

Abstract
Cocaine-induced changes in glutamatergic synaptic transmission in the ventral tegmental area (VTA) and the nucleus accumbens (NAc) play a key role in cocaine behavioral effects. Activation of ionotropic glutamate receptor NMDA receptor (NMDAR) in the VTA is critical for the development of cocaine psychomotor sensitization. However, the role of NMDAR in the NAc, a brain area critical for the expression of cocaine psychomotor sensitization, remains to be explored. Here, we show that repeated noncontingent cocaine injections increased NAc NMDAR subunits, NR1, NR2A, and NR2B 21 d, but not 1 d, after withdrawal from cocaine. These changes were associated with an increase in the GluR1 subunit of the AMPA receptor. We also found a time-dependent increase in extracellular signal-regulated kinase (ERK) activity which correlated with the increased expression of NMDAR subunits. Furthermore, the increase in GluR1 and ERK activity was blocked after inhibition of NR2B-containing NMDAR during the development of cocaine psychomotor sensitization or when the MEK (mitogen-activated protein/ERK kinase) inhibitor was microinjected into the NAc 21 d after withdrawal from cocaine. Together, these results suggest that the development of cocaine psychomotor sensitization triggers a delayed increase in the expression of NMDAR subunits in the NAc, which in turn enhances the activity of ERK. Enhanced ERK activity drives the increased expression of the GluR1 subunits, which increases the excitability of NAc neurons after prolonged withdrawal from cocaine and results in enduring expression of psychomotor sensitization.
Introduction
Repeated administration of psychostimulant drugs such as cocaine induces psychomotor sensitization, which is thought to contribute to the development of drug craving by enhancing the incentive motivational value of these drugs (Kalivas and Stewart, 1991; Robinson and Berridge, 1993). Among the enduring neuronal changes responsible for the expression of cocaine psychomotor sensitization, alterations in glutamate signaling and plasticity within the ventral tegmental area (VTA) and nucleus accumbens (NAc) play a critical role (Wolf, 1998; Kalivas, 2004). For example, microinjection of the NMDA receptor (NMDAR) antagonist MK-801 into the VTA prevented the development of cocaine psychomotor sensitization (Kalivas and Alesdatter, 1993), emphasizing the critical role of VTA–NMDAR.
Numerous studies have focused on cocaine-induced long-lasting molecular and synaptic changes in the NAc after withdrawal from cocaine (Kauer and Malenka, 2007). These changes involved the glutamatergic system and its downstream signaling such as the extracellular signal-regulated kinase (ERK) pathway (Girault et al., 2007; Thomas et al., 2008). Boudreau and Wolf (2005) have shown that cocaine psychomotor sensitization is associated with an increase in the surface expression of the GluR1 subunit of the AMPA receptor (AMPAR) in the NAc, 21 d but not 1 d after withdrawal from cocaine. Support for these findings comes from electrophysiological experiments demonstrating that in sensitized mice 14 d after the last cocaine injection, the ratio of AMPAR/NMDAR [an electrophysiological measure of long-term potentiation (LTP)] in NAc slices taken from these mice is increased, suggesting that prolonged cessation from drug administration enhances the synaptic strength in the NAc (Kourrich et al., 2007). Interestingly, both biochemical and electrophysiological changes were reversed when the sensitized rats received a single injection of cocaine (“challenge”), 21 d after withdrawal from cocaine (Boudreau et al., 2007; Kourrich et al., 2007). In addition, it was shown that in cocaine-sensitized rats, the increase in the surface expression of the GluR1 subunit paralleled an increase in the activity of ERK (Boudreau et al., 2007).
There is evidence for a role of ERK in cocaine behavioral effects (Lu et al., 2006; Girault et al., 2007), including psychomotor sensitization (Berhow et al., 1996; Valjent et al., 2004; Mattson et al., 2005). For example, inhibition of ERK by microinjection of PD98059 into the VTA before three daily cocaine injections blocked cocaine psychomotor sensitization (Pierce et al., 1999). Similarly, systemic injection of the mitogen-activated protein kinase/ERK kinase that selectively activates ERK (MEK) inhibitor SL327 before each cocaine injection prevented the development of cocaine psychomotor sensitization (Valjent et al., 2006). Unilateral intra-NAc infusions of the MEK inhibitor U0126 attenuated cocaine-induced ERK and cAMP response element-binding protein phosphorylation in cocaine-sensitized rats (Mattson et al., 2005). Together, these results emphasize the critical role of ERK signaling in the neuroadaptations underlying the development and expression of cocaine psychomotor sensitization. Here, we examined the hypothesis that cocaine-induced biochemical and electrophysiological changes are triggered by an increase in the expression profile of the NMDAR in the NAc. We found that prolonged but not short withdrawal from repeated cocaine injections increased the expression of NMDAR subunits in the NAc. Blockade of NMDAR during development of cocaine psychomotor sensitization or inhibition of ERK signaling in the NAc after prolonged withdrawal from cocaine blocked the increase in ERK activity and the expression of GluR1 in the NAc.
Materials and Methods
Animals.
Male Sabra rats, strain of the Hebrew University (Harlan Laboratories) were housed in groups of four with food and water available ad libitum. A 12 h light/dark cycle was used with the lights on at 7:00 A.M. All saline or cocaine injections and behavioral testing were performed between 10:00 A.M. and 4:00 P.M. All procedures were approved by the Institutional Animal Care and Use Committee of the Hebrew University of Jerusalem.
Repeated cocaine injections and behavioral analysis.
All rats were assigned to saline and cocaine treatment groups after a week of acclimation to their home cage environment. Two days before the first cocaine or saline injection, rats were habituated to the behavioral testing procedure by placement in photocell cages (MED Associates) for 30 min after saline injection. On the first day of treatment (day 1), rats were habituated to photocell cages for 20 min before injection of cocaine (15 mg/kg, i.p.) or saline (1 ml/kg, i.p.). Locomotor activity (total beam breaks) was measured for an additional 30 min. For the next 4 d (days 2–5), the same procedure was applied. For ifenprodil experiments, the same procedure was applied except that ifenprodil or vehicle was intraperitoneally injected after 20 min habituation, then cocaine or saline was injected 30 min later. Locomotion activity was measured for an additional 30 min. All rats were returned to their home cage for 21 d. On day 21, all rats were taken from their home cage and killed for biochemical analysis. Criteria for sensitization were based on the coefficient of variance (CV) of the day 5/day 1 beam break ratio in the saline group (CV = SD/mean) as described previously (Boudreau and Wolf, 2005). The CV provides a measure of variability within the saline group. A cocaine-injected rat was considered sensitized if its increase in activity over the course of cocaine treatment (day 5/day 1 beam break ratio) exceeded the CV of the saline. For this analysis, day 5/day 1 beam break ratios were calculated based on the first 30 min of activity after injection.
Preparation of brain homogenates and subcellular fractionation.
After the behavioral sessions, either 1 or 21 d after the last daily injection of cocaine or saline, rats were anesthetized with isoflurane and decapitated. Brains were rapidly removed, and a 2.5 mm coronal section containing the NAc was obtained. The NAc containing both the core and shell subregions was dissected bilaterally on an ice-cold platform and immediately transferred to liquid nitrogen in an Eppendorf tube. After the termination of the dissections, pools of bilateral NAc tissue blocks from two animals were merged to obtain enough material for subcellular fractionation. Tissue was then homogenized in homogenization buffer (320 mm sucrose, 10 mm Tris-HCl, pH 7.4, 1 mm EDTA, 1 mm EGTA, protease inhibitor mixture; Sigma). Subcellular fractionation was performed as described previously (Schumann et al., 2009). Briefly, homogenates were centrifuged at 1000 × g for 5 min at 4°C to remove nuclei and large debris. The supernatant was centrifuged at 10,000 × g to obtain a crude synaptosomal fraction and subsequently was lysed hypoosmotically and centrifuged at 25,000 × g to pellet a synaptosomal membrane fraction. Concurrently, the supernatant above the crude synaptosomal fraction was centrifuged at 165,000 × g to obtain a cytosolic fraction and a light membrane fraction. After each centrifugation, the resulting pellet was rinsed briefly with ice-cold homogenization buffer before subsequent fractionations were performed to avoid possible crossover contamination.
Western blot analysis.
Protein (50 μg) from brain homogenates was resolved by 10% SDS-PAGE and transferred to a nitrocellulose membrane. The membranes were incubated overnight at 4°C with appropriate primary antibodies and then incubated (1 h at room temperature) with appropriate IRDye conjugated fluorescent secondary antibodies (Rockland Immunochemicals). IRDye conjugates are optimized for the Odyssey Infrared Imaging System (LI-COR Biosciences). The bands were quantitatively analyzed by densitometry, with National Institutes of Health Image providing peak areas, and values were expressed as the percentage of control. The antibodies used in this study were as follows: anti-NR1 (Zymed Laboratories), anti-NR2B (Santa Cruz Biotechnologies), anti-NR2A and anti-GluR1 (Millipore Bioscience Research Reagents), anti-pERK and anti-ERK (Cell Signaling Technology), and anti-tubulin (Sigma).
Surgery and microinjection.
Rats were anesthetized with xylazine plus ketamine (0.15:0.85), and guide cannulae (28 gauge; Small Parts) were implanted bilaterally 2 mm above the NAc to minimize damage to the target site [the coordinates were as follows: 1.7 mm anterior to bregma, ±2.0 mm mediolateral, 4.6 mm ventral to the skull surface, according to the atlas of Paxinos and Watson (1998)]. One week after recovery, psychomotor sensitization was performed as described above. Drug or vehicle was infused using Hamilton syringes (Hamilton) over 2 min into the NAc at a pump-controlled rate of 0.250 μl per minute via injection cannulae extending 2 mm beyond the guide cannula tip. Injection cannulae were left in place for an additional 2 min to allow for drug absorption. U0126 (Calbiochem) was diluted to a concentration of 1 μg/μl in 5% DMSO and 6% Tween 80 in PBS, as described previously (Carnicella et al., 2008). A total of 0.5 μl of U0126 (1 μg/μl) or the appropriate vehicle per side was infused into the NAc 1 h before rats were killed. Locations of cannula were verified in 30 μm coronal sections stained with Cresyl Violet (see Fig. 6B).
Figure 6.

Inhibition of ERK activation abolished cocaine-induced increase in the expression of the GluR1 subunit in the NAc 21 d after withdrawal from cocaine. A, Rats were microinjected with the MEK inhibitor U0126 directly into the NAc once a day on days 20 and 21 after withdrawal from cocaine. One hour after the second microinjection, rats were immediately killed and NAc tissue blocks were dissected and treated as described in Figure 2. The levels of GluR1 and pERK2 were detected using specific antibodies and normalized to tubulin or total ERK2, respectively. The histogram depicts quantification of the protein levels of GluR1 subunits and the phosphorylation levels of ERK2 in the total homogenate fraction and synaptosomal membrane fraction. **p < 0.01, significant differences between cocaine vehicle and saline vehicle; #p < 0.05; ##p < 0.01 cocaine U0126 versus cocaine vehicle. B, Representative coronal section of cannula placement in the NAc. Drawings are adapted from the rat atlas of Paxinos and Watson (1998). Note that the guide cannula was placed 2 mm above the injection site. The arrow indicates the location of the guide cannula tip.
Statistical analysis.
Data are presented as mean ± SEM percentage of control. The data from the Western blot assays in Figures 2 and 3 were analyzed using one-way ANOVA followed by a post hoc Dunnett's test. In Figures 5 and 6, data were analyzed using 2 × 2 factorial design. Significant main effects and interactions (p < 0.05) from the factorial ANOVA were followed by simple ANOVA and post hoc Tukey test. For the behavioral tests, pairwise comparisons were done using independent t test. Two-way ANOVA model for repeated-measures followed by a post hoc Tukey test for multiple comparisons was applied to the data to test the time effect, cocaine effect, ifenprodil effect, and the interactions between them. The within-subject effects were assessed using the Greenhouse–Geisser test. The accepted value of significance for all tests was p < 0.05. Additionally, post hoc analyses are indicated by asterisks in the figures but are not described in the results.
Figure 2.
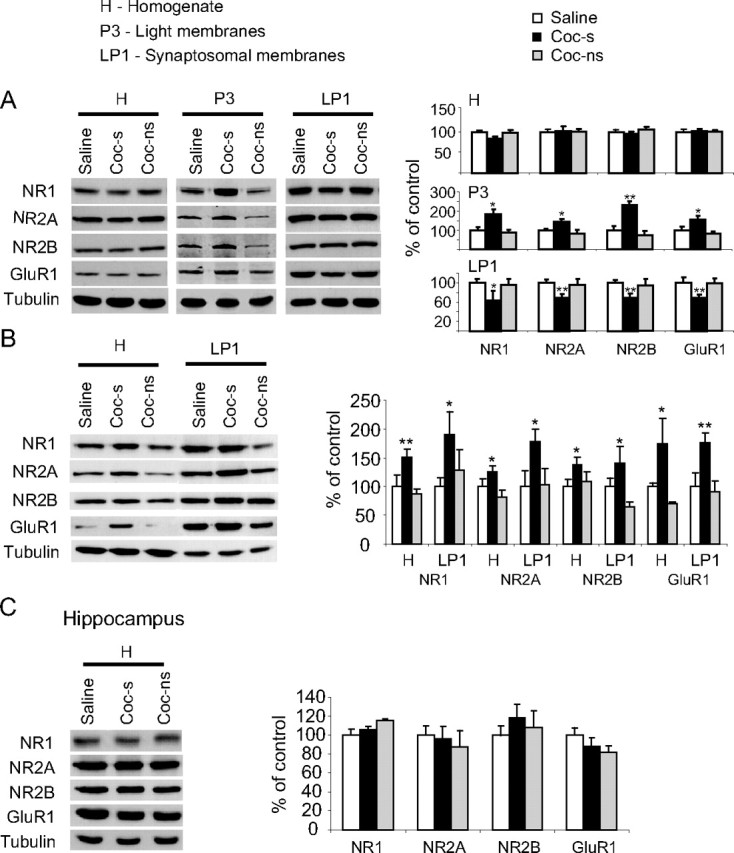
Repeated cocaine injections cause time-dependent alteration of NMDAR and GluR1 subunits in the NAc. A, B, One day (A) or 21 d (B) after withdrawal from cocaine, rats were killed, and NAc tissue blocks were dissected, homogenized, and subcellular fractionation was performed as described in Materials and Methods. Samples (40–50 μg) from saline (saline), cocaine-sensitized (coc-s), and cocaine-nonsensitized (coc-ns) rats were resolved by SDS-PAGE, and the levels of NR1, NR2A, NR2B, and GluR1 were detected using specific antibodies and normalized to tubulin. The bar histograms represent normalized levels of NMDAR and GluR1 subunits in the homogenate, synaptosomal, or light membranes (only for A) fractions, plotted as percentage of control ± SEM of four independent experiments (pools of bilateral NAc tissue blocks from 2 rats in each experiment). C, Hippocampal tissue blocks were analyzed as in A. Asterisks indicate significant differences between cocaine-treated (sensitized) and saline-treated rats (*p < 0.05; **p < 0.01).
Figure 3.
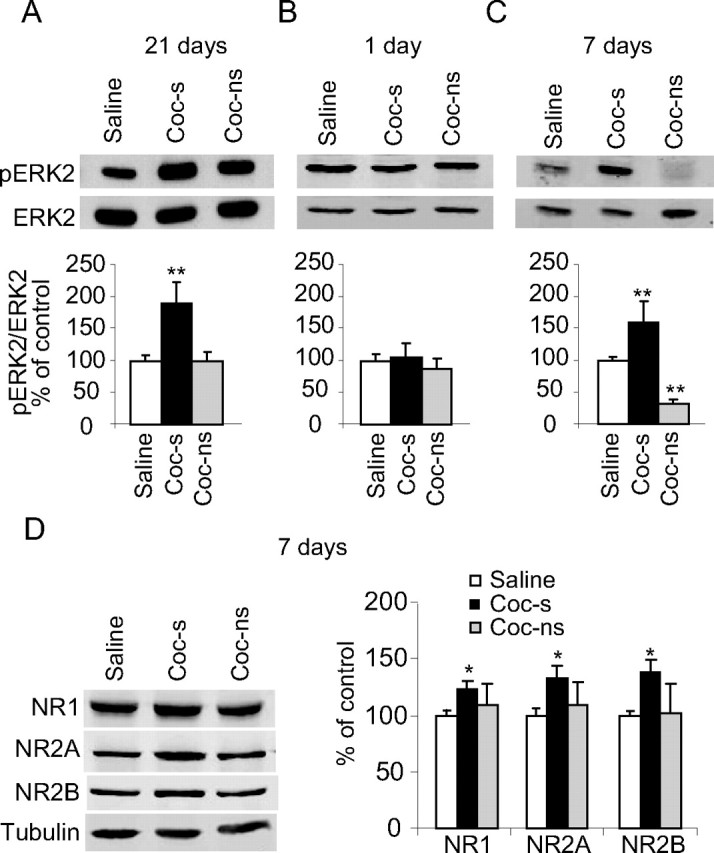
Time-dependent increase in ERK activity after withdrawal from cocaine. A, Rats were killed 21 d after withdrawal from cocaine. Membranes were probed with anti-phospho–p44/42 ERK and anti-p44/42 ERK antibodies. Histograms depict the level of ERK2 phosphorylation (phospho-p42) normalized to total ERK (total p42) from six independent experiments (pools of 2 rats in each experiment). Data are presented as mean ± SEM percentage of saline (saline vs cocaine sensitized, **p < 0.01). B, Rats were killed 1 d after withdrawal from cocaine. Histograms depict the level of ERK2 phosphorylation (normalized to total ERK) from four independent experiments (pools of 2 rats in each experiment). There was no significant difference between saline- and cocaine-injected rats. C, Rats were killed 7 d after withdrawal from cocaine. Histograms depict the level of ERK2 phosphorylation (normalized to total ERK) from four independent experiments (pools of 2 rats in each experiment). Data are presented as mean ± SEM percentage of control (saline vs cocaine sensitized or cocaine nonsensitized, **p < 0.01). D, Rats were killed 7 d after withdrawal from cocaine. Histograms depict the level of NMDAR subunits (normalized to total tubulin) from four independent experiments (pools of 2 rats in each experiment). Data are presented as mean ± SEM percentage of control (saline vs cocaine sensitized, **p < 0.01).
Figure 5.

Pretreatment with ifenprodil blocked cocaine-induced increase in NMDAR subunits, GluR1 expression, and ERK activity in the NAc 21 d after withdrawal from cocaine. All groups were killed 21 d after withdrawal from cocaine. The levels of NR1, NR2A-B, GluR1, and pERK2 were detected using specific antibodies and normalized to tubulin and total ERK, respectively. Histogram depicts quantification of the protein levels of NMDAR subunits, GluR1, and phosphorylation levels of ERK2 in the homogenate fraction. **p < 0.01—significant differences between cocaine-sensitized and saline; #p < 0.05; ##p < 0.01 cocaine plus ifenprodil versus cocaine plus saline. No significant differences were found between ifenprodil and saline.
Results
Repeated cocaine injections produce psychomotor sensitization in a subset of rats
To induce cocaine psychomotor sensitization, we paired repeated cocaine injections with exposure of the rats to a distinct test environment. After 2 d of saline injections to habituate the rats to the procedure, the immediate locomotor response to a fixed dose of cocaine (15 mg/kg) or saline (1 ml/kg) was measured. Experimental design is shown in Figure 1A. By applying the criteria of sensitization (CV) described in Materials and Methods, we found no significant difference between saline- and cocaine-treated rats (t(1,77) = −1.48, p = 0.142, t test). However, as shown in Figure 1A, it was apparent that only a subset of cocaine-treated rats had developed psychomotor sensitization. A highly significant group effect was found, F(2,76) = 8.29, p < 0.001, one-way ANOVA, when the sensitized group differed from the saline group (*p < 0.05) and from the nonsensitized group (**p < 0.01). These results were similar to previous studies (Churchill et al., 1999; Boudreau and Wolf, 2005), which reported a high proportion of nonsensitized Sprague Dawley rats, although different pairing protocols were used compared with the present study in which Sabra rats were daily injected in the locomotion chambers. The nonsensitized rats served as an additional control group in our biochemical analysis. As shown in Figure 1B, a typical development profile of psychomotor sensitization across the 5 d of treatment was found in cocaine-sensitized rats. The following statistically significant main effects were found: day (within subjects: F(3.31,208.67) = 4.51, p < 0.003); group (between subjects: F(2,63) = 72.56, p < 0.0001); day × group (F(6.62,208.67) = 12.72, p < 0.0001). Post hoc group differences within each time point are indicated by asterisks.
Figure 1.
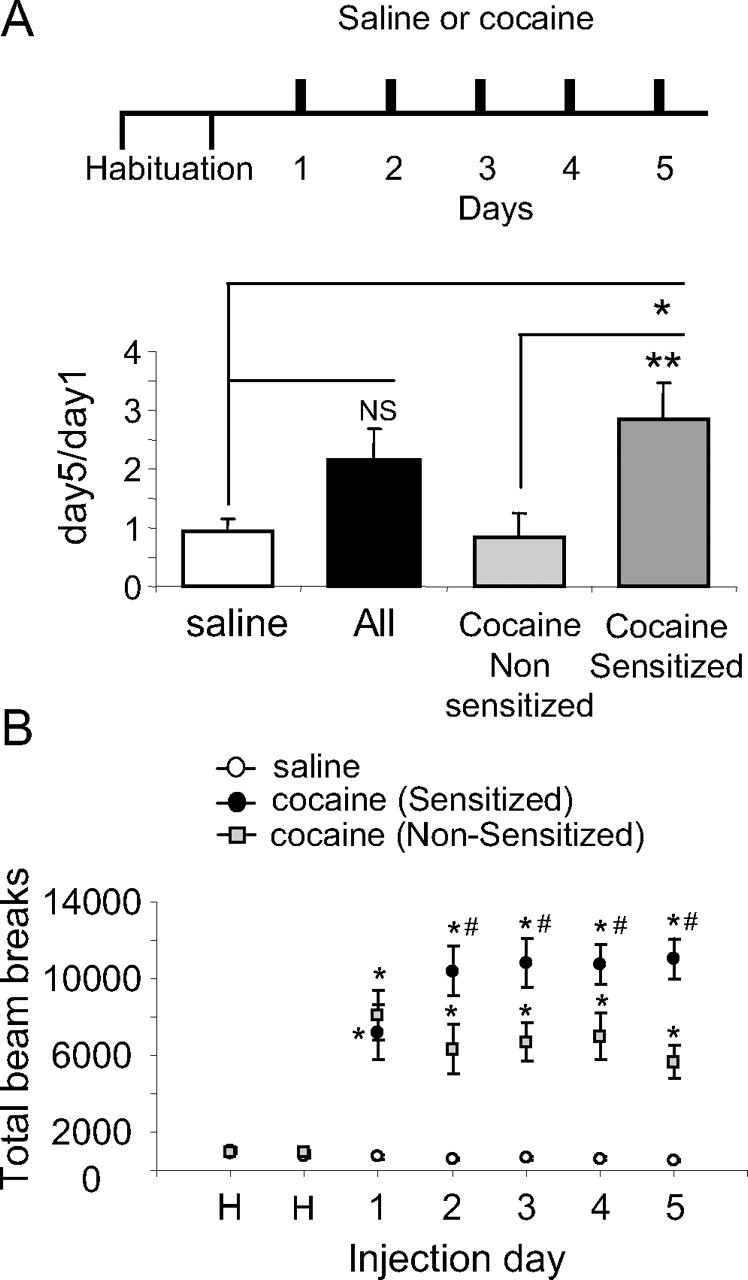
Repeated cocaine injections cause psychomotor sensitization in a subset of rats. A, Cocaine-treated rats were divided into sensitized (60%) and nonsensitized (40%) subgroups (for criteria, see Materials and Methods). The histogram depicts the day 5/day 1 mean ratios ± SEM of total beam breaks (n = 68, all subjects; n = 20, nonsensitized; n = 48, sensitized; n = 20, saline). *p < 0.05 cocaine sensitized versus saline; **p < 0.01 cocaine sensitized versus cocaine nonsensitized; NS, nonsignificant. B, Plot represents locomotor activity for cocaine-sensitized, cocaine-nonsensitized, and saline-treated rats receiving repeated cocaine (15 mg/kg, i.p) or saline (1 ml/kg, i.p) injections for 5 consecutive days. Data are presented as mean ± SEM of total beam breaks. Post hoc group differences within each time point are designated as follows: *different from saline; #different from nonsensitized.
Repeated injections of cocaine induced time-dependent alteration of NMDAR and GluR1 subunits in the NAc
We first determined whether the changes in GluR1 are accompanied by time-dependent alterations of the NMDAR in the NAc. For this purpose, the first group was killed 1 d after the last cocaine injection (day 6) and the second group 21 d later. Brain tissue from the NAc was dissected and homogenized. To determine the intracellular localization of these subunits, subcellular fractionation was performed, and synaptosomal membranes fraction was isolated as described previously (Schumann et al., 2009). As shown in Figure 2A, rats killed 1 d after withdrawal from cocaine did not show any change either in the NMDAR subunits or in GluR1 protein levels in the total homogenate (p > 0.5). However, one-way ANOVA revealed a significant decrease in all NMDAR subunits and GluR1 protein levels in the synaptosomal membranes in rats that developed psychomotor sensitization (coc-s) compared with saline controls (saline) (NR1: F(2,9) = 4.99, p < 0.05; NR2A: F(2,15) = 9.00, p < 0.01; NR2B: F(2,12) = 10.02, p < 0.01; GluR1: F(2,12) = 18.28, p < 0.01). In addition, we found an increase in these subunits in the light membrane fraction (representing the intracellular membranal content of the cells) of the cocaine-sensitized rats (NR1: F(2,6) = 10.33, p < 0.05; NR2A: F(2,6) = 9.03, p < 0.05; NR2B: F(2,6) = 45.50, p < 0.01; GluR1: F(2,6) = 12.78, p < 0.01) (Fig. 2). Since no change in protein levels of these subunits was evident in the total homogenates, these results suggest that NMDAR subunits and GluR1 were redistributed within the cells but not degraded in the NAc 1 d after withdrawal from cocaine. Next, we predicted that NMDAR expression is affected in a similar manner as the GluR1 (Boudreau and Wolf, 2005) after prolonged withdrawal from repeated cocaine injections. To test this hypothesis, brain tissue from the NAc of rats 21 d after withdrawal from cocaine was dissected and homogenized. Subcellular fractionation was then performed. One-way ANOVA revealed that cocaine-sensitized rats (but not nonsensitized rats), killed 21 d after withdrawal from cocaine, showed a significant increase in all NMDAR subunits (NR1: F(2,6) = 4.96, p < 0.05; NR2A: F(2,6) = 13.77, p < 0.01; NR2B: F(2,8) = 6.78, p < 0.05) and in GluR1 (GluR1: F(2,12) = 18.87, p < 0.01) in the synaptosomal membranes (Fig. 2B). Furthermore, in the cocaine-sensitized rats, we found an increase in total protein levels of all NMDAR subunits in the homogenates (NR1: F(2,12) = 10.78, p < 0.01; NR2A: F(2,6) = 5.67, p < 0.05; NR2B: F(2,9) = 8.16, p < 0.01) and in GluR1 (F(2,12) = 5.07, p < 0.05), indicating an increased protein synthesis of these subunits. To examine whether these changes are specific to the NAc, we repeated the experiments in hippocampal tissue punches. One-way ANOVA revealed no significant differences (p > 0.5) both in NMDAR and GluR1 subunits expression in the total homogenates (Fig. 2C) or the synaptosomal membranal fraction (data not shown). Together, these results suggest that prolonged withdrawal from cocaine increased the expression of NMDAR subunits in the NAc of sensitized rats in a region-specific manner.
Prolonged withdrawal from repeated cocaine injections is associated with time-dependent increases in ERK activity
Next, we studied whether the time-dependent increases in NMDAR expression in the NAc are associated with time-dependent increases in ERK activity. To test this hypothesis, NAc punches from cocaine-treated (sensitized and nonsensitized) and saline-treated rats were dissected 21 d after withdrawal from cocaine and homogenized. ERK phosphorylation was then detected and compared with total ERK. As shown in Figure 3A, cocaine-sensitized rats showed a significant increase in ERK2 phosphorylation (F(2,10) = 7.6, p < 0.01), indicating an increase in ERK activity. In contrast, no significant increase in ERK activity was observed in rats that failed to develop sensitization (p > 0.5).
This result led us to hypothesize that cocaine-induced increases in the expression of NMDARs leads to activation of the ERK signaling pathway, which in turn increases the expression and/or surface expression of the GluR1 subunit, as we previously found in a model of head injury in mice (Schumann et al., 2008). Therefore, we predicted that the time-dependent increase in NMDAR subunits expression correlates with time-dependent increases in ERK activity. To test this hypothesis, we assessed the time course of ERK activity after cessation of repeated cocaine injections, 1 or 7 d after withdrawal from cocaine. As shown in Figure 3B, the level of ERK2 phosphorylation in cocaine-sensitized rats was unchanged compared with saline-treated rats 1 d after withdrawal from cocaine (p > 0.5). However, as shown in Figure 3C at withdrawal day 7, we found a significant increase in ERK activity (phospho-ERK2) in cocaine-sensitized groups (F(2,9) = 7.27, p < 0.05), similar to the increase that we found after 21 d of withdrawal from cocaine.
If the increase in ERK activity indeed results from increased expression of NMDAR subunits, then we predicted that increased NMDAR occurs at a similar time course as ERK activation. We, therefore, measured NMDAR subunits membranal levels at withdrawal day 7 and found a significant increase in all NMDAR subunits protein levels in cocaine-sensitized but not in nonsensitized rats (NR1: F(2,6) = 7.20, p < 0.05; NR2A: F(2,6) = 17.83, p < 0.01; NR2B: F(2,6) = 6.35, p < 0.05) (Fig. 3D). Together, these findings demonstrate that ERK phosphorylation in the NAc is differentially regulated during drug-free withdrawal periods as recently reported (Kim and Kim, 2008), likely as a result of increased NMDAR expression.
Inhibition of NR2B-containing NMDARs attenuates the development of cocaine psychomotor sensitization
If cocaine-induced increase in NMDAR causes an increase in AMPAR expression via activation of the ERK signaling pathway, we speculated that inhibition of NMDAR will abolish both cocaine-induced increases in ERK phosphorylation and GluR1 protein levels on withdrawal day 21. To test this hypothesis, we first assessed the role of the NR2B subunit by using systemic injections of ifenprodil, which is representative of a class of NMDAR antagonists (phenylethanolamines) with high selectivity for NR2B-containing receptors (Kew and Kemp, 1998; Chenard and Menniti, 1999) and crosses the blood–brain barrier.
We first assessed the effects of ifenprodil on spontaneous locomotor activity. As illustrated in Figure 4A, acute injection of ifenprodil in different doses (10, 20 mg/kg) that were previously shown to effectively block morphine reinstatement (Ma et al., 2007) had no effect on the basal locomotor activity. However, at a higher dose (30 mg/kg), ifenprodil reduced spontaneous locomotor activity (Fig. 4A). Therefore, 10 mg/kg of ifenprodil was chosen as the optimal dose for examining the effects of NR2B blockade on the development of psychomotor sensitization. Four groups of rats were injected daily for 5 consecutive days in the locomotion chamber. Rats received cocaine (15 mg/kg) or saline (1 ml/kg) 30 min after pretreatment with ifenprodil or vehicle. Experimental design is shown in Figure 4B (for more details, see Materials and Methods). The locomotor response of rats treated with cocaine without ifenprodil pretreatment enhanced progressively, demonstrating psychomotor sensitization (Fig. 4B). In contrast, when ifenprodil (10 mg/kg) was injected 30 min before each cocaine injection, the locomotor response of the rats remained similar during the 5 d of treatment. The three-way ANOVA demonstrated significant main effects of day (F(2.77,94.28) = 10.94, p < 0.0001), cocaine (F(1,34) = 81.84, p < 0.0001), ifenprodil (F(1,34) = 7.04, p < 0.02), and significant interactions between cocaine and ifenprodil (F(1,34) = 6.46, p < 0.02), time and cocaine (F(2.77,94.28) = 13.83, p < 0.0001) and time and ifenprodil (F(2.77,94.28) = 5.68, p < 0.002). Together, these results indicate that inhibition of NR2B-containing NMDARs attenuated the development of cocaine psychomotor sensitization.
Figure 4.
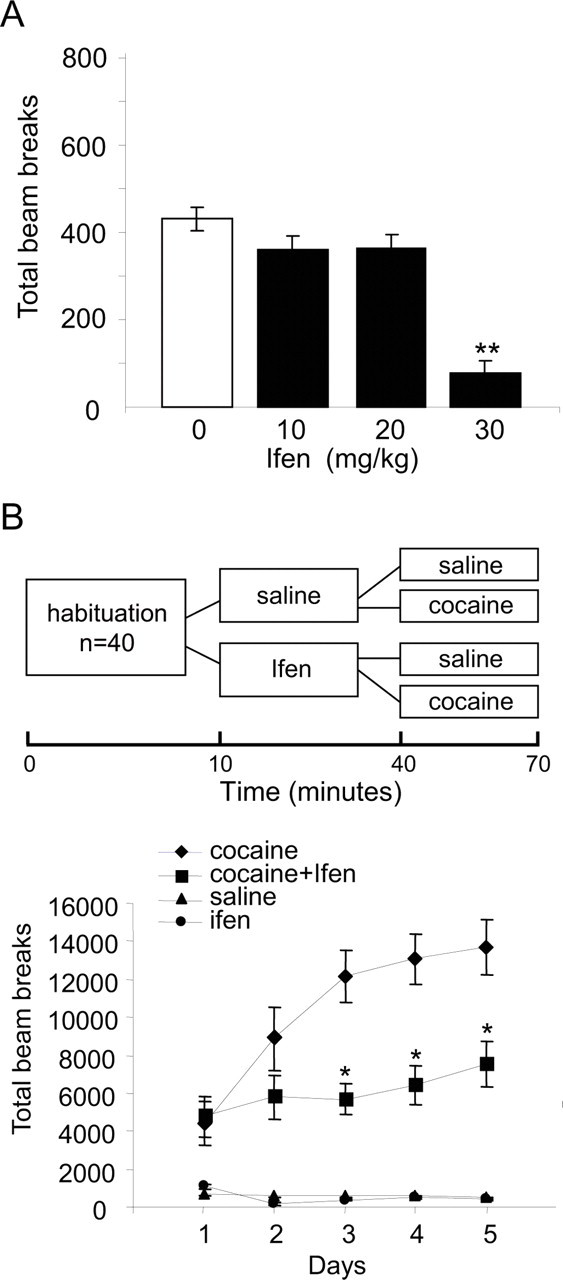
Ifenprodil prevents the development of cocaine locomotor sensitization. A, To determine the effects of ifenprodil on spontaneous locomotion, locomotor activity was measured during 60 min after acute intraperitoneal injection of vehicle (saline; 0) or the NR2B subunit-selective antagonist Ifenprodil at different doses (10, 20, and 30 mg/kg). B, Locomotor activity was measured during 5 consecutive days, and the treatment schedule is shown in the scheme. After 15 min of habituation to all rats (n = 40), rats were injected with vehicle (saline, n = 20) or ifenprodil (10 mg/kg, n = 20), and 30 min later, these two groups were divided so that half received cocaine (15 mg/kg) and half received saline (n = 10), and locomotor activity was measured for an additional 30 min. The results are presented as mean ± SEM of total beam breaks during 25 min after the last injections. *p < 0.001 cocaine plus ifenprodil versus cocaine plus saline.
Inhibition of NR2B-containing NMDARs abolished cocaine-induced increases in NMDAR expression, ERK phosphorylation, and GluR1 expression 21 d after withdrawal from cocaine
If cocaine-induced increase in NMDAR results in an increase in AMPAR expression via activation of the ERK signaling pathway, then inhibition of NMDAR should abolish both cocaine-induced increases in ERK phosphorylation and in GluR1 protein levels, 21 d after withdrawal from cocaine. As shown in Figure 5, we found that ifenprodil injection blocked cocaine-induced increases in NMDAR subunits, GluR1, and ERK2 phosphorylation. Data were analyzed using the factors of cocaine dose (0 and 15 mg/kg) and ifenprodil dose (0 and 10 mg/kg). The analysis of the data from the Western blot assays revealed significant interactions between cocaine dose and ifenprodil dose for NMDAR subunits (NR1: F(1,16) = 10.44, p < 0.01; NR2A: F(1,16) = 9.20, p < 0.01; NR2B: F(1,16) = 8.56, p < 0.01) (Fig. 5C), GluR1, and phosphorylated ERK (F(1,16) = 10.37, p < 0.01; and F(1,16) = 12.23, p < 0.01, respectively) (Fig. 5A,B). Post hoc group differences are indicated in Figure 5. These results indicate that NMDARs, and in particular its NR2B subunits, are necessary to induce psychomotor sensitization to cocaine and underlie some of the long-lasting neuroadaptations that occur in the NAc after withdrawal from the drug.
Inhibition of ERK abolished cocaine-induced increase in GluR1 expression 21 d after withdrawal from cocaine
Finally, we studied the involvement of the ERK signaling in cocaine-induced increase in GluR1 expression. To test this possibility, we blocked the activation of ERK1/2 by microinjecting the MEK inhibitor U0126 directly into the NAc. As shown in Figure 6, we found that inhibition of ERK activity by microinjections of the MEK inhibitor U0126 (Davies et al., 2000), given on days 20 and 21, blocked the increases in ERK phosphorylation and GluR1. Data were analyzed using the factors of cocaine dose (0 and 15 mg/kg) and U0126 dose (0 and 0.5 μg per side). The analysis of the data from the Western blot assays revealed a significant interaction between cocaine dose and U0126 dose for both GluR1 and phosphorylated ERK in total homogenates (F(1,16) = 10.35, p < 0.01; and F(1,16) = 15.55, p < 0.01, respectively) and the synaptosomal membrane fraction (F(1,16) = 19.97, p < 0.01; and F(1,16) = 20.50, p < 0.01, respectively). Post hoc group differences are indicated in Figure 6. Together, these results indicate that cocaine-induced increase in GluR1 expression is mediated by the ERK signaling pathway.
Discussion
Our results indicate that withdrawal from repeated cocaine injections induced time-dependent alterations in glutamate receptors and signaling pathways involved in their activity. First, we found an increase in NMDAR subunits and GluR1 expression in the NAc 21 d, but not 1 d, after withdrawal from cocaine. After 1 d of withdrawal, we did not observe any changes in NMDAR and GluR1 expression; however, these subunits were redistributed in NAc cells, as reflected by an increase in their intracellular expression. In parallel, ERK activity gradually increased after withdrawal from cocaine and was correlated with the time-dependent increases in NMDAR and GluR1. Inhibition of NR2B attenuated the development of cocaine psychomotor sensitization and abolished the cocaine-induced increases in GluR1 expression and ERK phosphorylation. Finally, intra-NAc injection of the MEK inhibitor U0126 blocked cocaine-induced increases in GluR1 expression. These findings suggest that alterations in NMDAR are part of the triggering mechanism which enhances the excitability of NAc neurons after prolonged withdrawal from cocaine, and these changes may be responsible for the increased behavioral response to cocaine (Baker et al., 2003) and cocaine-associated stimuli (Conrad et al., 2008).
Electrophysiological and biochemical evidence for synaptic plasticity in the NAc
Synaptic plasticity is required for neuroadaptations that result from a wide range of environmental stimuli. Emerging evidence suggests that addictive drugs elicit or modify synaptic plasticity in the mesolimbic system and have important behavioral consequences (Kauer and Malenka, 2007). NMDAR-dependent LTP and long-term depression (LTD) have been reported to occur in the NAc (Kombian and Malenka, 1994; Thomas et al., 2000; Schramm et al., 2002). To explore the changes in synaptic plasticity in the NAc after prolonged withdrawal from repeated cocaine administration, the ratio of AMPAR/NMDAR currents was used as an indicator of synaptic plasticity in vivo. Cocaine challenge given 10–14 d after withdrawal decreased this ratio in the NAc shell indicating the occurrence of LTD (Thomas et al., 2001). However, when measured before challenge, this ratio was dramatically increased, representing LTP (Kourrich et al., 2007). These results are inconsistent with our findings indicating an increase in NMDAR subunits and GluR1, and therefore, one can speculate that the electrophysiological ratio measured in the NAc shell will remain unchanged. However, several possibilities can explain this discrepancy. First, we did not differentiate between core/shell and the biochemical analysis includes both subregions of the NAc. Second, although GluR1 and NMDAR levels increased similarly, it is difficult to conclude from this quantitative comparison the exact stoichiometry of these changes that will be reflected as a change in this ratio, measured electrophysiologically. Therefore, it is possible that the relative increase in GluR1 is greater than that of the NMDAR subunits. Finally, the increase in NMDAR subunits seen biochemically may be a reflection of both synaptic and extrasynaptic receptors, whereas the electrophysiological recordings are made exclusively from synaptic receptors. The electrophysiological evidence was further supported by biochemical findings demonstrating an increase in the surface expression of GluR1 in the NAc 21 d after withdrawal from cocaine (Churchill et al., 1999; Boudreau and Wolf, 2005).
Our present results are different from those of Boudreau and Wolf (2005), who did not detect any change in GluR1 expression at this time point. However, in this study, total protein levels were determined as a sum of surface expression and intracellular pools after cross-linking with BS3, compared with our methods which measured total protein directly from the total homogenates. Moreover, using a homogenization procedure similar to ours, Churchill et al. (1999) reported that three weeks after withdrawal from cocaine, an increase in the amount of total NR1 and GluR1 was found in the NAc. Recently, it was shown that 21 d after withdrawal from cocaine, an increase in synaptic NMDAR subunits as well as postsynaptic scaffolding proteins in the synaptosomal membrane fraction was found (Ghasemzadeh et al., 2009). Together, these results indicate that increased excitability of NAc neurons is achieved by increased surface expression of GluR1; however, the mechanisms which trigger these events are unknown. Our results show that after withdrawal from cocaine, there is a parallel increase in GluR1 and NMDAR expression in the NAc. It is tempting to speculate that the activation of downstream signaling through NMDAR may mediate increased surface expression of GluR1 which represents LTP. An open question that remains to be explored is what happens to the NMDAR after a challenge cocaine injection, where both the increase in GluR1 and activation of ERK are normalized (Boudreau et al., 2007).
The ERK signaling pathway mediates the increase in GluR1 expression
Since calcium influx through the NMDAR activates the Ras/ERK pathway, and ERK in the brain is activated in response to various NMDAR-related stimuli, including LTP (English and Sweatt, 1996), ERK signaling pathway is a likely candidate to mediate the increase in GluR1. There is evidence for a role of ERK in cocaine behavioral effects (Lu et al., 2005, 2006; Girault et al., 2007), including psychomotor sensitization (Berhow et al., 1996; Valjent et al., 2004; Mattson et al., 2005, Boudreau et al., 2007). Inhibition of ERK activation abolishes the development of cocaine psychomotor sensitization and diminishes the rewarding effects of cocaine in a place preference paradigm (Pierce et al., 1999; Valjent et al., 2000, 2006; Miller and Marshall, 2005). In rats that self-administer cocaine, exposure to cocaine-related cues increases ERK phosphorylation in the central amygdala 30 but not 1 d after withdrawal from cocaine. Additionally, inhibition of ERK phosphorylation at this time decreases lever-pressing for cocaine cues (Lu et al., 2005). Our results suggest that prolonged withdrawal from repeated noncontingent cocaine exposure induces sustained activation of ERK, which was not triggered by environmental cues. We, therefore, speculate that the increased expression of the NMDAR is responsible for the sustained increase in ERK activity.
Similar to our results, Boudreau et al. (2007) demonstrated that, 14 d after withdrawal from cocaine, both ERK phosphorylation and GluR1 surface expression in the NAc are increased. Moreover, our results indicate that ERK signaling is directly responsible for the increase in GluR1 expression in the NAc. It is tempting to tie our biochemical data to expression of psychomotor sensitization. We predict that inhibition of NMDAR or ERK during development of sensitization or acutely before cocaine challenge will block expression. Although acute systemic injection of MEK inhibitor in mice failed to block the expression of sensitization (Valjent et al., 2006), direct NAc inhibition of ERK followed by cocaine challenge will reveal the role of ERK exclusively in this region.
NMDAR triggers cocaine-induced neuroadaptations after withdrawal
Previous studies have demonstrated that both systemic and direct VTA microinjection of NMDAR antagonist MK-801 prevents the development of cocaine psychomotor sensitization (Kalivas and Alesdatter, 1993). In addition, cocaine-induced LTP in the VTA is dependent on NMDAR activation, since coadministration of MK-801 with cocaine prevented the increase in AMPAR/NMDAR ratio (Ungless et al., 2001). These studies suggest that NMDAR in the VTA plays a critical role in the development of cocaine psychomotor sensitization and implicates a role for NMDAR-mediated signaling in the long-term changes that occur during repeated drug administration.
Previously, we have shown that acute cocaine exposure increases the expression and synaptic redistribution of NR2B-containing NMDAR in VTA slices (Schilström et al., 2006). Furthermore, using transgenic mice, overexpression of NR2B enhanced LTP (Tang et al., 1999), suggesting that activation of NR2B-containing NMDAR plays a critical role in triggering long-term alterations involved in synaptic plasticity. Although inhibition of NR2B by ifenprodil blocked both sensitization and biochemical adaptations in NMDAR, GluR1, and ERK seen 21 d after withdrawal from cocaine, the primary location in which the inhibition occurred remains unknown. One possibility is that acute activation of NMDAR in the VTA during the development phase triggers long-lasting biochemical changes in the NAc after withdrawal via multistep processes involving NMDAR downstream signaling. Another possibility is that activation of NR2B-containing NMDAR directly in the NAc is mediating the biochemical adaptations seen after prolonged withdrawal. Direct blockade of NR2B during development either in the VTA or the NAc will determine the brain region responsible for these changes.
One day after withdrawal from cocaine, we did not observe any changes in NMDAR and GluR1 expression; however, these subunits internalized into NAc neurons. As suggested previously (Boudreau et al., 2007), one potential mechanism for AMPAR and NMDAR internalization may be attributable to a transient surge in cocaine-induced glutamate levels that occurs only after repeated cocaine exposure (Pierce et al., 1996; Reid and Berger, 1996). Such excessive stimulation of AMPAR and NMDAR is known to rapidly internalize those receptors in a calcium-dependent manner (Carroll et al., 1999; Lissin et al., 1999; Beattie et al., 2000; Mangiavacchi and Wolf, 2004) and to lead to NMDAR-dependent LTD-like decreases in AMPAR-mediated synaptic currents (Carroll et al., 1999; Heynen et al., 2000). Together, our results demonstrate a causal link between NMDAR transmission at the early stage of cocaine psychomotor sensitization and the long-lasting neuroadaptations that persist for at least 3 weeks after withdrawal from the drug, including alterations in ERK signaling and GluR1 expression. Our findings confirm and extend previous studies on pronounced time-dependent molecular neuroadaptations in the NAc after withdrawal from cocaine.
Footnotes
This research was supported by the Israel Science Foundation (Grant No. 292/05; R.Y.) and the National Institute for Psychobiology in Israel founded by the Charles E. Smith family. R.Y. is affiliated with the David R. Bloom Center for Pharmacy and the Brettler Center for Research in Molecular Pharmacology and Therapeutics, School of Pharmacy, The Hebrew University of Jerusalem. We thank Dr. Tatyana Poltyrev for her critical help with the statistical analysis.
References
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW. Neuroadaptations in cystine–glutamate exchange underlie cocaine relapse. Nat Neurosci. 2003;6:743–749. doi: 10.1038/nn1069. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD [In Process Citation] Nat Neurosci. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Berhow MT, Hiroi N, Nestler EJ. Regulation of ERK (extracellular signal regulated kinase), part of the neurotrophin signal transduction cascade, in the rat mesolimbic dopamine system by chronic exposure to morphine or cocaine. J Neurosci. 1996;16:4707–4715. doi: 10.1523/JNEUROSCI.16-15-04707.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25:9144–9151. doi: 10.1523/JNEUROSCI.2252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnicella S, Kharazia V, Jeanblanc J, Janak PH, Ron D. GDNF is a fast-acting potent inhibitor of alcohol consumption and relapse. Proc Natl Acad Sci U S A. 2008;105:8114–8119. doi: 10.1073/pnas.0711755105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Lissin DV, von Zastrow M, Nicoll RA, Malenka RC. Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat Neurosci. 1999;2:454–460. doi: 10.1038/8123. [DOI] [PubMed] [Google Scholar]
- Chenard BL, Menniti FS. Antagonists selective for NMDA receptors containing the NR2B subunit. Curr Pharm Des. 1999;5:381–404. [PubMed] [Google Scholar]
- Churchill L, Swanson CJ, Urbina M, Kalivas PW. Repeated cocaine alters glutamate receptor subunit levels in the nucleus accumbens and ventral tegmental area of rats that develop behavioral sensitization. J Neurochem. 1999;72:2397–2403. doi: 10.1046/j.1471-4159.1999.0722397.x. [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- Ghasemzadeh MB, Mueller C, Vasudevan P. Behavioral sensitization to cocaine is associated with increased glutamate receptor trafficking to the postsynaptic density after extended withdrawal period. Neuroscience. 2009;159:414–426. doi: 10.1016/j.neuroscience.2008.10.027. [DOI] [PubMed] [Google Scholar]
- Girault JA, Valjent E, Caboche J, Hervé D. ERK2: a logical AND gate critical for drug-induced plasticity? Curr Opin Pharmacol. 2007;7:77–85. doi: 10.1016/j.coph.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Heynen AJ, Quinlan EM, Bae DC, Bear MF. Bidirectional, activity-dependent regulation of glutamate receptors in the adult hippocampus in vivo. Neuron. 2000;28:527–536. doi: 10.1016/s0896-6273(00)00130-6. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. Glutamate systems in cocaine addiction. Curr Opin Pharmacol. 2004;4:23–29. doi: 10.1016/j.coph.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Alesdatter JE. Involvement of N-methyl-D-aspartate receptor stimulation in the ventral tegmental area and amygdala in behavioral sensitization to cocaine. J Pharmacol Exp Ther. 1993;267:486–495. [PubMed] [Google Scholar]
- Kalivas PW, Stewart J. Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Brain Res Rev. 1991;16:223–244. doi: 10.1016/0165-0173(91)90007-u. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kew JN, Kemp JA. An allosteric interaction between the NMDA receptor polyamine and ifenprodil sites in rat cultured cortical neurones. J Physiol. 1998;512:17–28. doi: 10.1111/j.1469-7793.1998.017bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim JH. Time-dependent change of ERK phosphorylation levels in the nucleus accumbens during withdrawals from repeated cocaine. Neurosci Lett. 2008;436:107–110. doi: 10.1016/j.neulet.2008.02.068. [DOI] [PubMed] [Google Scholar]
- Kombian SB, Malenka RC. Simultaneous LTP of non-NMDA- and LTD of NMDA-receptor-mediated responses in the nucleus accumbens. Nature. 1994;368:242–246. doi: 10.1038/368242a0. [DOI] [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissin DV, Carroll RC, Nicoll RA, Malenka RC, von Zastrow M. Rapid, activation-induced redistribution of ionotropic glutamate receptors in cultured hippocampal neurons. J Neurosci. 1999;19:1263–1272. doi: 10.1523/JNEUROSCI.19-04-01263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Hope BT, Dempsey J, Liu SY, Bossert JM, Shaham Y. Central amygdala ERK signaling pathway is critical to incubation of cocaine craving. Nat Neurosci. 2005;8:212–219. doi: 10.1038/nn1383. [DOI] [PubMed] [Google Scholar]
- Lu L, Koya E, Zhai H, Hope BT, Shaham Y. Role of ERK in cocaine addiction. Trends Neurosci. 2006;29:695–703. doi: 10.1016/j.tins.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Ma YY, Chu NN, Guo CY, Han JS, Cui CL. NR2B-containing NMDA receptor is required for morphine-but not stress-induced reinstatement. Exp Neurol. 2007;203:309–319. doi: 10.1016/j.expneurol.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Mangiavacchi S, Wolf ME. Stimulation of N-methyl-D-aspartate receptors, AMPA receptors or metabotropic glutamate receptors leads to rapid internalization of AMPA receptors in cultured nucleus accumbens neurons. Eur J Neurosci. 2004;20:649–657. doi: 10.1111/j.1460-9568.2004.03511.x. [DOI] [PubMed] [Google Scholar]
- Mattson BJ, Bossert JM, Simmons DE, Nozaki N, Nagarkar D, Kreuter JD, Hope BT. Cocaine-induced CREB phosphorylation in nucleus accumbens of cocaine-sensitized rats is enabled by enhanced activation of extracellular signal-related kinase, but not protein kinase A. J Neurochem. 2005;95:1481–1494. doi: 10.1111/j.1471-4159.2005.03500.x. [DOI] [PubMed] [Google Scholar]
- Miller CA, Marshall JF. Molecular substrates for retrieval and reconsolidation of cocaine-associated contextual memory. Neuron. 2005;47:873–884. doi: 10.1016/j.neuron.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson CS. The rat brain in stereotaxic coordinates. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Born B, Adams M, Kalivas PW. Repeated intra-ventral tegmental area administration of SKF-38393 induces behavioral and neurochemical sensitization to a subsequent cocaine challenge. J Pharmacol Exp Ther. 1996;278:384–392. [PubMed] [Google Scholar]
- Pierce RC, Pierce-Bancroft AF, Prasad BM. Neurotrophin-3 contributes to the initiation of behavioral sensitization to cocaine by activating the Ras/Mitogen-activated protein kinase signal transduction cascade. J Neurosci. 1999;19:8685–8695. doi: 10.1523/JNEUROSCI.19-19-08685.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MS, Berger SP. Evidence for sensitization of cocaine-induced nucleus accumbens glutamate release. Neuroreport. 1996;7:1325–1329. doi: 10.1097/00001756-199605170-00022. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–291. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- Schilström B, Yaka R, Argilli E, Suvarna N, Schumann J, Chen BT, Carman M, Singh V, Mailliard WS, Ron D, Bonci A. Cocaine enhances NMDA receptor-mediated currents in ventral tegmental area cells via dopamine D5 receptor-dependent redistribution of NMDA receptors. J Neurosci. 2006;26:8549–8558. doi: 10.1523/JNEUROSCI.5179-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm NL, Egli RE, Winder DG. LTP in the mouse nucleus accumbens is developmentally regulated. Synapse. 2002;45:213–219. doi: 10.1002/syn.10104. [DOI] [PubMed] [Google Scholar]
- Schumann J, Alexandrovich GA, Biegon A, Yaka R. Inhibition of NR2B phosphorylation restores alterations in NMDA receptor expression and improves functional recovery following traumatic brain injury in mice. J Neurotrauma. 2008;25:945–957. doi: 10.1089/neu.2008.0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann J, Michaeli A, Yaka R. Src-protein tyrosine kinases are required for cocaine-induced increase in the expression and function of the NMDA receptor in the VTA. J Neurochem. 2009;108:697–706. doi: 10.1111/j.1471-4159.2008.05794.x. [DOI] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Malenka RC, Bonci A. Modulation of long-term depression by dopamine in the mesolimbic system. J Neurosci. 2000;20:5581–5586. doi: 10.1523/JNEUROSCI.20-15-05581.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Kalivas PW, Shaham Y. Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. Br J Pharmacol. 2008;154:327–342. doi: 10.1038/bjp.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Pages C, Besson MJ, Maldonado R, Caboche J. Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J Neurosci. 2000;20:8701–8709. doi: 10.1523/JNEUROSCI.20-23-08701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Pagès C, Hervé D, Girault JA, Caboche J. Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur J Neurosci. 2004;19:1826–1836. doi: 10.1111/j.1460-9568.2004.03278.x. [DOI] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Trzaskos JM, Girault JA, Hervé D. Role of the ERK pathway in psychostimulant-induced locomotor sensitization. BMC Neurosci. 2006;7:20. doi: 10.1186/1471-2202-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:679–720. doi: 10.1016/s0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]