

Abstract
Leptin-stimulated Stat3 activation in proopiomelanocortin (POMC)-expressing neurons of the hypothalamus plays an important role in maintenance of energy homeostasis. While Stat3 activation in POMC neurons is required for POMC expression, the role of elevated basal Stat3 activation as present in the development of obesity has not been directly addressed. Here, we have generated and characterized mice expressing a constitutively active version of Stat3 (Stat3-C) in POMC neurons (Stat3-CPOMC mice). On normal chow diet, these animals develop obesity as a result of hyperphagia and decreased POMC expression accompanied by central leptin and insulin resistance. This unexpected finding coincides with POMC-cell-specific, Stat3-mediated upregulation of SOCS3 expression inhibiting both leptin and insulin signaling as insulin-stimulated PIP3 (phosphatidylinositol-3,4,5 triphosphate) formation and protein kinase B (AKT) activation in POMC neurons as well as with the fact that insulin's ability to hyperpolarize POMC neurons is largely reduced in POMC cells of Stat3-CPOMC mice. These data indicate that constitutive Stat3 activation is not sufficient to promote POMC expression but requires simultaneous PI3K (phosphoinositide 3-kinase)-dependent release of FOXO1 repression. In contrast, upon exposure to a high-fat diet, food intake and body weight were unaltered in Stat3-CPOMC mice compared with control mice. Taken together, these experiments directly demonstrate that enhanced basal Stat3 activation in POMC neurons as present in control mice upon high-fat feeding contributes to the development of hypothalamic leptin and insulin resistance.
Introduction
Energy homeostasis is stringently regulated by neuronal populations of the arcuate nucleus (ARC) in the hypothalamus, which sense and integrate signals mediated by nutrients, cytokines, and hormones such as leptin (Plum et al., 2006a). In the ARC, leptin inhibits the orexigenic agouti-related peptide (AgRP)/neuropeptide Y (NPY)-producing neurons, while it activates the anorexigenic proopiomelanocortin (POMC) neurons (Elias et al., 1999; van den Top et al., 2004). Consistently, leptin-deficient ob/ob and leptin receptor-deficient db/db mice display increased levels of AgRP mRNA and reduced levels of POMC mRNA (Schwartz et al., 1997; Mizuno and Mobbs, 1999). Ablation of POMC neurons and loss of POMC-derived transmitters lead to obesity (Yaswen et al., 1999; Balthasar et al., 2004; Gropp et al., 2005), while ablation of AgRP neurons and loss of AgRP expression result in lean mice (Bewick et al., 2005; Gropp et al., 2005; Luquet et al., 2005). This underlines the importance of leptin-evoked AgRP and POMC neuron regulation in the control of energy homeostasis.
Leptin binding to the leptin receptor activates Stat3 (Bjørbaek et al., 1997; Banks et al., 2000), which regulates expression of target genes such as POMC and SOCS3 (Schindler and Darnell, 1995; Leaman et al., 1996). Ablating Stat3 in the CNS causes severe obesity with a concomitant decrease in POMC expression (Gao et al., 2004), while Stat3 inactivation specifically in POMC neurons results in mild obesity and decreased POMC expression, indicating Stat3 as a transcriptional activator of POMC expression (Xu et al., 2007). However, the Stat3-binding site in the POMC promoter overlaps with a FOXO1-binding element, and FOXO1 inhibits POMC transcription (Kitamura et al., 2006). Accordingly, POMC neuronal PDK-1 deficiency reduces POMC expression (Belgardt et al., 2008), and overactivation of PI3K (phosphoinositide 3 kinase) by disrupting PTEN in POMC neurons leads to increased POMC expression (Plum et al., 2006b).
While mutations affecting leptin or leptin receptor expression are rare in humans and rodents, leptin levels are dramatically increased in obese patients, indicating the existence of leptin resistance (Frederich et al., 1995). Numerous potential mechanisms to explain leptin resistance have been put forth, such as saturation of leptin transport across the blood–brain barrier (El-Haschimi et al., 2000) and the activation of inhibitory molecules such as SHP-2 and SOCS3 in the leptin signaling cascade (Bjørbaek et al., 1999, 2001; Münzberg et al., 2004). However, the contribution of altered basal leptin signaling and Stat3 activation has been less well investigated, although it was demonstrated that upon high-fat feeding, basal hypothalamic Stat3 phosphorylation is increased (Martin et al., 2006). To assess the role of activated basal Stat3 signaling in POMC-expressing neurons, we analyzed mice expressing a constitutively active version of Stat3 selectively in POMC neurons. Surprisingly, these mice exhibit mild obesity as a result of hyperphagia, elevated plasma leptin concentrations, and decreased hypothalamic POMC expression. The observed leptin and insulin resistance in this mouse model results from increased hypothalamic SOCS3 expression. Moreover, exposure to high-fat diet (HFD) abrogated the effect of Stat3 activation in POMC cells, as both Stat3-CPOMC and control mice became similarly obese. Thus, we conclude elevated basal hypothalamic Stat3 signaling leads to SOCS3 upregulation, which subsequently inhibits leptin and insulin signaling.
Materials and Methods
Animal care
Care of all animals was within institutional animal care committee guidelines, and all animal procedures were approved by local government authorities (Bezirksregierung Köln, Cologne, Germany) and were in accordance with National Institutes of Health guidelines. Mice were housed in groups of 3–5 at 22–24°C using a 12-h-light/12-h-dark cycle, with lights on at 7:00 A.M. Animals were either fed regular chow food [normal chow diet (NCD); Teklad Global Rodent 2018; Harlan) containing 53.5% carbohydrates, 18.5% protein, and 5.5% fat (12% of calories from fat) or an HFD (C1057; Altromin) containing 32.7% carbohydrates, 20% protein, and 35.5% fat (55.2% of calories from fat). Animals had ad libitum access to water at all times, and food was withdrawn only when required for experimental procedures. At 20 weeks of age, animals were killed using CO2.
Generation of Stat3-CPOMC and Stat3-C/CPOMC mice
Stat3-CFL/FL mice were crossed with POMC-Cre mice. Only animals from the same litter were compared with each other. Mice were genotyped by PCR, using genomic DNA isolated from tail tips. POMC-Cre primers were as follows: N16R, 5′-TGG CTC AAT GTC CTT CCT GG-3′; N57R, 5′-CAC ATA AGC TGC ATC GTT AAG-3′; AA03, 5′-GAG ATA TCT TTA ACC CTG ATC-3′. Stat3-C primers were as follows: Typ_forward, 5′-AAA GTC GCT CTG AGT TGT TAT C-3′; NeoRT, 5′-CGG ACC GCT ATC AGG ACA TA-3′; Typ_reverse, 5′-GAT ATG AAG TAC TGG GCT CTT-3′. Germline deletion by the POMC-Cre was excluded using the NeoRT primer located in the loxP-flanked stop cassette.
Body weight, food intake, body length, and body composition
Body weight was measured weekly. Food intake was measured once a week over 3 weeks in regular cages using racks. Daily food intake was calculated as the average intake of chow. Body fat content was measured in vivo by nuclear magnetic resonance using the minispec mq 7.5 (Bruker). At the end of the study, body length (naso–anal length) was determined directly after killing, and relevant organs were dissected and stored at −80°C until further preparation.
Protein extract preparation and Western blot analysis
To prepare ES cell lysates, 106 ES cells were stimulated with 30 U/ml leukemia inhibiting factor (LIF) (Chemicon) for 0, 30, and 60 min and dissolved in 20 μl of RIPA buffer [1× PBS, 1% NP-40, supplemented with protease inhibitor mixture (Roche)]. To separate the cytoplasmic protein fractions from nuclear extracts, 106 ES cells were resuspended in 20 μl of hypotonic solution [10 mm HEPES, pH 7.6, 10 mm KCl, 2 mm MgCl2, 0.1 mm EDTA, completed with protease inhibitor mixture (Roche)] and NP-40 was added to 1% after 10 min incubation on ice. After centrifugation, the supernatant containing the cytoplasmic fraction was recovered from the nuclear pellet and the nuclear pellet was washed in 15 μl of hypotonic buffer and dissolved in extraction buffer (50 mm HEPES, pH 8, 50 mm KCl, 300 mm NaCl, 0.1 mm EDTA, 10% glycerol). Western blot analyses were performed by standard methods using antibodies raised against Stat3 (#4904, Cell Signaling Technology), pStat3 (#9145 Cell Signaling Technology), α-tubulin (#T6074, Sigma) and LaminA/C (#s.c.-6215, Santa Cruz Biotechnology).
Electrophoretic mobility shift assay
Mice were intraperitoneally injected with either saline or leptin (5 mg/kg body weight) (Sigma-Aldrich) after overnight (16 h) fasting and killed after 30 min. Starved ES cells were stimulated with 30 U/ml LIF for 2 h. Nuclear extracts of hypothalamic tissue or ES cells were isolated as described above. Four micrograms of nuclear extracts was incubated at room temperature for 30 min with 2 μg of poly(dI-dC) (Pharmacia) and 0.5 ng of 32P-labeled Stat3 probe (#s.c.-2571, Santa Cruz Biotechnology), Stat3 mutant probe (#s.c.-2572, Santa Cruz Biotechnology), or SP1 probe (#s.c.-2502, Santa Cruz Biotechnology). Samples were separated by 5% PAGE overnight and visualized by autoradiography.
Transient transfection of ES cells and luciferase detection
Control and Stat3-C ES cells were cultured in growth medium containing high glucose (4.5 g/L) DMEM (PAA), 15% FCS (Biochrom), 1 mm sodium pyruvate (Invitrogen), 2 mm l-glutamine (Invitrogen), 1× nonessential amino acids (Invitrogen), 10 U/ml LIF, 0.1 mm 2-β-mercaptoethanol (Merck). ES cells (107) were transiently transfected with 30 μg of pSTAT3-TA-Luc (Clontech) and 10 μg of pRLnull vector (Promega) by electroporation. After transfection, ES cells were suspended in growth medium and plated into 24-well culture plates. Twenty-four hours after transfection, ES cells were washed with 1× PBS and incubated for 24 h with DMEM or growth medium supplemented with 30 U/ml LIF (Chemicon). ES cells were then washed in PBS, lysed in 100 μl of passive lysis buffer, and centrifuged at 4°C. Supernatant was assayed for Fluc activities using the Dual Luciferase Assay (Promega) according to the manufacturer's protocol. Each experiment was performed in triplicate, and each transfection was repeated four times.
RT-PCR
RNA isolation was performed using the RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. Total RNA was treated with RNase-free DNase (Promega) and 200 ng of RNA was reverse transcribed using EuroScript reverse transcriptase (Eurogentec). Stat3-C, endogenous Stat3, and GAPDH were amplified using the following specific primers: Stat3-C primer, Rosa512: 5′-GCC GTT CTG TGA GAC AG-3′; 3StatRT, 5′-AGG ACA TTG GAC TCT TGC AG-3′; endogenous Stat primer 5StatRT, 5′-CAG TCG GGC CTC AGC CC-3′; 3StatRT, 5′-AGG ACA TTG GAC TCT TGC AG-3′; GAPDH primer GAPDH 5′, 5′-ACC ACA GTC CAT GCC ATC AC-3′; and GAPDH 3′, 5′-TCC ACC ACC CTG TTG CTG TA-3′.
Analysis of RNA expression
Neuropeptide and SOCS3 mRNA expression was analyzed using quantitative real-time PCR. Two hundred nanograms of total RNA was reverse transcribed with EuroScript reverse transcriptase (Eurogentec) and amplified using TaqMan Universal PCR-Master Mix and NO AmpErase UNG with TaqMan Assay on Demand kits (Applied Biosystems). Relative expression of mRNA was determined using standard curves based on hypothalamic cDNA. Samples were adjusted for total RNA content either by hypoxanthine phosphoribosyltransferase or by glucuronidase β RNA quantitative PCR. Calculations were performed by a comparative method (2-ΔΔCT). Quantitative PCR was performed on an ABI-PRISM 7700 Sequence Detector (Applied Biosystems). Assays were linear over 4 orders of magnitude.
Glucose and insulin tolerance test
Glucose tolerance tests were performed with 16-h-fasted animals. After determination of fasting blood glucose levels, each animal received an intraperitoneal injection of 20% glucose (10 ml/kg) (DeltaSelect). Blood glucose levels were detected after 15, 30, 60, and 120 min. Insulin tolerance tests were performed with randomly fed mice. Animals were injected with 0.75 U/kg body weight of human regular insulin (Novo Nordisk) into the peritoneal cavity. Blood glucose levels were detected after 0, 15, 30, and 60 min.
Leptin sensitivity
Leptin sensitivity was examined by intraperitoneal injection of mice at the age of 15 weeks with saline twice a day for three consecutive days and subsequently with 2 mg/kg leptin (Sigma) twice a day for three consecutive days. Body weight and food intake were determined daily.
Analytical procedures
Blood glucose values were determined from whole venous blood using an automatic glucose monitor (GlucoMen; A. Menarini Diagnostics). Serum leptin and insulin concentrations were measured by ELISA using mouse standards according to the manufacturer's guidelines (mouse leptin ELISA, #90030, Crystal Chem; rat insulin ELISA, #INSKR020, Crystal Chem).
Immunohistochemistry
For staining of POMC neurons, Stat3-CPOMC mice were anesthetized and transcardially perfused with saline solution followed by 4% paraformaldehyde in 0.1 m PBS (pH 7.4). The brains were dissected, postfixed in 4% PFA at 4°C, transferred to 20% sucrose for 6 h, and frozen in Jung tissue-freezing medium (Leica Microsystems). Subsequently, 25-μm-thick free-floating coronal sections were dissected through the ARC using a freezing microtome (Leica). The sections were collected in PBS–azide, pH 7.4, and washed extensively to remove cryoprotectant. The sections were stained using anti-EGFP (enhanced green fluorescent protein) antibody (#A6455; Invitrogen/Molecular Probes).
For the staining of PIP3 (phosphatidylinositol-3,4,5 triphosphate) in POMC neurons, Stat3-CPOMC mice were crossed with ROSAArte26 reporter mice (Seibler et al., 2003). At the age of 12 weeks, 16-h-fasted animals were anesthetized and intravenously injected with 5 U of human regular insulin (Novo Nordisk) for 10 min. Mice were transcardially perfused with saline solution, and the dissected brains were frozen in tissue-freezing medium. The 7 μm coronal sections containing the ARC were stained with β-galactosidase (#555976; Cappel) and PIP3 (#Z-G345; Echelon) antibodies. Double fluorescence immunostainings were performed as previously described (Schubert et al., 2004). For quantitative analysis of PIP3 levels in POMC neurons, a total of 918 LacZ-positive neurons were counted in ARC slices of control (n = 5; 842 POMC neurons) and Stat3-CPOMC ROSAArte26 (n = 5; 824 POMC neurons) mice, and the amount of PIP3 was classified as described before (Plum et al., 2006b) as low (<5 immunoreactive PIP3 dots), moderate (5–10 immunoreactive PIP3 dots), or high (>10 immunoreactive PIP3 dots). Neurons positive for β-galactosidase were counted and marked digitally to prevent multiple counts, and PIP3 immunoreactivity was rated as described above. Results were expressed as percentage of POMC neurons, showing the respective PIP3 levels.
For the staining of pSTAT3 in POMC neurons, Stat3-CPOMC mice were crossed with ROSAArte26 reporter mice (Seibler et al., 2003). At the age of 10–12 weeks, 16-h-fasted animals were anesthetized and intraperitoneally injected with either saline or 1 mg/kg leptin (Sigma) for 30 min. Mice were transcardially perfused with saline solution, and the dissected brains were frozen in tissue-freezing medium. The 7 μm coronal sections containing the ARC were stained with β-galactosidase (#9361; Abcam) and pStat3 (#9145; Cell Signaling Technology) antibodies. Double fluorescence immunostainings were performed as previously described (Schubert et al., 2004). For quantitative analysis of pStat3-positive POMC neurons, a total of 2103 β-galactosidase-positive neurons were counted and digitally marked to prevent multiple counts. pStat3-positive POMC neurons were expressed as percentage of total POMC neurons.
For the staining of pAKT in POMC neurons, Stat3-CPOMC mice were crossed with ROSAArte26 reporter mice (Seibler et al., 2003). At the age of 10–14 weeks, 48-h-fasted animals were anesthetized and intravenously injected with either saline or 5 U of human regular insulin (Novo Nordisk) for 10 min. Mice were transcardially perfused with saline solution, and the dissected brains were postfixed in 4% PFA at 4°C, transferred to 20% sucrose for 6 h, and frozen in tissue-freezing medium. The 25 μm coronal sections containing the ARC were stained with β-galactosidase (#9361; Abcam) and pAKT (#4060; Cell Signaling Technology) antibodies. For quantitative analysis of pAKT-positive POMC neurons, a total of 2351 β-galactosidase-positive neurons were counted and digitally marked to prevent multiple counts. pAKT-positive POMC neurons were expressed as a percentage of total POMC neurons.
Combined histochemistry and in situ hybridization
For X-gal stainings, Stat3-CPOMC were mated with ROSAArte26 reporter mice (Seibler et al., 2003). At the age of 12 weeks, 16-h-fasted animals were anesthetized and transcardially perfused with saline followed by 4% paraformaldehyde in 0.1 m PBS (pH 7.4). The brains were dissected, postfixed in 4% PFA for 4 h, soaked in 20% sucrose overnight at 4°C, and frozen in tissue-freezing medium. Coronal sections (8 μm thick) containing the ARC were fixed for 10 min in cold formalin, washed three times with PBS, rinsed in distilled water, and subjected to X-gal staining overnight at 37°C [X-gal solution: 5 mm K3Fe(CN)6, 5 mm K4Fe(CN)6, 2 mm MgCl2, 1 mg/ml X-gal in PBS, pH 7.4]. After staining, sections were rinsed in distilled water and mounted or directly used for combined in situ hybridization. For the synthesis of the SOCS3 probe and POMC probe, fragments were amplified by PCR from mouse hypothalamic cDNA using primers containing a T7 promoter sequence or cloned into the T7 promoter containing pGEM-T easy Vector (Promega). Primers were as follows: SOCS3 primers, 5′-GGC GCG CCA CCA TGG TCA CCC ACA GCA AGT TTC C-3′, 5′-ATT TAA ATT AAA GTG GAG CAT CAT ACT G-3′); and POMC primers, 5′-ATG CCG AGA TTC TGC TAC AG-3′, 5′-TGC TGC TGT TCC TGG GGC-3′. The DNA fragment was transcribed into digoxigenin (DIG)-labeled RNA using the DIG RNA Labeling Kit (Roche). For combined SOCS3 in situ hybridization, X-gal-stained sections were washed with PBS and treated with proteinase K (0.25 μg/ml) for 10 min at 37°C. The sections were rinsed with glycine (2 mg/ml), placed into 4% PFA, washed with PBS, and then washed with 2× SSC. Prehybridization was performed for 5 h at 56°C in prehybridization buffer containing 50% formamide, 5× SSC, 1× Denhardt's solution, and 0.1% Tween 20. Hybridization was performed at 56°C overnight with 2 ng/μl digoxigenin-labeled RNA probe. After washing with 2× SSC, the sections were RNase digested for 1 h at 37°C and afterward washed with 0.1% SSC for 1 h at 55°C and then cooled to room temperature. The sections were blocked for 1 h and incubated with anti-digoxigenin antibody coupled to alkaline phosphatase (Roche) for 1 h. After washing, SOCS3 in situ hybridization was detected using Liquid Permanent Red (Dako) or BM Purple (Roche). For quantitative analysis of SOCS3-positive POMC neurons, a total of 3069 X-gal-positive neurons were counted in ARC slides of control (n = 3; 1486 POMC neurons) and Stat3-CPOMC ROSAArte26 (n = 3; 1583 POMC neurons) mice and marked digitally to prevent multiple counts. SOCS3-positive POMC neurons were expressed as the percentage of total POMC neurons. For quantitative analysis of X-gal-positive/POMC mRNA-positive neurons, a total of 2214 POMC mRNA-positive neurons were counted in ARC slides of control (n = 4; 1155 POMC mRNA-positive neurons) and Stat3-CPOMC ROSAArte26 (n = 4; 1059 POMC mRNA-positive neurons) mice and marked digitally to prevent multiple counts. X-gal-positive/POMC mRNA-positive neurons were expressed as the percentage of total POMC mRNA-positive neurons.
Electrophysiology
Animals and brain slice preparation.
Experiments were performed on brain slices from 18- to 28-d-old POMC-EGFP and STAT3-CPOMC POMC-EGFP mice that expressed EGFP selectively in POMC neurons (Cowley et al., 2001). The animals were anesthetized with halothane (SigmaAldrich) and subsequently decapitated. The brain was rapidly removed, and a block of tissue containing the hypothalamus was immediately cut out. Coronal slices (250–300 μm) containing the ARC were cut with a vibration microtome (HM-650 V; Thermo Scientific) under cold (4°C), carbogenated (95% O2 and 5% CO2), glycerol-based modified artificial CSF (GaCSF) to enhance the viability of neurons. GaCSF contained the following (in mm): 250 glycerol, 2.5 KCl, 2 MgCl2, 2 CaCl2, 1.2 NaH2PO4, 10 HEPES, 21 NaHCO3, 5 glucose adjusted to pH 7.2 (with NaOH), resulting in an osmolarity of ∼310 mOsm. Brain slices were transferred into carbogenated artificial CSF (aCSF). First, they were kept for 20 min in a 35°C “recovery bath” and then stored at room temperature (24°C) for at least 30 min before recording. aCSF contained the following (in mm): 125 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 1.2 NaH2PO4, 21 NaHCO3, 10 HEPES, and 5 glucose adjusted to pH 7.2 (with NaOH), resulting in an osmolarity of ∼310 mOsm. Slices were transferred to a recording chamber (∼3 ml volume) and continuously superfused with carbogenated aCSF at a flow rate of ∼2 ml · min−1. Insulin (200 nm; Sigma) and the KATP channel blocker tolbutamide (200 μm; Sigma) were bath applied via the superfusion system. The tolbutamide was dissolved in DMSO and added to the normal aCSF with a final DMSO concentration of 0.25%. The DMSO concentration had no obvious effect on the investigated neurons.
Perforated patch recordings.
Slices were transferred to a recording chamber (∼3 ml volume) and continuously superfused with carbogenated aCSF at a flow rate of ∼2 ml · min−1. Perforated patch recordings were performed using protocols modified from the studies by Horn and Marty (1988) and Akaike and Harata (1994). Electrodes with tip resistances between 3 and 5 MΩ were fashioned from borosilicate glass (0.86 mm inner diameter; 1.5 mm outer diameter; GB150-8P; Science Products) with a vertical pipette puller (PP-830; Narishige). Perforated patch recordings were performed with ATP- and GTP-free pipette solution containing the following (in mm): 128 K-gluconate, 10 KCl, 10 HEPES, 0.1 EGTA, 2 MgCl2 adjusted to pH 7.3 (with KOH), resulting in an osmolarity of ∼300 mOsm. ATP and GTP were omitted from the intracellular solution to prevent uncontrolled permeabilization of the cell membrane (Lindau and Fernandez, 1986). The patch pipette was tip filled with internal solution and back filled with amphotericin B-containing internal solution (∼200 μg · ml−1; Sigma) to achieve perforated patch recordings. Amphotericin B was dissolved in dimethyl sulfoxide (final concentration, 0.4–0.5%; DMSO; Sigma) as described by Rae et al. (1991) and was added to the modified pipette solution shortly before use. Experiments were performed at ∼31°C using an inline solution heater (Warner Instruments) operated by a temperature controller (Warner Instruments). During the perforation process, access resistance (Ra) was constantly monitored, and experiments were started after Ra had reached steady state (∼15–20 min) and the action potential amplitude was stable. Since we use an ATP-free pipette solution, a change to the whole-cell configuration would be obvious by a spontaneous hyperpolarization of the neuron due to KATP activation. Such experiments were rejected. Neurons in the ARC were visualized with a fixed-stage upright microscope (Olympus) using a 60× water-immersion objective (LUMplan FI/IR; 60×; 0.9 numerical aperture; 2 mm working distance; Olympus) with infrared-differential interference contrast (Dodt and Zieglgänsberger, 1990) and fluorescence optics. POMC neurons were identified by their EGFP fluorescence, which was visualized using a Chroma 41001 filter set (excitation, HQ480/40×; beam splitter, Q505LP; emission, HQ535/50 m; Chroma). Current-clamp recordings were performed with an EPC10 patch-clamp amplifier (HEKA) controlled by the PatchMaster software (version 2.32; HEKA). Data were sampled at intervals of 100 μs (10 kHz) and low-pass filtered at 2 kHz with a four-pole Bessel filter. The liquid junction potential between intracellular and extracellular solution was not compensated [14.6 mV; calculated with Patcher's Power Tools plug-in for Igor Pro 6 (Wavemetrics)].
Data analysis.
In agreement with previous studies, we found that the basic electrophysiological properties of POMC neurons and their insulin responsiveness (Parton et al., 2007) (Claret et al., 2007) were not homogeneous. Therefore, we used the 3 SD criterion and considered a neuron insulin responsive when the change in membrane potential before and during insulin application was >3 SD (Dhillon et al., 2006; Kloppenburg et al., 2007). For each neuron, the membrane potential averaged from 60 s intervals was taken as one data point. To determine the mean membrane potential with SD, five data points at stable membrane potentials before and during insulin application were averaged. Data analysis was performed with Igor Pro 6 (Wavemetrics) and SigmaStat (version 3.1; Systat Software).
Statistical methods
Data were analyzed for statistical significance using a two-tailed unpaired Student's t test.
Results
Generation of POMC neuron-specific Stat3-C mice
Stat3 is a latent transcription factor that mediates leptin- and cytokine-directed expression of target genes. In a previous study, a constitutive active mutant version of Stat3 (Stat3-C) was created by substituting two cysteine residues in the C-terminal loop of the SH2 domain of Stat3, thereby producing a mutant which spontaneously dimerizes upon formation of disulfide bonds, thus binding DNA to activate transcription (Bromberg et al., 1999). Moreover, the Stat3-C molecule had been demonstrated to function as constitutive transcriptional activator of Stat3 target genes in several studies (Shen et al., 2001; Niu et al., 2002; Leslie et al., 2006; Azare et al., 2007). We have generated ES cells and mice with a targeted insertion of this Stat3-C cDNA preceded by a loxP-flanked transcriptional stop cassette into the ubiquitously expressed ROSA26 locus (Fig. 1A). Cre-mediated recombination of the loxP-flanked stop cassette leads to expression of the Stat3-C transgene and EGFP in cell types expressing the Cre recombinase (Fig. 1A).
Figure 1.
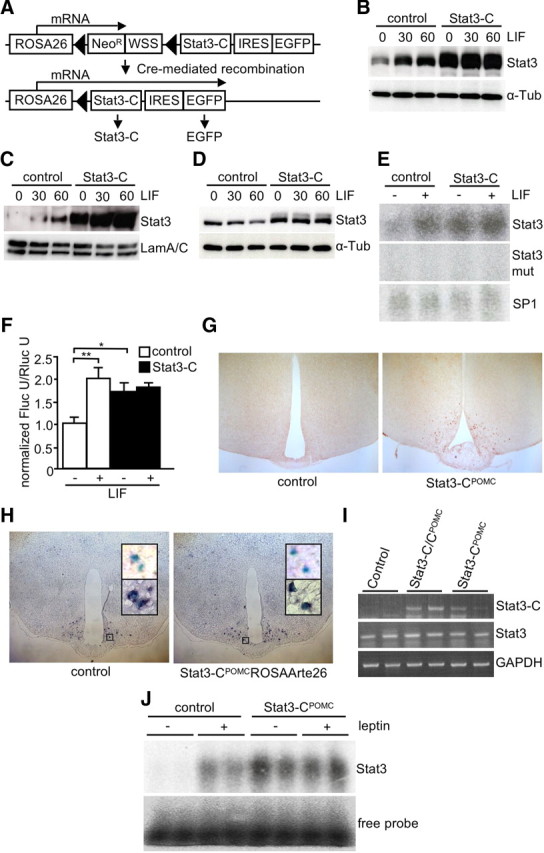
Generation of mice expressing Stat3-C in POMC neurons. A, Scheme of the Stat3-C transgene inserted into the ROSA26 locus. Cre-mediated recombination eliminates the loxP-flanked NeoR and Westphal stop sequence only in cell types expressing Cre, thereby allowing transcription of the bicistronic Stat3-C EGFP mRNA. Filled triangles, loxP sites; NeoR, neomycin resistance gene driven by the TK promoter; WSS, Westphal stop sequence; IRES, internal ribosome entry site. B, Western blot analysis with Stat3 and α-tubulin antibodies of whole-cell lysates from control and Stat3-C-expressing ES cells stimulated with LIF for 0, 30, and 60 min. C, Western blot analysis with Stat3 and LaminA/C antibodies of nuclear lysates from control and Stat3-C-expressing ES cells stimulated with LIF for 0, 30, and 60 min. D, Western blot analysis using Stat3 and α-tubulin antibodies of cytoplasmic lysates from control and Stat3-C-expressing ES cells stimulated with LIF for 0, 30, and 60 min. E, EMSA of nuclear extracts isolated from control and Stat3-C-expressing ES cells upon 2 h LIF stimulation using a radioactively labeled consensus sequence for Stat3, a mutant Stat3-consensus sequence and SP1-consensus sequence as loading control, respectively. F, Transcriptional activity of Stat3 and Stat3-C in control (white bars) and Stat3-C-expressing (black bars) ES cells transfected with pSTAT3-TA-Luc and pRL-null. ES cells were incubated with or without LIF 24 h before measurement of firefly luciferase (Fluc) and Renilla luciferase (Rluc) activity. G, To visualize Cre-mediated recombination, immunohistochemistry for EGFP was performed using brains of 12-week-old control and Stat3-CPOMC mice. H, Representative in situ hybridization using a POMC probe in hypothalamic neurons of ROSAArte26POMC and Stat3-CPOMC ROSAArte26 mice at the age of 10–14 weeks. Shown are X-gal-positive neurons before (top insets) and after (bottom insets) in situ hybridization. Blue, X-gal; purple, POMC mRNA. I, Hypothalamic expression of Stat3-C in Stat3-CPOMC mice shown by RT-PCR of control, Stat3-CPOMC, and Stat3-C/CPOMC mice by use of oligonucleotides detecting transgenic Stat3-C, endogenous Stat3, and GAPDH. J, EMSA of hypothalamic nuclear extracts isolated from control and Stat3-CPOMC mice after injection with saline or leptin using a radioactive labeled Stat3 probe.
To functionally validate this construct, we transfected Stat3-C ES cells with a Cre-expressing plasmid, thereby excising the stop cassette and activating expression of Stat3-C and EGFP. Western blot analyses revealed increased Stat3 expression in Cre-treated ES cells (Fig. 1B). Moreover, while Stat3 in control cells translocated from the cytoplasm to the nucleus only upon stimulation with LIF, the Stat3-C variant was already detectable in the nucleus, even in the absence of LIF stimulation (Fig. 1C,D). Furthermore, electrophoretic mobility shift assay (EMSA) using nuclear extracts from Stat-C-expressing and control ES cells revealed constitutive binding of Stat3-C to its consensus DNA sequence independent of LIF stimulation. The specificity of Stat3-C binding was addressed by using a mutated Stat3-consensus sequence with the same extracts, demonstrating that mutation of three nucleotides within the Stat3 probe abolishes recognition by endogenous Stat3 as well as Stat3-C (Fig. 1E). Moreover, transfection studies using a luciferase expression vector under the transcriptional control of Stat3-responsive elements revealed LIF-independent activation of luciferase expression in Stat3-C-expressing ES cells compared with wild-type ES cells (Fig. 1F). Thus, Cre-activated expression of the Stat3-C transgene results in its constitutive nuclear localization, target gene promoter occupancy, and specific activation of Stat3 target gene expression.
To investigate the effect of exaggerated Stat3-dependent signaling in POMC-expressing neurons, Stat3-CFL/FL mice were intercrossed with mice carrying the POMC-Cre transgene to obtain Stat3-CFL/+; POMC-Cre double transgenics, i.e., Stat3-CPOMC mice expressing Stat3-C selectively in POMC neurons, while Stat3-CFL/+ mice served as controls. Furthermore, to investigate a potential dose dependency of Stat3-dependent transcriptional regulation in POMC neurons, homozygous Stat3-C/CPOMC mice were produced by further intercrossing Stat3-CPOMC with Stat3-CFL/FL mice.
The POMC-specific expression of Stat3-C in the ARC was directly assessed by immunohistochemistry detecting the EGFP expressed from the IRES (internal ribosome entry site) included in the Stat3-C transgene. As expected, EGFP-positive neurons were only detectable in hypothalami of mice carrying both the Stat3-C and the POMC-Cre transgene (Fig. 1G). Consistent with the previous demonstration that the POMC-Cre mice used in this study indeed exhibit POMC-restricted recombination (Balthasar et al., 2004), X-gal staining for Cre-dependent expression of β-galactosidase from the ROSA26 locus and in situ hybridization for endogenous POMC mRNA revealed that 88 and 85% of cells expressing POMC mRNA also expressed the ROSA26 in controls and Stat3-CPOMC mice, respectively (Fig. 1H). These experiments support the notion that Stat3-C in Stat3-CPOMC mice is expressed in hypothalamic POMC neurons. Consistently, hypothalamic cDNA from Stat3- C/CPOMC and Stat3-CPOMC, but not from controls, showed the specific PCR product for Stat3-C when using oligonucleotides located in exon 1 of the ROSA26 and in the Stat3-C transcript in a PCR while expression of endogenous Stat3 and GAPDH served as controls (Fig. 1I). Furthermore, hypothalamic nuclear extracts isolated from Stat3-CPOMC mice showed constitutive binding of Stat3-C to its radioactively labeled consensus sequence in EMSA without dependence on the leptin signal (Fig. 1J). Taken together, these results demonstrate the functionality of the ROSA26 Stat3-C transgene in ES cells and POMC cells of mice in vivo.
Mild obesity in Stat3-CPOMC mice
To investigate the impact of Stat3-C signaling in POMC neurons on the regulation of energy homeostasis, we monitored body weight of male control, Stat3-CPOMC, and Stat3-C/CPOMC mice from weaning until 20 weeks of age. Stat3-CPOMC mice exhibited an ∼10% elevated body weight compared with control mice, an effect which appeared slightly but not significantly further aggravated by expressing Stat3-C from two alleles (Fig. 2A).
Figure 2.
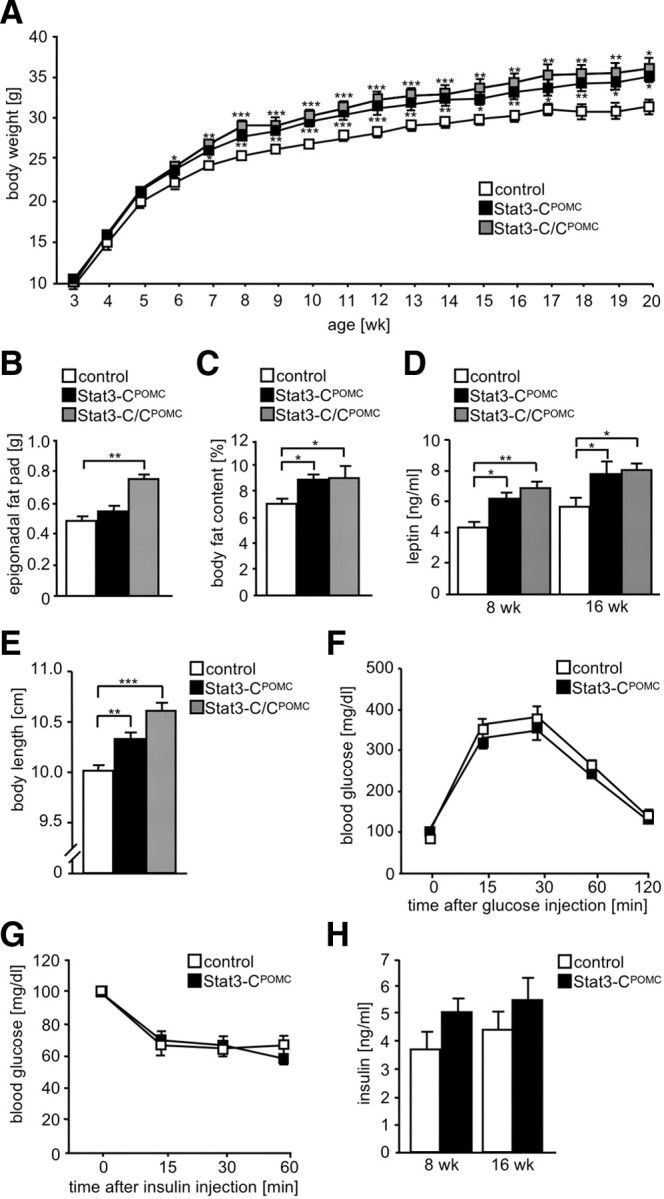
Mild obesity in Stat3-CPOMC mice. A, Body weight of control (white squares), Stat3-CPOMC (black squares), and Stat3-C/CPOMC (gray squares) mice was determined weekly (n = 10–18/genotype). B, Epigonadal fat pad weights of control (white bar), Stat3-CPOMC (black bar), and Stat3-C/CPOMC (gray bar) mice at the age of 20 weeks (n = 12–18/genotype). C, Whole-body fat content of control (white bar), Stat3-CPOMC (black bar), and Stat3-C/CPOMC (gray bar) mice at the age of 16 weeks was determined using nuclear magnetic resonance (n = 10–12/genotype). D, Serum leptin concentrations of control (white bars), Stat3-CPOMC (black bars), and Stat3-C/CPOMC (gray bars) mice at the age of 8 and 16 weeks were investigated by ELISA (n = 12–20/genotype). E, Body length of control (white bar), Stat3-CPOMC (black bar), and Stat3-C/CPOMC (gray bar) mice at the age of 20 weeks (n = 10–12/genotype). F, Glucose tolerance test of control (white squares) and Stat3-CPOMC (black squares) mice at the age of 12 weeks (n = 12–15/genotype). G, Insulin tolerance test of control (white squares) and Stat3-CPOMC (black squares) mice at the age of 13 weeks (n = 12–15/genotype). H, Serum insulin concentrations of control (white bars) and Stat3-CPOMC (black bars) at the age of 8 and 16 weeks were investigated by ELISA (n = 13–15/genotype). Displayed values are means ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 versus control.
To investigate whether the elevated body weight in Stat3-CPOMC mice is a consequence of increase in fat mass, we next examined the amount of epigonadal fat in control, Stat3-CPOMC, and Stat3-C/CPOMC mice. Consistent with the increased body weight, Stat3-C/CPOMC mice displayed significantly increased epigonadal fat pad masses at the age of 20 weeks compared with controls (Fig. 2B). Furthermore, increased adiposity in Stat3-CPOMC mice was confirmed by determining body fat composition using in vivo magnetic resonance spectrometry. Mean body fat content was significantly elevated to 9% in Stat3-CPOMC and in Stat3-C/CPOMC mice (Fig. 2C). Obesity in Stat3-CPOMC mice was accompanied by significantly increased plasma leptin concentrations (Fig. 2D).
Moreover, determination of body length revealed that Stat3-CPOMC and Stat3-C/CPOMC mice exhibit significantly increased body lengths compared with control mice (Fig. 2E), a phenomenon commonly found as an indicator of impaired melanocortin 4 receptor function.
To determine whether glucose homeostasis or insulin sensitivity is affected by expressing Stat3-C in POMC neurons, glucose and insulin tolerance tests were performed (Fig. 2F,G) and serum insulin concentrations were determined (Fig. 2H). These analyses revealed no differences in whole-body glucose homeostasis in Stat3-CPOMC mice compared with controls despite the mild obesity of the mutant mice. Taken together, these results indicate that constitutive activation of Stat3-dependent signaling in POMC-expressing neurons results in mild obesity with increases in body weight, fat mass, circulating leptin concentrations, and body length without overtly affecting whole-body insulin sensitivity.
Stat3-CPOMC mice exhibit increased food intake and decreased POMC expression
Increased body weight is a consequence of decreased energy expenditure or a result of increased food intake, or a combination of both. We have recently demonstrated that Stat3-CPOMC mice showed a tendency toward reduced energy expenditure compared with controls, although this effect did not reach statistical significance (Mesaros et al., 2008). To further analyze the mechanism underlying the mild obesity observed in Stat3-CPOMC mice, we directly measured food intake in these mice. Assessment of food intake revealed that Stat3-CPOMC as well as Stat3-C/CPOMC mice showed increased food intake compared with control mice (Fig. 3A). Furthermore, food intake was assessed after a 24 h fasting period (Fig. 3B). This analysis revealed a significantly increased refeeding response in both Stat3-CPOMC and Stat3-C/CPOMC compared with controls. Thus, constitutive Stat3 signaling in POMC neurons increases steady-state and refeeding-associated food intake.
Figure 3.

Stat3-CPOMC mice show increased food intake and defects in compensatory refeeding resulting from decreased POMC expression. A, Daily food intake of control (white bar), Stat3-CPOMC (black bar), and Stat3-C/CPOMC (gray bar) mice at the age of 10 weeks (n = 8–18/genotype). B, Compensatory 4 and 24 h refeeding after a 24 h fasting period of control (white bars), Stat3-CPOMC (black bars), and Stat3-C/CPOMC (gray bars) mice at the age of 16 weeks (n = 7–9/genotype). C, Relative hypothalamic expression of POMC, AgRP, and NPY under random-fed conditions of control (white bars), Stat3-CPOMC (black bars), and Stat3-C/CPOMC (gray bars) mice by use of quantitative real-time PCR (n = 8/genotype). D, X-gal stainings of brains isolated from ROSAArte26POMC and Stat3-CPOMCROSAArte26 mice at 12 weeks of age. Displayed values are means ± SEM. *p ≤ 0.05 versus control.
Ultimately, we determined hypothalamic mRNA expressions of neuropeptides involved in the regulation of food intake by real-time PCR. Surprisingly, hypothalamic expression of POMC was decreased by 50% in Stat3-CPOMC and Stat3-C/CPOMC mice when compared with controls, despite the fact that Stat3 serves as a transcriptional activator of POMC expression (Fig. 3C). Moreover, the relative expression of AgRP and NPY mRNA was unaltered in Stat3-CPOMC mice (Fig. 3C).
To determine whether the decreased hypothalamic POMC mRNA expression observed in Stat3-CPOMC results from reduced POMC neuron numbers, i.e., decreased POMC cell formation or increased POMC cell death, we directly compared the number of POMC neurons in control and Stat3-CPOMC mice. To this end, we used POMC-Cre mice carrying either alleles for Stat3-C and a lacZ reporter in the ROSA26 locus or the Cre-dependent lacZ reporter alone, the brains of which were stained for β-galactosidase activity (Fig. 3D). Counting X-gal-positive neurons revealed indistinguishable POMC neuron numbers in Stat3-CPOMC and control mice, as on average 495 POMC neurons in control and 546 POMC neurons in Stat3-CPOMC mice were identified in four coronal sections containing the ARC of five mice of each genotype. Taken together, these data indicate that the mild obesity observed in Stat3-CPOMC mice is a consequence of increased food intake resulting from reduced POMC expression in the absence of alterations in hypothalamic POMC cell number.
Stat3-CPOMC mice are leptin resistant and exhibit increased SOCS3expression
To experimentally address whether the mildly obese phenotype of Stat3-CPOMC mice is a consequence of leptin resistance, control, Stat3-CPOMC, and Stat3-C/CPOMC mice were injected with leptin and food intake was assessed. Leptin injection in control mice reduced food intake significantly by 20%, while leptin had no significant effect on food intake of Stat3-CPOMC and Stat3-C/CPOMC mice (Fig. 4A). Consistently, leptin injection induced phosphorylation of Stat3 in ARCs of control mice, while this response was clearly blunted in Stat3-CPOMC mice (Fig. 4B). Moreover, brains of leptin-injected ROSAArte26POMC and Stat3-CPOMC ROSAArte26 mice were stained with antibodies specific for phosphorylated Stat3 and β-galactosidase to determine leptin-induced Stat3 phosphorylation specifically in POMC neurons (Fig. 4C). This experiment revealed that 27% of POMC cells from control mice showed nuclear accumulation of phosphorylated Stat3 after leptin injection, in contrast to only 4% of POMC neurons in Stat3-CPOMC mice (Fig. 4D).
Figure 4.
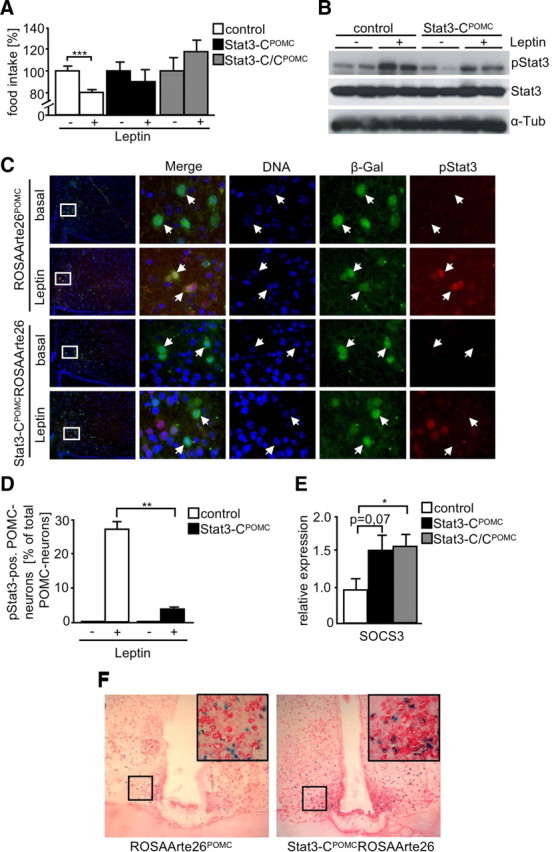
Stat3-C induces leptin resistance in POMC neurons by enforcing expression of SOCS3. A, Leptin sensitivity test of control (white bars), Stat3-CPOMC (black bars), and Stat3-C/CPOMC (gray bars) mice at the age of 17 weeks (n = 6–10/genotype). Mice were injected twice a day for three consecutive days with saline and subsequently with 2 mg/kg leptin for three consecutive days twice a day. Food intake of saline-injected control mice was set to 100%. B, Western blot analysis of ARC extracts from fasted and leptin-injected control and Stat3-CPOMC mice by use of antibodies against pStat3 and α-tubulin (α-Tub). C, Representative immunohistochemistry for pSTAT3 and β-gal in POMC neurons of fasted and leptin-injected ROSAArte26POMC and Stat3-CPOMC ROSAArte26 mice at the age of 10–12 weeks. Blue (DAPI), DNA; green, β-gal (POMC neurons); red, pStat3. D, Quantitation of pSTAT3-positive POMC neurons in hypothalamic sections of fasted and leptin-injected control (white bars) and Stat3-CPOMC ROSAArte26 (black bars) mice at the age of 10–12 weeks (n = 3–4/genotype). A total of 2103 POMC neurons were analyzed. E, Relative hypothalamic expression of SOCS3 under random-fed conditions of control (white bar), Stat3-CPOMC (black bar), and Stat3-C/CPOMC (gray bar) mice by use of quantitative real-time PCR (n = 8/genotype). F, Representative in situ hybridization using a SOCS3 probe in hypothalamic neurons of fasted ROSAArte26POMC and Stat3-CPOMC ROSAArte26 mice at the age of 12 weeks. Blue, X-gal (POMC neurons); red, SOCS3 mRNA. Displayed values are means ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 versus control.
Recently, it has been demonstrated that leptin signaling is regulated by a negative feedback mechanism involving the induction of SOCS3 expression, which involves SOCS3 binding to tyrosine residue 1138 of the leptin receptor thereby silencing the leptin signal (Dunn et al., 2005). To assess whether chronic Stat3 signaling in POMC neurons results in enhanced SOCS3 expression, we performed real-time PCR on mRNA isolated from hypothalami of control, Stat3-CPOMC, and Stat3-C/CPOMC mice. This analysis revealed a significant increase in SOCS3 expression in hypothalami of Stat3-CPOMC and Stat3-C/CPOMC mice despite the fact that approximately only 5% of hypothalamic neurons express POMC and therefore also Stat3-C (Fig. 4E).
To further directly address whether SOCS3 expression increases primarily in POMC neurons, we performed combined in situ hybridization/X-gal staining using a SOCS3 probe on brain slices from food-deprived Stat3-CPOMC and POMC-Cre mice expressing the lacZ reporter construct specifically in POMC neurons (Fig. 4F). This analysis revealed that in the fasted state, ∼32% of POMC neurons from control mice express SOCS3, while almost all (86%) Stat3-C-expressing POMC neurons express SOCS3 mRNA. These data suggest that chronic Stat3 signaling in POMC neurons diminishes whole-body leptin sensitivity in the presence of increased expression of the negative regulator SOCS3.
Increased SOCS3 expression in POMC neurons leads to central insulin resistance
Since SOCS3 has been demonstrated to inhibit not only cytokine, i.e., leptin, signaling but also insulin signaling via binding to the insulin receptor, we next aimed to investigate whether Stat3-induced SOCS3 expression not only results in leptin resistance but also affects insulin action in POMC neurons. To this end, reporter mice expressing lacZ specifically in POMC neurons (controls) or the reporter mice also expressing Stat3-C in POMC neurons were injected with insulin, and brains were subjected to double immunohistochemical analysis for PIP3 as well as β-galactosidase. Strikingly, while control mice showed ratios of low:medium:high PIP3-immunoreactive POMC neurons upon insulin stimulation comparable to those previously described after insulin treatment (Plum et al., 2006b), Stat3-CPOMC mice showed significantly elevated counts of low PIP3-immunoreactive POMC neurons accompanied with significantly decreased numbers of high PIP3-immunoreactive POMC neurons, indicating that Stat3-C expression inhibits insulin-stimulated PI3K activation in POMC neurons (Fig. 5A). Moreover using the same reporter mice, we determined insulin-stimulated AKT phosphorylation by double immunohistochemistry. While no activation of AKT was observed in POMC neurons of fasted animals, 23% of control POMC neurons exhibited insulin-stimulated AKT phosphorylation, an effect that was significantly reduced to 10% in Stat3-C-expressing POMC neurons (Fig. 5B).
Figure 5.
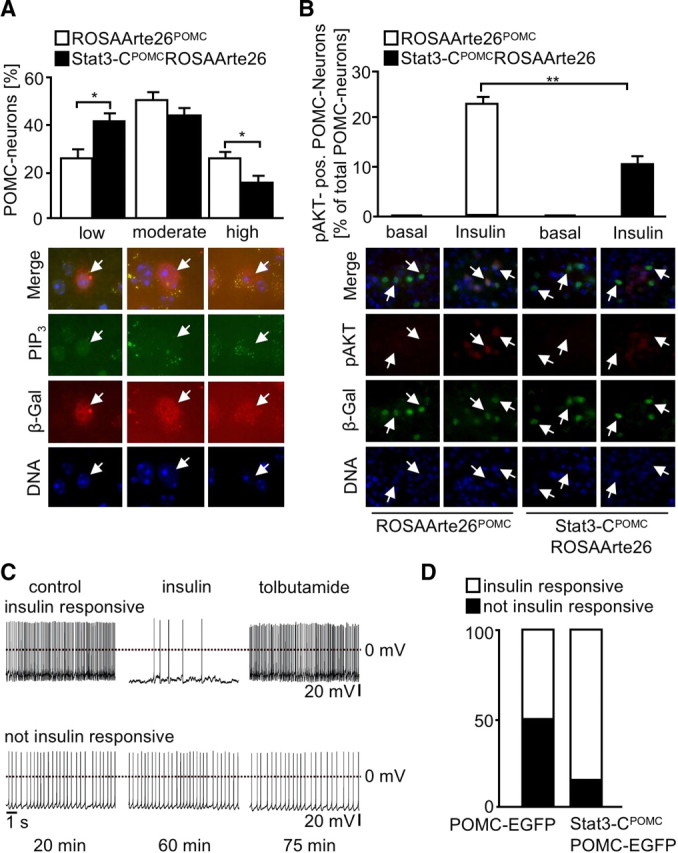
Increased SOCS3 expression in POMC neurons leads to central insulin resistance. A, Representative stainings and quantitation of PIP3 levels in ROSAArte26POMC (white bars) and Stat3-CPOMC ROSAArte26 (black bars) mice after intravenous injection of insulin (n = 5/genotype). A total of 918 POMC neurons were analyzed. PIP3 immunoreactivity was classified in low (<5 dots), intermediate (5–10 dots), and high (>10 dots). Blue (DAPI), DNA; red, β-gal (POMC neurons); green, PIP3. B, Representative stainings and quantitation of pAKT-positive (pAKT-pos.) POMC neurons in hypothalamic sections of fasted and insulin-injected control (white bars) and Stat3-CPOMC ROSAArte26 (black bars) mice at the age of 10–14 weeks (n = 5/genotype). A total of 2351 POMC neurons were analyzed. Blue (DAPI), DNA; green, β-gal (POMC neurons); red, pAKT. C, Representative perforated patch recordings of insulin-responsive POMC-EGFP neurons and POMC-EGFP neurons that were not insulin responsive. The traces represent sections of the recordings before and during bath application of 200 nm insulin (∼35 min after application start). In the insulin-responsive neurons, bath application of 200 μm tolbutamide reversed the insulin effect. The times below the traces indicated the duration after establishing the recording. D, The percentage of POMC-EGFP (5/10) and STAT3-CPOMC POMC-EGFP neurons (2/13) that responded to bath application of 200 nm insulin with a significant hyperpolarization. A neuron was determined to be insulin responsive when the hyperpolarization was >3 times the SD. Displayed values are means ± SEM. *p ≤ 0.05, **p ≤ 0.01 versus control.
Ultimately, we intercrossed control and Stat3-CPOMC mice with POMC-EGFP mice (Cowley et al. 2001) and performed perforated patch-clamp recordings of EGFP-expressing POMC neurons to determine their electrophysiological response to insulin. Figure 5C shows representative recordings of an insulin-responsive and a nonresponsive POMC neuron: application of tolbutamide reversed insulin's effect. Remarkably and in line with our previous experiments, 50% of control POMC neurons responded to insulin by hyperpolarization and silencing, while this effect was significantly reduced to 15% in Stat3-CPOMC mice (Fig. 5D). Taken together, there is clear evidence from three independent experimental approaches that expression of Stat3-C in POMC neurons results in POMC-cell-specific insulin resistance in Stat3-CPOMC mice.
Chronic Stat3 signaling has no effect under leptin-resistant conditions
To address whether enhanced Stat3 activation affects mice under leptin-resistant conditions, Stat3-CPOMC, Stat3-C/CPOMC, and control mice were exposed to HFD. However, Stat3-CPOMC and Stat3-C/CPOMC body weight was indistinguishable from that of control mice under those conditions (Fig. 6A). Accordingly, Stat3-CPOMC, Stat3-C/CPOMC, and control mice showed similar daily food intake (Fig. 6B). Consistent with this, epigonadal fat pad weights and body fat content were unaltered in Stat3-CPOMC and Stat3-C/CPOMC mice compared with controls (Fig. 6C,D). Furthermore, serum leptin levels were significantly increased under HFD compared with mice exposed to NCD but remained unchanged compared with controls exposed to HFD, at both 8 and 16 weeks of age (Fig. 6E). Moreover, no differences in body length were observed between control, Stat3-CPOMC, and Stat3-C/CPOMC mice (Fig. 6F).
Figure 6.

Chronic Stat3 signaling has no effect under leptin-resistant conditions. A, Body weight of control (white squares), Stat3-CPOMC (black squares), and Stat3-C/CPOMC (gray squares) mice on HFD was determined weekly (n = 8–12/genotype). B, Daily food intake of control (white bar), Stat3-CPOMC (black bar), and Stat3-C/CPOMC (gray bar) mice at the age of 10 weeks under HFD conditions (n = 8–14/genotype). C, Epigonadal fat pad weight of control (white bar), Stat3-CPOMC (black bar), and Stat3-C/CPOMC (gray bar) mice on HFD at the age of 20 weeks (n = 10–13/genotype). D, Body fat content of control (white bar), Stat3-CPOMC (black bar), and Stat3-C/CPOMC (gray bar) mice on HFD at the age of 20 weeks by nuclear magnetic resonance (n = 10–13/genotype). E, Serum leptin concentration of control (white bars), Stat3-CPOMC (black bars), and Stat3-C/CPOMC (gray bars) mice on HFD at the age of 8 and 16 weeks was determined by ELISA (n = 12–16/genotype). F, Body length of control (white bar), Stat3-CPOMC (black bar), and Stat3-C/CPOMC (gray bar) mice on HFD at the age of 20 weeks (n = 8–12/genotype). G, Relative hypothalamic expression of SOCS3 and POMC of control (white bars), Stat3-CPOMC (black bars), and Stat3-C/CPOMC (gray bars) mice under NCD (n = 12/genotype) and HFD (n = 4/genotype) conditions using quantitative real-time PCR. H, EMSA of nuclear extracts isolated from arcuate nuclei of four individual C57BL/6 mice exposed to NCD and HFD and from one Stat3-CPOMC mouse on NCD by use of radioactively labeled Stat3 and SP1 probes, respectively. Displayed values are means ± SEM. *p ≤ 0.05 versus control.
To investigate the effect of HFD on hypothalamic SOCS3 and POMC expression, quantitative real-time PCR was performed using mRNA isolated from hypothalami of control, Stat3-CPOMC, and Stat3-C/CPOMC mice exposed to NCD and HFD (Fig. 6G). This analysis revealed significantly increased mRNA expression of SOCS3 in control mice exposed to HFD when compared with NCD-fed controls, but HFD feeding attenuated the effect of Stat3-C expression on hypothalamic SOCS3 expression (Fig. 6G). Similarly, POMC expression was consistently reduced in all groups of mice when exposed to HFD feeding (Fig. 6G) to an extent similar to that observed in mice expressing Stat3-C in POMC neurons under NCD conditions.
To investigate whether elevated leptin in obesity translates into enhanced basal Stat3 activation in the ARC of control mice, we performed EMSA of random-fed C57BL/6 mice on NCD and HFD. Strikingly, obesity induced by HFD drastically increased basal Stat3 binding in the ARC to its consensus sequence compared with NCD, similarly as observed in Stat3-CPOMC mice (Fig. 6H).
These results clearly demonstrate that activated Stat3 signaling in POMC neurons has no additional effect on body weight, body fat content, serum leptin concentration, body length, and hypothalamic gene expression under conditions of hyperleptinemia induced by HFD feeding, indicating that during the course of HFD-induced central leptin and insulin resistance, Stat3 overactivation in POMC neurons is a pathophysiologically relevant component.
Discussion
The last decade has highlighted the crucial role for leptin and insulin signaling in the CNS to control body weight and glucose homeostasis (Friedman and Halaas, 1998; Elias et al., 1999; Brüning et al., 2000; Cohen et al., 2001; Obici et al., 2002; Dhillon et al., 2006; Könner et al., 2007; Koch et al., 2008; Myers et al., 2009). Comparative analysis of brain-restricted leptin receptor knock-out mice and leptin receptor knock-out mice with brain-restricted reconstitution of leptin receptor expression evidenced that the CNS is the major site mediating leptin's effects on energy and glucose homeostasis (Cohen et al., 2001; Kowalski et al., 2001). However, the leptin receptor is expressed in several regions of the brain, and only recent experiments with mice lacking the receptor in specific neuronal subpopulations have shed light on the relative contribution of leptin signaling in individual CNS sites (Balthasar et al., 2004; Dhillon et al., 2006). The primary sites of leptin action are first-order neurons such as POMC and AgRP neurons in the ARC of the hypothalamus, which are crucial in regulating food intake and energy expenditure. Various lines of evidence have demonstrated that leptin-activated Stat3 in POMC neurons mediates expression of POMC, thereby reducing food intake and increasing energy expenditure, and thus ultimately reducing adiposity (Balthasar et al., 2004; Xu et al., 2007). Furthermore, studies using pharmacological compounds blocking the MC4R have demonstrated the importance of the melanocortin system on the acute anorectic effect of centrally applied leptin in the regulation of food intake (da Silva et al., 2004). More recently, it was demonstrated that POMC expression depends on the coordinated activation of Stat3 binding to the POMC promoter with simultaneous insulin/leptin-stimulated PI3K-dependent release of FOXO1-mediated repression (Kitamura et al., 2006; Belgardt et al., 2008). Taken together, coordinated leptin and insulin action in POMC-expressing neurons play a key role in regulating energy homeostasis.
To further investigate the importance of Stat3 signaling in these neurons, we analyzed mice expressing a nuclear, constitutive active mutant of Stat3 exclusively in POMC neurons. Surprisingly however, Stat3-CPOMC mutant mice show mild obesity and hyperleptinemia as a consequence of increased food intake, mirroring the phenotypes of POMC neuron-restricted Stat3 and leptin receptor knock-out animals, respectively (Balthasar et al., 2004; Xu et al., 2007). Considering the possibility that this could also result from impaired POMC neuron function due to transgenic overexpression of Stat3-C, we favor the alternative that the increased food intake of Stat3-CPOMC mice appears as a consequence of decreased POMC expression resulting from increased Stat3-mediated expression of SOCS3, which was previously shown to function as a negative regulator in insulin and leptin signaling (Bjørbaek et al., 1998; Emanuelli et al., 2000). This negative feedback loop is predicted to mainly operate by specific interaction of SOCS3 with tyrosine residues 985 and 1138 of the leptin receptor, respectively, in turn inhibiting phosphorylation of Stat3 and thus expression of SOCS3, thereby blunting the Stat3-mediated signaling (Bjorbak et al., 2000; Dunn et al., 2005). In line with this, mice haploinsufficient for SOCS3 and mice deficient for SOCS3 selectively in POMC neurons show enhanced leptin sensitivity and improved glucose homeostasis, thus demonstrating the existence of such a negative feedback inhibitory pathway mediated by SOCS3 in vivo (Howard et al., 2004; Kievit et al., 2006).
However, our model provides important novel insights into the molecular consequences of HFD-induced SOCS3 expression. In our model, the mutant Stat3-C protein is constantly localized in the nucleus and acts as a transcriptional activator independent of tyrosine phosphorylation. Thus, if Stat3 is the main regulator of POMC expression, as predicted from the Stat-responsive element in the POMC promoter, POMC expression would not be expected to be downregulated even in the presence of increased SOCS3. Our experiments clearly indicate the existence of another SOCS3-sensitive, leptin- and/or insulin-induced signaling molecule required for POMC expression, even in the presence of fully activated Stat3. Indeed, we and others could recently demonstrate the importance of PI3K activation to release FOXO1-mediated repression of POMC expression (Kitamura et al., 2006; Belgardt et al., 2008). Consistently, expression of Stat3-C with subsequent SOCS3 induction in POMC neurons impaired PI3K activity specifically in POMC neurons of our mice. Therefore, our data indicate that in obesity the negative effects of increased SOCS3 levels on leptin and insulin signaling result not only in impaired Stat3 activation but also in decreased POMC expression as a consequence of impaired FOXO nuclear exclusion, which occupies the Stat3-binding sites in the POMC promoter, leading to the inaccessibility of Stat3 to activate POMC expression (Fig. 7). Moreover, our experiments directly demonstrate that FOXO1 removal is a prerequisite for functional Stat3 binding, even when expressed as a constitutively active mutant.
Figure 7.

Model of SOCS3-mediated leptin and insulin resistance. Increased Stat3-mediated SOCS3 expression as present in early hyperleptinemia leads to the combined feedback inhibition of leptin and insulin signaling. Decreased insulin signaling leads to inefficient FOXO phosphorylation, thereby occupying the POMC promoter and thus inhibiting binding of Stat3, which serves as a transcriptional activator for POMC expression.
Finally, our experiments directly demonstrate that initial overactivation of Stat3 signaling naturally contributes to the development of HFD-induced leptin resistance, as overactivation of Stat3 signaling in POMC neurons has no additional effect on mice exposed to HFD. Concordantly, we and others could demonstrate that in HFD-fed mice, Stat3 activation is indeed increased in the ARC (Martin et al., 2006), explaining that hypothalamic expression of SOCS3 is elevated in various rodent models of obesity. On the other hand, a more pronounced effect of HFD compared with that of Stat3 overactivation in POMC neurons supports the importance of leptin resistance in neurons other than POMC cells.
Ultimately, the observation that enhanced Stat3 signaling in obesity can initiate a vicious negative regulatory feedback circle to inhibit not only leptin but also insulin action in the CNS explains how initial weight loss restores leptin sensitivity by reducing circulating leptin concentrations as a driving force of elevated basal Stat3 activation and subsequent SOCS3-mediated leptin and insulin resistance (Rosenbaum et al., 2008).
Footnotes
This work was supported by grants from Zentrum für Molekulare Medizin der Universität zu Köln (to J.C.B.), the Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases (to F.T.W. and J.C.B.), and the Deutsche Forschungsgemeinschaft (1492-7) (to J.C.B.). We thank Gisela Schmall and Tanja Rayle for excellent secretarial assistance. We thank Klaus Rajewsky and Tomoo Okamura for experimental assistance and discussions.
References
- Akaike N, Harata N. Nystatin perforated patch recording and its applications to analyses of intracellular mechanisms. Jpn J Physiol. 1994;44:433–473. doi: 10.2170/jjphysiol.44.433. [DOI] [PubMed] [Google Scholar]
- Azare J, Leslie K, Al-Ahmadie H, Gerald W, Weinreb PH, Violette SM, Bromberg J. Constitutively activated Stat3 induces tumorigenesis and enhances cell motility of prostate epithelial cells through integrin beta 6. Mol Cell Biol. 2007;27:4444–4453. doi: 10.1128/MCB.02404-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, McGovern RA, Chua SC, Jr, Elmquist JK, Lowell BB. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004;42:983–991. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Banks AS, Davis SM, Bates SH, Myers MG., Jr Activation of downstream signals by the long form of the leptin receptor. J Biol Chem. 2000;275:14563–14572. doi: 10.1074/jbc.275.19.14563. [DOI] [PubMed] [Google Scholar]
- Belgardt BF, Husch A, Rother E, Ernst MB, Wunderlich FT, Hampel B, Klöckener T, Alessi D, Kloppenburg P, Brüning JC. PDK1 deficiency in POMC-expressing cells reveals FOXO1-dependent and -independent pathways in control of energy homeostasis and stress response. Cell Metab. 2008;7:291–301. doi: 10.1016/j.cmet.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Bewick GA, Gardiner JV, Dhillo WS, Kent AS, White NE, Webster Z, Ghatei MA, Bloom SR. Post-embryonic ablation of AgRP neurons in mice leads to a lean, hypophagic phenotype. FASEB J. 2005;19:1680–1682. doi: 10.1096/fj.04-3434fje. [DOI] [PubMed] [Google Scholar]
- Bjørbaek C, Uotani S, da Silva B, Flier JS. Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem. 1997;272:32686–32695. doi: 10.1074/jbc.272.51.32686. [DOI] [PubMed] [Google Scholar]
- Bjørbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell. 1998;1:619–625. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- Bjørbaek C, El-Haschimi K, Frantz JD, Flier JS. The role of SOCS-3 in leptin signaling and leptin resistance. J Biol Chem. 1999;274:30059–30065. doi: 10.1074/jbc.274.42.30059. [DOI] [PubMed] [Google Scholar]
- Bjørbaek C, Buchholz RM, Davis SM, Bates SH, Pierroz DD, Gu H, Neel BG, Myers MG, Jr, Flier JS. Divergent roles of SHP-2 in ERK activation by leptin receptors. J Biol Chem. 2001;276:4747–4755. doi: 10.1074/jbc.M007439200. [DOI] [PubMed] [Google Scholar]
- Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, Myers MG., Jr SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem. 2000;275:40649–40657. doi: 10.1074/jbc.M007577200. [DOI] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- Brüning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Müller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–2125. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, Clements M, Al-Qassab H, Heffron H, Xu AW, Speakman JR, Barsh GS, Viollet B, Vaulont S, Ashford ML, Carling D, Withers DJ. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest. 2007;117:2325–2336. doi: 10.1172/JCI31516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM. Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest. 2001;108:1113–1121. doi: 10.1172/JCI13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- da Silva AA, Kuo JJ, Hall JE. Role of hypothalamic melanocortin 3/4-receptors in mediating chronic cardiovascular, renal, and metabolic actions of leptin. Hypertension. 2004;43:1312–1317. doi: 10.1161/01.HYP.0000128421.23499.b9. [DOI] [PubMed] [Google Scholar]
- Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, Kenny CD, Christiansen LM, White RD, Edelstein EA, Coppari R, Balthasar N, Cowley MA, Chua S, Jr, Elmquist JK, Lowell BB. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. 2006;49:191–203. doi: 10.1016/j.neuron.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Dodt HU, Zieglgänsberger W. Visualizing unstained neurons in living brain slices by infrared DIC-videomicroscopy. Brain Res. 1990;537:333–336. doi: 10.1016/0006-8993(90)90380-t. [DOI] [PubMed] [Google Scholar]
- Dunn SL, Björnholm M, Bates SH, Chen Z, Seifert M, Myers MG., Jr Feedback inhibition of leptin receptor/Jak2 signaling via Tyr1138 of the leptin receptor and suppressor of cytokine signaling 3. Mol Endocrinol. 2005;19:925–938. doi: 10.1210/me.2004-0353. [DOI] [PubMed] [Google Scholar]
- El-Haschimi K, Pierroz DD, Hileman SM, Bjørbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest. 2000;105:1827–1832. doi: 10.1172/JCI9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- Emanuelli B, Peraldi P, Filloux C, Sawka-Verhelle D, Hilton D, Van Obberghen E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem. 2000;275:15985–15991. doi: 10.1074/jbc.275.21.15985. [DOI] [PubMed] [Google Scholar]
- Frederich RC, Hamann A, Anderson S, Löllmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1:1311–1314. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, Fu XY. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci U S A. 2004;101:4661–4666. doi: 10.1073/pnas.0303992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, Plum L, Balthasar N, Hampel B, Waisman A, Barsh GS, Horvath TL, Brüning JC. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci. 2005;8:1289–1291. doi: 10.1038/nn1548. [DOI] [PubMed] [Google Scholar]
- Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J Gen Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjørbaek C, Flier JS. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med. 2004;10:734–738. doi: 10.1038/nm1072. [DOI] [PubMed] [Google Scholar]
- Kievit P, Howard JK, Badman MK, Balthasar N, Coppari R, Mori H, Lee CE, Elmquist JK, Yoshimura A, Flier JS. Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells. Cell Metab. 2006;4:123–132. doi: 10.1016/j.cmet.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Feng Y, Kitamura YI, Chua SC, Jr, Xu AW, Barsh GS, Rossetti L, Accili D. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med. 2006;12:534–540. doi: 10.1038/nm1392. [DOI] [PubMed] [Google Scholar]
- Kloppenburg P, Zipfel WR, Webb WW, Harris-Warrick RM. Heterogeneous effects of dopamine on highly localized, voltage-induced Ca2+ accumulation in identified motoneurons. J Neurophysiol. 2007;98:2910–2917. doi: 10.1152/jn.00660.2007. [DOI] [PubMed] [Google Scholar]
- Koch L, Wunderlich FT, Seibler J, Könner AC, Hampel B, Irlenbusch S, Brabant G, Kahn CR, Schwenk F, Brüning JC. Central insulin action regulates peripheral glucose and fat metabolism in mice. J Clin Invest. 2008;118:2132–2147. doi: 10.1172/JCI31073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Könner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, Xu C, Enriori P, Hampel B, Barsh GS, Kahn CR, Cowley MA, Ashcroft FM, Brüning JC. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5:438–449. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Kowalski TJ, Liu SM, Leibel RL, Chua SC., Jr Transgenic complementation of leptin-receptor deficiency. I. Rescue of the obesity/diabetes phe notype of LEPR-null mice expressing a LEPR-B transgene. Diabetes. 2001;50:425–435. doi: 10.2337/diabetes.50.2.425. [DOI] [PubMed] [Google Scholar]
- Leaman DW, Leung S, Li X, Stark GR. Regulation of STAT-dependent pathways by growth factors and cytokines. FASEB J. 1996;10:1578–1588. [PubMed] [Google Scholar]
- Leslie K, Lang C, Devgan G, Azare J, Berishaj M, Gerald W, Kim YB, Paz K, Darnell JE, Albanese C, Sakamaki T, Pestell R, Bromberg J. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res. 2006;66:2544–2552. doi: 10.1158/0008-5472.CAN-05-2203. [DOI] [PubMed] [Google Scholar]
- Lindau M, Fernandez JM. IgE-mediated degranulation of mast cells does not require opening of ion channels. Nature. 1986;319:150–153. doi: 10.1038/319150a0. [DOI] [PubMed] [Google Scholar]
- Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–685. doi: 10.1126/science.1115524. [DOI] [PubMed] [Google Scholar]
- Martin TL, Alquier T, Asakura K, Furukawa N, Preitner F, Kahn BB. Diet-induced obesity alters AMP kinase activity in hypothalamus and skeletal muscle. J Biol Chem. 2006;281:18933–18941. doi: 10.1074/jbc.M512831200. [DOI] [PubMed] [Google Scholar]
- Mesaros A, Koralov SB, Rother E, Wunderlich FT, Ernst MB, Barsh GS, Rajewsky K, Brüning JC. Activation of Stat3 signaling in AgRP neurons promotes locomotor activity. Cell Metab. 2008;7:236–248. doi: 10.1016/j.cmet.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Mizuno TM, Mobbs CV. Hypothalamic agouti-related protein messenger ribonucleic acid is inhibited by leptin and stimulated by fasting. Endocrinology. 1999;140:814–817. doi: 10.1210/endo.140.2.6491. [DOI] [PubMed] [Google Scholar]
- Münzberg H, Flier JS, Bjørbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- Myers MG, Jr, Münzberg H, Leinninger GM, Leshan RL. The geometry of leptin action in the brain: more complicated than a simple ARC. Cell Metab. 2009;9:117–123. doi: 10.1016/j.cmet.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, Heller R, Ellis LM, Karras J, Bromberg J, Pardoll D, Jove R, Yu H. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–2008. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med. 2002;8:1376–1382. doi: 10.1038/nm1202-798. [DOI] [PubMed] [Google Scholar]
- Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, Elmquist JK, Cowley MA, Lowell BB. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007;449:228–232. doi: 10.1038/nature06098. [DOI] [PubMed] [Google Scholar]
- Plum L, Belgardt BF, Brüning JC. Central insulin action in energy and glucose homeostasis. J Clin Invest. 2006a;116:1761–1766. doi: 10.1172/JCI29063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plum L, Ma X, Hampel B, Balthasar N, Coppari R, Münzberg H, Shanabrough M, Burdakov D, Rother E, Janoschek R, Alber J, Belgardt BF, Koch L, Seibler J, Schwenk F, Fekete C, Suzuki A, Mak TW, Krone W, Horvath TL, Ashcroft FM, Brüning JC. Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J Clin Invest. 2006b;116:1886–1901. doi: 10.1172/JCI27123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Rosenbaum M, Sy M, Pavlovich K, Leibel RL, Hirsch J. Leptin reverses weight loss-induced changes in regional neural activity responses to visual food stimuli. J Clin Invest. 2008;118:2583–2591. doi: 10.1172/JCI35055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler C, Darnell JE., Jr Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu Rev Biochem. 1995;64:621–651. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- Schubert M, Gautam D, Surjo D, Ueki K, Baudler S, Schubert D, Kondo T, Alber J, Galldiks N, Küstermann E, Arndt S, Jacobs AH, Krone W, Kahn CR, Brüning JC. Role for neuronal insulin resistance in neurodegenerative diseases. Proc Natl Acad Sci U S A. 2004;101:3100–3105. doi: 10.1073/pnas.0308724101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MW, Seeley RJ, Woods SC, Weigle DS, Campfield LA, Burn P, Baskin DG. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes. 1997;46:2119–2123. doi: 10.2337/diab.46.12.2119. [DOI] [PubMed] [Google Scholar]
- Seibler J, Zevnik B, Küter-Luks B, Andreas S, Kern H, Hennek T, Rode A, Heimann C, Faust N, Kauselmann G, Schoor M, Jaenisch R, Rajewsky K, Kühn R, Schwenk F. Rapid generation of inducible mouse mutants. Nucleic Acids Res. 2003;31:e12. doi: 10.1093/nar/gng012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Devgan G, Darnell JE, Jr, Bromberg JF. Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc Natl Acad Sci U S A. 2001;98:1543–1548. doi: 10.1073/pnas.041588198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Top M, Lee K, Whyment AD, Blanks AM, Spanswick D. Orexigen-sensitive NPY/AgRP pacemaker neurons in the hypothalamic arcuate nucleus. Nat Neurosci. 2004;7:493–494. doi: 10.1038/nn1226. [DOI] [PubMed] [Google Scholar]
- Xu AW, Ste-Marie L, Kaelin CB, Barsh GS. Inactivation of signal transducer and activator of transcription 3 in proopiomelanocortin (Pomc) neurons causes decreased pomc expression, mild obesity, and defects in compensatory refeeding. Endocrinology. 2007;148:72–80. doi: 10.1210/en.2006-1119. [DOI] [PubMed] [Google Scholar]
- Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med. 1999;5:1066–1070. doi: 10.1038/12506. [DOI] [PubMed] [Google Scholar]