

Abstract
Activity-dependent regulation of synaptic inputs in neurons is controlled by highly compartmentalized and dynamic dendritic calcium signaling. Among multiple Ca2+ mechanisms operating in neuronal dendrites, voltage-sensitive Ca2+ channels (VSCCs) represent a major source of Ca2+ influx; however, their use-dependent implication, regulation, and function in different types of central neurons remain widely unknown. Using two-photon microscopy to probe Ca2+ signaling in dendrites of hippocampal oriens/alveus interneurons, we found that intense synaptic activity or local activation of mGluR5 induced long-lasting potentiation of action potential evoked Ca2+ transients. This potentiation of dendritic Ca2+ signaling required mGluR5-induced intracellular Ca2+ release and PKC activation and was expressed as a selective compartmentalized potentiation of L-type VSCCs. Thus, in addition to mGluR1a-dependent synaptic plasticity, hippocampal interneurons in the feedback inhibitory circuit demonstrate a novel form of mGluR5-induced dendritic plasticity. Given an implication of L-type VSCCs in the induction of Hebbian LTP at interneuron excitatory synapses, their activity-dependent regulation may represent a powerful mechanism for regulating synaptic plasticity.
Introduction
One of the key elements in regulating synaptic efficacy is intracellular calcium. At many synapses, the spatio-temporal profile of postsynaptic Ca2+ elevations determines the direction of synaptic modifications. High-resolution Ca2+ imaging allowed detailed characterization of cellular and molecular mechanisms of postsynaptic Ca2+ signaling involved in plasticity induction in many central neurons. Moreover, it became apparent that being a master of synaptic communication, Ca2+ signaling itself is highly dynamic, undergoing activity-dependent modifications. Indeed, several forms of activity-dependent plasticity of Ca2+ sources and extrusion mechanisms have been reported recently (Yasuda et al., 2003; Scheuss et al., 2006; Sobczyk and Svoboda, 2007), calling attention to the idea of Ca2+-dependent regulation of synaptic plasticity or “metaplasticity.”
Local circuit inhibitory interneurons represent heterogeneous populations of GABAergic cells subserving specialized network functions. Controlling many stages of information processing in principal neurons, from dendritic integration to spike initiation, these network gatekeepers have developed a large repertoire of subcellular mechanisms allowing for multiple forms of synaptic modifications at their excitatory synapses. Importantly, a number of specific postsynaptic Ca2+ mechanisms are engaged in inhibitory interneurons to dynamically tune their network activity. For instance, Ca2+-permeable AMPA receptor and metabotropic glutamate receptor 1a (mGluR1a), coexpressed by interneurons in the feedback inhibitory circuit, provide specific Ca2+ mechanisms able to induce two distinct forms of long-term potentiation (LTP; anti-Hebbian and Hebbian, respectively) within the same excitatory synapse (Perez et al., 2001; Topolnik et al., 2005; Lamsa et al., 2007). Although a Hebbian form of LTP in CA1 oriens/alveus (O/A) interneurons is critically controlled by mGluR1a Ca2+ signaling (Topolnik et al., 2005, 2006), the requirement of postsynaptic depolarization for its induction suggests a potential involvement of other Ca2+ sources, including voltage-sensitive Ca2+ channels (VSCCs). Being the largest source of dendritic Ca2+ influx (Sabatini and Svoboda, 2000), VSCCs control the induction of several forms of synaptic plasticity and are themselves subject of activity- and Ca2+-dependent regulation (for review, see Catterall, 2000), modulating synaptic plasticity (Yasuda et al., 2003). Although different types of VSCCs are expressed in O/A interneurons (Vinet and Sík, 2006) and should be readily activated because of a highly reliable propagation of action potentials (APs) in dendrites of these cells (Martina et al., 2000), the contribution of VSCCs to interneuron dendritic Ca2+ signaling has received little attention. Thus, activity-dependent regulation of VSCCs as well as its significance for interneuron synaptic plasticity remain unknown. Here, we investigated whether Ca2+ transients evoked in O/A interneuron dendrites by APs, and thus mediated by the activation of VSCCs, can be modulated in an activity-dependent manner. We report that AP-evoked Ca2+ signaling undergoes a form of long-lasting potentiation that is expressed as a selective increase in L-type VSCC function. This potentiation occurs in specific dendritic microdomains expressing mGluR5 and is controlled by mGluR5-mediated intracellular Ca2+ release and PKC activation. This novel form of interneuron dendritic plasticity may provide an activity-dependent mechanism for regulating synaptic LTP.
Materials and Methods
Slice preparation and electrophysiology.
Hippocampal slices (300 μm thick) were prepared from 2- to 3-week-old Sprague Dawley rats as described previously (Topolnik et al., 2006) in accordance with the Université de Montréal and Université Laval animal welfare guidelines. During experiments, slices were perfused with standard artificial CSF (ACSF) containing (mm) 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 MgSO4, 2 CaCl2, and 10 glucose, saturated with 95%O2/5%CO2, pH 7.4. CA1 O/A interneurons were visually identified using infrared differential interference contrast or Dodt infrared scanning gradient contrast and two-photon excitation microscopy. Whole-cell patch-clamp recordings were performed in most experiments in current-clamp (CC) mode to monitor action potential-evoked Ca2+ transients (AP–CaTs). Some experiments were performed in voltage clamp (VC) to record synaptically evoked CaTs and to examine Hebbian LTP of synaptic currents (see below). Holding membrane potential was −60 mV in both configurations. In CC experiments, recording electrodes (4–5 MΩ) were filled with (in mm) 130 KMeSO3, 2 MgCl2, 10 diNa-phosphocreatine, 10 HEPES, 2 ATPTris, 0.4 GTPTris, and 0.2 Ca2+-sensitive indicator Oregon green-488-BAPTA-1-hexapotassium salt (OGB-1; kD = 170 nm) (pH 7.2–7.3; 290 mOsm) or a combination of Ca2+-insensitive dye Alexa 594 [red (R), 0.01 mm] with a Ca2+-sensitive indicator Fluo-5F [green (G), 0.15 mm; kD = 2.3 μm] (Invitrogen). In VC experiments, a Cs+-based patch solution containing 0.5 Fluo-4FF (kD = 9.7 μm) was used (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). Biocytin (0.2%) was routinely added to the patch solutions (supplemental Methods, available at www.jneurosci.org as supplemental material).
AP–CaTs were evoked by somatic depolarization (0.05–0.1 nA; 200–250 ms). High frequency synaptic stimulation consisted of 100 Hz stimulation for 1 s given three times at an interval of 30 s (100 μs duration, 25–100 μA intensity) via a pipette filled with ACSF and positioned near the dye-filled dendrite. mGluR1/mGluR5 agonist (S)-3,5-dihydroxyphenylglycine (DHPG); 100 μm was applied repetitively (three times at an interval of 1 min) via a pipette connected to a pressure application system (PicoSpritzer II; Parker Instrumentation) and positioned ∼10 μm above the dye-filled dendrite. Hebbian LTP at interneuron excitatory synapses was induced in VC experiments by pairing a theta-burst synaptic stimulation (TBS) with postsynaptic depolarization (Perez et al., 2001) (supplemental Methods, available at www.jneurosci.org as supplemental material). Data acquisition (filtered at 2–3 kHz, digitized at 10 kHz) was performed using Clampex 10.2 (Molecular Devices). Pharmacological agents were purchased from Tocris, Sigma, or Ascent Scientific.
Two-photon Ca2+ imaging.
Postsynaptic Ca2+ imaging was performed using two imaging systems: (1) laser-scanning microscope 510 equipped with one-channel non-descanned detector (Carl Zeiss) and based on a mode-locked Ti:Sapphire laser operating at 800 nm wavelength (Mira900 pumped by 5W Verdi; Coherent) or (2) Leica TCS SP5 equipped with two-channel external detector allowing for simultaneous collection of green and red fluorescence (Leica Microsystems) and coupled with Chameleon-Ultra-II laser (Coherent) tuned to the same wavelength. A long-range water-immersion objective (40×, NA 0.8) was used in both cases. AP–CaTs (average of 3–4 responses) were acquired at 1–5 min intervals by scanning a line (300–500 Hz) across the dendrite and analyzed using LSM 510 or Leica LAS AF SP5 software (supplemental Methods, available at www.jneurosci.org as supplemental material).
Results
To probe for dendritic VSCCs in interneurons, we imaged Ca2+ transients evoked by action potentials (AP–CaTs). CA1 O/A interneurons, recorded in the whole-cell current-clamp mode, were filled with a Ca2+-sensitive indicator OGB-1 or a combination of Ca2+ insensitive Alexa 594 (to visualize cell morphology) and a lower-affinity Ca2+ dye Fluo-5F (Fig. 1A,B). Morphological reconstruction of biocytin-labeled neurons confirmed that 17 of 38 processed neurons had axon projected to stratum lacunosum-moleculare (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material), indicating that a population of the recorded interneurons were O-LM (oriens-lacunosum moleculare) cells. In the remaining 21 cells, only a short segment of axon was visualized suggesting its damage during the slicing procedure. APs-CaTs had a maximal amplitude in proximal dendritic sites and slightly decreased with the distance from the soma (40 μm: 87.5 ± 10.8% ΔF/F; 140 μm: 43.3 ± 10.7% ΔF/F; p > 0.05; n = 6; ANOVA) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). To ensure optimal voltage control during somatic voltage-clamp recordings (Williams and Mitchell, 2008) and diffusional equilibration of the Ca2+ indicators (Yasuda et al., 2004), we imaged AP–CaTs in proximal dendrites (60 ± 10 μm), starting at least 20 min after establishing a whole-cell recording. Given that in inhibitory interneurons, single APs produce dendritic CaTs of a very small amplitude often below the detection level (Goldberg et al., 2003; Rozsa et al., 2004), we used constant short somatic bursts of APs (4–5) to reliably induce dendritic CaTs with a good signal-to-noise ratio (Fig. 1B). AP–CaTs recorded in control were stable for at least 30 min (n = 8) (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). AP–CaTs were significantly reduced by a low concentration of Ni2+ (50 μm; acting mostly at R/T-type VSCCs), T-type VSCC antagonist NNC 55–0396 (10 μm), L-type VSCC blocker nifedipine (10 μm), P/Q-type VSCC antagonist ω-agatoxin IVA (AgTx, 250 nm), N-type VSCC antagonist ω-conotoxin GVIA (CTx, 250 nm) and intracellular store inhibitor ryanodine (30 μm) but were insensitive to R-type antagonist SNX-482 (0.3 μm) (supplemental Fig. 2B–D,F, available at www.jneurosci.org as supplemental material; see also Fig. 3), indicating that T-, L-, N- and P/Q-types of VSCCs as well as Ca2+ release from ryanodine-sensitive stores contribute to dendritic AP–CaTs. When a mixture of blockers (Ni2+, nifedipine, AgTx, CTx, and ryanodine) was applied, AP–CaTs were almost completely blocked (supplemental Fig. 2E,F, available at www.jneurosci.org as supplemental material).
Figure 1.
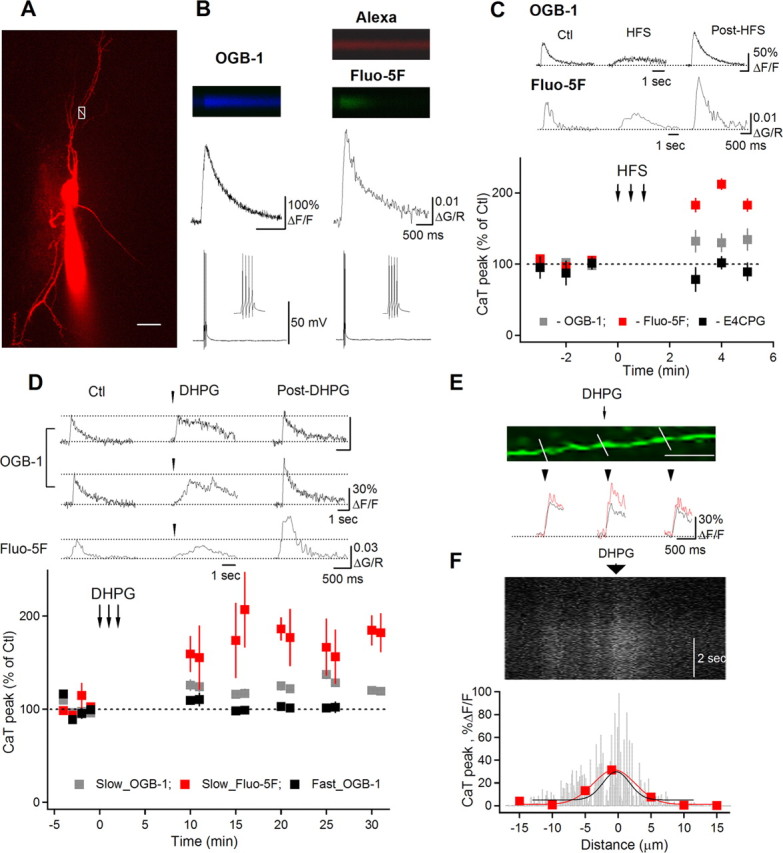
Potentiation of AP–CaTs in interneuron dendrites. A, Two-photon image of an O/A interneuron (maximal projection of a Z-stack) filled with Alexa 594 and Fluo-5F. White box with a line across the dendrite indicate the position of the line scan to measure AP–CaTs shown in B (right). Scale bar, 20 μm. B, Line scan images and associated AP–CaTs obtained with OGB-1 (left column) or Fluo-5F (right column). C, Representative AP–CaTs evoked before (Ctl) and after high frequency synaptic stimulation (middle traces; HFS) (post-HFS) within the same dendritic region and a summary plot, indicating a significant post-HFS potentiation of AP–CaTs, reported with both high- (n = 5) and low-affinity (n = 4) Ca2+ dyes. HFS-induced AP–CaT potentiation was prevented by the mGluR1/mGluR5 antagonist E4CPG (500 μm; n = 5). D, AP–CaTs obtained before (Ctl) and 15 min after DHPG application (100 μm; post-DHPG) using OGB-1 (top and middle rows) or Fluo-5F (bottom row). Plot below summarizes group data of DHPG effect for cells with fast (OGB-1, n = 3) or slow DHPG-induced Ca2+ responses (OGB-1, n = 4; Fluo-5F, n = 5). E, Magnified image of a dendrite with lines indicating locations for AP–CaT measurements. Scale bar, 10 μm. Traces below show AP–CaTs obtained from these locations in control (black) and after DHPG application (red). F, Line scan image collected along the middle part of the dendrite shown in E and demonstrating a slow DHPG Ca2+ response with corresponding spatial profile histogram (black fit). For comparison, the spatial profile of AP–CaT potentiation (red) obtained from this region is shown superimposed. All AP–CaTs represent the average of three consecutive responses.
Figure 3.
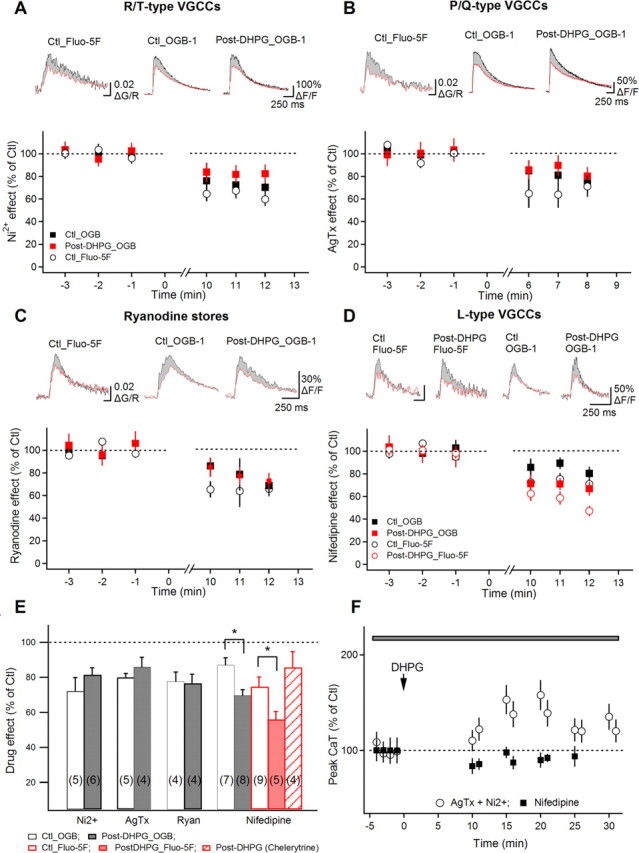
AP–CaT potentiation is associated with enhanced L-type VSCC function. A–D, Summary plots of the reduction by Ni2+ (A; 50 μm), ω-agatoxin (AgTx, 250 nm; B), ryanodine (C; 30 μm), and nifedipine (D; 10 μm) of APs-CaT amplitude (in percentage of predrug control) assessed in control (black symbols) and after DHPG-induced AP–CaT potentiation (red symbols; in different cells) with low- and high-affinity dyes. Traces above are average AP–CaTs (n = 3 traces) obtained from representative recordings in control (black) and after blocker application (red). Shaded region indicates the AP–CaT area eliminated by the corresponding blocker in each condition. E, Summary bar graph showing drug effects in control and after DHPG-induced AP–CaT potentiation. The increase in nifedipine sensitivity of AP–CaTs after DHPG was not observed in the presence of PKC blocker chelerythrine. F, Summary plot of AP–CaT amplitude versus time, demonstrating block of DHPG-induced AP–CaT modulation in nifedipine (n = 5), but not in T- and P/Q-type VSCC antagonists (Ni2+ and AgTx, respectively; n = 5).
We examined whether AP–CaTs in interneuron dendrites undergo activity-dependent modifications and whether mGluR1/mGluR5, highly expressed in O/A interneurons (Lujan et al., 1996) and capable of modulating VSCCs (Sahara and Westbrook, 1993; Catterall, 2000), are involved in regulation of AP–CaTs. We first tested if synaptic activation of mGluR1/mGluR5 during high-frequency stimulation (HFS; 100 Hz; 1 s; in bicuculline, DNQX, and AP5) (Topolnik et al., 2005) has any effect on dendritic AP–CaTs. After HFS, a significant potentiation of AP–CaTs was reported by both high- and low-affinity Ca2+ indicators (OGB-1: 132.6 ± 15% of control, *p < 0.05, n = 5; Fluo-5F: 192.7 ± 8.5% of control, *p < 0.05, n = 4) (Fig. 1C). Furthermore, AP–CaT potentiation was not observed after HFS in the presence of the mGluR1/mGluR5 antagonist E4CPG (500 μm; p > 0.05; n = 4) (Fig. 1C), suggesting that synaptic activation of mGluR1/mGluR5 induces potentiation of AP–CaTs, a novel form of interneuron dendritic plasticity.
To investigate whether mGluR1/mGluR5 are sufficient in regulating AP–CaTs, we assessed if local micropressure application of the receptor agonist (S)-3,5-DHPG to interneuron dendrite can also modulate AP–CaTs (Fig. 1D). Consistent with our previous results, application of DHPG produced two temporally distinct dendritic Ca2+ transients with either fast- (time-to-peak less than 1 s) or slow-rising (time-to-peak exceeding 1 s) Ca2+ profiles (Fig. 1D) (Topolnik et al., 2006). Interestingly, AP–CaT amplitude remained unchanged in dendritic microdomains that demonstrated fast-rising Ca2+ transient to DHPG application (n = 3) (Fig. 1D). In contrast, dendritic segments, showing slow-rising Ca2+ response to DHPG application, demonstrated significant potentiation of AP–CaTs, which persisted for at least 25 min after induction (OGB-1: 119.7 ± 4.8% of control, *p < 0.05, n = 8; Fluo-5F: 172.4 ± 24.3% of control; *p < 0.05, n = 5) (Fig. 1D). This AP–CaT potentiation was not associated with changes in somatic AP generation since local DHPG application did not affect the properties of somatic APs [AP amplitude control (Ctl): 104.0 ± 1.4 mV, post-DHPG: 104.6 ± 1.7 mV; AP duration Ctl: 1.2 ± 0.1 ms, post-DHPG: 1.2 ± 0.1 ms; AP threshold Ctl: −46.4 ± 1.0 ms, post-DHPG: −47.8 ± 1.1 ms; n = 8]. Given that fast-rising DHPG-induced Ca2+ response is associated with mGluR1a activation, whereas slow-rising Ca2+ signal is mediated by mGluR5 (Topolnik et al., 2006), these data point to a compartmentalized mGluR5-dependent plasticity of AP Ca2+ signaling.
To further estimate the spatial extent of AP–CaT potentiation, we compared a degree of AP–CaT potentiation along the dendrite (Fig. 1E,F). Our data showed that AP–CaTs were locally boosted in dendritic sites that demonstrated slow DHPG-induced Ca2+ response (DHPG-site: 128.3 ± 7.2% of Ctl; p < 0.01; 20 μm away: 104.9 ± 9.5% of Ctl; p > 0.05; n = 5) (Fig. 1E). Furthermore, in four cells, the lateral spread of DHPG-evoked slow Ca2+ responses was estimated at 15.53 ± 0.43 μm (σ value of Gaussian fit) (Fig. 1F, black line) and AP–CaT potentiation was spatially localized to 14.5 ± 0.3 μm (σ value of Gaussian fit) (Fig. 1F, red line). These data indicate a correlation between the spatial extent of slow mGluR5-mediated Ca2+ response and AP–CaT potentiation. Additionally, increasing the concentration of Ca2+ buffers (OGB-1 plus 1 mm BAPTA) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) in the patch solution blocked AP–CaT potentiation, suggesting its Ca2+ dependence. Thus, selective potentiation of AP–CaTs in dendritic compartments expressing mGluR5 but not mGluR1a highlights a specific role for mGluR5 in a novel form of interneuron dendritic plasticity.
To investigate the induction mechanisms of AP–CaT potentiation, we further examined DHPG-induced potentiation in presence of selective mGluR1a (LY367385, 100 μm) or mGluR5 [2-methyl-6-(phenylethynyl)-pyridine (MPEP), 5 μm] antagonists (Fig. 2A). Our data showed that AP–CaT potentiation was not affected by LY367385 (n = 5) (Fig. 2A) but was prevented by MPEP (n = 6) (Fig. 2A). Furthermore, MPEP, but not LY367385, also blocked AP–CaT potentiation induced by HFS (91.1 ± 8.1% of control AP–CaTs; p > 0.05; n = 5). These results clearly indicate that the induction of AP–CaT potentiation requires mGluR5 and not mGluR1a activation.
Figure 2.
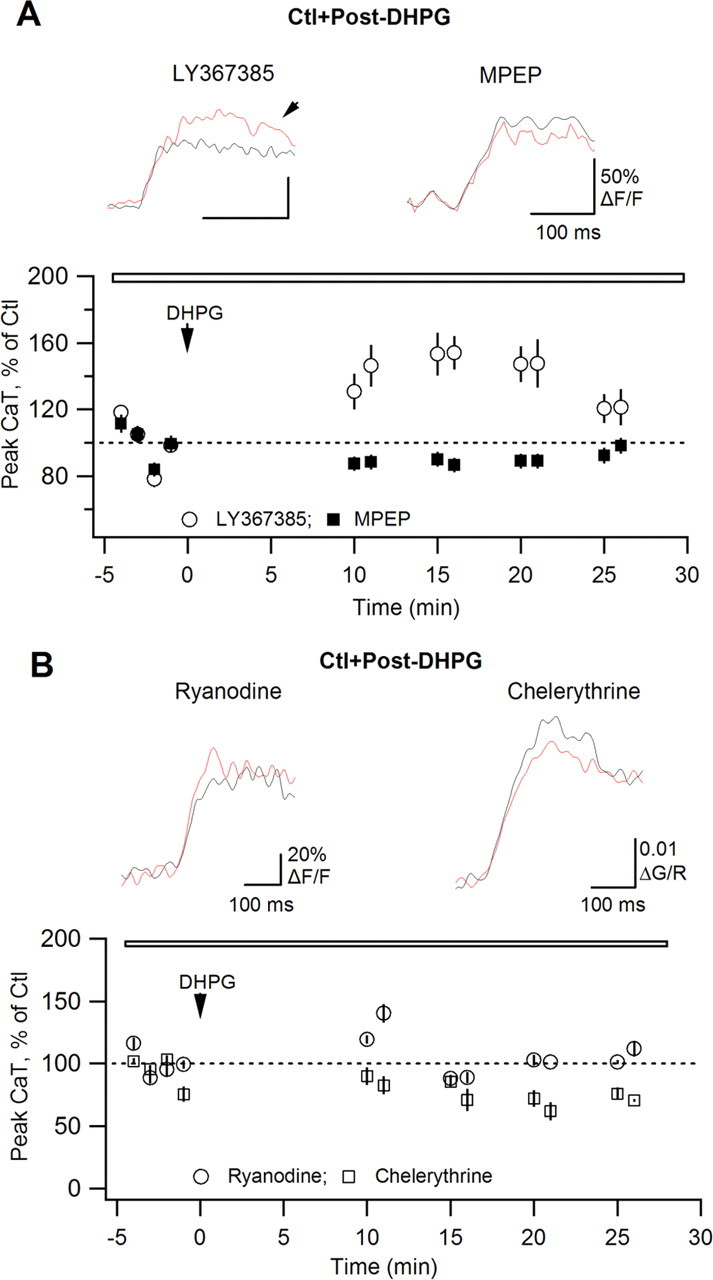
AP–CaT potentiation requires mGluR5, ryanodine-sensitive Ca2+ release and PKC activation. A, B, Summary plots of CaT peak amplitude versus time demonstrating the effects of DHPG on AP–CaTs in the presence of the mGluR1a antagonist LY367385 (A; 100 μm; n = 5), the mGluR5 antagonist MPEP (A; 5 μm; n = 5), the Ca2+ store inhibitor ryanodine (B; 30 μm; n = 5), and of PKC blocker chelerythrine (B; 10 μm; n = 4). Traces above are average expanded AP–CaTs (n = 3 traces) obtained in control (black) and after DHPG application (red).
mGluR5 activation is linked to Gq-protein-dependent phospholipase C, IP3 production, and intracellular Ca2+ release as well as DAG-dependent PKC activation. In O/A interneurons, mGluR5 activation is coupled to intracellular Ca2+ release via ryanodine-sensitive Ca2+ stores (Woodhall et al., 1999). To determine whether ryanodine-sensitive stores might be required for the induction of AP–CaT potentiation, we tested DHPG-induced modulation of AP–CaTs in the presence of ryanodine (Fig. 2B). Our data showed that bath application of ryanodine (30 μm) throughout the experiment prevented the induction of AP–CaT potentiation (n = 5) (Fig. 2B), suggesting that ryanodine-sensitive Ca2+ release is involved in this plasticity. Furthermore, DHPG-induced potentiation of AP–CaTs was prevented by inclusion of the PKC blocker chelerythrine (10 μm) in the intracellular recording solution (n = 4) (Fig. 2B). Together, these results demonstrate that AP–CaT potentiation requires mGluR5-dependent Ca2+ release and PKC activation.
Because mGluR5-mediated and PKC-dependent modulation may affect a subset of VSCCs as well as ryanodine-sensitive stores (Catterall, 2000; Yang et al., 2005), we next examined whether T-, L- and P/Q-types of VSCCs or ryanodine-sensitive Ca2+ stores were modulated after mGluR5 activation. We tested the relative contribution of these Ca2+ sources to AP–CaTs in control conditions and after DHPG-induced potentiation (different cells) (Fig. 3). The contribution of T- and P/Q-types of VSCCs as well as ryanodine-sensitive stores to AP–CaTs was not significantly changed after DHPG-induced potentiation (Fig. 3A–C,E). However, nifedipine sensitivity of AP–CaTs was significantly smaller in control conditions (OGB-1, decrease to 85.3 ± 5.8% of control AP–CaTs, n = 7; Fluo-5F, decrease to 74.8 ± 5.4% of control AP–CaTs, n = 9) than after DHPG-induced potentiation (OGB-1, decrease to 70.2 ± 3.8% of control AP–CaTs, n = 8; *p < 0.05; Fluo-5F, decrease to 56.0 ± 4.5% of control AP–CaTs, n = 5; *p < 0.05) (Fig. 3D,E), revealing an increased contribution of L-type VSCCs to AP–CaTs during DHPG-induced potentiation. Moreover, DHPG-induced increase in contribution of L-type VSCCs to AP–CaTs was not observed with chelerythrine present in the intracellular recording solution (Fig. 3E). These data suggest that mGluR5-induced potentiation of AP–CaTs is expressed selectively as an increased contribution of L-type VSCCs attributable to phosphorylation by PKC. If this is the case, then DHPG-induced AP–CaT potentiation should still be observed in the presence of other VSCC antagonists. Indeed, local application of DHPG in the presence of Ni2+ and AgTx produced potentiation of AP–CaTs (137.8 ± 12.3% of control AP–CaTs; *p < 0.05; n = 5) (Fig. 3F), indicating that AP–CaT potentiation occurs independently of T- and P/Q-types of VSCCs. However, potentiation was occluded by nifedipine (post-DHPG AP–CaTs were 93 ± 7.6% of control AP–CaTs; p > 0.05; n = 5) (Fig. 3F). Together, these data demonstrate that mGluR5-induced AP–CaT potentiation is likely attributable to PKC phosphorylation of L-type VSCCs.
Finally, we examined the role of L-type VSCCs in plasticity at O/A interneuron synapses. A Hebbian LTP is induced at interneuron excitatory synapses by pairing TBS with postsynaptic depolarization (Perez et al., 2001). First, we established that L-type VSCCs contribute in part to Ca2+ rises evoked by the Hebbian induction protocol. Using the lower affinity dye Fluo-4FF, we found that the Ca2+ transients elicited by the TBS pairing protocol were significantly reduced by nifedipine (CaT peak amplitude 65.2 ± 2.4% of control; n = 5; p < 0.05) (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). Next, we determined that L-type VSCCs are necessary for the induction of Hebbian LTP in O/A interneurons. In control conditions, the pairing protocol induced LTP of EPSCs lasting at least 30 min, associated with a decrease in failure rate and an increase in EPSC potency (p < 0.05; n = 7) (supplemental Fig. 4B,E, available at www.jneurosci.org as supplemental material). Nifedipine did not affect basal synaptic transmission (n = 3; data not shown) but prevented LTP induction (p < 0.05; n = 5) (supplemental Fig. 4C,E, available at www.jneurosci.org as supplemental material), uncovering an important role of L-type VSCCs in Hebbian LTP. Hebbian LTP was not affected by the mGluR5 antagonist MPEP (supplemental Fig. 4D,E, available at www.jneurosci.org as supplemental material), confirming that mGluR5 is not required for its induction. Overall, these data demonstrate that Ca2+ influx via L-type VSCCs occurs during the pairing protocol, contributing to LTP induction. Given that mGluR5 does not play a direct role in the induction of Hebbian LTP, activity-dependent mGluR5-induced potentiation of L-type VSCCs might provide a mean of regulating synaptic plasticity in O/A interneurons.
Discussion
Our results provide new insights into mechanisms and plasticity of dendritic Ca2+ signaling in hippocampal interneurons. O/A interneurons express a large repertoire of dendritic VSCCs, which together with ryanodine-sensitive Ca2+ stores contribute to AP Ca2+ signaling. Such abundance of dendritic Ca2+ sources may be required for specific Ca2+-dependent functions, from regulating synaptic efficacy and local release of retrograde messengers to modulating ion conductances and dendritic excitability, ensuring reliable interneuron operation in dynamic environment. Furthermore, AP-evoked Ca2+ signaling is subject to activity-dependent compartmentalized potentiation which is induced by mGluR5 and involves ryanodine-sensitive stores, PKC activation and selective increase in L-type VSCC contribution. These results reveal a novel form of interneuron dendritic plasticity triggered by intense synaptic activity. Given that L-type VSCCs are involved in the induction of Hebbian LTP at excitatory synapses of these interneurons, such long-lasting modulation of L-type VSCC function may provide a potential mechanism for regulating interneuron synaptic plasticity.
Selective mGluR5 activation is responsible for potentiation of L-type VSCCs, resulting in an enhancement of AP Ca2+ signaling. Indeed, AP–CaT potentiation was compartmentalized to specific dendritic microdomains, likely dependent on a site-specific distribution of mGluR5 (Topolnik et al., 2006). Moreover, activation of mGluR5 alone was sufficient for AP–CaT potentiation, consistent with previous demonstrations of mGluR5-induced modulation of L-type VSCCs (Sosa and Gleason, 2004; Yamamoto et al., 2005). Furthermore, this form of potentiation required Ca2+ release from ryanodine-sensitive stores. Ryanodine receptors were shown to physically interact with L-type VSCCs (Chavis et al., 1996), thus being responsible for direct conformational changes and modulation of channel function. Conversely, L-type VSCCs are necessary for store loading and function (Chavis et al., 1996), suggesting a constant bidirectional dialogue between internal stores and L-type channels. In our conditions, mGluR5 activation, by triggering internal Ca2+ release, results in potentiation of L-type VSCCs function, presumably via channel phosphorylation or membrane expression. Interestingly, long-lasting modifications of L-type channel function may require a specific profile of intracellular Ca2+ signal. Indeed, AP–CaT potentiation was compartmentalized to the same dendritic microdomain as mGluR5-induced slow, but not mGluR1-mediated fast, Ca2+ signals. Importantly, mGluR5- and Ca2+-dependent potentiation of L-type VSCC required PKC activation. These data are consistent with L-type VSCC regulation via PKC phosphorylation. Indeed, PKC was shown to form a macromolecular complex with the α(1c) subunit of Cav1.2, leading to channel phosphorylation and upregulation (Yang et al., 2005).
L-type VSCCs are high-voltage activated, large conductance channels mediating sustained Ca2+ influx (Magee and Johnston, 1995). In neurons, they subserve many functions from the modulation of AP firing to gene expression and neuronal survival. Our results uncover a novel role of L-type VSCCs in interneuron excitatory synapses: they provide a necessary component of the Ca2+ signal during the LTP induction pairing protocol, in addition to mGluR1a-activated Ca2+ signaling via TRP channels (Topolnik et al., 2006). Use-dependent modification of any of these Ca2+ mechanisms may represent a potential tool for regulating synaptic plasticity, enabling metaplasticity at interneuron excitatory synapses. Thus, mGluR5-induced potentiation of dendritic Ca2+ signaling may act as a priming stimulus for synaptic LTP induction.
Together, our data show that in O/A interneuron dendrites, mGluR1 and mGluR5 are linked to distinct signaling cascades and play specific physiological roles: whereas mGluR1 is primarily involved in the induction of synaptic plasticity, mGluR5 acts as a regulator of L-type VSCC function, and by this means, may modify synaptic plasticity. Interestingly, mGluR5 may be preferentially activated during seizures and contribute to epileptiform activity in O/A interneurons (Sanon et al., 2005). Thus, mGluR5-induced long-lasting modifications in dendritic signaling may be relevant to functional changes in inhibitory circuits in epilepsy.
Footnotes
This work was supported by the Canadian Institutes of Health Research (Operating Grant 10848 to J.-C.L.), Fonds de la Recherche en Santé du Québec (FRSQ; Groupe de Recherche sur le Système Nerveux Central), Savoy Foundation (research grant to L.T.), and Natural Sciences and Engineering Research Council of Canada (NSERC Discovery grant to L.T.). J.-C.L. is recipient of the Canada Research Chair in Cellular and Molecular Neurophysiology. L.T. is recipient of the University Faculty Award from NSERC. J.G.P. was supported by a Fellowship of the FRSQ. We thank Richard Robitaille for expert help with two-photon microscopy.
References
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Chavis P, Fagni L, Lansman JB, Bockaert J. Functional coupling between ryanodine receptors and L-type calcium channels in neurons. Nature. 1996;382:719–722. doi: 10.1038/382719a0. [DOI] [PubMed] [Google Scholar]
- Goldberg JH, Tamas G, Yuste R. Ca2+ imaging of mouse neocortical interneuron dendrites: Ia type K+ channels control action potential backpropagation. J Physiol. 2003;551:49–65. doi: 10.1113/jphysiol.2003.042580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamsa KP, Heeroma JH, Somogyi P, Rusakov DA, Kullmann DM. Anti-Hebbian long-term potentiation in the hippocampal feedback inhibitory circuit. Science. 2007;315:1262–1266. doi: 10.1126/science.1137450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan R, Nusser Z, Roberts JD, Shigemoto R, Somogyi P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Synaptic activation of voltage-gated channels in the dendrites of hippocampal pyramidal neurons. Science. 1995;268:301–304. doi: 10.1126/science.7716525. [DOI] [PubMed] [Google Scholar]
- Martina M, Vida I, Jonas P. Distal initiation and active propagation of action potentials in interneuron dendrites. Science. 2000;287:295–300. doi: 10.1126/science.287.5451.295. [DOI] [PubMed] [Google Scholar]
- Perez Y, Morin F, Lacaille JC. A hebbian form of long-term potentiation dependent on mGluR1a in hippocampal inhibitory interneurons. Proc Natl Acad Sci U S A. 2001;98:9401–9406. doi: 10.1073/pnas.161493498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozsa B, Zelles T, Vizi ES, Lendvai B. Distance-dependent scaling of calcium transients evoked by backpropagating spikes and synaptic activity in dendrites of hippocampal interneurons. J Neurosci. 2004;24:661–670. doi: 10.1523/JNEUROSCI.3906-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Svoboda K. Analysis of calcium channels in single spines using optical fluctuation analysis. Nature. 2000;408:589–593. doi: 10.1038/35046076. [DOI] [PubMed] [Google Scholar]
- Sahara Y, Westbrook GL. Modulation of calcium currents by a metabotropic glutamate receptor involves fast and slow kinetic components in cultured hippocampal neurons. J Neurosci. 1993;13:3041–3050. doi: 10.1523/JNEUROSCI.13-07-03041.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanon N, Carmant L, Lacaille J-C. Interneuron subtype-specific activation of mGluR1/5 during epileptiform activity in rat hippocampal slices. Soc Neurosci Abstr. 2005;31:433–14. [Google Scholar]
- Scheuss V, Yasuda R, Sobczyk A, Svoboda K. Nonlinear [Ca2+] signaling in dendrites and spines caused by activity-dependent depression of Ca2+ extrusion. J Neurosci. 2006;26:8183–8194. doi: 10.1523/JNEUROSCI.1962-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobczyk A, Svoboda K. Activity-dependent plasticity of the NMDA-receptor fractional Ca2+ current. Neuron. 2007;53:17–24. doi: 10.1016/j.neuron.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Sosa R, Gleason E. Activation of mGluR5 modulates Ca2+ currents in retinal amacrine cells from the chick. Vis Neurosci. 2004;21:807–816. doi: 10.1017/S0952523804216017. [DOI] [PubMed] [Google Scholar]
- Topolnik L, Congar P, Lacaille JC. Differential regulation of mGluR- and AMPAR-mediated dendritic Ca2+ signals by pre- and postsynaptic activity in hippocampal interneurons. J Neurosci. 2005;25:990–1001. doi: 10.1523/JNEUROSCI.4388-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topolnik L, Azzi M, Morin F, Kougioumoutzakis A, Lacaille JC. mGluR1/5 subunit-specific Ca2+ signalling and long-term potentiation in rat hippocampal oriens/alveus interneurons. J Physiol. 2006;575:115–131. doi: 10.1113/jphysiol.2006.112896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinet J, Sík A. Expression pattern of voltage-dependent calcium channel subunits in hippocampal inhibitory neurons in mice. Neuroscience. 2006;143:189–212. doi: 10.1016/j.neuroscience.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Williams SR, Mitchell SJ. Direct measurement of somatic voltage clamp errors in central neurons. Nat Neurosci. 2008;11:790–798. doi: 10.1038/nn.2137. [DOI] [PubMed] [Google Scholar]
- Woodhall G, Gee CE, Robitaille R, Lacaille JC. Membrane potential and intracellular Ca2+ oscillations activated by mGluRs in hippocampal stratum oriens/alveus interneurons. J Neurophysiol. 1999;81:371–382. doi: 10.1152/jn.1999.81.1.371. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sakagami Y, Sugiura S, Inokuchi K, Shimohama S, Kato N. Homer 1a enhances spike-induced calcium influx via L-type calcium channels in neocortex pyramidal cells. Eur J Neurosci. 2005;22:1338–1348. doi: 10.1111/j.1460-9568.2005.04278.x. [DOI] [PubMed] [Google Scholar]
- Yang L, Liu G, Zakharov SI, Morrow JP, Rybin VO, Steinberg SF, Marx SO. Ser1928 is a common site for Cav1.2 phosphorylation by protein kinase C isoforms. J Biol Chem. 2005;280:207–214. doi: 10.1074/jbc.M410509200. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Sabatini BL, Svoboda K. Plasticity of calcium channels in dendritic spines. Nat Neurosci. 2003;6:948–955. doi: 10.1038/nn1112. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Nimchinsky EA, Scheuss V, Pologruto TA, Oertner TG, Sabatini BL, Svoboda K. Imaging calcium concentration dynamics in small neuronal compartments. Sci STKE. 2004;219:pl5. doi: 10.1126/stke.2192004pl5. [DOI] [PubMed] [Google Scholar]