

Abstract
Altered amyloid precursor protein (APP) processing leading to increased production and oligomerization of Aβ may contribute to Alzheimer's disease (AD). Understanding how APP processing is regulated under physiological conditions may provide new insights into AD pathogenesis. Recent reports demonstrate that excitatory neural activity regulates APP metabolism and Aβ levels, although understanding of the molecular mechanisms involved is incomplete. We have investigated whether NMDA receptor activity regulates APP metabolism in primary cultured cortical neurons. We report that a pool of APP is localized to the postsynaptic compartment in cortical neurons and observed partial overlap of APP with both NR1 and PSD-95. NMDA receptor stimulation increased nonamyloidogenic α-secretase-mediated APP processing, as measured by a 2.5-fold increase in cellular α-C-terminal fragment (C83) levels after glutamate or NMDA treatment. This increase was blocked by the NMDA receptor antagonists d-AP5 and MK801 but not by the AMPA receptor antagonist CNQX or the L-type calcium channel blocker nifedipine, was prevented by chelation of extracellular calcium, and was blocked by the α-secretase inhibitor TAPI-1. Cotreatment of cortical neurons with bicuculline and 4-AP, which stimulates glutamate release and activates synaptic NMDA receptors, evoked an MK801-sensitive increase in C83 levels. Furthermore, NMDA receptor stimulation caused a twofold increase in the amount of soluble APP detected in the neuronal culture medium. Finally, NMDA receptor activity inhibited both Aβ1-40 release and Gal4-dependent luciferase activity induced by β-γ-secretase-mediated cleavage of an APP-Gal4 fusion protein. Altogether, these data suggest that calcium influx through synaptic NMDA receptors promotes nonamyloidogenic α-secretase-mediated APP processing.
Introduction
Alzheimer's disease (AD) is a progressive dementia characterized by extracellular deposits of amyloid β (Aβ) in senile plaques and intracellular neurofibrillary tangles comprising hyperphosphorylated tau assembled into paired helical filaments (Selkoe, 2001). In the early stages of AD, the most common symptom is short-term memory loss, which is thought to result from a failure of specific neuronal networks of the hippocampus and cortex that are involved in memory functions (Selkoe, 2002) and use glutamate as the major excitatory neurotransmitter (Ozawa et al., 1998). A recently updated version of the amyloid cascade hypothesis of AD proposes that soluble Aβ oligomers are responsible for the early cognitive decline in AD (Walsh and Selkoe, 2007), which emerged as a result of experiments showing that soluble Aβ oligomers block learning and memory processes in rodents (Lambert et al., 1998; Walsh et al., 2002; Wang et al., 2002; Lesné et al., 2006). It has been proposed that AD may be an Aβ-mediated synaptic failure (Selkoe, 2002; Marcello et al., 2008), and experimental evidence suggests that excitatory glutamatergic synapses may be the major target of soluble Aβ oligomers (Lacor et al., 2004, 2007). Furthermore, soluble Aβ oligomers have been reported to disrupt postsynaptic glutamate receptor trafficking and signaling (Kamenetz et al., 2003; Wang et al., 2004; Roselli et al., 2005; Snyder et al., 2005; Hsieh et al., 2006; Shankar et al., 2007; Abbott et al., 2008).
Aβ is produced when the amyloid precursor protein (APP) is cleaved by β- and γ-secretase, but there is also a non-Aβ-forming pathway in cells involving α-secretase (Wilquet and De Strooper, 2004). Within the brain, Aβ is found in its normal soluble form throughout life, and evidence suggests that APP metabolism and Aβ levels are closely correlated with neural activity in animals (Fazeli et al., 1994; Cirrito et al., 2005, 2008) and humans (Buckner et al., 2005). It has recently been demonstrated that synaptic activity modulates a readily releasable pool of Aβ from nerve terminals (Cirrito et al., 2005). Alternatively, electrical depolarization, protein kinase C activation, and stimulation of muscarinic M1 acetylcholine receptors or mGluR1α glutamate receptors have all been reported to promote nonamyloidogenic α-secretase cleavage of APP (Nitsch et al., 1992, 1993, 1997, 2000; Caputi et al., 1997; Caccamo et al., 2006). In primary cultures of hippocampal neurons, stimulation of NMDA receptors increases trafficking of the candidate α-secretase ADAM10 to the postsynaptic membrane (Marcello et al., 2007); thus, NMDA receptor activity may also enhance nonamyloidogenic APP processing. However, it has previously been reported that chronic NMDA receptor activation decreases α-secretase-mediated APP processing and increases Aβ production in cultured cortical neurons (Lesné et al., 2005).
We have investigated whether NMDA receptor activity regulates APP processing in primary cultured cortical neurons by analyzing APP C-terminal fragments (CTFs), soluble APP (sAPP) and Aβ1-40 levels, and cleavage of a reporter APP protein. We report that synaptic NMDA receptor activity stimulates nonamyloidogenic α-secretase-mediated APP processing and inhibits Aβ production and release.
Materials and Methods
Antibodies
Rabbit polyclonal antibody (pAb) CT20 raised against residues 676–695 of human APP (APP695 numbering) has been described previously (Perkinton et al., 2004); APP C-terminal, phospho-APP (Thr668), and phospho-ERK1/ERK2 (Thr202/Tyr204) rabbit polyclonal Abs were purchased from Cell Signaling Technology; mouse monoclonal APP Ab 13-M raised against a 21 aa sequence in the N-terminal domain of human APP, and which is identical in mouse and rat APP, was purchased from Alpha Diagnostic International; mouse monoclonal APP Ab 22C11 that recognizes amino acids 66–81 in the N terminus of APP, and MAP2 rabbit polyclonal antibody were purchased from Millipore Bioscience Research Reagents; PSD-95 monoclonal Ab (mAb) was purchased from Calbiochem; ERK2 (C-14) rabbit polyclonal antibody was purchased from Santa Cruz Biotechnology; β-tubulin III rabbit pAb and synaptophysin (clone SVP-38) mouse mAb were purchased from Sigma ; glutamate receptor NMDAR1 (clone 54.1) mAb was purchased from BD Biosciences; synaptophysin 1 rabbit polyclonal Ab was purchased from Synaptic Systems; AlexaFluor 350, AlexaFluor 488, and AlexaFluor 594 secondary Abs were purchased from Invitrogen; and HRP-conjugated secondary Abs were purchased from Millipore Bioscience Research Reagents. Unless otherwise indicated, primary Abs were used for immunoblotting at the following dilutions: APP CT20 (1:20,000), APP C-terminal and phospho-APP (Thr668) (1:2000), APP 13-M (1:2000), ERK2 (1:2000), phospho-ERK1/ERK2 (1:2500), synaptophysin 1 (1:2000), and β-tubulin III (1:2000).
Compounds
l-Glutamate, NMDA, d-AP5, (+)-MK801, CNQX, EGTA, Nifedipine, and L685,458 were purchased from Sigma; N-[N-(3,5-difluorophenacetyl)-l-alanyl]-(S)-phenylglycine t-butyl ester (DAPT) and Boc-D-FMK were purchased from Calbiochem; 4-AP and (+)-bicuculline were purchased from Axxora; TAPI-1 was purchased from Peptides International; Complete Protease Inhibitor Cocktail tablets were purchased from Roche Applied Science; and the small molecule BACE1 inhibitor, Compound 3 (C3), was kindly provided by Merck Research Laboratories.
CNQX, DAPT, TAPI-1, C3, (+)-bicuculline, L685,458, and Boc-D-FMK were made up as 1000× concentrated stocks in DMSO (Sigma). All other compounds were made up as 1000× concentrated stocks in ultrapure water.
Plasmids
pRC-CMV vector containing a cDNA encoding for human APP695 fused in-frame at its C terminus via a 5 glycine hinge to the yeast transcription factor Gal4 containing both the DNA-binding and activation domains (APP695-Gal4) was kindly provided by Prof. Tommaso Russo (Department of Biochemistry Federico II, University of Naples, Naples, Italy). pRC-CMV vector containing a cDNA encoding for human APP695 fused in-frame at its C terminus to the Gal4 DNA-binding domain (APP695-Gal4DBD) was created using pRC-CMV vector containing the cDNA encoding for APP695-Gal4 as a template and introducing a stop codon immediately after the Gal4 DNA-binding domain using the QuikChange Site-Directed Mutagenesis kit according to the manufacturer's instructions (Stratagene). pFR-Luciferase reporter vector containing the firefly (Photinus pyralis) luciferase gene under the control of a synthetic promoter consisting of five tandem repeats of the yeast GAL4 activation sequence upstream of a minimal TATA box, and phRL-thymidine kinase (TK) vector containing the sea pansy (Renilla reniformis) luciferase gene under the control of the HSV (herpes simplex virus)-TK promoter, were from Promega. Plasmid containing a cDNA encoding for human Fe65 has been described previously (Perkinton et al., 2004). Plasmid containing a cDNA encoding for human APP695 fused in-frame at its C terminus to enhanced green fluorescent protein (APP–GFP), which was created by cloning of the human APP695 coding sequence in which the stop codon had been removed, into pEGFP-N1 vector (Clontech) as a SalI fragment, was kindly provided by Prof. Chris Miller (Medical Research Council Centre for Neurodegeneration Research, Institute of Psychiatry, London, UK).
Primary neuronal culture
Primary cortical neuronal cultures were prepared as described previously (Perkinton et al., 1999). Briefly, cortices were dissected from embryonic day 15–16 Swiss mouse embryos (Harlan) and mechanically dissociated using a fire-polished glass Pasteur pipette in HBSS (Ca2+- and Mg2+-free). Neurons were plated into Nunc multiwell tissue culture plates that had been coated previously with 20 μg/ml poly-d-lysine (Sigma) and were maintained in neurobasal medium without phenol red, supplemented with B-27, 2 mm glutamine, 100 μg/ml streptomycin, and 60 μg/ml penicillin (Invitrogen), at 37°C in a humidified atmosphere of 95% air and 5% CO2. Cultures were used after 9–14 d in vitro (DIV) and, under these conditions, were ∼97–99% neuronal, as judged by β-tubulin III staining. Glial elements were <2%, as judged by GFAP staining.
Neuronal transfections
APP-GFP plasmid (1 μg) was transfected into primary cortical neurons cultured on glass coverslips in 12-well plates (2 × 105 cells per well) at 8 DIV using Lipofectamine 2000 (1 μl) (Invitrogen) according to the manufacturer's instructions. Neurons were analyzed 24 h after transfection.
Immunofluorescence and image acquisition
Primary cortical neurons at 14 DIV, cultured on glass coverslips, were fixed at room temperature in PBS, pH 7.4, containing 4% paraformaldehyde for 10 min. For labeling of cell surface APP, neurons were incubated in blocking buffer (PBS, pH 7.4, containing 5% skimmed milk) for 15 min. For all other immunofluorescence labeling, neurons were incubated in blocking permeabilization buffer (PBS, pH 7.4, containing 5% skimmed milk, 0.1% Triton X-100) for 15 min. Primary Abs: APP CT20 (1:1500 dilution), 22C11 for labeling cell surface APP (1:500 dilution), MAP2 (1:1000 dilution), NR1 (clone 54.1) (1:150 dilution), PSD-95 (1:100 dilution), and synaptophysin (1:150 dilution) were applied in Ab buffer (PBS, pH 7.4, containing 1% skimmed milk) for 2 h, followed by application of AlexaFluor 350 (1:1000 dilution), AlexaFluor 488 (1:2000 dilution), or AlexaFluor 594 (1:2000 dilution) secondary Abs in Ab buffer for 1 h. Data were only acquired from healthy nonapoptotic neurons as judged by assessment of nuclear morphology with 4′,6-diamidino-2-phenylindole (DAPI) stain. Incubation of neurons with secondary Abs in the absence of primary Abs produced a very weak diffuse staining of cell bodies that did not overlap with the primary Ab-specific staining (data not shown). In multiple label experiments, the labeling patterns were the same as those seen with single labeling. Multichannel fluorescence (DAPI–FITC–Texas Red filter set) images were captured using AxioVision (release 4.6) imaging software and either a Plan-Apochromat 20× 0.75 NA objective or a Plan-Apochromat 63× 1.4 NA oil objective fitted to an ApoTome workstation (Axioplan 2 imaging microscope with Apotome slider module, motorized focus, and an AxioCam MRm cooled monochrome digital camera set at 1388 × 1040 pixel resolution) (Carl Zeiss). Channels were imaged sequentially to eliminate bleed-through. Z-stack images were captured at 200 nm depth intervals, Z-series projections were acquired using ImageJ software (NIH), and multichannel image overlays were obtained using Adobe Photoshop 7.0 (Adobe Systems).
Detection of APP695 and APP CTFs
To detect APP695 and APP CTFs, primary cortical neurons cultured in six-well plates (2 × 106 cells per well) for 9 DIV were treated (as detailed in the figure legends), washed once with PBS, pH 7.4, and lysed immediately in 100 μl of SDS-PAGE sample buffer (62.5 mm Tris, pH 6.8, 2% SDS, 5% 2-mercaptoethanol, 10% glycerol, and 0.0025% bromophenol blue), and then boiled for 5 min. For APP695 detection, samples were resolved by 8% Tris-glycine SDS-PAGE, and for APP CTFs detection, samples were resolved by 16.5% Tris-tricine SDS-PAGE (cathode buffer: 0.1 m Tris, 0.1 m tricine, 0.1% SDS; anode buffer: 0.2 m Tris-HCl, pH 8.9). After gel electrophoresis, proteins were transferred to 0.45 μm nitrocellulose (GE Healthcare) for APP695 detection or 0.2 μm Immobilon polyvinylidene difluoride (PVDF) membranes (Millipore) for APP CTFs detection. Immunoblotting for APP695 was performed using primary polyclonal APP Abs CT20 (1:20,000 dilution) or phospho-APP (Thr668) (1:2000 dilution), and HRP-conjugated goat-anti-rabbit IgG secondary Ab (1:20,000 dilution). Immunoblotting for APP CTFs was performed using primary polyclonal APP Abs CT20 (1:75,000 dilution), or APP C-terminal (Cell Signaling Technology) (1:10,000 dilution), or phospho-APP (Thr668) (1:15,000 dilution), and HRP-conjugated goat-anti-rabbit IgG secondary Ab (1:200,000 dilution). APP695 bands were detected using the ECL system (GE Healthcare), and APP CTFs bands were detected using the ECL Advance system (GE Healthcare), followed by exposure to Hyperfilm ECL according to the manufacturer's instructions (GE Healthcare).
Activation of synaptic and extrasynaptic NMDA receptors in primary cultured cortical neurons
To activate synaptic NMDA receptors (“synaptic protocol”), the conditioned neurobasal medium of primary cultured cortical neurons at 10 DIV was removed, and neurons were exposed to fresh neurobasal medium containing 1 mm 4-AP and 25 μm bicuculline for 15 min. DMSO or water vehicle was added to wells not receiving treatments. This protocol has previously been reported to selectively stimulate synaptic NMDA receptors in large populations of neurons in culture (Hardingham et al., 2002).
To activate extrasynaptic NMDA receptors (“extrasynaptic protocol”), the conditioned neurobasal medium of primary cultured cortical neurons at 10 DIV was removed, and neurons were exposed to fresh neurobasal medium containing 1 mm 4-AP, 25 μm bicuculline, and 5 μm MK801 for 5 min to activate and block synaptic NMDA receptors. Then, neurons were washed four times with fresh neurobasal medium to remove unbound MK801, followed by addition of neurobasal medium containing either water vehicle (control) or 50 μm NMDA for 15 min. This protocol has previously been reported to selectively stimulate extrasynaptic NMDA receptors (Hardingham et al., 2002; Ivanov et al., 2006).
Detection of sAPP
For detection of sAPP in the neuronal culture medium, primary neurons cultured in six-well plates (2 × 106 cells per well) for 9 DIV were treated as detailed in the legend to Figure 8, followed by removal of the neuronal culture medium into tubes containing Complete Protease Inhibitor Cocktail. Samples were then centrifuged at 20,000 × g for 15 min at 4°C to remove cell debris and boiled for 5 min in SDS-PAGE sample buffer. Samples (15 μl) were resolved by 8% SDS-PAGE and transferred to 0.2 μm Immobilon PVDF membranes (Millipore). Immunoblotting was performed using primary mAb APP 13-M (1:15,000 dilution) and HRP-conjugated goat-anti-mouse IgG secondary Ab (1:50,000 dilution), and immunoreactive bands were detected using the ECL Plus detection system and Hyperfilm ECL according to the manufacturer's instructions (GE Healthcare).
Figure 8.
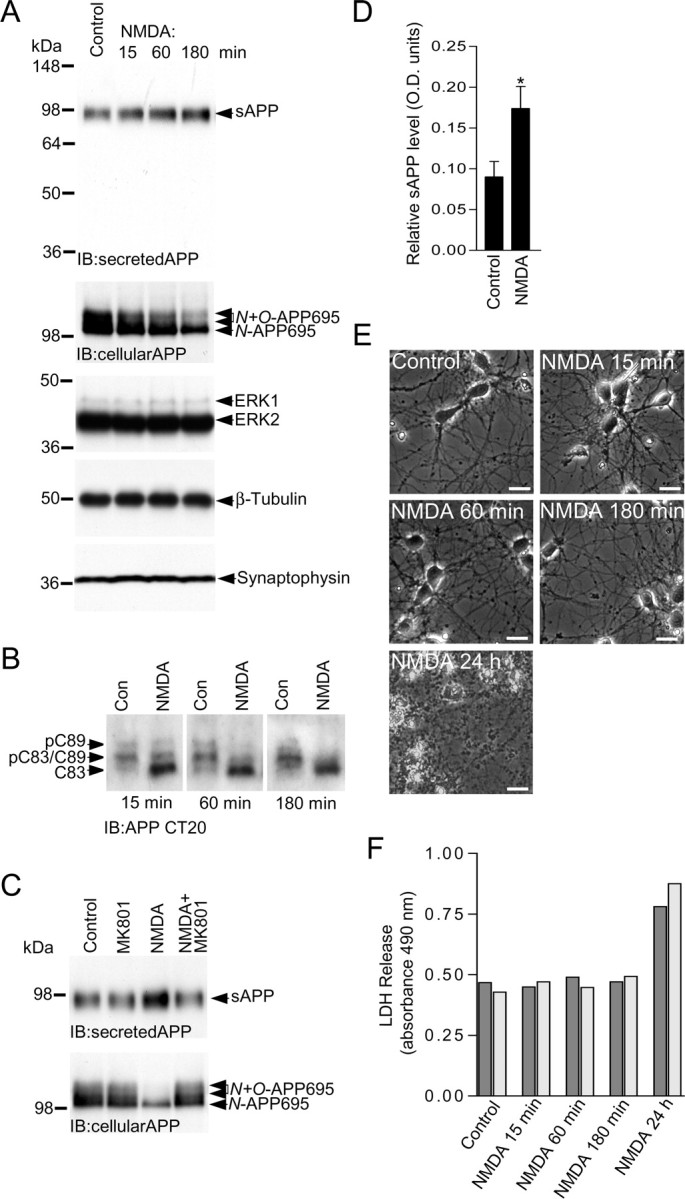
NMDA receptor activity stimulates sAPP release from primary cultured cortical neurons. A, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control) or 50 μm NMDA for 15, 60, and 180 min followed by immunoblotting of samples of the neuronal culture medium with APP N-terminal Ab 13-M to detect secreted APP, and immunoblotting of neuronal lysates with APP CT20 (cellular APP), ERK1/ERK2, β-tubulin, and synaptophysin Abs. B, Primary cultured cortical neurons at 10 DIV were treated with vehicle (Con) or 50 μm NMDA for 15, 60, and 180 min followed by immunoblotting of neuronal lysates with APP Ab CT20 to detect APP CTFs. C, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control), 2 μm MK801, 50 μm NMDA, or 50 μm NMDA in the presence of 2 μm MK801 (NMDA+MK801) for 180 min followed by immunoblotting of samples of the neuronal culture medium with APP N-terminal Ab 13-M to detect secreted APP, and immunoblotting of neuronal lysates with APP Ab CT20 to detect cellular APP. D, sAPP levels in the neuronal culture medium under basal (control) conditions and after treatment with NMDA for 180 min were analyzed by ECL protein band densitometry using calibrated ImageJ software. Each column is the mean ± SEM of four independent experiments (n = 4; *p < 0.05, control vs NMDA, unpaired two-tailed Student's t test). E, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control) or 50 μm NMDA for 15 min, 60 min, 180 min, and 24 h, and then photographed under phase contrast microscopy (320× magnification). Scale bars, 20 μm. F, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control) or 50 μm NMDA for 15 min, 60 min, 180 min, and 24 h, and the levels of LDH in the neuronal cell culture medium (LDH release) determined as described in Materials and Methods. All treatments were performed in duplicate as indicated by dark gray and light gray bars.
Assessment of neurotoxicity: lactate dehydrogenase release
Protocol 1.
Primary cortical neurons cultured in 12-well plates (0.5 × 106 cells per well) for 9 DIV were treated with vehicle (control) or 50 μm NMDA for 15 min, 60 min, 180 min, and 24 h as detailed in the legend to Figure 8. At the end of each time point, a sample of the neuronal culture medium was removed, and neurotoxicity was evaluated by measuring the amount of lactate dehydrogenase (LDH) released by dead and dying cells using the CytoTox 96 nonradioactive cytotoxicity assay according to the manufacturer's instructions (Promega), as described previously (Molina-Holgado et al., 2008). Absorbance was measured at 490 nm using a VersaMax microplate reader. Background LDH release (neuronal culture medium alone) was subtracted from the experimental values.
Protocol 2.
To assess the effect of acute NMDA or 4-AP/bicuculline treatments on neuronal viability 24 h later (delayed neurotoxicity), the conditioned neuronal culture medium from primary cortical neurons at 9 DIV cultured in 12-well plates (0.5 × 106 cells per well) was removed and replaced with conditioned medium containing either 50 μm NMDA for 5 min, or 1 mm 4-AP and 25 μm bicuculline for 15 min. After drug washout, neurons were returned to the incubator in conditioned neurobasal medium for 24 h, followed by removal of samples of the medium to evaluate LDH release using the CytoTox 96 nonradioactive cytotoxicity assay according to the manufacturer's instructions (Promega).
Phase contrast microscopy
Morphological assessment of the effect of NMDA on neuronal viability was made by phase contrast microscopy. Images were captured with a Zeiss AxioCam MRm cooled monochrome digital camera set at 1388 × 1040 pixel resolution and AxioVision (release 4.6) imaging software, using an Achrostigmat LD 32× 0.4 NA Ph1 objective, fitted to a Zeiss Axiovert S100 microscope (Carl Zeiss).
Mouse Aβ1-40 ELISA
Determination of Aβ1-40 release.
To determine the effect of NMDA receptor activity on Aβ1-40 release from primary mouse neurons, the conditioned medium from primary cortical neurons at 10 DIV cultured in 12-well plates (0.5 × 106 cells per well) was removed, and neurons were washed twice with warm (37°C) PBS, pH 7.4, to remove Aβ that had accumulated with time in culture. Then, 300 μl of fresh, warm (37°C) neurobasal medium without phenol red, supplemented with B-27, containing vehicle or drug treatments as detailed in the legend to Figure 9, was added to the neurons, which were then returned to the cell culture incubator for 6 h. After this period, the neurobasal medium (300 μl) was transferred to Eppendorf tubes containing Complete Protease Inhibitor Cocktail and centrifuged at 100,000 × g for 30 min at 4°C. Samples (200 μl) were then added to a mouse/rat Aβ1-40 ELISA plate (code no. 27720; Immuno-Biological Laboratories) and processed for detection of Aβ1-40 according to the manufacturer's instructions (Immuno-Biological Laboratories). Mouse Aβ1-40 levels were calculated from a mouse/rat Aβ1-40 standard curve.
Figure 9.

NMDA receptor activity modulates Aβ1-40 release from primary cultured neurons. A, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control) or 2 μm DAPT for 6 h, and the neuronal culture medium was removed and processed for measurement of Aβ1-40 levels using a mouse/rat Aβ1-40 ELISA kit as detailed in Materials and Methods. Each column is the mean ± SEM of four independent treatments (n = 4; **p < 0.01, control vs DAPT, unpaired two-tailed Student's t test). B, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control) or 50 μm NMDA for 6 h, and the neuronal culture medium was removed and processed for measurement of Aβ1-40 levels using a mouse/rat Aβ1-40 ELISA kit as detailed in Materials and Methods. Each column is the mean ± SEM of 18 independent treatments (n = 18; *p < 0.05, control vs NMDA, unpaired two-tailed Student's t test). C, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control) or 50 μm NMDA for 6 h and processed for measurement of intracellular Aβ1-40 levels using a mouse/rat Aβ1-40 ELISA kit as detailed in Materials and Methods. Each column is the mean ± SEM of six independent treatments (n = 6; *p < 0.05, control vs NMDA, unpaired two-tailed Student's t test). D, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control), 100 μm d-AP5, or 2 μm MK801 for 6 h, and the neuronal culture medium was removed and processed for measurement of Aβ1-40 levels using a mouse/rat Aβ1-40 ELISA kit as detailed in Materials and Methods. Each column is the mean ± SEM of nine independent treatments (n = 9; **p < 0.01, control vs d-AP5 and control vs MK801, one-way ANOVA with Bonferroni post test). E, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control), 2 μm DAPT, 50 μm NMDA, 100 μm d-AP5, or 2 μm MK801 for 6 h followed by immunoblotting of neuronal lysates with APP CT20, ERK2 and β-tubulin Abs. A single representative immunoblot is shown.
Determination of intracellular Aβ1-40 levels.
Neurons used for determination of Aβ1-40 release (see above) were washed twice with PBS, pH 7.4, and solubilized in 200 μl of ice-cold modified RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.5% sodium deoxycholate, 0.5% Triton X-100, 0.1% SDS, 1 mm EGTA, plus Complete Protease Inhibitor Cocktail) and left on ice for 1 h. Samples were then centrifuged at 100,000 × g for 30 min at 4°C, and supernatants (100 μl) were added to a mouse/rat Aβ1-40 ELISA plate (code no. 27720; Immuno-Biological Laboratories) and processed for detection of Aβ1-40 according to the manufacturer's instructions (Immuno-Biological Laboratories). Mouse Aβ1-40 levels were calculated from a mouse/rat Aβ1-40 standard curve.
Dual-Glo luciferase reporter gene activity assay for quantification of γ-secretase-mediated cleavage of a human APP695-Gal4 fusion protein
pFR-Luciferase reporter gene, APP695-Gal4, APP695-Gal4DBD, and Fe65 plasmids (all 0.5 μg) were transfected into primary cortical neurons at 8 DIV cultured in 12-well plates (2 × 105 cells per well), using Lipofectamine 2000 (1 μl/well) (Invitrogen), as detailed in the legend to Figure 10. All wells were cotransfected with phRL-TK plasmid (0.5 μg), which constitutively expresses moderate levels of Renilla luciferase. Transfection mixes containing lipid and DNA were prepared in Opti-MEM I reduced serum medium (Invitrogen) by vortexing for 1 s and leaving for 25 min. Neuronal cultures were removed from the incubator, and transfection mixes (150 μl/well) were added dropwise onto the neuronal culture medium, after which, neuronal cultures were returned to the incubator. Neurons were treated with various compounds (as detailed in the legend to Figure 10) and processed 24 h after transfection for quantification of firefly luciferase reporter gene and constitutive Renilla luciferase expression as described previously (Perkinton et al., 2004). Briefly, neurons were lysed with Glo Lysis Buffer (40 μl/well) (Promega), and the Dual-Glo luciferase activity assay was performed according to the manufacturer's instructions (Promega). Luciferase signals were captured using a Veritas microplate luminometer (Turner BioSystems). Firefly luciferase reporter activity was normalized using the constitutive Renilla luciferase activity, which helps to differentiate between specific and nonspecific cellular responses and also controls for transfection efficiencies across experiments.
Figure 10.
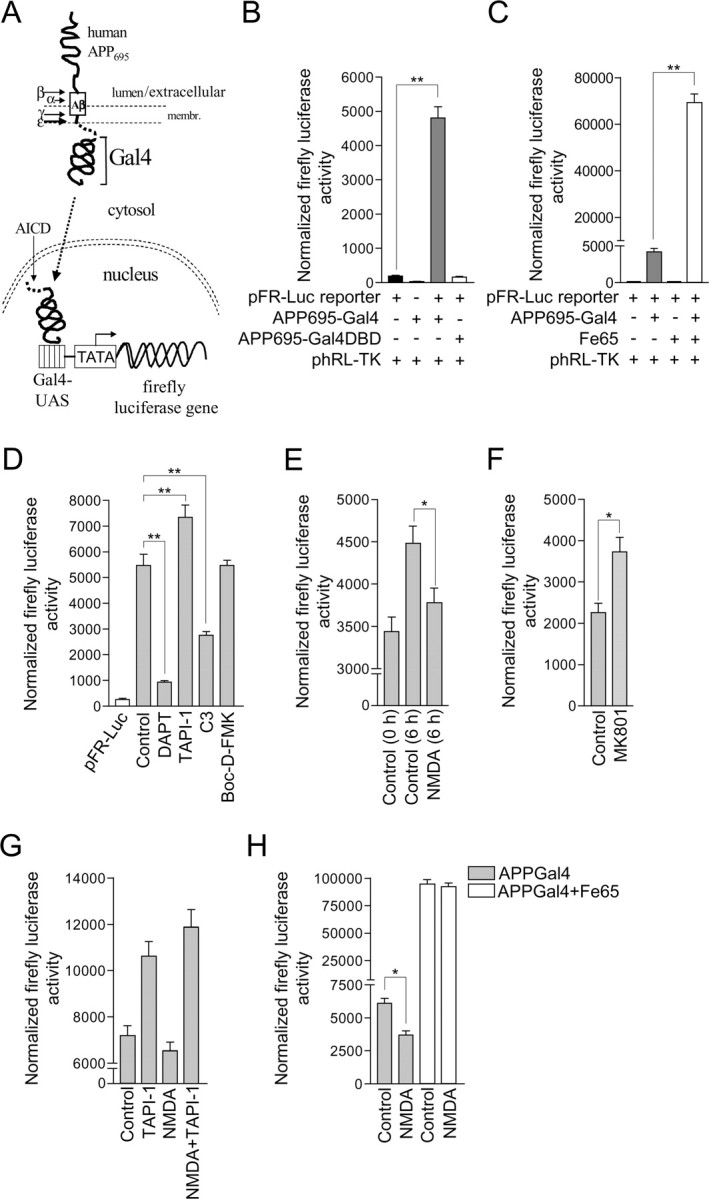
NMDA receptor stimulation inhibits AICD-Gal4-driven luciferase reporter gene activity in primary cultured cortical neurons. A, Schematic representation of a cell-based reporter gene assay for γ-secretase-mediated cleavage of APP in primary cultured cortical neurons. The assay uses a reporter APP protein consisting of human APP695 fused at its C terminus to the yeast transcription factor Gal4. After proteolytic processing of exogenously expressed APP695-Gal4 by α-secretase and/or β-secretase, cleavage by γ-secretase at the ε-site, produces an AICD-Gal4 fragment that can translocate to the nucleus in which Gal4 induces transcription of a transfected Gal4-dependent firefly luciferase reporter gene. Firefly luciferase expression is quantified by performing a Dual-Glo luciferase activity assay as detailed in Materials and Methods. B, Primary cultured cortical neurons at 8 DIV were transfected with pFR-Luc firefly luciferase reporter gene plasmid alone and in combination with plasmids encoding for APP695-Gal4 or APP695-Gal4DBD. All cells were cotransfected with phRL-TK plasmid that constitutively expresses moderate levels of Renilla luciferase. Dual-Glo luciferase activity assays were performed 24 h after transfection for quantification of firefly and Renilla luciferase expression. Firefly luciferase reporter activity was normalized using the constitutive Renilla luciferase activity. Each column is the mean ± SEM of 12 separate transfections (n = 12; **p < 0.01, pFR-Luc reporter vs pFR-Luc reporter+APP695-Gal4, one-way ANOVA with Bonferroni post test). C, Primary cultured cortical neurons at 8 DIV were transfected with pFR-Luc firefly luciferase reporter gene plasmid alone and in combination with plasmids encoding for APP695-Gal4 or Fe65. All cells were cotransfected with phRL-TK plasmid that constitutively expresses moderate levels of Renilla luciferase. Dual-Glo luciferase activity assays were performed 24 h after transfection for quantification of firefly and Renilla luciferase expression. Firefly luciferase reporter activity was normalized using the constitutive Renilla luciferase activity. Each column is the mean ± SEM of 12 separate transfections (n = 12; **p < 0.01, pFR-Luc reporter+APP695-Gal4 vs pFR-Luc reporter+APP695-Gal4+Fe65, one-way ANOVA with Bonferroni post test). D, Primary cultured cortical neurons at 8 DIV were treated with vehicle (control), 10 μm DAPT, 50 μm TAPI-1, 10 μm C3, or 20 μm Boc-d-FMK for 30 min before cotransfection with pFR-Luc firefly luciferase reporter gene, APP695-Gal4 and phRL-TK plasmids. Inhibitors were present throughout the course of the experiment. Dual-Glo luciferase activity assays were performed 24 h after transfection for quantification of firefly and Renilla luciferase expression. Firefly luciferase reporter activity was normalized using the constitutive Renilla luciferase activity. Inhibitor treatments did not alter the low level basal luciferase reporter activity arising from transfection of neurons with pFR-Luc reporter gene plasmid alone (data not shown). Each column is the mean ± SEM of 12 separate transfections (n = 12; **p < 0.01, control vs DAPT, control vs TAPI-1 control vs C3, one-way ANOVA with Bonferroni post test). E, Primary cultured cortical neurons at 8 DIV were cotransfected with pFR-Luc firefly luciferase reporter gene, APP695-Gal4, and phRL-TK plasmids. To determine whether NMDA receptor activity altered firefly luciferase reporter gene transcription over a 6 h time period, firefly and Renilla luciferase activity measurements were made in a subset of neurons 24 h after transfection (control 0 h), and at the same time, neurons were treated with vehicle (control) or 50 μm NMDA, and left for an additional 6 h followed by firefly and Renilla luciferase activity measurements. Firefly luciferase reporter activity was normalized using the constitutive Renilla luciferase activity. This experimental protocol is made possible because over the time course of the experiment, luciferase expression is cumulative and stable. Each column is the mean ± SEM of 56 separate transfections (n = 56; *p < 0.05, control vs NMDA, unpaired two-tailed Student's t test). F, Primary cultured cortical neurons at 8 DIV were cotransfected with pFR-Luc firefly luciferase reporter gene, APP695-Gal4 and phRL-TK plasmids. Twenty-four hours after transfection, neurons were treated with vehicle (control) or 2 μm MK801 for 6 h, followed by quantification of firefly and Renilla luciferase expression by performing Dual-Glo luciferase activity assays. Firefly luciferase reporter activity was normalized using the constitutive Renilla luciferase activity. Each column is the mean ± SEM of 56 separate transfections (n = 56; *p < 0.05, control vs MK801, unpaired two-tailed Student's t test). G, Primary cultured cortical neurons at 8 DIV were cotransfected with pFR-Luc firefly luciferase reporter gene, APP695-Gal4 and phRL-TK plasmids. Twenty-four hours after transfection, neurons were treated with vehicle (control), 50 μm TAPI-1, 50 μm NMDA, or 50 μm NMDA in the presence of 50 μm TAPI-1 (NMDA+TAPI-1) for 6 h, followed by quantification of firefly and Renilla luciferase expression by performing Dual-Glo luciferase activity assays. Firefly luciferase reporter activity was normalized using the constitutive Renilla luciferase activity. Each column is the mean ± SEM of 36 separate transfections. H, Primary cultured cortical neurons at 8 DIV were transfected with pFR-Luc firefly luciferase reporter gene plasmid in combination with APP695-Gal4 plasmid, or APP695-Gal4 and Fe65 plasmids. All cells were cotransfected with phRL-TK plasmid. Twenty-four hours after transfection, neurons were treated with vehicle (control) or 50 μm NMDA for 6 h, followed by quantification of firefly and Renilla luciferase expression by performing Dual-Glo luciferase activity assays. Firefly luciferase reporter activity was normalized using the constitutive Renilla luciferase activity. Each column is the mean ± SEM of 12 (APPGal4) or 24 (APPGal4+Fe65) separate transfections (*p < 0.05, APPGal4, control vs NMDA, unpaired two-tailed Student's t test).
Quantification and statistics
Immunoblot Hyperfilm ECL bands were quantified by scanning into NIH ImageJ software at a resolution of 1200 dots per inch using an Epson Perfection V700 Photo flatbed scanner fitted with a Transparency Unit, and the mean background-corrected optical density (OD) of each band was interpolated from an OD calibration curve created using an OD step tablet. Only Hyperfilm exposures that gave band OD values that were within the linear range of the OD calibration curve were used for statistical analysis.
Mean data ± SEM were graphed using GraphPad Prism software. Immunoblot, Aβ1-40 ELISA, and Dual-Glo luciferase activity assay data were analyzed by one-way ANOVA with Bonferroni's post test, or by two-tailed Student's t tests, using GraphPad Instat software. Differences between experimental treatments were considered to be statistically significant when p < 0.05.
Results
APP is trafficked to both presynaptic and postsynaptic compartments and is expressed in glutamatergic neurons
It has recently been reported that under steady-state conditions APP is sorted equally to the axonal and somatodendritic compartment in primary neurons (Back et al., 2007). We examined the distribution pattern of endogenous APP and the trafficking of APP-GFP in primary cultured cortical neurons. We performed double immunofluorescence labeling for cell surface APP and the dendritic marker MAP2. Using APP Ab 22C11, which recognizes amino acids 66–81 in the N-terminal extracellular domain of APP, we observed a discrete punctate pattern of surface APP staining along the length of MAP2-positive dendrites (Fig. 1A–A**), suggesting that APP is trafficked to the dendrites of primary cultured cortical neurons. However, on the basis of these data, we could not rule out the possibility that the APP-immunopositive puncta we observed along the length of dendrites were actually presynaptic in origin. To address this, we performed double immunofluorescence labeling for APP and the presynaptic marker synaptophysin. We found little evidence of APP colocalizing with synaptophysin in dendritic regions but instead observed numerous APP-positive puncta that were apposed to synaptophysin-immunopositive puncta (Fig. 1B–B**), which strongly suggests that there is a pool of APP that is trafficked to postsynaptic sites. In addition, analysis of cultured cortical neurons expressing APP-GFP revealed that APP-GFP is trafficked to both axons and dendrites (Fig. 1C). Axonal APP-GFP strongly colocalized with synaptophysin (Fig. 1C–C**, bottom panel), suggesting that APP is trafficked to presynaptic nerve terminals or boutons. In support of this finding, it has previously been demonstrated that a large proportion of endogenous axonal APP colocalizes with synaptophysin (Sabo et al., 2003). However, of particular relevance to our study, we observed that at numerous sites along dendrites, APP-GFP appeared to be apposed to synaptophysin-immunoreactive puncta (Fig. 1D–D**, boxes 1–6), which correlates well with the labeling patterns of endogenous APP and synaptophysin in dendritic regions (Fig. 1B–B**).
Figure 1.
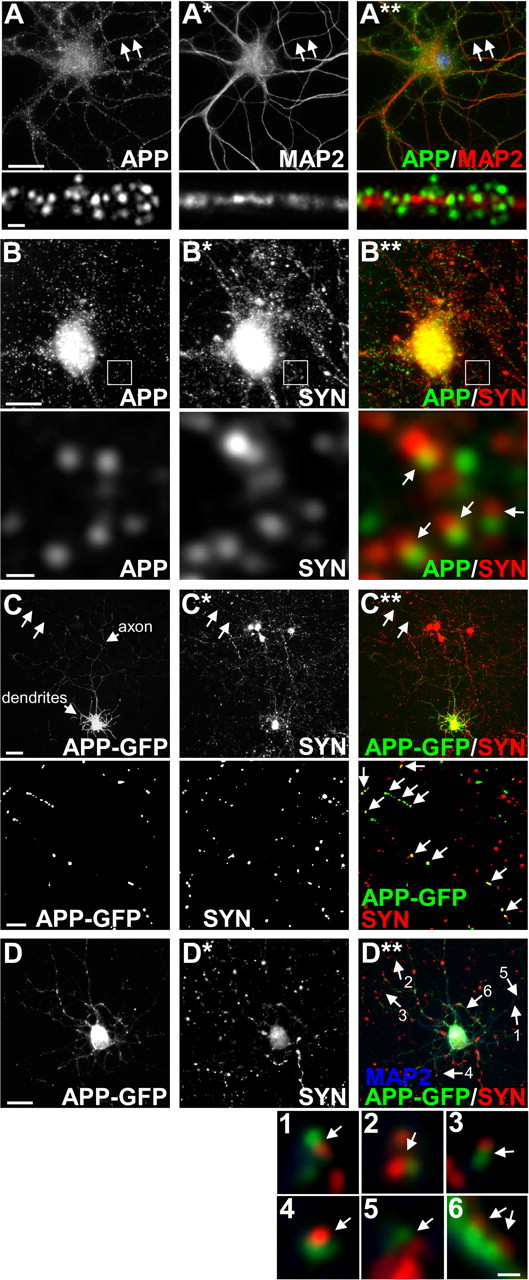
APP is trafficked to both presynaptic and postsynaptic compartments in primary cortical neurons. A–B**, Double immunofluorescence staining of primary cultured cortical neurons at 14 DIV. Neurons were fixed and double immunostained for APP and MAP2 (A–A**), and APP and the presynaptic marker synaptophysin (SYN) (B–B**). A**, Image overlay showing localization of cell surface APP to MAP2-positive dendrites. Cell surface APP was labeled with mouse monoclonal APP Ab 22C11 that recognizes the N terminus of APP, followed by cell permeabilization and labeling of MAP2 with a MAP2 rabbit pAb. Bottom, Higher-magnification image of area indicated by white arrows in top panel. B**, Image overlay showing a pool of cell surface APP is apposed to SYN-immunoreactive puncta, suggesting that APP is trafficked to synapses in primary cortical neurons. Cell surface APP was labeled with mouse monoclonal APP Ab 22C11 that recognizes the N terminus of APP, followed by cell permeabilization and labeling of SYN with a SYN rabbit pAb. Examples of APP-positive puncta adjacent to SYN are shown in the bottom panel (white arrows), which is a higher-magnification image of the area indicated by the white box in the top panel. C–C** and D–D**, A plasmid encoding for APP-GFP (1 μg) was transfected into primary cortical neurons at 8 DIV using Lipofectamine 2000 (1 μl). Immunofluorescence labeling for SYN was performed 24 h after transfection. Only neurons expressing low to moderate levels of APP-GFP were analyzed. Images were captured from areas in which there was only a single neuron expressing APP-GFP (when using a 20× objective). C–C**, Image series showing colocalization of axonal APP-GFP with SYN, suggesting that a pool of APP is trafficked to presynaptic nerve terminals in primary cortical neurons. C, Top, Low-magnification image of a neuron expressing APP-GFP; axonal and dendritic arborizations are indicated. C**, Image overlay showing colocalization of axonal APP-GFP with SYN. Bottom, Higher-magnification image of the axon region indicated by two white arrows in the top panel. Discrete APP-GFP and SYN-positive puncta are visible; colocalization of APP-GFP with SYN is indicated by numerous white arrows in bottom panel. D–D**, Image series showing dendritic APP-GFP apposed to SYN-immunoreactive puncta, suggesting that a pool of APP-GFP is trafficked to synapses in primary cortical neurons. D**, Image overlay showing a pool of dendritic APP-GFP is apposed to SYN-immunoreactive puncta. Neurons were also immunostained for MAP2 to reveal dendrites (blue). Examples of APP-GFP puncta adjacent to SYN are shown in panels 1–6, which are higher-magnification images of the dendritic regions indicated by numbered arrows in D**. Scale bars: A, B, 20 μm (top), 1 μm (bottom); C, 50 μm (top), 5 μm (bottom); D, 25 μm (top), 1 μm (panels 1–6).
Considering that our primary aim was to study whether glutamatergic neuronal activity regulates APP processing, we next assessed whether APP was expressed in glutamatergic neurons. Z-series projections of primary cortical neurons double labeled for APP and the NR1 subunit of the NMDA receptor revealed that APP and NR1 partially overlap in intracellular compartments (Fig. 2A–A**) and somatodendritic compartments (Fig. 2B–B**). Partial overlap of APP and NR1 immunofluorescence was also observed in neuritic processes (Fig. 2C–C**). In addition, double immunofluorescence labeling for APP and the NMDA receptor scaffolding protein PSD-95 revealed overlap of APP and PSD-95 in intracellular compartments (Fig. 2D–D**) and in neuritic processes (Fig. 2E–E**).
Figure 2.
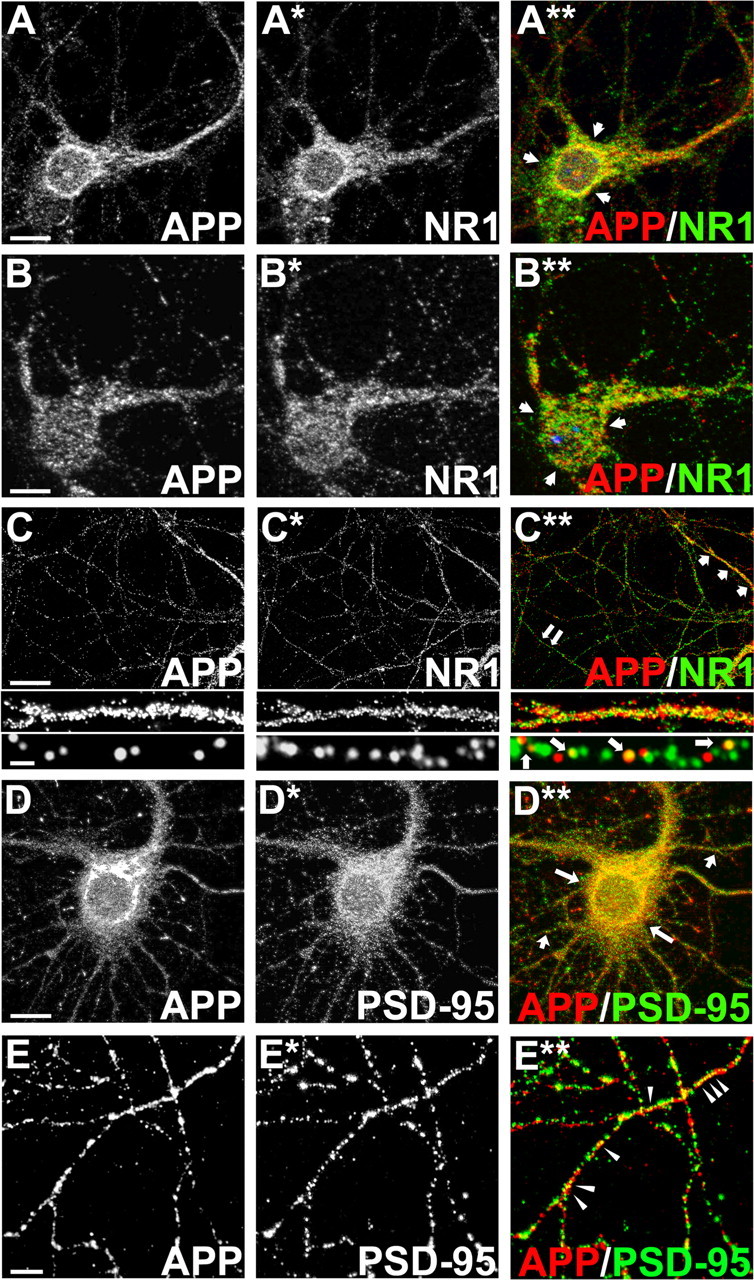
APP is expressed in primary cortical glutamatergic neurons. A–E**, Double immunofluorescence staining of primary cultured cortical neurons at 14 DIV. Neurons were fixed and double immunostained for APP and NR1 (A–A**, B–B**, C–C**), and APP and PSD-95 (D–D**, E–E**). A**, Mid Z-stack image overlay showing partial overlap of APP with NR1 in the intracellular compartment, as indicated by white arrows. B**, Top Z-stack image overlay showing partial overlap of APP with NR1 in the somatodendritic compartment, as indicated by white arrows. C**, Image overlay showing partial overlap of APP with NR1 in neuritic processes. Middle, Higher-magnification image of area indicated by three white arrows (top right of top panel). Bottom, Higher-magnification image of area indicated by two white arrows (bottom left of top panel). D**, Image overlay showing partial overlap of APP with PSD-95, as indicated by white arrows. E**, Image overlay showing partial overlap of APP with PSD-95 in neuritic processes, as indicated by white arrowheads. Scale bars: A, B, 10 μm; C, 20 μm (top), 1 μm (bottom); D, 10 μm; E, 5 μm.
Altogether, our data suggest that a pool of APP is expressed postsynaptically in which it partially overlaps with NR1 and PSD-95. In support of our findings, it has previously been demonstrated using immunoelectron microscopy that a minor proportion of APP is localized to the postsynaptic density region of asymmetrical synapses in rat cortical neurons (Shigematsu et al., 1992).
APP695 is the major APP isoform in primary cultured cortical neurons
APP is a type 1 transmembrane protein, and there are three major isoforms containing 695, 751, or 770 aa residues, termed APP695, APP751, and APP770, respectively (Wilquet and De Strooper, 2004). They are generated by alternative splicing of mRNA derived from a single gene. Under normal physiological conditions, APP695 is the major isoform expressed in neurons, and expression of APP751 and APP770 is barely detectable (Buxbaum et al., 1998a). APP undergoes extensive posttranslational modifications including N- and O-linked glycosylation, phosphorylation, and tyrosine sulfation (Weidemann et al., 1989; Buxbaum et al., 1990; Oltersdorf et al., 1990). We prepared cell lysates from primary cultured mouse cortical neurons and performed SDS-PAGE and immunoblotting with APP Ab CT20 that is specific for the C-terminal residues 676–695 of APP695, and which will detect all full-length unprocessed cellular APP isoforms. We observed three discrete polypeptide bands running just above the 98 kDa molecular weight marker. Based on previous findings (Buxbaum et al., 1998a), we assigned the lowest and major band as N-linked immature APP695, and the two slower migrating bands as N- and O-linked mature forms of APP695 (Fig. 3A). We did not detect any other higher molecular weight bands with APP Ab CT20; thus, the major isoform of APP in primary mouse cortical neuronal cultures appears to be APP695.
Figure 3.
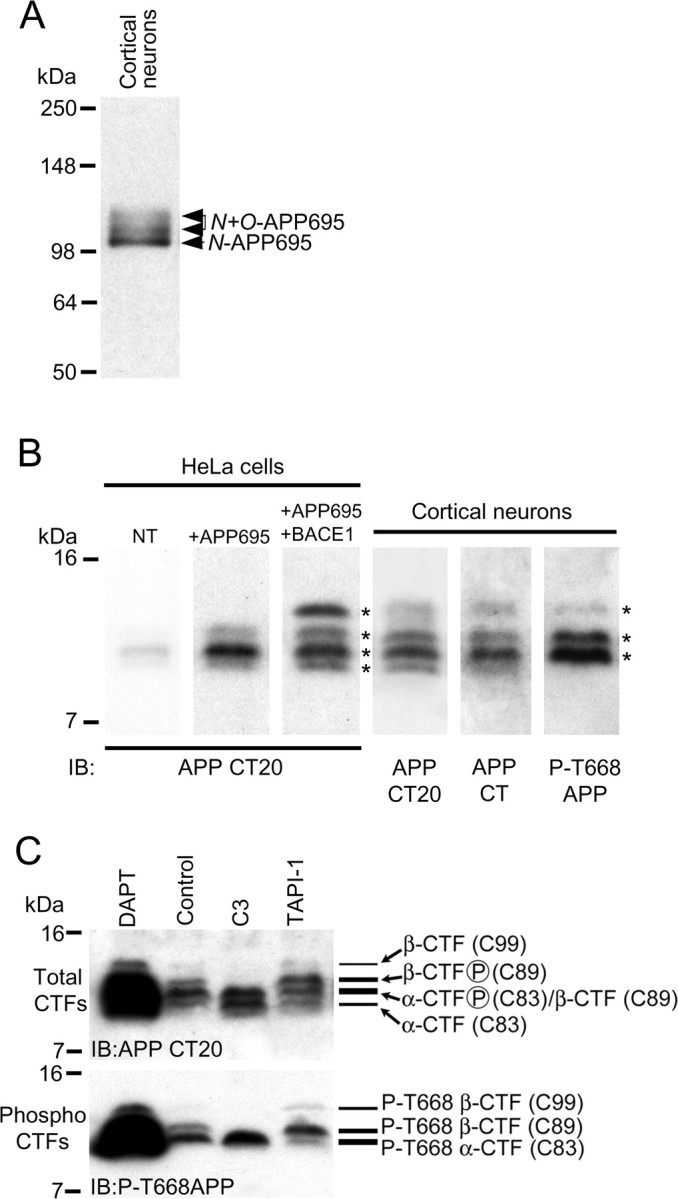
Characterization of APP and APP CTFs in primary cultured cortical neurons. A, Lysate prepared from primary cultured cortical neurons at 10 DIV was immunoblotted with APP Ab CT20. Bands representing N-linked (immature) and N+O-linked (mature) APP695 are indicated. B, Cell extracts prepared from nontransfected HeLa cells (NT), HeLa cells transfected with a plasmid encoding for APP695 (+APP695), or HeLa cells cotransfected with plasmids encoding for APP695 and BACE1 (+APP695+BACE1), were resolved by 16.5% Tris-tricine SDS-PAGE and immunoblotted with APP Ab CT20. Cell extracts prepared from primary cultured cortical neurons at 10 DIV were resolved by 16.5% Tris-tricine SDS-PAGE and immunoblotted with APP Ab CT20, a commercial APP C-terminal Ab (APP CT), or phospho-APP (thr668) Ab (P-T668APP). APP CTF bands are indicated by asterisks. C, Primary cultured cortical neurons at 10 DIV were treated with DMSO (control), 2 μm DAPT, 10 μm BACE1 inhibitor C3, or 50 μm TAPI-1 for 3 h followed by immunoblotting of cell extracts with APP Ab CT20 to detect total (unphosphorylated and phosphorylated) APP CTFs (top) or phospho-APP (Thr668) Ab (P-T668APP) to detect phospho-CTFs (bottom).
Characterization of APP CTFs in mouse primary cultured cortical neurons
In neurons, cleavage of APP by α-secretase results in the formation of a membrane-embedded CTF 83 aa in length, termed C83, and cleavage of APP by β-secretase results in the formation of membrane-embedded CTFs 89 or 99 aa in length, termed C89 and C99, respectively (Buxbaum et al., 1998a; Kimberly et al., 2005). These CTFs, which range in size from ∼8 to 14 kDa, can be phosphorylated on specific amino acid residues including Thr668 and Tyr682 (APP695 numbering) (Suzuki et al., 1994; Standen et al., 2001; Zambrano et al., 2001; Tarr et al., 2002), however, the functional consequence of CTF phosphorylation has not been fully resolved. After cleavage by α-secretase or β-secretase, it is thought that CTFs undergo complex intramembraneous processing by a multiprotein γ-secretase complex that minimally includes presenilin, PEN-2, APH-1, and nicastrin, which generates a cytosolic APP intracellular domain (AICD) fragment (Wolfe, 2006).
We reasoned that APP CTFs would provide a sensitive readout to study secretase-dependent regulation of APP processing since they are generated in cells directly after cleavage of APP by α-secretase and β-secretase. First, we characterized the pattern of APP CTFs in primary mouse cortical neurons. We analyzed APP CTFs using 16.5% Tris-tricine SDS-PAGE, followed by immunoblotting using C-terminally directed APP pAbs that should recognize total (phosphorylated and nonphosphorylated) CTF levels: (1) an in-house APP Ab, APP CT20, raised against residues 676–695 of APP695; and (2) a commercially available APP pAb raised against residues surrounding threonine 668 (APP CT). We observed four discrete bands in the 7–16 kDa molecular weight marker range (Fig. 3B, asterisks) that appeared to run at the correct molecular weight of APP CTFs, as judged by running our cortical neuron samples alongside lysate samples prepared from HeLa cells overexpressing APP695, or APP695 and BACE1 (Fig. 3B). Both APP Abs revealed identical band patterns, strongly suggesting that these bands represent APP CTFs. Furthermore, it is known that APP CTFs exist as a mixture of phosphorylated and nonphosphorylated forms, and we examined the pattern of phosphorylated CTFs in primary cortical neurons by performing immunoblotting using a C-terminally directed APP pAb specific for the phosphorylated Thr668 residue of APP (P-T668APP), and which will detect phosphorylated APP CTFs. We observed three discrete bands in the 7–16 kDa marker range (Fig. 3B, asterisks).
Regarding our data with APP Ab CT20, and based on previous observations (Buxbaum et al., 1998a; Kimberly et al., 2005), we concluded that the faint lowest molecular weight band is nonphosphorylated C83 generated from α-secretase activity; the next and noticeably stronger band is a mixture of phospho-C83 and β-secretase-generated nonphosphorylated C89; the third band is phospho-C89; and the faint highest molecular weight band represents either nonphosphorylated or phosphorylated C99 generated from β-secretase cleavage. With respect to our data obtained using the APP polyclonal antibody specific for the phosphorylated Thr668 residue of APP (P-T668APP), we concluded that the lowest molecular weight phospho-CTF band was phospho-C83; the middle band was phospho-C89; and the faint highest molecular weight band was phospho-C99.
We also characterized the nature of APP CTFs in primary mouse cortical neurons using a range of APP secretase inhibitors. We found that treatment of cortical neuronal cultures with the nontransition state γ-secretase inhibitor, DAPT, massively and specifically increased the immunoreactive signal detected between 7 and 16 kDa compared with the control signal (Fig. 3C, DAPT vs control). It has previously been reported that inhibition of γ-secretase with DAPT leads to accumulation of α-secretase and β-secretase-generated CTFs (Dovey et al., 2001; Sastre et al., 2001), and thus, our data with DAPT also confirmed that the APP-immunoreactive bands we detected between 7 and 16 kDa were bona fide α-CTF and β-CTF. However, to strengthen our conclusions regarding the pattern of CTFs in primary cortical neurons, we performed experiments with the small-molecule cell-permeable β-secretase BACE1 inhibitor, Merck C3 (Stachel et al., 2004) and the broad-spectrum cell-permeable α-secretase (ADAM) inhibitor, TAPI-1 (Hooper et al., 1997; Slack et al., 2001), and then compared the pattern of CTFs to the control pattern. We found that treatment of neurons with C3 markedly altered the total CTF and phospho-CTF pattern (Fig. 3C). It is noticeable in Fig. 3C that when compared with the control sample, the upper two total CTF bands are largely absent in samples prepared from neurons treated with C3, and the upper two phospho-CTF bands were similarly absent in C3-treated neurons. Additionally, when compared with control samples, C3 treatment increased the levels of the two lowest molecular weight total CTF bands and the lowest phospho-CTF band. C3 is reported to be a highly specific small-molecule BACE1 inhibitor (Stachel et al., 2004); thus, our C3 data strongly suggest that the upper two total CTF and phospho-CTF bands are generated from BACE1 cleavage of APP and represent C89 and C99 CTFs, and thus, it is likely that the lowest molecular weight total CTF and phospho-CTF band is C83 generated from α-secretase activity. As a point of interest, our observed increase in C83 levels in the presence of C3 also suggests that in neurons, blocking BACE1-mediated cleavage of APP leads to increased α-secretase cleavage of APP. Alternatively, treatment of neurons with the α-secretase inhibitor, TAPI-1, also markedly altered the total CTF and phospho-CTF pattern (Fig. 3C). It can be seen from Figure 3C, and especially evident in the phospho-CTF immunoblot, that when compared with the control sample, TAPI-1 treatment markedly decreased the levels of the lower CTF band, and increased the levels of the upper two CTF bands. These results lend further support to our conclusion that the lowest CTF band is α-secretase-generated C83, and the upper two CTF bands are β-secretase-generated C89 and C99. Furthermore, our observed increase in C89 and C99 levels in the presence of TAP1–1 further confirms that blocking one secretase processing pathway appears to redirect APP processing down the alternative secretase-processing pathway in primary cultured cortical neurons.
Glutamate increases α-CTF (C83) levels in primary cultured cortical neurons
Having demonstrated that a pool of APP is localized to the postsynaptic region of glutamatergic cortical neurons, and after establishing the pattern of APP CTFs in primary mouse cortical neuronal cultures, we performed experiments to determine whether application of the major excitatory neurotransmitter l-glutamate to the cortical cultures altered the pattern of APP CTFs. We treated cortical neurons with glutamate (20 μm) for 15 min and prepared samples for detection of APP CTFs. We found that glutamate treatment caused a 2.5-fold increase in α-secretase-generated C83 levels compared with control levels (Fig. 4A,D) and also led to a marked increase in phospho-C83 levels (Fig. 4A). Our observed difference in C83 levels between control and glutamate-treated neurons was not the result of differences in the total levels of protein loaded onto gels, because the total levels of full-length APP695 in control and glutamate-treated cultures were the same (Fig. 4A). After analysis of β-secretase-generated C89 and phosphoC89 levels, we failed to find any difference between control and glutamate-treated cultures (Fig. 4A,D), suggesting that glutamate treatment was only altering the levels of C83. To assess whether our observed glutamate-evoked increase in C83 levels was due to stimulation of NMDA-type glutamate receptors, we treated cortical neurons with glutamate in the absence or presence of the competitive and selective NMDA receptor antagonist d-AP5. We found that whereas d-AP5 had no noticeable effect on control CTF levels, it completely blocked the glutamate-evoked increase in C83 levels (Fig. 4B), strongly suggesting that our observed glutamate effects were NMDA receptor driven. We also found that MK801, a noncompetitive open-channel blocker of the NMDA receptor, prevented the glutamate-mediated increase in C83 levels (Fig. 4C,D), lending additional support to our conclusion that glutamate was having its effects on APP CTF levels through stimulation of NMDA-type glutamate receptors.
Figure 4.

Glutamate increases α-CTF (C83) levels in primary cultured cortical neurons. A, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control) or 20 μm glutamate for 15 min followed by immunoblotting of neuronal lysates with APP Abs CT20 or phospho-APP (thr668) (P-T668APP). B, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control), 100 μm d-AP5, 20 μm glutamate, or 20 μm glutamate in the presence of 100 μm d-AP5 (Glut.+D-AP5) for 15 min followed by immunoblotting of neuronal lysates with APP Ab CT20. C, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control), 2 μm MK801, 2 mm EGTA, 20 μm glutamate, 20 μm glutamate in the presence of 2 μm MK801 (Glut.+MK801), or 20 μm glutamate in the presence of 2 mm EGTA (Glut.+EGTA) for 15 min followed by immunoblotting of neuronal lysates with APP Ab CT20. D, C83 levels in cortical neurons treated with vehicle (control), MK801, EGTA, glutamate, glutamate in the presence of MK801 (Glut.+MK801), and glutamate in the presence of EGTA (Glut.+EGTA), and C89 levels in cortical neurons treated with vehicle (control) or glutamate, were analyzed by ECL protein band densitometry using calibrated ImageJ software. Each column is the mean ± SEM of six independent experiments (n = 6; ***p < 0.001, control vs glutamate, one-way ANOVA with Bonferroni post test).
When activated, a primary function of NMDA receptors is to allow the influx of extracellular Na+ and Ca2+ into dendritic spines (Malinow et al., 1994). Essentially, the NMDA receptor is an ionotropic signaling receptor, and it is the influx of Ca2+ through NMDA receptors that is responsible for triggering numerous cell signaling events involved in synaptic plasticity, and learning and memory processes (Bliss and Collingridge, 1993). We assessed the role of extracellular Ca2+ in our glutamate-evoked increase in C83 levels in cortical neurons. We found that treatment of neurons with glutamate in the absence of extracellular free Ca2+, which was chelated by addition of EGTA to the neurobasal medium, failed to elicit an increase in C83 levels above control levels (Fig. 4C,D), suggesting that extracellular Ca2+ is required for glutamate-evoked modulation of C83 levels. Treatment of neurons with EGTA alone did not alter basal (control) APP CTF levels (Fig. 4C,D). Furthermore, none of the drug treatments altered the levels of full-length APP695 (Fig. 4A–C).
NMDA receptor activity increases α-CTF (C83) levels in primary cultured cortical neurons
We confirmed that glutamate was acting at NMDA receptors to modulate APP C83 levels by directly stimulating NMDA receptors using the specific synthetic agonist NMDA. Treatment of neurons with NMDA led to an increase in C83 levels similar to that seen with glutamate, and we determined the pharmacological concentration range over which NMDA was exerting its effect on C83 levels to be between 10 and 100 μm (Fig. 5A), which is typical for activation of native NMDA receptors in vitro (Perkinton et al., 2002). We next determined the time course of increased C83 levels in response to 50 μm NMDA treatment and observed increased C83 levels as early as 5 min after application of NMDA (Fig. 5B). Treatment of neurons with NMDA for 5 min did not induce neurotoxicity, which was assessed 24 h later using an LDH release assay [absorbance units (490 nm): control, 0.75 ± 0.016; 50 μm NMDA, 0.77 ± 0.01; n = 8]. LDH release occurs after cell plasma membrane damage or rupture and is a widely used marker of cytotoxicity.
Figure 5.

NMDA receptor activity increases α-CTF (C83) levels in primary cultured cortical neurons. A, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control) or 1, 3, 10, 30, and 100 μm NMDA for 15 min followed by immunoblotting of neuronal lysates with APP Ab CT20. B, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control) or 50 μm NMDA for 5, 10, 20, 40, and 60 min followed by immunoblotting of neuronal lysates with APP antibody CT20. C, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control), 2 μm MK801, 50 μm NMDA, or 50 μm NMDA in the presence of 2 μm MK801 (NMDA+MK801) for 15 min followed by immunoblotting of neuronal lysates with APP Abs CT20 or phospho-APP (thr668) (P-T668APP). D, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control), 100 μm d-AP5, 50 μm NMDA, or 50 μm NMDA in the presence of 100 μm d-AP5 (NMDA+D-AP5) for 15 min followed by immunoblotting of neuronal lysates with APP Ab CT20. E, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control), 2 mm EGTA, 50 μm NMDA, or 50 μm NMDA in the presence of 2 mm EGTA (NMDA+EGTA) for 15 min followed by immunoblotting of neuronal lysates with APP Ab CT20. F, C83 levels in cortical neurons treated with vehicle (control), MK801, EGTA, NMDA, NMDA in the presence of MK801 (NMDA+MK801), and NMDA in the presence of EGTA (NMDA+EGTA), and C89 levels in cortical neurons treated with vehicle (control) or NMDA, were analyzed by ECL protein band densitometry using calibrated ImageJ software. Each column is the mean ± SEM of six independent experiments (n = 6; **p < 0.01, control vs NMDA, one-way ANOVA with Bonferroni post test). G, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control), 50 μm CNQX, 10 μm nifedipine, 50 μm NMDA, 50 μm NMDA in the presence of 50 μm CNQX (NMDA+CNQX), or 50 μm NMDA in the presence of 10 μm nifedipine (NMDA+Nifedipine) for 15 min followed by immunoblotting of neuronal lysates with APP Ab CT20. H, Primary cultured cortical neurons at 10 DIV or 14 DIV were treated with vehicle (Con) or 50 μm NMDA for 15 min followed by immunoblotting of neuronal lysates with APP Ab CT20.
Similar to glutamate, NMDA treatment evoked a 2-fold to 2.5-fold increase in C83 levels, and this effect was blocked by the NMDA receptor antagonists MK801 (Fig. 5C,F) and d-AP5 (Fig. 5D), strongly suggesting that NMDA receptor channel activity was mediating the modulatory effect on C83 CTF levels. Our observed difference in C83 levels between control and NMDA-treated neurons was not the result of differences in the total levels of protein loaded onto gels, because the total levels of full-length APP695 in control and NMDA-treated cultures were the same (Fig. 5A–E). NMDA treatment also increased the levels of phospho-C83 (Fig. 5C). However, C89 levels were not altered by treatment of cortical neurons with NMDA for 15 min (Fig. 5A,F). The NMDA-evoked increase in C83 levels was dependent on extracellular Ca2+, because chelation of extracellular free Ca2+ by addition of EGTA to the neurobasal medium blocked the NMDA-evoked increase in C83 levels (Fig. 5E,F).
Our results with EGTA strongly indicated that Ca2+ influx through NMDA receptors was responsible for modulating C83 CTF levels. However, to rule out the possibility that NMDA receptor activation might be increasing APP C83 levels via secondary Ca2+ influx through AMPA-type glutamate receptors, kainate-type glutamate receptors, or L-type voltage-sensitive Ca2+ channels, we performed experiments with NMDA in the absence or presence of the selective AMPA/kainate receptor antagonist CNQX, or the L-type voltage-sensitive Ca2+ channel blocker nifedipine. We found that the NMDA-evoked increase in C83 levels was not diminished by blocking AMPA/kainate glutamate receptors or L-type voltage-sensitive Ca2+ channels (Fig. 5G), strongly suggesting that the effect of NMDA on C83 levels was due to the influx of Ca2+ through NMDA receptors.
It has previously been reported that the levels of phosphorylated APP CTFs in primary cortical neurons increase with time in culture (Kimberly et al., 2005), and in agreement with this, we noticed that there was an increase in the basal levels of phospho-C83, phospho-C89, and phospho-C99 in 14 DIV cortical cultures compared with 9–10 DIV cultures (Fig. 5H, control). However, despite this altered basal CTF pattern at 14 DIV, we found that treatment of cortical neurons with 50 μm NMDA for 15 min still increased the levels of C83 CTF compared with control levels (Fig. 5H), which indicates that NMDA receptor modulation of APP CTF levels is not restricted to a single time point in culture.
Finally, none of the drug treatments altered the levels of full-length APP695 (Fig. 5A–H).
In conclusion, our experimental results with glutamate and NMDA suggest that over a short timescale of minutes, NMDA receptor activity selectively enhances α-secretase-mediated APP processing and consequently increases C83 levels.
The α-secretase inhibitor TAPI-1 blocks the NMDA-receptor-mediated increase in C83 levels
C83 CTFs are formed from cleavage of APP by α-secretase, and in neurons, the disintegrin metalloproteases ADAM10 and/or ADAM17 are strongly believed to be the α-secretases that mediate nonamyloidogenic constitutive and regulated APP cleavage (Buxbaum et al., 1998b; Lammich et al., 1999). To test the involvement of ADAM metalloprotease activity in our observed NMDA-receptor-evoked increase in C83 levels, we conducted experiments with the widely used broad-spectrum ADAM inhibitor TAPI-1, which has previously been reported to block constitutive and muscarinic-receptor-stimulated sAPPα release in HEK 293 cells expressing M3 muscarinic receptors (Slack et al., 2001). Furthermore, our results showing that TAPI-1 blocks α-CTF and phospho-α-CTF (C83) production in cortical neurons (Fig. 3C) confirmed that TAPI-1 inhibits α-secretase-mediated APP cleavage. We found that pretreatment of cortical neuronal cultures with TAPI-1 blocked the NMDA-receptor-mediated increase in C83 levels (Fig. 6A,B). Contrastingly, treatment of cortical cultures with the small molecule BACE1 inhibitor, C3, did not significantly alter the NMDA-evoked increase in C83 levels (Fig. 6A,B). These results indicate that our observed NMDA-receptor-evoked increase in C83 levels is dependent on α-secretase (ADAM) activity. Treatment of cortical neurons with secretase inhibitors did not alter the levels of full-length APP695 over the 15 min time course of the experiment (Fig. 6A).
Figure 6.
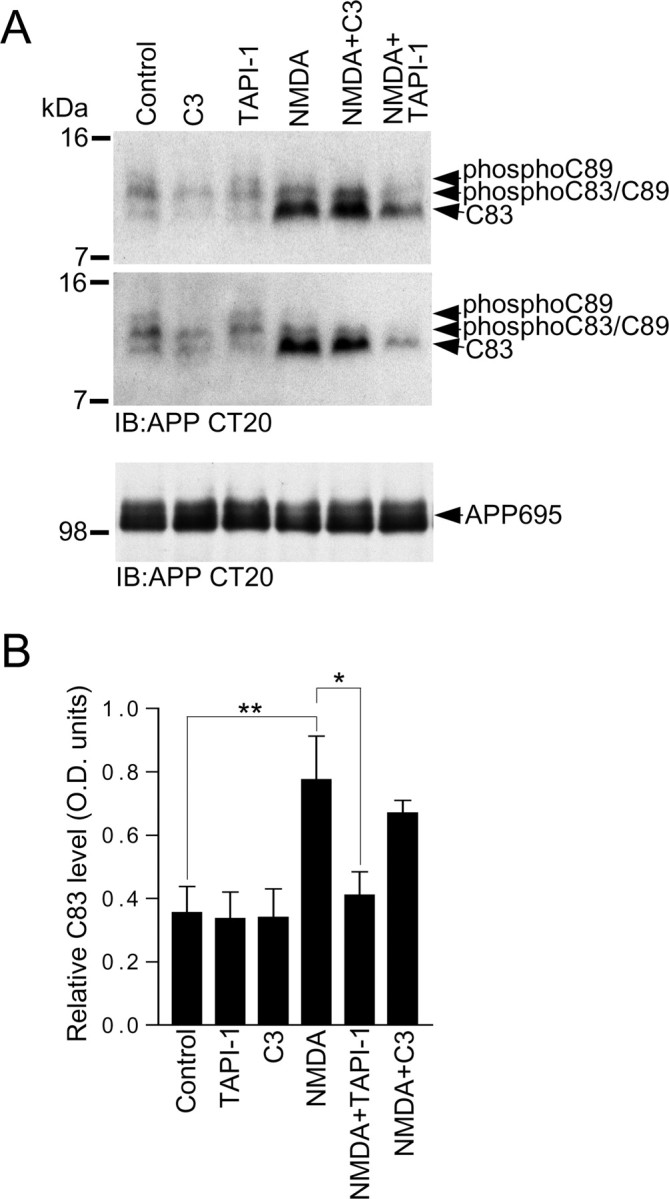
The α-secretase inhibitor TAPI-1 blocks the NMDA-receptor-mediated increase in C83 levels. A, Primary cortical neurons at 10 DIV were treated with vehicle (control), 10 μm BACE1 inhibitor C3, 50 μm TAPI-1, 50 μm NMDA, 50 μm NMDA in the presence of 10 μm C3 (NMDA+C3), or 50 μm NMDA in the presence of 50 μm TAPI-1 (NMDA+TAPI-1) for 15 min followed by immunoblotting of neuronal lysates with APP antibody CT20. Two immunoblots generated from independent experiments are shown. B, C83 levels were analyzed by ECL protein band densitometry using calibrated ImageJ software. Each column is the mean ± SEM value of four independent experiments (n = 4; **p < 0.01, control vs NMDA, *p < 0.05, NMDA vs NMDA+TAPI-1, one-way ANOVA with Bonferroni post test).
Synaptic NMDA receptor activity increases α-CTF (C83) levels in primary cultured cortical neurons
Application of NMDA agonist directly into the neurobasal medium (bath application) results in the simultaneous activation of the total pool of NMDA receptors (synaptic and extrasynaptic) in primary cultured cortical neurons. Activation of synaptic NMDA receptors has been implicated in physiological processes such as long-term potentiation induction, cAMP response element (CRE)-binding protein (CREB) activation, and synaptic plasticity, whereas extrasynaptic NMDA receptor activity is thought to occur as a result of pathophysiological processes (Hardingham and Bading, 2003). Our data with bath application of NMDA and glutamate does not discriminate between these two pools of NMDA receptors. To determine whether our observed NMDA-receptor-mediated modulation of APP processing was due to activation of synaptic or extrasynaptic NMDA receptors, we performer a series of experiments to activate synaptic and extrasynaptic NMDA receptors separately. Activation of synaptic NMDA receptors can be achieved after bath application of the K+ channel blocker 4-AP and the GABAA receptor blocker bicuculline, which together increase glutamate-driven activity of the neuronal network in the cortical cultures. This pharmacologically induced network disinhibition has previously been reported to selectively stimulate synaptic NMDA receptors in large populations of neurons in culture (Hardingham et al., 2002). We confirmed that 4-AP/bicuculline treatment increases synaptic neuronal activity by measuring ERK activity, which has previously been shown to be increased during bicuculline treatment in primary cultured hippocampal neurons (Hardingham et al., 2001; Ivanov et al., 2006). To measure ERK activation, we coapplied 4-AP and bicuculline for 15 min and then performed immunoblotting of cortical lysates with an anti-ACTIVE ERK Ab that detects ERK when it is dually phosphorylated on Thr202 and Tyr204 residues of ERK1 and on Thr183 and Tyr185 residues of ERK2. We have previously reported that phosphorylation on these residues positively correlates with ERK activation (Perkinton et al., 1999, 2002). In line with previous reports, we found that increasing synaptic activity in the neuronal cultures with 4-AP and bicuculline led to robust ERK phosphorylation compared with control levels (Fig. 7A, top panel). Our observed difference in phospho-ERK1 and phospho-ERK2 levels between control and 4-AP/bicuculline-treated neurons was not due to differences in the total levels of protein loaded onto gels, because the total levels of ERK1 and ERK2 in control and 4-AP/bicuculline-treated cultures were the same (Fig. 7A, bottom panel). Furthermore, similar to previous reports (Hardingham et al., 2002), treatment of cortical neurons with 4-AP/bicuculline for 15 min did not induce any neurotoxicity, which was assessed 24 h later using an LDH release assay (data not shown).
Figure 7.

Synaptic NMDA receptor activity increases C83 levels in primary cultured cortical neurons. A, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control) or 1 mm 4-AP and 25 μm bicuculline (4-AP/Bic) for 15 min followed by immunoblotting of neuronal lysates with phospho-ERK1/ERK2 (pERK1/pERK2) and ERK1/ERK2 Abs. B, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control) or 1 mm 4-AP and 25 μm bicuculline (4-AP/Bic) for 15 min followed by immunoblotting of neuronal lysates with APP antibody CT20. C, Extrasynaptic NMDA receptor activation protocol: primary cultured cortical neurons at 10 DIV were treated with 1 mm 4-AP, 25 μm bicuculline and 5 μm MK801 for 5 min to activate and block synaptic NMDA receptors, followed by washout of unbound MK801 and bath application of vehicle (control) or 50 μm NMDA for 15 min; synaptic NMDA receptor activation protocol: primary cultured cortical neurons were treated with 2 μm MK801, 1 mm 4-AP and 25 μm bicuculline (4-AP/Bic), or 1 mm 4-aminopyridine and 25 μm bicuculline in the presence of 2 μm MK801 (4-AP/Bic+MK801) for 15 min. Neuronal lysates were prepared and immunoblotted with APP Ab CT20 to detect APP CTFs and APP695, and also with phospho-ERK1/ERK2 (pERK1/pERK2) and ERK1/ERK2 antibodies. D, C83 levels in cortical neurons treated with vehicle (control), MK801, 4-AP/Bic, and 4-AP/Bic in the presence of MK801 (4-AP/Bic+MK801) were analyzed by ECL protein band densitometry using calibrated ImageJ software. Each column is the mean ± SEM value of five independent experiments (n = 5; *p < 0.05, control vs 4-AP/Bic, one-way ANOVA with Bonferroni post test). E, Primary cultured cortical neurons at 10 DIV were treated with vehicle (control), 1 mm 4-AP and 25 μm bicuculline (4-AP/Bic), 100 μm d-AP5, or 1 mm 4-aminopyridine and 25 μm bicuculline in the presence of 100 μm d-AP5 (4-AP/Bic+D-AP5) for 15 min followed by immunoblotting of neuronal lysates with APP Ab CT20 to detect CTFs. Neuronal lysates were also immunoblotted with phospho-ERK1/ERK2 (pERK1/pERK2) and ERK1/ERK2 Abs.
We then analyzed the same samples for APP CTF levels and detected a robust increase in α-secretase-generated C83 in 4-AP/bicuculline-treated neuronal cultures compared with control vehicle-treated cultures (Fig. 7B). These data suggest that increasing synaptic activity stimulates α-secretase-mediated APP cleavage. Our observed difference in C83 levels between control and 4-AP/bicuculline-treated neurons was not due to differences in the total levels of protein loaded onto gels, because the total levels of full-length APP695 in control and 4-AP/bicuculline-treated cultures were the same (Fig. 7B).
Having established that 4-AP/bicuculline treatment increases α-CTF (C83) levels in cultured cortical neurons, we investigated the involvement of synaptic NMDA receptors by performing 4-AP/bicuculline experiments in the absence or presence of the NMDA receptor blocker MK801. Interestingly, our observed 4-AP/bicuculline-evoked increase in C83 levels was prevented by the presence of MK801 in the stimulation solution [Fig. 7C (synaptic protocol), D], suggesting that synaptic NMDA receptor activity is coupled to increased α-secretase-mediated APP processing. Additional analysis of these same cortical lysates revealed that 4-AP/bicuculline treatment did not alter the levels of full-length APP695 over the 15 min time course of the experiment (Fig. 7C, second panel), and also that 4-AP/bicuculline treatment stimulated ERK phosphorylation in a strictly MK801-sensitive manner (Fig. 7C, third panel, synaptic protocol), confirming that synaptic NMDA receptors were being activated during the 4-AP/bicuculline treatment. In addition, we found that 4-AP/bicuculline-evoked increases in C83 levels and phospho-ERK levels were strongly inhibited by the competitive selective NMDA receptor antagonist d-AP5 (Fig. 7E).
Together, these data demonstrate that 4-AP/bicuculline treatment increases synaptic activity in cultured cortical neurons and that this increased synaptic activity can stimulate APP cleavage and C83 CTF production through the activation of synaptic NMDA receptors.
Having established that synaptic NMDA receptor activity increases C83 levels, we performed experiments to isolate and activate the extrasynaptic NMDA receptor population in cultured cortical neurons to determine whether they also increase C83 levels. To selectively activate extrasynaptic NMDA receptors, we first inactivated synaptic NMDA receptors using MK801 in conjunction with 4-AP/bicuculline treatment for 5 min. MK801 is an open channel blocker and only binds to activated NMDA receptors. Therefore, NMDA receptors activated by 4-AP/bicuculline treatment (those at synapses) will be blocked, whereas extrasynaptic NMDA receptors will be left unblocked. After extensive washout of unbound MK801, bath application of NMDA will now selectively stimulate the extrasynaptic pool of NMDA receptors (synaptic NMDA receptors have been blocked with MK801). This protocol has previously been published by independent laboratories (Hardingham et al., 2002; Ivanov et al., 2006). Using this protocol, we found that bath application of NMDA did not stimulate C83 levels (Fig. 7C, extrasynaptic protocol). Additionally, activation of extrasynaptic NMDA receptors failed to stimulate ERK1/2 phosphorylation (Fig. 7C, third panel, extrasynaptic protocol), which is in agreement with previous reports (Ivanov et al., 2006). These data suggest that extrasynaptic NMDA receptors do not modulate α-secretase-mediated APP processing.
NMDA receptor activity stimulates sAPP release from primary cultured cortical neurons
Cleavage of APP by α-secretase produces a membrane-embedded CTF, termed C83. At the same time, a large soluble ectodomain fragment known as sAPPα is generated, which is secreted into the extracellular space. Our observation that NMDA receptor activity increases the levels of C83 in neurons suggested that activation of NMDA receptors may also increase sAPP release. To test this, we stimulated NMDA receptors for varying time periods followed by removal of samples of the conditioned neuronal culture medium to analyze sAPP levels, which was performed by immunoblotting. Immunoblotting was performed using an APP Ab raised against a 21 aa sequence in the N-terminal ectodomain of human APP that detects human, mouse, and rat sAPP. We observed a single broad band running on SDS-PAGE gels just below the 98 kDa molecular weight marker (Fig. 8A), which corresponds to the correct molecular weight for sAPP. It is worth noting that uncleaved full-length APP695 runs above the 98 kDa molecular weight marker on SDS-PAGE gels (Fig. 3A). Furthermore, we did not detect any full-length APP immunoreactivity in our neuronal medium samples, as judged by immunoblotting with a C-terminally directed APP Ab, CT20, which detects APP695 but not sAPP (data not shown). We noticed that the basal levels of sAPP in the conditioned neuronal culture medium were quite high, presumably due to accumulation of sAPP with time in culture, and we were unable to detect any significant change in sAPP levels above baseline (control) after a brief 15 min period of NMDA receptor activity (Fig. 8A). Therefore, we treated cortical cultures with NMDA for longer time periods and found that there was a noticeable increase in the levels of sAPP in the conditioned neuronal culture medium at 60 and 180 min of NMDA treatment compared with control (Fig. 8A). Stimulation of NMDA receptors for 180 min produced an ∼2-fold increase in the levels of sAPP detected in the neuronal culture medium (Fig. 8D). This increase mirrors the twofold increase in C83 CTF levels that we observed after NMDA receptor stimulation (Fig. 5F). We also found that the levels of C83 were still increased after 60 and 180 min of NMDA treatment compared with control levels (Fig. 8B). Furthermore, we noticed that while the levels of sAPP increased with prolonged NMDA receptor activation, the levels of mature glycosylated cellular APP695 appeared to decrease (Fig. 8A, second panel). This decrease may be the result of the increased ectodomain shedding (sAPP release) of mature cellular APP695 after prolonged NMDA receptor activation. However, it has previously been reported that prolonged NMDA receptor activity can exert neurotoxic effects on primary cultured neurons (Skaper et al., 2001), and we also observed neurotoxicity after treatment of cortical neurons with NMDA for 24 h (Fig. 8E,F). However, at all time points tested here, compared with control, the levels of two major neuronal proteins ERK2 and β-tubulin and the synaptic marker synaptophysin were not altered after NMDA treatment (Fig. 8A), and NMDA treatment did not induce any morphological changes to the neuronal network (Fig. 8E), nor did it alter LDH release from the neuronal cultures (Fig. 8F).
Finally, we found that the increase in sAPP release and decrease in cellular mature APP695 after NMDA treatment were prevented by blocking NMDA receptors with MK801 (Fig. 8C).
Altogether, these data strongly suggest that NMDA receptor activity increases sAPP release from primary cultured cortical neurons.
NMDA receptor activity modulates Aβ1-40 release from primary cultured cortical neurons
Aβ is produced intracellulary in vesicles by a two-step cleavage process that involves an initial cleavage by BACE1 to produce C89 or C99 CTFs, followed by cleavage of these CTFs by γ-secretase to produce Aβ (Vassar and Citron, 2000). A significant proportion of the intracellular pool of Aβ is released into the extracellular space (Haass and Selkoe, 1993). Our results showing that NMDA receptor activity increases α-secretase-generated C83 and sAPP release prompted us to investigate what effect NMDA receptor activity has on Aβ release by measuring the levels of Aβ1-40 in the neuronal culture medium using an Aβ1-40 ELISA kit that specifically detects endogenous mouse and rat Aβ1-40 (code no. 27720; Immuno-Biological Laboratories). Aβ release experiments were conducted over a period of 6 h after replacing the conditioned neuronal culture medium with fresh neurobasal medium containing vehicle or drug treatments. Before examining the effect of NMDA receptor stimulation on Aβ release, to be sure that the Aβ1-40 we detected in the neuronal culture medium was actually due to the release of Aβ1-40 from cortical neurons, we analyzed the levels of Aβ1-40 in the neuronal medium after treatment of neuronal cultures with the cell permeable γ-secretase inhibitor DAPT. DAPT has been widely reported to block Aβ production and release. We found that treatment with DAPT caused a 92% decrease in the amount of Aβ1-40 detected in the neuronal culture medium compared with control (Fig. 9A), which strongly suggests that the Aβ1-40 we were detecting in the neuronal culture medium was Aβ1-40 released from cortical neurons. We then studied what effect stimulation of NMDA receptors had on Aβ1-40 release. Treatment of cortical neurons with NMDA caused a 25% decrease in the amount of Aβ1-40 detected in the neuronal culture medium compared with control, from 68.7 ± 5.8 to 51.8 ± 5.7 pg/ml (Fig. 9B). Furthermore, we studied what effect treatment of neuronal cultures with NMDA had on intracellular Aβ1-40 levels to assess whether our observed NMDA-receptor-dependent reduction in Aβ1-40 release was the result of NMDA-receptor-mediated inhibition of Aβ production. We found that NMDA treatment caused a statistically significant decrease in intracellular Aβ1-40 levels (Fig. 9C), which suggests that NMDA receptor activity leads to a decrease in Aβ1-40 production. Interestingly, we also found that treatment of cortical neurons with the NMDA receptor antagonists, d-AP5 and MK801, caused a robust increase in the amount of Aβ1-40 detected in the neuronal culture medium (Fig. 9D), which suggests that tonic NMDA receptor activity in the cortical cultures modulates Aβ1-40 release. Finally, analysis of the neuronal cultures used for Aβ1-40 release experiments showed that the protein levels of APP695 and two abundant neuronal proteins, ERK2 and β-tubulin, were identical across experimental treatments, as judged by immunoblotting (Fig. 9E), which indicates that our observed alterations in Aβ release were not due to differences in cell number or drug-induced cytotoxicity. Altogether, these data indicate that during periods of NMDA receptor activity, the production and release of Aβ1-40 is attenuated.
NMDA receptor stimulation inhibits AICD-Gal4-driven luciferase reporter gene activity in primary cultured cortical neurons
Aβ is generated from cleavage of APP by β- and γ-secretase. We wished to pursue an alternative experimental approach to confirm our ELISA data showing that NMDA receptor activity inhibits Aβ1-40 release. Therefore, we developed a cell-based reporter gene assay for γ-secretase-mediated cleavage of APP in primary cultured cortical neurons that takes advantage of the finding that the AICD generated after γ-secretase-mediated cleavage of APP can form a tripartite transcriptionally active complex in the nucleus with the APP-binding protein Fe65 and the histone acetlytransferase Tip60 (Cao and Südhof, 2001; Cupers et al., 2001; Gao and Pimplikar, 2001; Kimberly et al., 2001; Perkinton et al., 2004; Konietzko et al., 2008). The assay uses a reporter APP protein consisting of human APP695 fused at its C terminus to the yeast transcription factor Gal4 containing both the DNA-binding and activation domains (APP695-Gal4). During proteolytic processing of exogenously expressed APP695-Gal4 by α- and/or β-secretase, cleavage by γ-secretase at the ε-site produces an AICD-Gal4 fragment that can translocate to the nucleus. Once in the nucleus, Gal4 specifically induces transcription of a transfected UAS-luciferase reporter gene (pFR-Luc) by virtue of its transactivation domain and specific binding to a Gal4-UAS promoter via its DNA-binding domain (Fig. 10A). Firefly luciferase expression can be quantified by performing luciferase activity assays. Thus, although this assay system ultimately relies on translocation of AICD to the nucleus, it does not actually monitor AICD-mediated transactivation ability and functions purely as a reporter of APP processing by secretases.
Before studying whether stimulation of NMDA receptors regulates AICD-Gal4-driven luciferase activity, we validated the reporter assay in primary cortical neurons. We found that cortical neurons transfected with pFR-Luc reporter gene plasmid alone produced a low level of basal luciferase activity, and neurons transfected with a plasmid encoding for APP695-Gal4 had no detectable luciferase activity (Fig. 10B). However, when cortical neurons were cotransfected with both pFR-Luc and APP695-Gal4 plasmids, we detected 20–25 times more luciferase activity than neurons transfected with pFR-Luc plasmid alone (Fig. 10B). Cotransfection of neurons with pFR-Luc plasmid and an APP695-Gal4 plasmid containing a cDNA encoding for APP695-Gal4 containing the Gal4 DNA-binding domain but lacking the Gal4 activation domain (APP695-Gal4DBD) produced the same level of luciferase activity as neurons transfected with pFR-Luc plasmid alone (Fig. 10B). We verified by immunoblotting that APP695-Gal4 and APP695-Gal4DBD fusion proteins were expressed at similar levels in cells (data not shown). These data demonstrate that our observed firefly luciferase activity is strictly Gal4 dependent.
It has previously been reported that the AICD is a highly labile fragment that is stabilized when complexed with Fe65 (Kimberly et al., 2001); however, a more recent study has shown that overexpression of Fe65 stimulates both liberation of AICD from full-length APP and luciferase reporter gene transcription driven by an APP-Gal4VP16 fusion protein (Wiley et al., 2007). We examined whether Fe65 regulates APP695-Gal4-driven luciferase activity in primary cortical neurons and found that neurons cotransfected with plasmids encoding for APP695-Gal4 and Fe65 produced 15–20 times more luciferase activity than neurons transfected with APP695-Gal4 plasmid alone (Fig. 10C).
Next, we inhibited endogenous α-, β-, and γ-secretase activities in neurons transfected with pFR-Luc and APP695-Gal4 plasmids to determine the relative contribution of each secretase activity toward the total luciferase activity. Treatment of cortical neurons with the γ-secretase inhibitor DAPT reduced the total luciferase activity by ∼85% (Fig. 10D), which suggests that the majority of the Gal4-driven luciferase activity is dependent on γ-secretase activity, as previously reported (Karlström et al., 2002; Gianni et al., 2003; Liao et al., 2004; Zhang et al., 2007). We obtained a similar result using an alternative γ-secretase inhibitor, L685,458 (data not shown). The lack of complete inhibition of luciferase activity by γ-secretase inhibitors has previously been reported and discussed (Liao et al., 2004). The primary cleavage of APP by either α- or β-secretase to generate C83 or C89/99 CTFs, respectively, must occur before γ-secretase can cleave APP and produce the AICD. We found that treatment of neurons with the α-secretase (ADAM) inhibitor TAPI-1 significantly stimulated luciferase activity, whereas treatment of neurons with the small molecule β-secretase (BACE1) inhibitor, C3, robustly decreased luciferase activity by ∼60% (Fig. 10D). These results, together with our biochemical data showing that C3 blocks C89/99 formation and stimulates C83 levels, and TAPI-1 blocks C83 formation and increases C89/C99 levels (Fig. 3C), strongly suggest that in primary cortical neurons the β-secretase cleavage pathway of APP preferentially mediates AICD nuclear signaling, and also that this assay system preferentially monitors amyloidogenic (Aβ-forming) APP processing by β- and γ-secretase.
It has been reported that the cytoplasmic domain of APP can be cleaved by caspases, which generates a C-terminal APP fragment, termed C31 (Lu et al., 2000); however, treatment of cortical neurons with the broad-spectrum caspase inhibitor, Boc-D-FMK, did not alter the total luciferase activity (Fig. 10D), demonstrating that caspase cleavage of APP-Gal4 does not contribute to the luciferase activity.
Our finding that this assay system preferentially monitors the Aβ-forming pathway in neurons indicated that it would provide the alternative experimental approach that we were seeking to validate our ELISA data showing that NMDA receptor activity inhibits Aβ1-40 release. Although lipid-based transfection rates in primary cultured neurons are significantly lower than cell lines, we were confident that the plasmids used for monitoring γ-secretase-mediated cleavage of APP would be transfected into primary cortical neurons containing functional NMDA receptors. This assumption was based on our finding that NMDA treatment evokes phosphorylation of endogenous CREB (Ser133) in ∼85–90% of primary cortical neurons, as judged by immunofluorescence (data not shown), thus indicating that the vast majority of primary cultured cortical neurons contain functional NMDA receptors. In addition, we have previously observed that treatment of primary cortical neurons with NMDA robustly stimulates luciferase activity arising from a transfected CRE-luciferase reporter construct (M. S. Perkinton and R. J. Williams, unpublished observations), further indicating that the plasmids used in the present study would be transfected into neurons containing functional NMDA receptors.
We found that treatment of primary cortical neurons with NMDA decreased the total firefly luciferase activity when compared with control activity (Fig. 10E). NMDA treatment had no significant effect on basal luciferase activity arising from the firefly luciferase reporter alone (pFR-Luc reporter: control, 194 ± 15.5 relative luminescence units; NMDA-treated, 185.7 ± 13 relative luminescence units, n = 56), nor did it significantly alter the constitutive Renilla luciferase activity that was used to normalize firefly luciferase activity (phRL-TK: control, 43,729 ± 3256 relative luminescence units; NMDA-treated, 46,587 ± 3173 relative luminescence units, n = 56). These results demonstrate that NMDA treatment inhibits AICD-Gal4-driven firefly luciferase reporter gene transcription. In addition, we found that treatment of cortical neurons with the NMDA receptor blocker MK801 stimulated firefly luciferase activity (Fig. 10F), suggesting that tonic endogenous NMDA receptor activity in the cortical cultures reduces AICD-Gal4-driven events. The NMDA-evoked decrease in total firefly luciferase reporter activity was prevented when α-secretase activity was inhibited by treatment of neurons with TAPI-1 (Fig. 10G), suggesting that α-secretase activity was required for the NMDA effect (see also Fig. 6). Finally, we found that NMDA treatment did not inhibit AICD-Gal4-driven luciferase reporter activity in cortical neurons overexpressing both APP695-Gal4 and Fe65 (Fig. 10H).
Discussion
Nonamyloidogenic α-secretase-mediated proteolytic processing of APP is regulated by neurotransmitters and hormones, classically acting through G-protein-coupled cell surface receptors (Mills and Reiner, 1999); however, we report here that ionotropic NMDA receptor activity enhances nonamyloidogenic APP processing in primary cortical neurons, as illustrated by our finding that calcium influx through NMDA receptors causes a 2- to 2.5-fold increase in α-secretase-generated C83 levels. We also report that NMDA receptor activity increases sAPP release and decreases both Aβ1-40 production and release.
A pool of APP is localized to the postsynaptic compartment in primary cortical glutamatergic neurons
We performed double immunofluorescence labeling for APP and the dendritic marker MAP2, for APP and synaptophysin, for APP and the NR1 subunit, and for APP and the postsynaptic protein PSD-95. We observed a punctate pattern of cell surface APP staining along MAP2-positive dendrites, and at numerous sites, APP appeared to be apposed to synaptophysin-immunoreactive puncta. We obtained similar results when we analyzed the dendritic pattern of exogenously expressed APP-GFP. Furthermore, NR1 and PSD-95 label the postsynaptic density of glutamatergic neurons, and we observed partial overlap of APP with NR1 and PSD-95. Together, these data indicate that APP is expressed in primary cortical glutamatergic neurons and a pool of this APP is trafficked to postsynaptic sites in which NMDA receptors are expressed. The biological function of postsynaptically localized APP remains to be determined, but we now report that its cleavage and metabolism may be subject to regulation by excitatory synaptic activity (see below).
NMDA receptor activity regulates C83 CTF levels
To study regulation of the proteolytic processing of APP, we analyzed α- and β-secretase-mediated APP CTF production. We reasoned that since CTFs are the direct products of α-secretase and β-secretase activity they would provide a dynamic readout of changes in cellular APP cleavage. We characterized the pattern of APP CTFs in primary cortical neurons using α-secretase, β-secretase, and γ-secretase inhibitors and concluded that they exist as a mixture of phosphorylated and nonphosphorylated α-secretase-generated C83 and β-secretase-generated C89/C99 fragments. We found that brief bath application of glutamate or NMDA to primary cortical cultures for as little as 5 min increased α-CTF (C83) levels, whereas the levels of β-CTF (C89) were unaltered. NMDA-evoked increases in C83 levels were blocked by the NMDA receptor antagonists d-AP5 and MK801 but not by CNQX or nifedipine, were prevented by chelating extracellular calcium with EGTA, and were blocked by TAPI-1. TAPI-1 is known to be a general ADAM inhibitor, blocking a range of ADAMs including ADAM10 and ADAM17 (Hooper et al., 1997; Slack et al., 2001), which have been reported to be the most likely α-secretases in neurons (Buxbaum et al., 1998b; Lammich et al., 1999). Thus, our CTF data strongly suggests that a short period of NMDA receptor activity stimulates α-secretase-mediated APP cleavage.
Synaptically activated NMDA receptors are involved in modulating APP processing
We found that increasing excitatory synaptic activity in the cortical cultures, using the GABAA receptor antagonist bicuculline and the K+ channel blocker 4-AP, increased C83 levels. This stimulation protocol has been reported to selectively activate the synaptic pool of NMDA receptors in primary cultured neurons (Hardingham et al., 2002; Ivanov et al., 2006) and thus serves a valuable means by which to study physiologically relevant synaptic NMDA receptor function in vitro. We found that 4-AP/bicuculline-evoked increases in C83 levels were blocked by both MK801 and d-AP5, indicating that synaptic NMDA receptors are capable of stimulating α-secretase-mediated APP cleavage and C83 production. In contrast, we found that stimulation of extrasynaptic NMDA receptors did not alter C83 levels.
NMDA receptor activity stimulates sAPP release
α-Secretase cleaves full-length APP to produce a small membrane-embedded C-terminal fragment, termed C83, and at the same time a large soluble ectodomain fragment, termed sAPPα, is produced and released into the extracellular space. Our finding that NMDA receptor activity increases C83 levels indicated to us that NMDA receptor activation should also increase sAPPα release. We detected a twofold increase in the levels of sAPP in the neuronal culture medium after treatment of cortical neurons with NMDA. This increase was blocked by MK801, thus, demonstrating that NMDA receptor activity stimulates sAPPα release from cortical neurons.
NMDA receptor activity modulates β-/γ-secretase-mediated APP cleavage and Aβ release
We studied whether our observed NMDA-receptor-mediated increase in nonamyloidogenic APP cleavage impacted on amyloidogenic APP processing, by measuring endogenous Aβ1-40 production and release from primary cortical neurons, and we found that NMDA receptor stimulation caused a significant decrease both in Aβ1-40 release and in intracellular Aβ1-40 levels compared with control. Furthermore, treatment of cortical neurons with d-AP5 and MK801 increased Aβ1-40 release, which suggests that tonic NMDA receptor activity in the cortical cultures regulates Aβ1-40 production.
We developed a luciferase reporter gene activity assay for quantification of γ-secretase-mediated cleavage of a human APP695-Gal4 fusion protein in cultured cortical neurons, and demonstrated, through the use of specific α-secretase and β-secretase inhibitors, that AICD-Gal4-mediated firefly luciferase reporter gene transcription may be preferentially driven by β-/γ-secretase-mediated cleavage in neurons. It has recently been reported that ablation of one BACE1 gene in mice strongly reduces AICD levels although the levels of all other APP cleavage fragments including C83 are unchanged (Ma et al., 2007). Thus, the β-secretase cleavage pathway of APP could be a primary route for AICD generation in neurons.
Our finding that the APP cleavage luciferase reporter assay system preferentially monitors the Aβ-forming pathway in cortical neurons indicated to us that it would provide a useful alternative experimental approach to validate our ELISA data showing that NMDA receptor activity decreases Aβ1-40 release, and as predicted, stimulation of NMDA receptors inhibited AICD-Gal4-driven luciferase reporter gene activity.
We also showed that coexpression of Fe65 negated the inhibitory effect of NMDA on AICD-Gal4-driven luciferase activity, and this may be explained by the recent finding that overexpression of Fe65 isoforms robustly stimulates AICD levels by increasing both the rate of α-CTF (C83) and β-CTF (C99) production and the rate of γ-secretase-mediated cleavage of its immediate substrates (C83 and C99) (Wiley et al., 2007).
In summary, our data suggests that during periods of NMDA receptor activity, amyloidogenic (Aβ-forming) processing appears to be reduced.
A mechanism of action for NMDA-receptor-mediated regulation of APP processing
Our findings contrast to a previous report that NMDA receptor activation inhibits sAPPα release and stimulates Aβ production in primary cortical neurons (Lesné et al., 2005). This apparent discrepancy may be explained by the fact that Lesné et al. chronically stimulated NMDA receptors for 24 h before harvesting neurons for analysis of APP fragments. Thus, it is possible that these authors were reporting effects that were not necessarily physiologically relevant but instead reflected a more pathophysiological situation, or, as the authors concluded, their observed effects were due to transcriptional changes in the APP isoforms expressed in the cultures after prolonged (24 h) NMDA receptor stimulation. It is unlikely that our observed increase in C83 levels after a 5–15 min period of synaptic NMDA receptor activity would be due to transcriptional changes.
Recently, it has been demonstrated that synaptic activity increases extracellular Aβ levels, which may involve a change in APP processing (Kamenetz et al., 2003), or be due to the direct stimulation of a readily releasable pool of Aβ present at nerve terminals (Cirrito et al., 2005, 2008), although the involvement of NMDA receptors was not tested. We think it is unlikely that our data demonstrating NMDA-receptor-mediated inhibition of Aβ release is due to the direct modulation of a readily releasable pool of presynaptic Aβ. Alternatively, we propose that NMDA-receptor-mediated inhibition of Aβ release is a secondary event resulting from increased α-secretase-mediated cleavage of APP in somatodendritic compartments. Considering that it has previously been reported that there is competition between the candidate α-secretases ADAM10 and ADAM17 and β-secretase for APP cleavage (Skovronsky et al., 2000), an increase in α-secretase-mediated APP cleavage during NMDA receptor stimulation could limit the amount of APP available for BACE1 cleavage, thus, attenuating Aβ production and release. In support of our proposal, it has recently been reported that the glutamate receptor subunit trafficking protein synapse-associated protein-97 drives the α-secretase ADAM10 to dendritic spines upon NMDA receptor activation in primary hippocampal neurons (Marcello et al., 2007).
Conclusion
We have studied whether stimulation of NMDA receptors, which are key molecular complexes involved in synaptic plasticity and learning and memory processes, influences APP metabolism, and our findings suggest that synaptic NMDA receptor activity promotes APP processing down the nonamyloidogenic α-secretase-generated pathway, while at the same time dampening down Aβ production and release.
Although a pathophysiological link between AD, altered APP processing, and NMDA receptor dysfunction has not been reported, our findings suggest that perturbation of NMDA-receptor-dependent glutamatergic synaptic transmission by currently unknown mechanisms, could alter APP metabolism and contribute to the rising Aβ levels that are thought to be centrally involved in AD. Greater understanding of the molecular mechanisms by which excitatory synaptic activity regulates APP processing may further contribute to our understanding of the pathogenesis of AD.
Footnotes
This work was supported by grants from the Alzheimer's Society and Research into Aging to M.S.P.
References
- Abbott JJ, Howlett DR, Francis PT, Williams RJ. Abeta(1–42) modulation of Akt phosphorylation via α7 nAChR and NMDA receptors. Neurobiol Aging. 2008;29:992–1001. doi: 10.1016/j.neurobiolaging.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Back S, Haas P, Tschäpe JA, Gruebl T, Kirsch J, Müller U, Beyreuther K, Kins S. β-Amyloid precursor protein can be transported independent of any sorting signal to the axonal and dendritic compartment. J Neurosci Res. 2007;85:2580–2590. doi: 10.1002/jnr.21239. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, Mintun MA. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Gandy SE, Cicchetti P, Ehrlich ME, Czernik AJ, Fracasso RP, Ramabhadran TV, Unterbeck AJ, Greengard P. Processing of Alzheimer β/A4 amyloid precursor protein: modulation by agents that regulate protein phosphorylation. Proc Natl Acad Sci U S A. 1990;87:6003–6006. doi: 10.1073/pnas.87.15.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Thinakaran G, Koliatsos V, O'Callahan J, Slunt HH, Price DL, Sisodia SS. Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci. 1998a;18:9629–9637. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor α converting enzyme is involved in regulated α-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998b;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, LaFerla FM. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49:671–682. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Cao XW, Südhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- Caputi A, Barindelli S, Pastorino L, Cimino M, Buxbaum JD, Cattabeni F, Di Luca M. Increased secretion of the amino-terminal fragment of amyloid precursor protein in brains of rats with a constitutive up-regulation of protein kinase C. J Neurochem. 1997;68:2523–2529. doi: 10.1046/j.1471-4159.1997.68062523.x. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupers P, Orlans I, Craessaerts K, Annaert W, De Strooper B. The amyloid precursor protein (APP)-cytoplasmic fragment generated by γ-secretase is rapidly degraded but distributes partially in a nuclear fraction of neurones in culture. J Neurochem. 2001;78:1168–1178. doi: 10.1046/j.1471-4159.2001.00516.x. [DOI] [PubMed] [Google Scholar]
- Dovey HF, John V, Anderson JP, Chen LZ, de Saint Andrieu P, Fang LY, Freedman SB, Folmer B, Goldbach E, Holsztynska EJ, Hu KL, Johnson-Wood KL, Kennedy SL, Kholodenko D, Knops JE, Latimer LH, Lee M, Liao Z, Lieberburg IM, Motter RN, et al. Functional gamma-secretase inhibitors reduce β-amyloid peptide levels in brain. J Neurochem. 2001;76:173–181. doi: 10.1046/j.1471-4159.2001.00012.x. [DOI] [PubMed] [Google Scholar]
- Fazeli MS, Breen K, Errington ML, Bliss TV. Increase in extracellular NCAM and amyloid precursor protein following induction of long-term potentiation in the dentate gyrus of anaesthetized rats. Neurosci Lett. 1994;169:77–80. doi: 10.1016/0304-3940(94)90360-3. [DOI] [PubMed] [Google Scholar]
- Gao Y, Pimplikar SW. The γ-secretase-cleaved C-terminal fragment of amyloid precursor protein mediates signaling to the nucleus. Proc Natl Acad Sci U S A. 2001;98:14979–14984. doi: 10.1073/pnas.261463298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni D, Zambrano N, Bimonte M, Minopoli G, Mercken L, Talamo F, Scaloni A, Russo T. Platelet-derived growth factor induces the β-γ-secretase-mediated cleavage of Alzheimer's amyloid precursor protein through a Src-Rac-dependent pathway. J Biol Chem. 2003;278:9290–9297. doi: 10.1074/jbc.m211899200. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Cellular processing of β-amyloid precursor protein and the genesis of amyloid β-peptide. Cell. 1993;75:1039–1042. doi: 10.1016/0092-8674(93)90312-e. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003;26:81–89. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H. A calcium microdomain near NMDA receptors: on switch for ERK-dependent synapse-to-nucleus communication. Nat Neurosci. 2001;4:565–566. doi: 10.1038/88380. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hooper NM, Karran EH, Turner AJ. Membrane protein secretases. Biochem J. 1997;321:265–279. doi: 10.1042/bj3210265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Physiol. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Karlström H, Bergman A, Lendahl U, Näslund J, Lundkvist J. A sensitive and quantitative assay for measuring cleavage of presenilin substrates. J Biol Chem. 2002;277:6763–6766. doi: 10.1074/jbc.C100649200. [DOI] [PubMed] [Google Scholar]
- Kimberly WT, Zheng JB, Guénette SY, Selkoe DJ. The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem. 2001;276:40288–40292. doi: 10.1074/jbc.C100447200. [DOI] [PubMed] [Google Scholar]
- Kimberly WT, Zheng JB, Town T, Flavell RA, Selkoe DJ. Physiological regulation of the β-amyloid precursor protein signaling domain by c-Jun N-terminal kinase JNK3 during neuronal differentiation. J Neurosci. 2005;25:5533–5543. doi: 10.1523/JNEUROSCI.4883-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konietzko U, Goodger ZV, Meyer M, Kohli BM, Bosset J, Lahiri DK, Nitsch RM. Co-localization of the amyloid precursor protein and Notch intracellular domains in nuclear transcription factories. Neurobiol Aging. Advance online publication. 2008. [DOI] [PMC free article] [PubMed]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid β oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated α-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné S, Ali C, Gabriel C, Croci N, MacKenzie ET, Glabe CG, Plotkine M, Marchand-Verrecchia C, Vivien D, Buisson A. NMDA receptor activation inhibits alpha-secretase and promotes neuronal amyloid-β production. J Neurosci. 2005;25:9367–9377. doi: 10.1523/JNEUROSCI.0849-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Liao YF, Wang BJ, Cheng HT, Kuo LH, Wolfe MS. Tumor necrosis factor-α, interleukin-1β, and interferon-γ stimulate γ-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem. 2004;279:49523–49532. doi: 10.1074/jbc.M402034200. [DOI] [PubMed] [Google Scholar]
- Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- Ma H, Lesné S, Kotilinek L, Steidl-Nichols JV, Sherman M, Younkin L, Younkin S, Forster C, Sergeant N, Delacourte A, Vassar R, Citron M, Kofuji P, Boland LM, Ashe KH. Involvement of β-site APP cleaving enzyme 1 (BACE1) in amyloid precursor protein-mediated enhancement of memory and activity-dependent synaptic plasticity. Proc Natl Acad Sci U S A. 2007;104:8167–8172. doi: 10.1073/pnas.0609521104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Otmakhov N, Blum KI, Lisman J. Visualizing hippocampal synaptic function by optical detection of Ca2+ entry through the N-methyl-d-aspartate channel. Proc Natl Acad Sci U S A. 1994;91:8170–8174. doi: 10.1073/pnas.91.17.8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcello E, Gardoni F, Mauceri D, Romorini S, Jeromin A, Epis R, Borroni B, Cattabeni F, Sala C, Padovani A, Di Luca M. Synapse-associated protein-97 mediates α-secretase ADAM10 trafficking and promotes its activity. J Neurosci. 2007;27:1682–1691. doi: 10.1523/JNEUROSCI.3439-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcello E, Epis R, Di Luca M. Amyloid flirting with synaptic failure: towards a comprehensive view of Alzheimer's disease pathogenesis. Eur J Pharmacol. 2008;585:109–118. doi: 10.1016/j.ejphar.2007.11.083. [DOI] [PubMed] [Google Scholar]
- Mills J, Reiner PB. Regulation of amyloid precursor protein cleavage. J Neurochem. 1999;72:443–460. doi: 10.1046/j.1471-4159.1999.0720443.x. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado F, Gaeta A, Francis PT, Williams RJ, Hider RC. Neuroprotective actions of deferiprone in cultured cortical neurones and SHSY-5Y cells. J Neurochem. 2008;105:2466–2476. doi: 10.1111/j.1471-4159.2008.05332.x. [DOI] [PubMed] [Google Scholar]
- Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science. 1992;258:304–307. doi: 10.1126/science.1411529. [DOI] [PubMed] [Google Scholar]
- Nitsch RM, Farber SA, Growdon JH, Wurtman RJ. Release of amyloid beta-protein precursor derivatives by electrical depolarization of rat hippocampal slices. Proc Natl Acad Sci U S A. 1993;90:5191–5193. doi: 10.1073/pnas.90.11.5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsch RM, Deng A, Wurtman RJ, Growdon JH. Metabotropic glutamate receptor subtype mGluR1α stimulates the secretion of the amyloid β-protein precursor ectodomain. J Neurochem. 1997;69:704–712. doi: 10.1046/j.1471-4159.1997.69020704.x. [DOI] [PubMed] [Google Scholar]
- Nitsch RM, Deng M, Tennis M, Schoenfeld D, Growdon JH. The selective muscarinic M1 agonist AF102B decreases levels of total Aβ in cerebrospinal fluid of patients with Alzheimer's disease. Ann Neurol. 2000;48:913–918. [PubMed] [Google Scholar]
- Oltersdorf T, Ward PJ, Henriksson T, Beattie EC, Neve R, Lieberburg I, Fritz LC. The Alzheimer amyloid precursor protein: identification of a stable intermediate in the biosynthetic/degradative pathway. J Biol Chem. 1990;265:4492–4497. [PubMed] [Google Scholar]
- Ozawa S, Kamiya H, Tsuzuki K. Glutamate receptors in the mammalian central nervous system. Prog Neurobiol. 1998;54:581–618. doi: 10.1016/s0301-0082(97)00085-3. [DOI] [PubMed] [Google Scholar]
- Perkinton MS, Sihra TS, Williams RJ. Ca2+-permeable AMPA receptors induce phosphorylation of cAMP response element-binding protein through a phosphatidylinositol 3-kinase-dependent stimulation of the mitogen-activated protein kinase signaling cascade in neurons. J Neurosci. 1999;19:5861–5874. doi: 10.1523/JNEUROSCI.19-14-05861.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ. Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurones. J Neurochem. 2002;80:239–254. doi: 10.1046/j.0022-3042.2001.00699.x. [DOI] [PubMed] [Google Scholar]
- Perkinton MS, Standen CL, Lau KF, Kesavapany S, Byers HL, Ward M, McLoughlin DM, Miller CC. The c-Abl tyrosine kinase phosphorylates the Fe65 adaptor protein to stimulate Fe65/amyloid precursor protein nuclear signaling. J Biol Chem. 2004;279:22084–22091. doi: 10.1074/jbc.M311479200. [DOI] [PubMed] [Google Scholar]
- Roselli F, Tirard M, Lu J, Hutzler P, Lamberti P, Livrea P, Morabito M, Almeida OF. Soluble β-amyloid1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J Neurosci. 2005;25:11061–11070. doi: 10.1523/JNEUROSCI.3034-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo. J Neurosci. 2003;23:5407–5415. doi: 10.1523/JNEUROSCI.23-13-05407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, Teplow DB, Haass C. Presenilin-dependent γ-secretase processing of β-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2001;2:835–841. doi: 10.1093/embo-reports/kve180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigematsu K, McGeer PL, McGeer EG. Localization of amyloid precursor protein in selective postsynaptic densities of rat cortical neurons. Brain Res. 1992;592:353–357. doi: 10.1016/0006-8993(92)91697-d. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Facci L, Strijbos PJ. Neuronal protein kinase signaling cascades and excitotoxic cell death. Ann N Y Acad Sci. 2001;939:11–22. doi: 10.1111/j.1749-6632.2001.tb03606.x. [DOI] [PubMed] [Google Scholar]
- Skovronsky DM, Moore DB, Milla ME, Doms RW, Lee VM. Protein kinase C-dependent α-secretase competes with β-secretase for cleavage of amyloid-β precursor protein in the trans-Golgi network. J Biol Chem. 2000;275:2568–2575. doi: 10.1074/jbc.275.4.2568. [DOI] [PubMed] [Google Scholar]
- Slack BE, Ma LK, Seah CC. Constitutive shedding of the amyloid precursor protein ectodomain is up-regulated by tumour necrosis factor-α converting enzyme. Biochem J. 2001;357:787–794. doi: 10.1042/0264-6021:3570787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-β. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Stachel SJ, Coburn CA, Steele TG, Jones KG, Loutzenhiser EF, Gregro AR, Rajapakse HA, Lai MT, Crouthamel MC, Xu M, Tugusheva K, Lineberger JE, Pietrak BL, Espeseth AS, Shi XP, Chen-Dodson E, Holloway MK, Munshi S, Simon AJ, Kuo L, et al. Structure-based design of potent and selective cell-permeable inhibitors of human β-secretase (BACE-1) J Med Chem. 2004;47:6447–6450. doi: 10.1021/jm049379g. [DOI] [PubMed] [Google Scholar]
- Standen CL, Brownlees J, Grierson AJ, Kesavapany S, Lau KF, McLoughlin DM, Miller CCJ. Phosphorylation of thr(668) in the cytoplasmic domain of the Alzheimer's disease amyloid precursor protein by stress-activated protein kinase 1b (Jun N-terminal kinase-3) J Neurochem. 2001;76:316–320. doi: 10.1046/j.1471-4159.2001.00102.x. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Oishi M, Marshak DR, Czernik AJ, Nairn AC, Greengard P. Cell cycle-dependent regulation of the phosphorylation and metabolism of the alzheimer amyloid precursor protein. EMBO J. 1994;13:1114–1122. doi: 10.1002/j.1460-2075.1994.tb06360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarr PE, Roncarati R, Pelicci G, Pelicci PG, D'Adamio L. Tyrosine phosphorylation of the β-amyloid precursor protein cytoplasmic tail promotes interaction with Shc. J Biol Chem. 2002;277:16798–16804. doi: 10.1074/jbc.M110286200. [DOI] [PubMed] [Google Scholar]
- Vassar R, Citron M. Aβ-generating enzymes: recent advances in β- and γ-secretase research. Neuron. 2000;27:419–422. doi: 10.1016/s0896-6273(00)00051-9. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. A β oligomers—a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL. Soluble oligomers of β amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924:133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid β-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidemann A, König G, Bunke D, Fischer P, Salbaum JM, Masters CL, Beyreuther K. Identification, biogenesis, and localization of precursors of Alzheimer's disease A4 amyloid protein. Cell. 1989;57:115–126. doi: 10.1016/0092-8674(89)90177-3. [DOI] [PubMed] [Google Scholar]
- Wiley JC, Smith EA, Hudson MP, Ladiges WC, Bothwell M. Fe65 stimulates proteolytic liberation of the β-amyloid precursor protein intracellular domain. J Biol Chem. 2007;282:33313–33325. doi: 10.1074/jbc.M706024200. [DOI] [PubMed] [Google Scholar]
- Wilquet V, De Strooper B. Amyloid-β precursor protein processing in neurodegeneration. Curr Opin Neurobiol. 2004;14:582–588. doi: 10.1016/j.conb.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Wolfe MS. The γ-secretase complex: membrane-embedded proteolytic ensemble. Biochemistry. 2006;45:7931–7939. doi: 10.1021/bi060799c. [DOI] [PubMed] [Google Scholar]
- Zambrano N, Bruni P, Minopoli G, Mosca R, Molino D, Russo C, Schettini G, Sudol M, Russo T. The β-amyloid precursor protein APP is tyrosine-phosphorylated in cells expressing a constitutively active form of the Abl protoncogene. J Biol Chem. 2001;276:19787–19792. doi: 10.1074/jbc.M100792200. [DOI] [PubMed] [Google Scholar]
- Zhang C, Khandelwal PJ, Chakraborty R, Cuellar TL, Sarangi S, Patel SA, Cosentino CP, O'Connor M, Lee JC, Tanzi RE, Saunders AJ. An AICD-based functional screen to identify APP metabolism regulators. Mol Neurodegener. 2007;2:15. doi: 10.1186/1750-1326-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]