

Abstract
Neuronal motility relies on actin treadmilling. In addition to regulating cytoskeletal dynamics in the cytoplasm, actin modulates nuclear gene expression. We present a hitherto unappreciated cross talk of actin signaling with gene expression governing neuronal motility. Toward this end, we used a novel approach using mutant actins either favoring (G15S) or inhibiting (R62D) F-actin assembly. Overexpressing these mutant actins in mouse hippocampal neurons not only modulated growth-cone function but also neurite elongation, which was ambiguous by traditional pharmacological interference. G15S actin enhanced neurite outgrowth and filopodia number. In contrast, R62D reduced neurite length and impaired growth-cone filopodia formation. Growth-cone collapse induced by ephrin-As, a family of repulsive axon guidance molecules, is impaired upon R62D expression, resulting in perseverance of ring-shaped F-actin filaments. R62D-induced phenotypes strongly resemble neurons lacking SRF (Serum Response Factor). SRF controls gene transcription of various actin isoforms (e.g., Actb, Acta1) and actin-binding proteins (e.g., Gsn) and is the archetypical transcription factor to study actin interplay with transcription. We show that neuronal motility evoked by these actin mutants requires SRF activity. Further, constitutively active SRF partially rescues R62D-induced phenotypes. Conversely, actin signaling regulates neuronal SRF-mediated gene expression. Notably, a nucleus-resident actin (R62DNLS) also regulates SRF's transcriptional activity. Moreover, R62DNLS decreases neuronal motility similar to the cytoplasmic R62D actin mutant although R62DNLS has no access to cytoplasmic actin dynamics. Thus, herein we provide first evidence that neuronal motility not only depends on cytoplasmic actin dynamics but also on the availability of actin to modulate nuclear functions such as gene transcription.
Introduction
During brain development, neurons pass through various phases of morphological differentiation encompassing neurite elongation, neurite differentiation in axon and dendrites, and directional navigation of growth cones toward postsynaptic targets. Actin is a key cytoskeletal component involved in many of these processes (Dent and Gertler, 2003; Pak et al., 2008). Filamentous F-actin, enriched in growth cones, is instrumental for sculpting growth-cone shape and processing of directional information by guidance cues (Gallo and Letourneau, 2004; Kalil and Dent, 2005). Contrastingly, the evidence for actin function in neurite elongation, mostly by using pharmacological inhibitors, is conflicting and here microtubules are thought to confer the driving elongation force (Dent and Gertler, 2003; Pak et al., 2008).
In addition to regulation of cytoplasmic cytoskeletal dynamics, a little appreciated property of actin signaling is modulation of gene expression so far only reported for non-neuronal cells. Actin modulates the transcriptional machinery by a nuclear-resident pool (Grummt, 2006) or by controlling nuclear translocation of MAL (MTRF-A), a Myocardin family transcription factor (Pipes et al., 2006; Posern and Treisman, 2006). In non-neuronal cells, F-actin polymerization favors MAL nuclear import, whereas G-actin prevents translocation (Miralles et al., 2003; Kuwahara et al., 2005; Vartiainen et al., 2007). Nuclear MAL cooperates with Serum Response Factor (SRF), which is a key event during cellular differentiation of many processes (Posern and Treisman, 2006; Miano et al., 2007). SRF activity is regulated by actin/MAL interaction and in turn, SRF transcriptionally controls many genes encoding Actin isoforms (Actb, Actg, Acta2) and actin-binding proteins (ABPs; e.g., Gsn) (Posern and Treisman, 2006; Sun et al., 2006). Recently, Srf mouse mutants revealed SRF's crucial function during cell migration, neurite outgrowth, axonal pathfinding, and synapse function (Alberti et al., 2005; Ramanan et al., 2005; Etkin et al., 2006; Knöll et al., 2006; Wickramasinghe et al., 2008).
Here we analyzed neuronal actin signaling by a novel approach using actin mutant isoforms known to favor or preclude actin polymerization. The actin mutants G15S and S14C favor F-actin assembly, yet differ with regard to their ABP interaction. S14C unlike G15S binds to e.g., the actin-severing protein Cofilin (Posern et al., 2004; Sarmiere and Bamburg, 2004). In opposite to G15S and S14C, R62D is excluded from F-actin thereby increasing monomeric G-actin (Posern et al., 2002, 2004). Employing these actin mutants, we uncovered a function for actin signaling in growth-cone motility modulated by ephrin-A guidance cues. Moreover these actin mutants affect neurite elongation, an actin function which by pharmacological actin interference was ambiguous. We demonstrate that actin-dependent modulation of the cytoskeleton requires nuclear SRF gene activity, as revealed by Srf−/− neurons. Conversely, cytoplasmic (R62D and G15S) and nuclear (R62DNLS) neuronal actin control SRF activity. Notably, R62D actin (cytoplasmic and nuclear) phenocopies SRF-deficient neuronal morphology providing evidence for an intricate cross talk of the nuclear and cytoplasmic compartment to shape neuronal morphology.
Materials and Methods
Neuronal cultures.
For conditional Srf mutants, see Alberti et al. (2005) and Knöll et al. (2006). P1–P2 hippocampal neurons were cultured on PLL/laminin-coated coverslips in NMEM/B27 medium for 1–2 d. Electroporation was performed with 3 μg of DNA (either actin mutant, or, as control, GFP or empty vector; Amaxa). For growth-cone collapse, cultures were incubated for 30 min at 37°C with 1 μg/ml ephrin-A5-Fc (R&D Systems) or Fc preclustered for 30 min at room temperature (rt) with 10 μg/ml anti-human IgG Fc-specific antibodies.
Immunocytochemistry.
We used mouse anti-FLAG (1:300; Sigma), rabbit anti-SRF (1:100; Santa Cruz), rabbit anti-MAL [a gift from Dr. R. Treisman, Cancer Research UK, London, UK], and rabbit anti-MAP2 antibodies (1:500; Millipore Bioscience Research Reagents). Texas-red or Alexa660-conjugated phalloidin (1:100, Invitrogen) was applied along with secondary Alexa488-, Alexa546-, or Alexa660-conjugated antibodies (1:1000). Images were recorded on a confocal microscope (Zeiss Axiovert 200 M) using Zeiss AxioCam and Axiovision software and processed using Adobe Photoshop. Objectives included 20×/0.8 Plan-Apochromat, 40×/1.3 Plan-Neofluar, 63×/1.4 Plan-Apochromat, and 100×/1.3 Plan-Neofluar.
Electron microscopy.
Cultures were pretreated in PEM buffer (100 mm PIPES, pH 6.8, 1 mm EGTA, 1 mm MgCl2) including 1% Triton X-100, 4.2% sucrose, 10 mm taxol, 10 mm phalloidin, and 0.2% glutaraldehyde for 4.5 min. Subsequently, cultures were washed for 5 min in PEM containing 1 mm taxol, 1 mm phalloidin, and 1% sucrose followed by fixation for 15 min with 2% glutaraldehyde/1% sucrose in 0.1 m sodium-cacodylate buffer, pH 7.3. After three PBS washes, we followed a published protocol (Svitkina and Borisy, 1998). The final rotary-shadowing was replaced with platinum and carbon by sputter coating with a 4 nm layer of gold-palladium. Cells were treated with 0.1% tannic acid in H2O at rt for 20 min, washed with H2O, and incubated with 0.1% aqueous uranyl acetate at 4°C for 20 min. After dehydration in a graded ethanol series, samples were critical-point-dried from CO2, sputter coated with a layer of 4 nm gold/palladium (Bal-Tec MED 010), and examined at 20 kV accelerating voltage in a Hitachi S-800 field emission scanning electron microscope.
Reporter gene assay.
We used 2 × 105 P2–P5 cerebellar neurons/well of a 24-well plate grown in NMEM/B27 medium overnight. Cells were transfected with Lipofectamine (Invitrogen) in OPTIMEM (Invitrogen) for 4 h according to manufacturer. We used 0.45 μg of SRF reporter gene construct (3DA.Luc) (Posern et al., 2002), 0.1 μg of β-galactosidase vector, 0.1–0.8 μg of actin expression vector, and 4 ng of SRF-VP16 (Schratt et al., 2002) per transfection. Luciferase activity was normalized to galactosidase 24 h after transfection.
Rho activity assay.
We used the Rhotekin-GST (Reid et al., 1996) (a gift from I. Just, Medizinische Hochschule Hannover, Hannover, Germany) to pull down active GTP-bound Rho. Rho-GST was bacterially expressed and purified via glutathione Sepharose (Bio-Rad). Lysates of P14 wild-type and Srf mutant cortices were incubated with the Rho-GST beads for 30 min at 4°C. Samples were resolved by 12.5% SDS-PAGE and blotted with an antibody recognizing RhoA, RhoB, and RhoC (Millipore).
Biochemistry.
Lysates of mock-electroporated E17 cortical neurons or electroporated with 3 μg of R62D or R62DNLS were analyzed using goat anti-MAL (1:1500; Santa Cruz), rabbit anti-MAL (a kind gift from Dr. R. Treisman), or rabbit anti-SRF antibodies (1:500; Santa Cruz). Lysis buffer consisted of 50 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 1 mm PMSF, 1 mm sodium-vanadate, 1 mm sodium-fluoride, 1 mm sodium pyrophosphate, and 1× protease inhibitor (Roche).
Statistics.
For neurite length the largest distance between neurites of one neuron was measured using Axiovision software. For growth-cone collapse, all growth cones of one neuron were scored: for percentage of full-growth-cone collapse, see Figure 3B as reference; for F-actin rings see Figure 3J as reference. Statistical analysis was performed using two-tailed t test with *, **, and *** indicating p < 0.05, 0.01, and 0.001, respectively. Error bars represent SD.
Figure 3.

R62D actin phenocopies Srf mutant growth cones. Wild-type (A–H) and SRF-deficient growth cones (I–P) were stimulated with Fc (control) or ephrin-A5-Fc. A–H, Neurons incubated with Fc (A) elaborated growth cones with individual filopodia, as do neurons expressing G15S (C) and S14C (E). Both G15S (D) and S14C (F) allowed for full growth-cone collapse upon ephrin-A5 treatment as in control (B). Contrastingly, R62D (G) induced aberrant growth-cone structures lacking filopodia. After ephrin-A5 addition, R62D growth cones did not collapse yet retain actin rings (H) very reminiscent of Srf-mutant growth cones after ephrin incubation (J, L, N, P). I–P, Srf-mutant growth cones typically did not contain filopodia (I) and retained F-actin rings upon ephrin-A treatment (J). Expression of G15S (K, L) or S14C (M, N) was not sufficient to rescue SRF ablation. Contrastingly, R62D induced, in absence of ephrin-A5, already F-actin rings (O), which cannot be further augmented by ephrin-A addition (P). Q, Quantification of the percentage of growth cones being fully collapsed as depicted in B, D, and F) R, The percentage of growth cones with F-actin rings as shown in O, J, L, N, and P is depicted. In Q and R, statistical significance is given by comparing corresponding Fc with eA5-Fc samples. Scale bar: (in A) A–P, 2 μm.
Results
SRF regulates growth-cone filopodia and filament bundling in neurites
We used platinum replica electron microscopy (EM) (Svitkina and Borisy, 1998) to uncover novel SRF-evoked neuronal morphological alterations. Neurons were directly subjected to EM (Fig. 1A,B,E,F,I,J,M,N) or simultaneously fixed and stabilized, while extracting soluble components. The latter allowed detailed views on the cytoskeleton underlying the plasma membrane (Fig. 1C,D,G,H,K,L,O,P).
Figure 1.
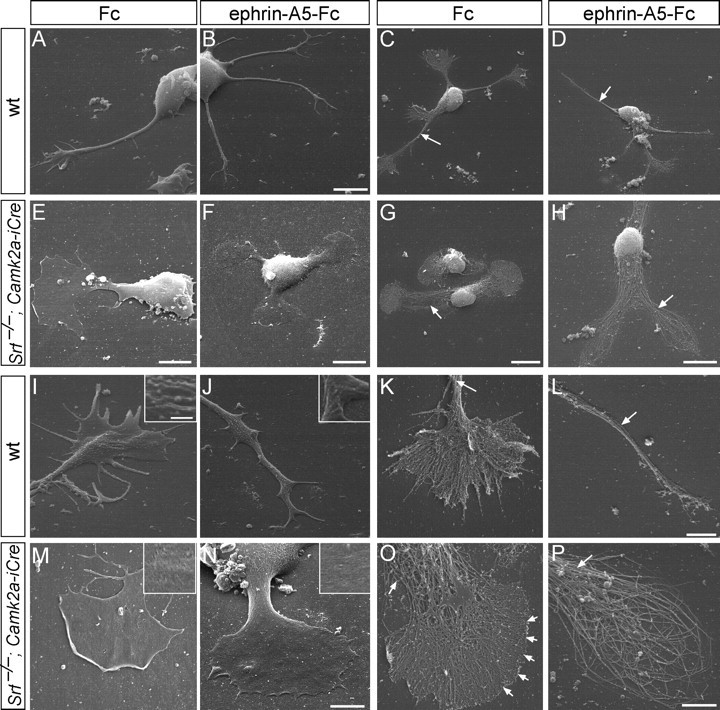
Growth-cone inflation and absence of filopodia in SRF-deficient neurons. Neurons were fixed (A, B, E, F, I, J, M, N) or peeled off the plasma membrane (C, D, G, H, K, L, O, P) followed by platinum replica EM. A–D, Wild-type neurons protruded multiple long neurites terminated by growth cones with several filopodia (A, C), which after ephrin-A treatment collapsed (B, D). Note the tight filament bundling in the neurite shaft (C, D, arrows). E–H, Srf−/− neurons were stunted in growth, typically bipolar in shape and growth-cone area almost equaled cell-body area. Ephrin-A did not collapse growth cones (F, H) and produced ring-shaped filaments (H; see P). I–L, Wild-type growth cones contained filopodia separated by lamellipodia (E, K). Ephrin-A incubation reduced growth-cone complexity (J, L). Arrows in K and L point at tightly bundled filaments. The plasma membrane contained many ruffles and vesicle-like structure (J, K, insets). M–P, SRF-deficient growth cones (M, O) were rounded up and inflated with no distinguishable filopodia (O, arrowheads). Ephrin-A5 did not break down the cytoskeleton completely and resulted in ring-like filaments (N, P). Arrows in O and P point at loosened filament bundles. The plasma membrane appeared smooth (M, O, insets). Scale bars: A, B, 6 μm; C, D, G, 10 μm; E, 5 μm; F, H, 8 μm; I–K, M–P, 3 μm; L, 2 μm; insets, 500 nm.
Wild-type neurons elaborated several neurites terminated by growth cones with many veil-like lamellipodia and finger-like filopodia (Fig. 1A,C,K,I). Induction of growth-cone collapse by the repulsive axon guidance molecule ephrin-A5 (Knöll and Drescher, 2002; Pasquale, 2005) resulted in break down of the growth-cone cytoarchitecture (Fig. 1B,D,J,L). Ephrin-A addition in Srf mutants (Fig. 1F,H,N,P) did not collapse growth cones yet retained ring-like filaments consisting of both F-actin and microtubules as before (Knöll et al., 2006). Srf−/− neurons (Fig. 1E–H) were shortened and growth-cone area almost equaled the cell-body area (Fig. 1E–H) (Knöll et al., 2006).
Here we report for the first time an almost complete lack of filopodia in Srf−/− growth cones (Fig. 1E–H). In fact, growth-cone borders appeared sharp producing a rather precise curvature (Fig. 1O, arrows). In wild type, filaments frequently ran from the center to the periphery entering filopodia (Fig. 1K). In Srf mutants (Fig. 1O), filaments were unable to form distinct filopodia indicating that lamellipodial networks take over interspaces which are cleared in wild type. Also, wild-type plasma membrane contained ruffles and vesicular-like structures (Fig. 1J, inset), which were absent in SRF-deficient neurons (Fig. 1M,N, insets). In wild-type neurites, filaments were bundled (Fig. 1C,D,K,L, arrows), whereas in Srf mutants, the neurite shaft lacked filament compaction (Fig. 1G,H,O,P, arrows).
Actin modulates neuronal morphology in an SRF-dependent manner
SRF is the archetypical transcription factor for studying actin interplay with gene expression (Posern and Treisman, 2006; Miano et al., 2007). Hence we asked, how interfering with actin treadmilling affects neuronal architecture and to what extent actin signaling requires SRF activity. For this, we used actin point mutations such as G15S and S14C favoring F-actin polymerization. R62D actin instead cannot be incorporated into F-actin, thereby raising G-actin levels (Posern et al., 2002, 2004).
First, we assessed neurite outgrowth upon actin mutant overexpression (Fig. 2 and supplemental Fig. 1, 3, available at www.jneurosci.org as supplemental material). In wild-type neurons G15S increased (Fig. 2C), S14C slightly reduced (Fig. 2E), and R62D strongly reduced (Fig. 2G) neurite length, compared with control (Fig. 2I). In keeping with G15S and S14C (Fig. 2C,E) favoring actin polymerization, both colocalized with phalloidin (recognizing F-actin). Contrastingly, R62D was largely excluded from the growth cone, thus accumulating complementarily to growth-cone F-actin in the neurite shaft and cell body [Figs. 2G (arrow), 3G; supplemental Figs. 1, 3, available at www.jneurosci.org as supplemental material), Notably, wild-type neurons expressing R62D strongly phenocopy Srf−/− neuron morphology (Fig. 2, compare G with B; Fig. 1). To test whether SRF is targeted by actin signaling, we asked, whether actin mutants can modulate neurite outgrowth in Srf−/− neurons. Neither actin mutant affected neurite length in Srf mutants (Fig. 2D,F,H; see also Fig. 2I), suggesting that actin modulation required SRF activity. In fact, R62D appeared to enhance SRF-induced phenotypes (Fig. 2, compare H with B), as revealed by ring-like growth-cone structures normally only visible in Srf mutants upon ephrin-A addition (Fig. 3).
Figure 2.

Actin signaling affects neurite outgrowth. Wild-type (A, C, E, G) or SRF-deficient (B, D, F, H) were stained for F-actin (phalloidin), anti-FLAG to visualize actin mutants, and anti-MAP2 antibodies (C–H). Arrows point at individual growth cones. A, C, E, G, Compared with mock-electroporated (A), G15S actin (C) increased and S14C (E) slightly and R62D (G) strongly decreased neurite length. In F-actin-stabilizing mutants G15S and S14C (C, E), FLAG and phalloidin staining merged. Contrastingly, R62D was mostly excluded from the F-actin-rich growth cone (G, arrow). Notably, R62D-expressing neurons (G) resembled SRF-deficient neurons (see B, D, F, H). B, D, F, H, SRF-deficient neurons (B) were shorter than wild-type neurons (see A). Actin signaling by G15S (D), S14C (F), or R62D (H) failed to rescue neurite length. I, Quantification of neurite length. The average neurite length of wild-type control electroporated neurons (see A) was set to 100%. Scale bar: (in H) A–H, 10 μm.
R62D actin inhibits growth-cone filopodia formation
Next, we investigated the consequences of actin mutants on growth-cone morphology (Fig. 3). Wild-type growth cones contained filopodia and lamellipodia (Fig. 3A), a situation not altered by S14C (Fig. 3E). G15S increased filopodia number (ctr: 2.5 ± 0.3 filopodia/growth cone; G15S: 4 ± 0.7; p < 0.05) (Fig. 3C). R62D actin instead prevented filopodia formation in wild-type neurons (Fig. 3G) thus resembling Srf−/− growth cones (Fig. 3I). Notably, R62D induced this growth-cone modification although being itself largely excluded from the growth cone and mainly remaining outside, in the neurite shaft (Fig. 3G). G15S and S14C localized to the growth cone (Fig. 3C,E). Ephrin-A5 addition to wild-type neurons resulted in growth-cone collapse and reduced F-actin content (Gallo et al., 2002; Pandithage et al., 2008) in control (Fig. 3B) and G15S- (Fig. 3D) and S14C- (Fig. 3F) expressing neurons (Fig. 3Q). Contrastingly, R62D blocked ephrin-A5 stimulated F-actin depolymerization and preserved F-actin rings reminiscent of Srf−/− growth cones upon ephrin addition (Figs. 1H,P, 3J; quantified in Fig. 3R).
As with neurite length (Fig. 2), neither G15S (Fig. 3K) nor S14C (Fig. 3M) reconstituted filopodia formation in SRF-deficient neurons. R62D induced already in wild-type growth cones ring-like filaments upon ephrin-A5 treatment resembling Srf−/− growth cones stimulated with ephrins (Fig. 3, compare J, O).
Nuclear and cytoplasmic actin signaling target neuronal gene expression
We showed above that actin requires SRF for modulating neuronal motility. Next we asked, whether actin itself modulates SRF-directed transcription in neurons (Fig. 4).
Figure 4.
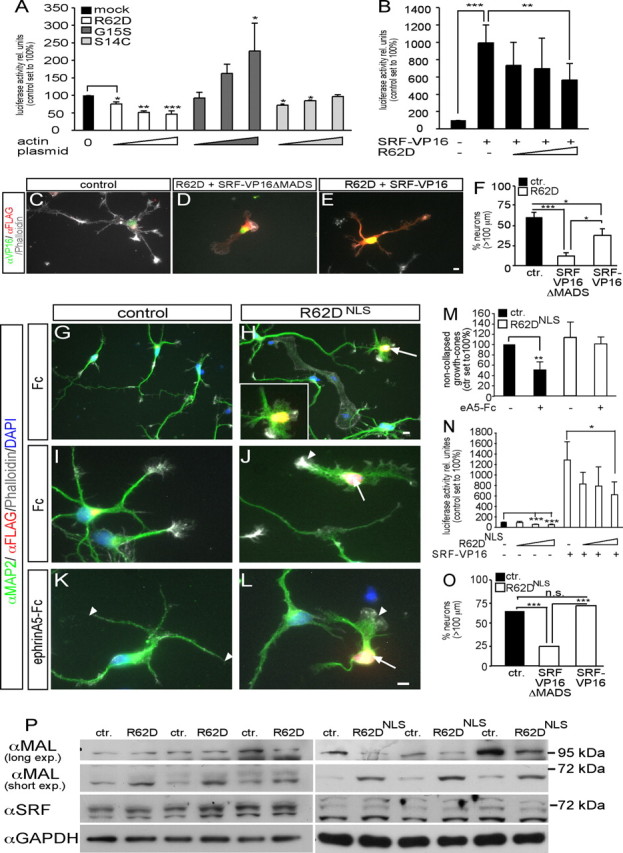
Actin signaling modulates neuronal gene expression. A, R62D decreased, G15S increased, and S14C did not influence SRF luciferase activity in neurons. B, R62D antagonized constitutively active SRF (SRF-VP16)-induced luciferase activity. C–F, SRF-VP16 (E) partially rescued R62D-decreased neurite length in contrast to SRF-VP16ΔMADS (D) used as control (quantified in F). G–M, R62D expression, confined to the nucleus (R62DNLS), was recognized by its FLAG tag. R62DNLS-positive neurons (H, J, L, arrows) showed severe structural abnormalities including decreased neurite length and lack of growth-cone filopodia. In control neurons (K), growth cones were collapsed by ephrin-A5 (K, arrowheads). Contrastingly, ephrin-A5 failed to collapse a growth cone (L, arrowhead) of an R62DNLS-expressing neuron (L, arrow; quantified in M). N, R62DNLS decreased SRF luciferase activity and counteracted SRF-VP16 enhanced gene activity (see B). O, SRF-VP16 fully rescued R62DNLS-decreased neurite length. P, Cytoplasmic R62D and R62DNLS modulated expression of MAL, but not SRF. R62DNLS decreased expression of a 100 kDa MAL protein, but upregulated a MAL variant of ∼70 kDa. Cytoplasmic R62D increased abundance of the 70 kDa MAL protein, but not of 100 kDa MAL. Triplicate culture of three different mice for R62D and R62DNLS are shown. Scale bars (C–E, G–L), 5 μm.
In SRF-dependent reporter gene assays with neurons R62D decreased, whereas G15S enhanced luciferase activity in line with non-neuronal cells (Posern et al., 2002, 2004). Contrastingly, S14C, incapable of binding the SRF cofactor MAL, did not stimulate neuronal SRF activity (Fig. 4A). Additionally, R62D reduced luciferase activity evoked by constitutively active SRF-VP16 (Fig. 4B) (Schratt et al., 2002).
Next, we investigated as to what extent actin shapes neuronal morphology by modulating gene expression in comparison to its cytoplasmic cytoskeletal function. In case R62D-induced phenotypes are not only exerted by modulating cytoplasmic actin dynamics but also by inhibition of SRF-directed gene activity, introduction of SRF-VP16 should be able to partially rescue these phenotypes (Fig. 4C–F). Indeed, SRF-VP16 strongly, but not completely, rescued R62D-decreased neurite length (Fig. 4E), in contrast to SRF-VP16 lacking the DNA-binding domain (SRF-VP16ΔMADS, control) (Fig. 4D,F). To test more directly whether actin governs neuronal motility via modulating gene transcription, we used a nuclear-resident actin mutant containing a nuclear localization sequence (R62DNLS). Indeed, R62DNLS exclusively localized to the nucleus and had no access to cytoplasmic cytoskeletal dynamics. R62DNLS similarly to cytoplasmic R62D (Fig. 2) decreased neurite length (Fig. 4H) (Ctr.: 131.3 ± 7.5 μm; R62DNLS: 71.3 ± 3.8 μm; p ≤ 0.001). Further, R62DNLS growth cones (Fig. 4H,J,L) similarly to cytoplasmic R62D (Figs. 3G, 4D) did not protrude filopodia compared with control (Fig. 4G,I). Ephrin-A induced growth-cone collapse was also impaired in R62DNLS-expressing neurons (Fig. 4L) compared with control (Fig. 4K,M; compare arrowheads). However, F-actin rings (Fig. 3H) were not as frequently observed as with cytoplasmic R62D. As with cytoplasmic R62D, R62DNLS decreased SRF gene activity (Fig. 4N). We tested, whether SRF-VP16 could rescue R62DNLS-decreased neurite length (Fig. 4O). Indeed, SRF-VP16 could fully rescue R62DNLS-decreased neurite length (Fig. 4O) unlike the cytoplasmic R62D mutant (Fig. 4C–F). Thus, actin impinges on neuron morphology not only by cytoplasmic cytoskeletal dynamics, but also by a nuclear gene expression function.
Finally, we addressed how actin might exert its impact on gene expression. Both, cytoplasmic R62D and R62DNLS modulated expression of MAL (Fig. 4P). Expression of a 100 kDa MAL protein was suppressed by R62DNLS, but not by cytoplasmic R62D. Strikingly, a MAL variant of ∼65 kDa, which was most abundant in neurons, was consistently upregulated by both R62D and R62DNLS. SRF expression in contrast was unaltered, as was full-length MAL. This is to our knowledge the first data on actin modulating protein level of a transcriptional regulator.
Discussion
Actin-dependent modulation of neuron morphology
Here we studied neuronal actin function by a novel approach using actin mutants rather than pharmacological interference. This strategy revealed that actin sculpts growth-cone morphology and modulates neurite length. Our data suggest that simply raising the F-actin content is not sufficient to increase neurite length, as G15S only and not S14C, which is likewise favoring F-actin assembly, increased neurite length (Fig. 2). This points at an important role of actin-associated molecules in which these actin mutants differ. G15S but not S14C acts independent of RhoA, a known neurite extension inhibitor (Knöll et al., 2006) whose activity is elevated in Srf mutants (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Additionally, G15S does not, unlike S14C, bind to the actin-severing protein Cofilin (Posern et al., 2004; Sarmiere and Bamburg. 2004), whose activity is impaired in Srf mutants (Alberti et al., 2005). Therefore G15S incorporated into F-actin might protect from Cofilin severing, thereby increase filament stability and enhance neurite length and filopodia number (Figs. 2, 3). In opposite to G15S, R62D decreased neurite length and filopodia number (Figs. 2–4). R62D cannot be incorporated into F-actin and increases the free G-actin pool (Posern et al., 2002, 2004). We noted that R62D is largely excluded from the F-actin-rich growth cone (Figs. 2G, 3G, 4C). Thus, these two pools of spatially and functionally distinct actin might antagonize and thereby decrease neurite elongation. In sum, our data suggest that F-actin (not associated with e.g., RhoA and Cofilin; i.e., G15S) favors, whereas G-actin (R62D) antagonizes neurite outgrowth and growth-cone filopodia formation.
Actin modulates neuronal morphology by nuclear gene expression
A key finding of this report is that actin-directed neuronal motility is not only executed by modulating cytoplasmic microfilament dynamics but in concert with nuclear gene expression. First, actin signaling requires SRF, as modulation of neuronal morphology by all actin mutants failed in Srf mutants. Second, R62D expression mimicked Srf−/− phenotypes, indicating genetic interaction in an actin–SRF signaling pathway. These phenotypes included reduced neurite length, broadened neurite shaft, lack of growth-cone filopodia, impaired ephrin-A stimulated growth-cone collapse. R62D induced these phenotypes in part by blocking SRF gene activity, as coexpression of SRF-VP16 alleviated severity of R62D phenotypes (Fig. 4C–F). As SRF-VP16 rescued cytoplasmic R62D-decreased neurite length to a considerable, yet not complete extent, this suggests that cytoplasmic R62D does impinge on neuronal motility also by its “non-nuclear” function, i.e., modulating cytoplasmic actin dynamics. Third, we used a nuclear-resident actin mutant (R62DNLS) without any cytoplasmic access to demonstrate modulation of neuron motility by nuclear actin. Similar to cytoplasmic R62D (Fig. 2), R62DNLS decreased neurite outgrowth (Fig. 4). In contrast to the cytoplasmic R62D mutant (see above), SRF-VP16 could rescue R62DNLS-decreased neurite length entirely. In sum, these data show that R62D actin inhibits neuronal motility by both, impinging on coordinated cytoplasmic actin treadmilling and also by inhibiting nuclear SRF gene transcription.
How would actin influence gene expression? Nuclear actin directly influences the core transcription machinery (Grummt, 2006). Additionally, actin dynamics regulate MAL nuclear translocation (Posern and Treisman, 2006). However in neurons, MAL appears constitutively nuclear (Kalita et al., 2006) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), arguing for a different mechanism. Our data show that actin adjusts MAL expression rather than localization (Fig. 4P) and this might impinge on SRF activity.
How is SRF ensuring dynamic regulation of the neuronal cytoskeleton? Activity of the actin-severing proteins gelsolin and cofilin is reduced in Srf mutants and might thereby disable actin filament turn-over (Alberti et al., 2005). Triple Mena/Vasp/Evl mutant neurons (Kwiatkowski et al., 2007), key molecules of filopodia formation (Drees and Gertler, 2008), resemble Srf−/− neurons suggesting that Ena/Vasp genes might be potential SRF target genes or upstream regulators.
Footnotes
B.K. is supported by the Deutsche Forschungsgemeinschaft Emmy Noether program, SFB 446, the Schram-Stiftung, and young investigator grants from Tübingen University. We thank Sofia Anastasiadou for excellent work during her laboratory rotation. Daniela Sinske provided excellent technical help throughout. We thank A. Wizenmann for discussions on the manuscript.
The authors declare no competing financial interests.
References
- Alberti S, Krause SM, Kretz O, Philippar U, Lemberger T, Casanova E, Wiebel FF, Schwarz H, Frotscher M, Schütz G, Nordheim A. Neuronal migration in the murine rostral migratory stream requires serum response factor. Proc Natl Acad Sci U S A. 2005;102:6148–6153. doi: 10.1073/pnas.0501191102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent EW, Gertler FB. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron. 2003;40:209–227. doi: 10.1016/s0896-6273(03)00633-0. [DOI] [PubMed] [Google Scholar]
- Drees F, Gertler FB. Ena/VASP: proteins at the tip of the nervous system. Curr Opin Neurobiol. 2008;18:53–59. doi: 10.1016/j.conb.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etkin A, Alarcón JM, Weisberg SP, Touzani K, Huang YY, Nordheim A, Kandel ER. A role in learning for SRF: deletion in the adult forebrain disrupts LTD and the formation of an immediate memory of a novel context. Neuron. 2006;50:127–143. doi: 10.1016/j.neuron.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Gallo G, Letourneau PC. Regulation of growth cone actin filaments by guidance cues. J Neurobiol. 2004;58:92–102. doi: 10.1002/neu.10282. [DOI] [PubMed] [Google Scholar]
- Gallo G, Yee HF, Jr, Letourneau PC. Actin turnover is required to prevent axon retraction driven by endogenous actomyosin contractility. J Cell Biol. 2002;158:1219–1228. doi: 10.1083/jcb.200204140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grummt I. Actin and myosin as transcription factors. Curr Opin Genet Dev. 2006;16:191–196. doi: 10.1016/j.gde.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Kalil K, Dent EW. Touch and go: guidance cues signal to the growth cone cytoskeleton. Curr Opin Neurobiol. 2005;15:521–526. doi: 10.1016/j.conb.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Kalita K, Kharebava G, Zheng JJ, Hetman M. Role of megakaryoblastic acute leukemia-1 in ERK1/2-dependent stimulation of serum response factor-driven transcription by BDNF or increased synaptic activity. J Neurosci. 2006;26:10020–10032. doi: 10.1523/JNEUROSCI.2644-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöll B, Drescher U. Ephrin-As as receptors in topographic projections. Trends Neurosci. 2002;25:145–149. doi: 10.1016/s0166-2236(00)02093-2. [DOI] [PubMed] [Google Scholar]
- Knöll B, Kretz O, Fiedler C, Alberti S, Schütz G, Frotscher M, Nordheim A. Serum response factor controls neuronal circuit assembly in the hippocampus. Nat Neurosci. 2006;9:195–204. doi: 10.1038/nn1627. [DOI] [PubMed] [Google Scholar]
- Kuwahara K, Barrientos T, Pipes GC, Li S, Olson EN. Muscle-specific signaling mechanism that links actin dynamics to serum response factor. Mol Cell Biol. 2005;25:3173–3181. doi: 10.1128/MCB.25.8.3173-3181.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski AV, Rubinson DA, Dent EW, Edward van Veen J, Leslie JD, Zhang J, Mebane LM, Philippar U, Pinheiro EM, Burds AA, Bronson RT, Mori S, Fässler R, Gertler FB. Ena/VASP is required for neuritogenesis in the developing cortex. Neuron. 2007;56:441–455. doi: 10.1016/j.neuron.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Miano JM, Long X, Fujiwara K. Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol. 2007;292:C70–C81. doi: 10.1152/ajpcell.00386.2006. [DOI] [PubMed] [Google Scholar]
- Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329–342. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- Pak CW, Flynn KC, Bamburg JR. Actin-binding proteins take the reins in growth cones. Nat Rev Neurosci. 2008;9:136–147. doi: 10.1038/nrn2236. [DOI] [PubMed] [Google Scholar]
- Pandithage R, Lilischkis R, Harting K, Wolf A, Jedamzik B, Lüscher-Firzlaff J, Vervoorts J, Lasonder E, Kremmer E, Knöll B, Lüscher B. The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J Cell Biol. 2008;180:915–929. doi: 10.1083/jcb.200707126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462–475. doi: 10.1038/nrm1662. [DOI] [PubMed] [Google Scholar]
- Pipes GC, Creemers EE, Olson EN. The myocardin family of transcriptional coactivators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006;20:1545–1556. doi: 10.1101/gad.1428006. [DOI] [PubMed] [Google Scholar]
- Posern G, Treisman R. Actin' together: serum response factor, its cofactors and the link to signal transduction. Trends Cell Biol. 2006;16:588–596. doi: 10.1016/j.tcb.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Posern G, Sotiropoulos A, Treisman R. Mutant actins demonstrate a role for unpolymerized actin in control of transcription by serum response factor. Mol Biol Cell. 2002;13:4167–4178. doi: 10.1091/mbc.02-05-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posern G, Miralles F, Guettler S, Treisman R. Mutant actins that stabilise F-actin use distinct mechanisms to activate the SRF coactivator MAL. EMBO J. 2004;23:3973–3983. doi: 10.1038/sj.emboj.7600404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan N, Shen Y, Sarsfield S, Lemberger T, Schütz G, Linden DJ, Ginty DD. SRF mediates activity-induced gene expression and synaptic plasticity but not neuronal viability. Nat Neurosci. 2005;8:759–767. doi: 10.1038/nn1462. [DOI] [PubMed] [Google Scholar]
- Reid T, Furuyashiki T, Ishizaki T, Watanabe G, Watanabe N, Fujisawa K, Morii N, Madaule P, Narumiya S. Rhotekin, a new putative target for Rho bearing homology to a serine/threonine kinase, PKN, and rhophilin in the rho-binding domain. J Biol Chem. 1996;271:13556–13560. doi: 10.1074/jbc.271.23.13556. [DOI] [PubMed] [Google Scholar]
- Sarmiere PD, Bamburg JR. Regulation of the neuronal actin cytoskeleton by ADF/cofilin. J Neurobiol. 2004;58:103–117. doi: 10.1002/neu.10267. [DOI] [PubMed] [Google Scholar]
- Schratt G, Philippar U, Berger J, Schwarz H, Heidenreich O, Nordheim A. Serum response factor is crucial for actin cytoskeletal organization and focal adhesion assembly in embryonic stem cells. J Cell Biol. 2002;156:737–750. doi: 10.1083/jcb.200106008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Chen G, Streb JW, Long X, Yang Y, Stoeckert CJ, Jr, Miano JM. Defining the mammalian CArGome. Genome Res. 2006;16:197–207. doi: 10.1101/gr.4108706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Borisy GG. Correlative light and electron microscopy of the cytoskeleton of cultured cells. Methods Enzymol. 1998;298:570–592. doi: 10.1016/s0076-6879(98)98045-4. [DOI] [PubMed] [Google Scholar]
- Vartiainen MK, Guettler S, Larijani B, Treisman R. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science. 2007;316:1749–1752. doi: 10.1126/science.1141084. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe SR, Alvania RS, Ramanan N, Wood JN, Mandai K, Ginty DD. Serum response factor mediates NGF-dependent target innervation by embryonic DRG sensory neurons. Neuron. 2008;58:532–545. doi: 10.1016/j.neuron.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]