

Abstract
The 42-aa-long β-amyloid protein—Aβ1-42—is thought to play a central role in the pathogenesis of Alzheimer's disease (AD) (Walsh and Selkoe, 2007). Data from AD brain (Shankar et al., 2008), transgenic APP (amyloid precursor protein)-overexpressing mice (Lesné et al., 2006), and neuronal cultures treated with synthetic Aβ peptides (Lambert et al., 1998) indicate that self-association of Aβ1-42 monomers into soluble oligomers is required for neurotoxicity. The function of monomeric Aβ1-42 is unknown. The evidence that Aβ1-42 is present in the brain and CSF of normal individuals suggests that the peptide is physiologically active (Shoji, 2002). Here we show that synthetic Aβ1-42 monomers support the survival of developing neurons under conditions of trophic deprivation and protect mature neurons against excitotoxic death, a process that contributes to the overall neurodegeneration associated with AD. The neuroprotective action of Aβ1-42 monomers was mediated by the activation of the PI-3-K (phosphatidylinositol-3-kinase) pathway, and involved the stimulation of IGF-1 (insulin-like growth factor-1) receptors and/or other receptors of the insulin superfamily. Interestingly, monomers of Aβ1-42 carrying the Arctic mutation (E22G) associated with familiar AD (Nilsberth et al., 2001) were not neuroprotective. We suggest that pathological aggregation of Aβ1-42 may also cause neurodegeneration by depriving neurons of the protective activity of Aβ1-42 monomers. This “loss-of-function” hypothesis of neuronal death should be taken into consideration when designing therapies aimed at reducing Aβ burden.
Introduction
The dominant hypothesis about the pathogenesis of Alzheimer's disease (AD) states that β-amyloid protein (Aβ1-42) aggregates into toxic species able to disrupt synaptic function and eventually leading to neuronal loss (Walsh and Selkoe, 2007). In Tg2576 mice expressing a human APP (amyloid precursor protein) variant linked to AD, a 56 kDa soluble Aβ assembly (Aβ*56) disrupts memory (Lesné et al., 2006). In the AD brain, both Aβ1-42 monomers and dimers have been isolated (Klyubin et al., 2008; Shankar et al., 2008), and dimers have been shown to impair synaptic plasticity in mouse hippocampal slices (Shankar et al., 2008). Different from native Aβ1-42 dimers and cell-secreted or synthetic oligomers, which are neurotoxic (Lambert et al., 1998; Walsh et al., 2002; Klyubin et al., 2008; Shankar et al., 2008), Aβ1-42 monomers are devoid of neurotoxicity in a number of different studies. We wondered whether Aβ1-42 monomers could instead act to support neuronal survival. Aβ1-42 is found in the CSF of nondemented individuals (Shoji, 2002), and has been implicated as a physiological regulator of synaptic activity (Kamenetz et al., 2003). There is indirect evidence that Aβ1-42 might be neuroprotective: (1) concentrations of Aβ1-42 in the cerebral interstitial fluid of patients with acute brain injury increase as their neurological status improves and fall as their neurological status declines (Brody et al., 2008); (2) addition of Aβ1-42 to cultured neurons enhances glucose uptake and metabolism via the induction of hypoxia-inducible factor-1α (Soucek et al., 2003). We now provide a direct demonstration that Aβ1-42 is neuroprotective, and that neuroprotection is a distinct feature of peptide monomers. In addition, we demonstrate that Aβ1-42 monomers activate the phosphatidylinositol-3-kinase (PI-3-K) pathway, which is a major survival pathway in neurons (Franke et al., 1997).
Materials and Methods
Aβ peptide preparation and analysis.
Aβ1-42, Aβ42-1, and Aβ1-40 were purchased from Bachem Distribution Services. Aβ1-42 and Aβ42-1 with the Arctic mutation E22G were obtained from Innovagen. Aβ peptides were dissolved in trifluoroacetic acid (TFA) at a concentration of 1 mg/ml and sonicated for 10 min. TFA was removed by gentle streaming of argon. Peptides were then dissolved in 1,1,1,3,3,3-hexa-fluoro-2-propanol (HFIP) and incubated at 37°C for 1 h. Following argon streaming, peptides were dissolved again in HFIP, lyophilized, and then resuspended in 5 mm anhydrous dimethyl sulfoxide (DMSO) before dilution to 100 μm in ice-cold cell culture medium DMEM-F12. All peptide suspensions, with the exception of the Arctic peptides, were allowed to oligomerize overnight at 4°C, and different molecular weight-sized fractions were isolated by filtration through cutoff filters (50, 30, and 10 kDa). The Arctic peptides, which rapidly aggregate (Nilsberth et al., 2001), were incubated for 15 min at room temperature before filtration. The peptide content of the recovered fractions was quantified by Bradford reagent. All samples were frozen briefly in liquid nitrogen and stored at −20°C until use.
Samples were examined by SDS-PAGE (12%) followed by Coomassie blue staining. For dot blot analysis, all different Aβ1-42 samples (0.6 μg of each) were spotted onto a nitrocellulose membrane. The membrane was first probed with anti-oligomer A11 antibody (Biosource, 1:1000), and then reprobed with the mouse anti Aβ1-42 antibody (The Genetic Company, clone G2-13, 1:200) selective for C-terminal of Aβ1-42. Horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology, 1:5000) were used, and signals were visualized using the enhancing chemiluminescence detection system (PerkinElmer LAS).
Atomic force microscopy (AFM) images were collected by using dynamic scanning force microscopy in air, using a Multimode/Nanoscope IIIa (Digital Instruments) and etched-silicon probes (Nanosensors) with a pyramidal-shaped tip having a nominal curvature of 10 nm and a nominal internal angle of 35° (Pignataro et al., 2002). Sampling of protein structures was randomly performed on at least three sample regions with area of ∼2 μm2. Each distribution histogram has been obtained by collecting from 500 to 1000 elements. Particles heights were measured by using the Nanoscope IIIA software, then gathered inside single datasets and statistically elaborated by Origin 8.
Culture preparation.
Cultures of pure cortical neurons were obtained from rats at embryonic day 15 as described previously (Copani et al., 1999). Cultures of mixed cortical cells, containing both neurons and glia, were obtained from rats at embryonic day 17 and grown onto poly-d-lysine coated 16 mm multiwell vessels (4 × 105 cells/well) as described previously (Copani et al., 1991). Mature cultures (14–16 d in vitro) were used for the study.
Assessment of viability in pure neuronal culture.
Aβ1-42 peptides were applied to mature neuronal cultures between 8 and 12 d and maintained in the growing medium as long as necessary. For measurements of neuronal survival, assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) reduction assay, Aβ1-42 peptides were maintained up to 48 h. In the case of insulin deprivation, Aβ1-42 monomers were added once and kept for 1 week.
Assessment of NMDA toxicity in culture.
Both pure and mixed cortical cultures at maturation were exposed to 300 μm NMDA for 10 min at room temperature in a HEPES-buffered salt solution. Neuronal toxicity was examined 24 h later by light microscopy and quantified after staining with trypan blue (0.4% for 5 min). Stained neurons were counted from three-random fields/well. Lactate dehydrogenase release into the medium was also measured as described previously (Copani et al., 1991). Peptide monomers were added in combination with NMDA. In some experiments, Aβ1-42 monomers were added 24 h before the excitotoxic pulse or soon after the pulse and kept into the maintenance medium for 24 h. Where required, LY 294002 or UO126 (both at a concentration of 10 μm) were applied 30 min before the NMDA pulse. AG1024 (100 nm) and picropodophyllin (500 nm) were applied 15 min before the excitotoxic pulse.
Western blot analysis.
Western blot analysis was performed on total cell extracts (30 μg/lane) from cultures of pure cortical neurons (Copani et al., 1999) treated with Aβ1-42 monomers (0.1 μm) for 10 min. Primary antibodies were as follows: rabbit anti-p(ser 9)-GSK-3β, rabbit anti-p(ser 473)-AKT, rabbit anti-AKT, and rabbit anti-β-catenin (all at 1:1000 dilution, Cell Signaling Technology). Other primary antibodies were as follows: rabbit anti-Bcl-2 (1:200 Santa Cruz Biotechnology), rabbit anti-p(tyr 1179)-IRS1 (1:500, Millipore), rabbit anti-p(ser 302)-IRS1 (1:1000, Cell Signaling Technology), rabbit anti-IRS1 (1:1000, Millipore), and mouse anti-β-actin (1:500 Sigma-Aldrich). Specific hybridization signals were obtained by using horseradish peroxidase-conjugated secondary antibodies, followed by the enhancing chemiluminescence detection system (PerkinElmer LAS). Goat anti-rabbit antibodies labeled with IRDye 680 or IRDye 800 (1:10,000 dilution, LI-COR Biosciences) were used for IRS1 immunoblots, and hybridization signals were detected with the Odyssey Infrared Imaging System (LI-COR Biosciences). Western blot data were quantified by densitometric analysis of the hybridization signals in three different blots per experiment.
Liquid chromatography electrospray ionization mass spectrometry and matrix-assisted laser desorption ionization TOF/TOF analysis.
Liquid chromatography electrospray ionization mass spectrometry (LC-ESI-MS) analysis was performed on culture-derived peptide solutions, i.e., 1 μm Aβ1-42 monomers in a HEPES-buffered salt solution, using a Thermo-Finnigan LCQ Deca XP instrument. The chromatographic analysis, coupled with ESI-MS detection, was performed on a Zorbax 300SB-C3 (2.1 × 150 mm, 5 μm particle size) column. For matrix-assisted laser desorption ionization (MALDI) TOF/TOF analysis, the sample was separated with reverse-phase (RP) LC using a monolithic capillary column [200 μm inner diameter × 5 cm, made of PS-DVB (polystyrenedivinylbenzene polymer thermostatted at 60°C)] and spotted on the MALDI plate using an Ultimate HPLC system (LC Packing/Dionex) coupled with a Probot Micro Fraction collector (LC Packing/Dionex). The spotted samples were analyzed by a MALDI TOF/TOF tandem mass spectrometer (ABI 4800 Proteomics Analyzer, Applied Biosystems). Both MS and MS/MS data were acquired with a Nd:YAG laser, with a 200 Hz repetition rate.
Results
We obtained monomers and larger size oligomers (>50 kDa) from synthetic Aβ1-42 by a modification of Lambert's protocol (Lambert et al., 1998). At the AFM, the monomer fraction consisted of small globules of ∼1.3 nm in height with little variation in size (Fig. 1A). The various forms of Aβ aggregation were further characterized by SDS-PAGE and dot blot analysis with the A11 antibody, which detects oligomers but not monomers (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). As opposed to >50 kDa oligomers (0.1 μm), monomers of Aβ1-42 (0.1–1 μm) were not toxic to pure cultures of rat cortical neurons (Fig. 1D,E), but rather increased the expression of the antiapoptotic protein, Bcl-2 (Adams and Cory, 1998) (Fig. 1C). We therefore tested the action of Aβ1-42 monomers on different paradigms of neuronal death. We first assessed the effect of monomers on neurons that spontaneously degenerated when insulin was removed from the growing medium. Addition of Aβ1-42 monomers (0.1 μm) to the growing medium completely rescued neurons from death by trophic deprivation (Fig. 2A). We searched for an action of the Aβ1-42 monomers on survival pathways that are stimulated by insulin, such as the extracellular regulated kinase (ERK1/2) pathway and the PI-3-K pathway (Avruch, 1998). Interestingly, Aβ1-42 monomers had no effect on ERK1/2 phosphorylation but activated the PI-3-K pathway, as shown by an enhanced phosphorylation of Akt (Fig. 2B). Aβ1-42 monomers also enhanced Ser9 phosphorylation (inhibition) of the Akt substrate, glycogen-synthase kinase-3β (GSK-3β) (Fig. 2B). Inhibition of GSK-3β promotes cell survival through a variety of mechanisms including a reduced degradation of β-catenin, which then translocates into the nucleus and activates the transcription of protective genes (Willert and Nusse, 1998). As expected, intracellular levels of β-catenin showed a rapid and substantial increase in response to Aβ1-42 monomers (Fig. 2B). The PI-3-K inhibitor, LY294002 (10 μm) (Vlahos et al., 1994), reduced the rescuing effect of Aβ1-42 monomers on insulin-deprived neurons (Fig. 2A). We extended the study to a model of excitotoxic neuronal death, a process that contributes to the overall neurodegeneration in AD and other CNS disorders (Hynd et al., 2004). We first challenged pure neuronal cultures with the excitotoxin, NMDA (300 μm for 10 min), obtaining a small extent of neuronal death that was abrogated by coincubation with Aβ1-42 monomers (supplemental Table 1, available at www.jneurosci.org as supplemental material). We moved to mixed cultures of cortical cells (containing both neurons and astrocytes), which were more responsive to NMDA neurotoxicity. In these cultures, >50 kDa Aβ1-42 oligomers were slightly toxic per se, and amplified NMDA toxicity at concentrations of 100 nm. A potentiation of NMDA toxicity was also observed at concentrations of oligomers (10 nm) that were per se devoid of toxicity (Fig. 3A). In contrast, Aβ1-42 monomers were neuroprotective in the 30–100 nm concentration range (Fig. 3B), and exhibited highly protective effects not only when combined with NMDA, but also when applied before or after the NMDA pulse (Fig. 3B). The latter evidence excludes a direct interaction between Aβ1-42 monomers and NMDA receptors. Monomers of Aβ1-40 were also fully protective against NMDA toxicity, whereas monomers of the reverse peptide, Aβ42-1 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), protected to a lesser extent even at a concentration of 1 μm (Fig. 3C). We extended the study to monomers of Aβ (either 1-42 or 42-1), carrying the Arctic mutation (E22G) associated with familiar AD (Nilsberth et al., 2001) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Interestingly, monomers of the two Arctic Aβ peptides lacked any neuroprotective activity (Fig. 3C). Neuroprotection by Aβ1-42 monomers against NMDA toxicity was again sensitive to the PI-3-K inhibitor, LY294002, but not to the ERK kinase inhibitor, UO126 (Favata et al., 1998). Similarly, LY294002 reduced neuroprotection produced by monomeric Aβ1-40 and Aβ42-1 (Fig. 3D). We also tested the activity of compound AG1024, which behaves as an inhibitor of all members of the insulin receptor superfamily, including the type-1 receptor for insulin-like growth factor-1 (IGF-1), and the activity of picropodophyllin (PPP), which is a selective inhibitor of the IGF-1 receptor (Vasilcanu et al., 2004). Both compounds mimicked the action of LY294002 in reducing neuroprotection by monomeric Aβ1-42 and Aβ42-1 (Fig. 3E). AG1024 antagonized the phosphorylation of AKT and IRS1 (insulin receptor substrate 1) promoted by monomeric Aβ1-42, further indicating that the peptide effect involved the activation of IGF-1/insulin receptors (Fig. 3F).
Figure 1.

Aβ1-42 monomers: separation, characterization, and lack of neurotoxicity in culture. A, B, AFM images of low-mass (<10 kDa, A) and high-mass (>50 kDa, B) Aβ1-42 species isolated from a single suspension by cutoff filters. The respective frequencies of species in the two samples are shown on the right side. The monomer fraction consisted primarily of small globules 1.3 nm in height (mean ± SD: 1.36 ± 0.42, n = 365). In contrast, the oligomer fraction consisted of larger globules 13 nm in height (mean ± SD: 13.5 ± 10.8, n = 207). In B, the asterisk indicates structures derived from the aggregation of several oligomer species which were excluded from the statistics. C, Representative Western blot image of Bcl-2 bands in control cultures (C) and cultures treated with <10 kDa Aβ1-42 for 6 h. β-Actin bands are shown for control of loading. Quantitation of Bcl-2/β-actin ratios was as follows: control (C) = 0.7 ± 0.1; <10 kDa Aβ1-42 = 1.2 ± 0.03* (means ± SEM of three independent experiments; *significantly different from control at p < 0.05 by Student's t test). Viability of pure cortical neurons, as measured by MTT assay, following 48 h treatment with different Aβ1-42 fractions (all at 0.1 μm) or different concentrations of <10 kDa Aβ1-42, is shown in D and E, respectively. Values are means ± SEM of eight determinations from two independent experiments. *p < 0.05 (one-way ANOVA + Fisher's LSD) compared with control (CTRL).
Figure 2.
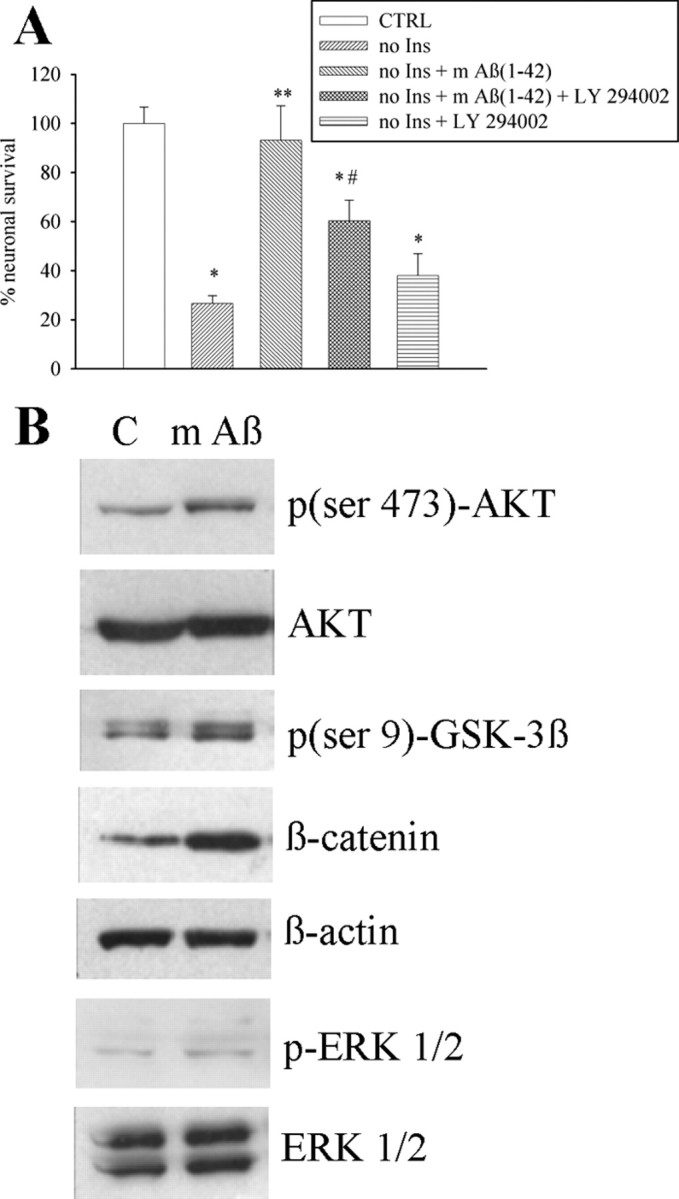
Aβ1-42 monomers support neuronal survival via the activation of the PI-3-K pathway. A, Viability of pure cortical neurons, as measured by MTT assay, insulin-deprived since plating for 1 week (no Ins), in the absence or presence of 0.1 μm Aβ1-42 monomers [m Aβ(1-42)]. Where required, the PI-3-K inhibitor, LY294002 (10 μm), was added two times (at plating and after 48 h). Values are means ± SEM of six determinations from two independent experiments. *,**,#Different from control (CTRL) (*), no Ins (**), or no Ins + m Aβ1-42 (#) at p < 0.05 by one-way ANOVA + Fisher's LSD test. B, Representative Western blot images of the PI-3-K-activated form of the serine-threonine kinase AKT [p(ser 473)-AKT], of the corresponding inactivated GSK-3β [p(ser 9)-GSK-3β], and of β-catenin levels in control cultures (C) and cultures treated with 0.1 μm monomeric Aβ1-42 (m Aβ) for 10 min. Levels of phosphorylated ERK1/2 (pERK1/2) in the same cultures are also shown. Total AKT levels, β-actin bands, or total ERK levels are shown for control of loading. Quantitation of p(ser 473)-AKT/AKT ratios was as follows: control (C) = 0.26 ± 0.04; mAβ1-42 = 0.65 ± 0.1* (means ± SEM of three independent experiments; *significantly different from control at p < 0.05 by Student's t test). Quantitation of p(ser 9)-GSK-3β/GSK-3β ratios was as follows: control (C) = 0.88 ± 0.15; mAβ1-42 = 1.44 ± 0.08* (means ± SEM of three independent experiments; *significantly different from control at p < 0.05 by Student's t test). Quantitation of β-catenin/β-actin ratios was as follows: control (C) = 0.47 ± 0.05; mAβ1-42 = 1.3 ± 0.09* (means ± SEM of three independent experiments; *significantly different from control at p < 0.05 by Student's t test). Quantitation of pERK 1/2/ERK 1/2 ratios was as follows: control (C) = 0.6 ± 0.08; mAβ1-42 = 0.55 ± 0.12. (means ± SEM of three independent experiments).
Figure 3.

Aβ1-42 monomers protect neurons against NMDA toxicity. A, NMDA-induced toxicity in mixed cortical cultures is potentiated by oligomeric Aβ1-42 [oAβ(1-42)] and (B) prevented by monomeric Aβ1-42 [mAβ(1-42)]. Toxicity was induced by a 10 min pulse with 300 μm NMDA and assessed by trypan blue staining (set to 100%) 24 h later. In the prepulse condition, Aβ1-42 monomers were applied 24 h before the NMDA pulse and cultures were extensively washed before the experiment. In the postpulse condition, Aβ1-42 monomers were applied soon after the excitotoxic pulse and kept for 24 h into the medium. Both in A and in B, values are means ± SEM of 9–18 determinations from three-six independent experiments. A, *Significantly different from the respective control (CTRL) at p < 0.05 (one-way ANOVA + Fisher's LSD test). B, *Significantly different from NMDA at p < 0.001 (one-way ANOVA + Fisher's LSD test). C, NMDA-induced toxicity in mixed cortical cultures is attenuated by the monomeric forms of Aβ1-42, Aβ1-40, and Aβ42-1, but not by Aβ1-42 or Aβ42-1 containing the Arctic mutation (m arcAβ). Values are means ± SEM of 12–18 determinations from three-six independent experiments. *Significantly different from NMDA at p < 0.001 (one-way ANOVA + Fisher's LSD test). D, The PI-3-K inhibitor LY 294002 (10 μm) prevents the neuroprotective activity of monomeric forms of Aβ1-42, Aβ1-40, and Aβ42-1. Values are means ± SEM of 8 determinations from two independent experiments. *,#Significantly different from NMDA (*) or from the respective Aβ conditions (#) at p < 0.05 (one-way ANOVA + Fisher's LSD test). [UO126] = 10 μm. E, The selective inhibitor of the insulin receptor superfamily, AG1024, and the preferential IGF-1 receptor inhibitor, picropodophyllin (PPP), prevent the neuroprotective activity of monomeric forms of Aβ1-42 and Aβ42-1. Values are means ± SEM of eight determinations from two independent experiments. *,#Significantly different from NMDA (*) or from the respective Aβ conditions (#) at p < 0.05 (one-way ANOVA + Fisher's LSD test). F, Representative immunoblots of p(ser 473)-AKT, p(tyr 1179)-IRS1, and p(ser 302)-IRS1 in pure neuronal cultures treated with 0.1 μm monomeric Aβ1-42 [mAβ(1-42)] for 5 min both in the absence and in the presence of 100 nm AG1024. Quantitation of p(ser 473)AKT/AKT ratios was as follows: control (C) = 0.5 ± 0.04; mAβ1-42 = 1.37 ± 0.1*; mAβ1-42 + AG1024 = 0.7 ± 0.08**; AG1024 = 0.55 ± 0.06 [means ± SEM of three independent experiments; *,**significantly different from control (*) or mAβ1-42 alone (**) at p < 0.05 by one-way ANOVA + Fisher's LSD test]. Quantitation of p(tyr 1179)-IRS1/IRS1 ratios was as follows: control (C) = 0.42 ± 0.12; mAβ1-42 = 1.31 ± 0.03*; mAβ1-42 + AG1024 = 0.63 ± 0.02**; AG1024 = 0.65 ± 0.08 [means ± SEM of three independent experiments; *,**significantly different from control (*) or mAβ1-42 alone (**) at p < 0.05 by one-way ANOVA + Fisher's LSD test]. Quantitation of p(ser 302)-IRS1/IRS1 ratios was as follows: control (C) = 0.39 ± 0.09; mAβ1-42 = 0.85 ± 0.06*; mAβ1-42 + AG1024 = 0.28 ± 0.11**; AG1024 = 0.46 ± 0.08 [means ± SEM of three independent experiments; *,**significantly different from control (*) or mAβ1-42 (**) alone at p < 0.05 by one-way ANOVA + Fisher's LSD test].
Discussion
Our data show that synthetic monomers of Aβ are able to support the survival of neurons developing under conditions of trophic deprivation, and also to protect mature neurons against excitotoxic death. The prosurvival effect of Aβ1-42 monomers was mediated by the PI-3-K pathway, which required the activation of IGF-1/insulin receptors. Accordingly, IRS1, which is a direct target of the IGF-1/insulin receptor tyrosine kinase, was phosphorylated on tyr 1179 within 5 min of exposure to monomeric Aβ. Monomeric Aβ also increased IRS1 phosphorylation on a serine residue (ser 302 in mice), which is required for an efficient insulin receptor signaling (Giraud et al., 2004). It is possible that Aβ1-42 monomers facilitate the activation of IGF-1/insulin receptors by locally produced IGF-1 or, alternatively, that Aβ monomers bind to IGF-1/insulin receptors, as already shown for Aβ oligomers (Xie et al., 2002; Townsend et al., 2007).
Aβ1-42 monomers might oligomerize in the culture medium into peptide species that might contribute to neuroprotection. However, this is unexpected when the peptide is coapplied with NMDA during the brief excitotoxic pulse because of the slow kinetic of Aβ1-42 self-association in vitro (Kusumoto et al., 1998). LC-ESI mass spectrometry did not detect any Aβ1-42 species (i.e., neither monomers nor oligomers) in the extracellular medium of cultures exposed to 1 μm monomeric Aβ1-42 for the time of the excitotoxic pulse. The more sensitive MALDI TOF mass spectrometry also failed to detect the peptide within the lower detection limit of 0.1 nm (data not shown). Monomeric Aβ has been shown to adsorb quickly and reversibly to lipid bilayers (Kremer and Murphy, 2003), and, therefore, Aβ1-42 monomers might have rapidly deposited on the plasma membranes in our cultures. Although we cannot exclude that membrane-bound Aβ1-42 oligomers contribute to neuroprotection, this is unlikely because concentrations of oligomers as low as 10 nm did not attenuate but rather amplified excitotoxic death.
Our evidence that Aβ1-42 monomers are neuroprotective is in line with the demonstration that inhibition of the Aβ-synthesizing enzymes, β- or γ-secretase, reduces neuronal viability (Plant et al., 2003). It is tempting to speculate that aggregation of Aβ1-42, as occurs in the AD brain, might deplete Aβ1-42 monomers, thus depriving neurons of a trophic support. The “loss-of-function” hypothesis of neuronal death must consider that monomers of Aβ1-40, which predominates over Aβ1-42 (Gregory and Halliday, 2005), were also neuroprotective. The possibility that Aβ1-40 monomers are also depleted in AD is suggested by the lower CSF levels of Aβ1-40 in mild cognitive impairment patients with a more rapid cognitive decline (Hansson et al., 2007). Data obtained with Aβ42-1 and Aβ peptides carrying the Arctic mutation (either 1-42 or 42-1) provide some insights into the relationship between peptide structure and neuroprotection. AFM analysis showed that monomers of Aβ1-42 and Aβ42-1 have comparable dimensions, which is indicative of similar folding properties. This may explain the partial neuroprotective activity of Aβ42-1. In contrast, the substitution of a charged with a neutral amino acid (E22G) in the central portion of the Arctic peptides might have caused a substantial change in polypeptide conformational features [i.e., a more bent structure as suggested by AFM imaging (see supplemental Fig. 2, available at www.jneurosci.org as supplemental material) (see also Rodziewicz-Motowidło et al., 2008)] associated with a complete loss of neuroprotective activity. This, combined with the rapid aggregation kinetics of the Arctic peptide (Nilsberth et al., 2001; Rodziewicz-Motowidło et al., 2008), might explain the aggressive and early-onset AD associated with this mutation.
Finally, the likeliness that Aβ1-42 monomers subserve a prosurvival function in the brain implies that therapeutic strategies aimed at targeting Aβ should spare forms endowed with physiological functions.
Footnotes
Support was provided by Italian Ministry for University and Research (FIRB RBNE03PX83 and FIRB RBIN04L28Y to E.R. and PRIN 2007 to A.C.) and by a University of Catania research grant to A.C.
References
- Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- Avruch J. Insulin signal transduction through protein kinase cascades. Mol Cell Biochem. 1998;182:31–48. [PubMed] [Google Scholar]
- Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, Zipfel GJ, Holtzman DM. Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science. 2008;321:1221–1224. doi: 10.1126/science.1161591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copani A, Koh JY, Cotman CW. Beta-amyloid increases neuronal susceptibility to injury by glucose deprivation. Neuroreport. 1991;2:763–765. doi: 10.1097/00001756-199112000-00008. [DOI] [PubMed] [Google Scholar]
- Copani A, Condorelli F, Caruso A, Vancheri C, Sala A, Giuffrida Stella AM, Canonico PL, Nicoletti F, Sortino MA. Mitotic signaling by beta-amyloid causes neuronal death. FASEB J. 1999;13:2225–2234. [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- Giraud J, Leshan R, Lee Y-H, White MF. Nutrient-dependent and insulin-stimulated phosphorylation of insulin receptor substrate-1 on serine 302 correlates with increased insulin signaling. J Biol Chem. 2004;279:3447–3454. doi: 10.1074/jbc.M308631200. [DOI] [PubMed] [Google Scholar]
- Gregory GC, Halliday GM. What is the dominant Abeta species in human brain tissue? A review. Neurotox Res. 2005;7:29–41. doi: 10.1007/BF03033774. [DOI] [PubMed] [Google Scholar]
- Hansson O, Zetterberg H, Buchhave P, Andreasson U, Londos E, Minthon L, Blennow K. Prediction of Alzheimer's disease using the CSF Abeta42/Abeta40 ratio in patients with mild cognitive impairment. Dement Geriatr Cogn Disord. 2007;23:316–320. doi: 10.1159/000100926. [DOI] [PubMed] [Google Scholar]
- Hynd MR, Scott HL, Dodd PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer's disease. Neurochem Int. 2004;45:583–595. doi: 10.1016/j.neuint.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JJ, Murphy RM. Kinetics of adsorption of β-amyloid peptide Aβ(1-40) to lipid bilayers. J Biochem Biophys Methods. 2003;57:159–169. doi: 10.1016/s0165-022x(03)00103-9. [DOI] [PubMed] [Google Scholar]
- Kusumoto Y, Lomakin A, Teplow DB, Benedek GB. Temperature dependence of amyloid beta-protein fibrillization. Proc Natl Acad Sci U S A. 1998;95:12277–12282. doi: 10.1073/pnas.95.21.12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Näslund J, Lannfelt L. The ‘Arctic’ APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation. Nat Neurosci. 2001;4:887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- Pignataro B, Chi L, Gao S, Anczykowski B, Niemeyer C, Adler M, Fuchs H. Dynamic scanning force microscopy study of self-assembled DNA-protein nanostructures. Appl Phys A Mater Sci Process. 2002;74:447–452. [Google Scholar]
- Plant LD, Boyle JP, Smith IF, Peers C, Pearson HA. The production of amyloid beta peptide is a critical requirement for the viability of central neurons. J Neurosci. 2003;23:5531–5535. doi: 10.1523/JNEUROSCI.23-13-05531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodziewicz-Motowidło S, Juszczyk P, Sikorska E, Spodzieja M, Kołodziejczyk AS. The Arctic mutation alters helix length and type in the 11–28 beta-amyloid peptide monomer-CD, NMR and MD studies in an SDS micelle. J Struct Biol. 2008 doi: 10.1016/j.jsb.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji M. Cerebrospinal fluid Abeta40 and Abeta42: natural course and clinical usefulness. Front Biosci. 2002;7:d997–d1006. doi: 10.2741/A826. [DOI] [PubMed] [Google Scholar]
- Soucek T, Cumming R, Dargusch R, Maher P, Schubert D. The regulation of glucose metabolism by HIF-1 mediates a neuroprotective response to amyloid beta peptide. Neuron. 2003;39:43–56. doi: 10.1016/s0896-6273(03)00367-2. [DOI] [PubMed] [Google Scholar]
- Townsend M, Mehta T, Selkoe DJ. Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J Biol Chem. 2007;282:33305–33312. doi: 10.1074/jbc.M610390200. [DOI] [PubMed] [Google Scholar]
- Vasilcanu D, Girnita A, Girnita L, Vasilcanu R, Axelson M, Larsson O. The cyclolignan PPP induces activation loop-specific inhibition of tyrosine phosphorylation of the insulin-like growth factor-1 receptor. Link to the phosphatidyl inositol-3 kinase/Akt apoptotic pathway. Oncogene. 2004;23:7854–7862. doi: 10.1038/sj.onc.1208065. [DOI] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. A beta oligomers—a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Willert K, Nusse R. β-Catenin: a key mediator of Wnt signaling. Curr Opin Genet Dev. 1998;8:95–102. doi: 10.1016/s0959-437x(98)80068-3. [DOI] [PubMed] [Google Scholar]
- Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R. Alzheimer's beta amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci. 2002;22:RC221. doi: 10.1523/JNEUROSCI.22-10-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]