed] [Google Scholar]
- Schuman EM, Madison DV. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science. 1991;254:1503–1506. doi: 10.1126/science.1720572. [DOI] [PubMed] [Google Scholar]
- Selig DK, Segal MR, Liao D, Malenka RC, Malinow R, Nicoll RA, Lisman JE. Examination of the role of cGMP in long-term potentiation in the CA1 region of the hippocampus. Learn Mem. 1996;3:42–48. doi: 10.1101/lm.3.1.42. [DOI] [PubMed] [Google Scholar]
- Serulle Y, Zhang S, Ninan I, Puzzo D, McCarthy M, Khatri L, Arancio O, Ziff EB. A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron. 2007;56:670–688. doi: 10.1016/j.neuron.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, Kandel ER. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell. 1996;87:1015–1023. doi: 10.1016/s0092-8674(00)81796-1. [DOI] [PubMed] [Google Scholar]
- Son H, Lu YF, Zhuo M, Arancio O, Kandel ER, Hawkins RD. The specific role of cGMP in hippocampal LTP. Learn Mem. 1998;5:231–245. [PMC free article] [PubMed] [Google Scholar]
- Szabadits E, Cserép C, Ludányi A, Katona I, Gracia-Llanes J, Freund TF, Nyíri G. Hippocampal GABAergic synapses possess the molecular machinery for retrograde nitric oxide signaling. J Neurosci. 2007;27:8101–8111. doi: 10.1523/JNEUROSCI.1912-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A, Hirsch DJ, Glatt CE, Ronnett GV, Snyder SH. Carbon monoxide: a putative neural messenger. Science. 1993;259:381–384. doi: 10.1126/science.7678352. [DOI] [PubMed] [Google Scholar]
- Williams JH, Li YG, Nayak A, Errington ML, Murphy KP, Bliss TV. The suppression of long-term potentiation in rat hippocampus by inhibitors of nitric oxide synthase is temperature and age dependent. Neuron. 1993;11:877–884. doi: 10.1016/0896-6273(93)90117-a. [DOI] [PubMed] [Google Scholar]
- Zhuo M, Hu Y, Schultz C, Kandel ER, Hawkins RD. Role of guanylyl cyclase and cGMP-dependent protein kinase in long-term potentiation. Nature. 1994;368:635–639. doi: 10.1038/368635a0. [DOI] [PubMed] [Google Scholar]
- Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]