

Abstract
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder, caused by a polyglutamine expansion in the huntingtin protein (htt). Increasing evidence suggests that transglutaminase (TGase) plays a critical role in the pathophysiology of HD possibly by stabilizing monomeric, polymeric and aggregated htt. We previously reported that in HEK293 and SH-SY5Y cells expression of a calmodulin (CaM)-fragment, consisting of amino acids 76-121 of CaM, decreased binding of CaM to mutant htt, TGase-modified htt and cytotoxicity associated with mutant htt and normalized intracellular calcium release. In this study, an adeno-associated virus (AAV) that expresses the CaM-fragment was injected into the striatum of HD transgenic R6/2 mice. The CaM-fragment significantly reduced body weight loss and improved motor function as indicated by improved rotarod performance, longer stride length, lower stride frequency, fewer low mobility bouts and longer travel distance than HD controls. A small but insignificant increase in survival was observed in R6/2 mice with CaM-fragment expression. Immunoprecipitation studies show that expression of the CaM-fragment reduced TGase-modified htt in the striatum of R6/2 mice. The percentage of htt-positive nuclei and the size of intranuclear htt aggregates were reduced by the CaM-fragment without striatal volume changes. The effects of CaM-fragment appear to be selective, as activity of another CaM-dependent enzyme, CaM-dependent kinase II, was not altered. Moreover, inhibition of TGase-modified htt was substrate-specific since overall TGase activity in the striatum was not altered by treatment with the CaM-fragment. Together, these results suggest that disrupting CaM–htt interaction may provide a new therapeutic strategy for HD.
Introduction
Patients with Huntington's disease (HD) present symptoms such as chorea, irregular gait, reduced motor coordination and weight loss (Harper, 1991). HD is a progressive and fatal neurological disorder caused by expansion of CAG repeats in HTT gene, conferring a toxic gain of function to the huntingtin (htt) protein (Huntington's Disease Collaborative Research Group, 1993). It is characterized neuropathologically by intranuclear inclusions and cytoplasmic aggregates composed of htt with an expanded polyglutamine domain (DiFiglia et al., 1997; Ross et al., 1998). Currently, there are no effective treatments to prevent or slow the progression of the disease.
Increasing evidence indicates that transglutaminases (TGases) may contribute to the pathophysiology of HD (Cooper et al., 1999; Karpuj et al., 2002a). TGases, a family of Ca2+-dependent enzymes, catalyze a covalent bond between peptide-bound glutamine residues and either lysine-bound peptide residues or mono- or polyamines (Folk et al., 1980; Griffin et al., 2002). In cell culture, mutant htt is an excellent substrate for TGases (Gentile et al., 1998; Kahlem et al., 1998; Zainelli et al., 2005). The mRNA, protein and enzymatic activity of TGases are elevated in HD brain (Karpuj et al., 1999; Lesort et al., 1999; Zainelli et al., 2003). TGase 2 ablation in HD transgenic mice resulted in a drastic reduction in ε(γ-glutamyl) lysine bond levels and neuronal death in the cortex and striatum (Mastroberardino et al., 2002). Administration of the TGase inhibitor, cystamine, extended survival and improved motor performance in HD transgenic mice (Karpuj et al., 2002b).
Calmodulin (CaM) increased TGase activity in human erythrocyte (Billett and Puszkin, 1991), platelets and chicken gizzard (Puszkin and Raghuraman, 1985). CaM activates a host of enzymes upon Ca2+ binding (Cheung, 1982) and associates with mutant htt as demonstrated by affinity purification (Bao et al., 1996) and immunoprecipitation (Zainelli et al., 2004). Moreover, CaM colocalizes with htt and TGase 2 in HD intranuclear inclusions (Zainelli et al., 2004). Since a CaM inhibitor decreased TGase-catalyzed cross-linking of htt in cells (Zainelli et al., 2004), we tested the ability of fragments of CaM to interrupt the interaction of CaM and mutant htt and decrease the deleterious effects of TGase in HEK-293 cells (Dudek et al., 2008). A CaM-fragment, containing amino acids 76–121, reduced binding of CaM to mutant htt in vitro and in HEK-293 cells. Adeno-associated virus (AAV)-mediated expression of the CaM-fragment significantly decreased TGase-modified htt, and mutant htt-associated cytotoxicity in differentiated SH-SY5Y cells, a neuroblastoma cell line, which stably express mutant htt. Importantly, CaM-fragment did not alter the total activity of TGase or another CaM-dependent enzyme, CaM kinase II, suggesting that CaM-fragment has specific effects on the CaM–htt interaction (Dudek et al., 2009). In the present study, we assessed the therapeutic potential of CaM-fragment in the R6/2 mouse model of HD. R6/2 transgenic mice express exon 1 of the human HD gene with an increased CAG repeat. Consequently, they develop a progressive neurological phenotype and pathological changes that resemble many features of HD (Mangiarini et al., 1996; Li et al., 2000). Survival, body weight, motor performance and neuropathological features were monitored to determine whether AAV-mediated expression of the CaM-fragment has beneficial effects on the course of HD in R6/2 mouse model.
Materials and Methods
Animals
Male R6/2 transgenic mice (with 100–115 CAG repeats in the transgene) and wild-type littermate mice were purchased from Jackson Laboratories at 6 weeks of age. A reduction in the number of CAG repeats from the original ∼154–159 to 100–115 was found in 2006, along with a delay in the age of onset and a decrease in the severity of the neurological phenotype (Jackson Laboratories, http://jaxmice.jax.org/strain/002810.html). The delayed onset of the neurological phenotype suggests that treatment at 7 weeks of age should be sufficient to determine the treatment effect. The mice had free access to food and water in an environment controlled for temperature and humidity and a 12 h light/dark cycle. The behavioral tests were conducted in the light part of the cycle. All procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals as approved by the University of Kansas Institutional Animal Care and Use Committee.
AAV injections
Recombinant AAV serotype 2 vectors were generated as described in our previous studies (Dudek et al., 2009). Three resultant vectors expressed each of the following: GFP and the CaM-fragment (containing amino acids 76-121 of calmodulin), GFP and a scrambled peptide (containing the same amino acids of the CaM-fragment but in a randomly scrambled sequence), and GFP alone. The titers of final AAV products ranged from 2 to 5 × 1014 vector genomes per ml. Bilateral striatal injections of GFP-expressing AAV were performed in 7-week-old mice [1 μl per side with 200 nl/min infusion rate at coordinates of anterior/posterior 0.5 mm, lateral/medial ±2 mm and dorsal/ventral −3 mm relative to bregma (Franklin and Paxinos, 1997)]. Mice were randomly divided into five groups (11–14 mice for each group) with a defined genotype and treatment: CaM-HD (R6/2 mice injected with AAV expressing the CaM-fragment), Vec-HD (R6/2 mice injected with empty vector AAV expressing GFP alone), Scr-HD (R6/2 mice injected with AAV expressing a scrambled version of the CaM-fragment), CaM-WT (wild-type mice injected with AAV expressing CaM-fragment) and Vec-WT (wild-type mice injected with empty vector AAV).
Body weight and survival
Body weights were measured twice a week. Survival was checked twice daily at 8 h intervals. Brains were removed after death and snap frozen in 2-methylbutane/dry ice bath and stored at −80°C.
Behavioral tests
Three behavioral tests were performed between 10 and 14 weeks of age at weekly intervals. Gait dynamics, locomotion, and rotarod performance were examined every Friday, Sunday, and Wednesday, respectively.
Gait dynamics.
The DigiGait imaging system (Mouse Specifics) was used for gait analyses as previously described (Hampton et al., 2004). Briefly, mice were placed on a motorized transparent treadmill belt moving at a speed of 14 cm/s. Digital images of paw placement were recorded at 150 Hz through a video camera mounted below the animal. The proprietary software analyzed the resulting digital images to generate a set of periodic waveforms that described the paw area and movement of each limb relative to the treadmill belt through consecutive strides. Gait data were pooled from all four paws and used to determine numerous gait dynamic measures including stride length and frequency, variability of stance width and paw area at peak stance.
Locomotion.
To accurately measure locomotor behaviors including the total distance traveled and the number of low mobility bouts (defined as remaining continuously in a virtual circle of 15 mm radius for 10 s), a force-plate actometer (constructed in Dr. Fowler's laboratory, University of Kansas) was used as described previously (Fowler et al., 2001). Mice were placed in a 28 cm × 28 cm force-plate actometer for a 30-min recording once a week. When the animal moves on the plate, its movements are sensed by four supporting force transducers positioned at the corners of the plate. The signals are processed by specialized computer algorithms written in house to yield measurements of travel distance and bouts of low mobility.
Rotarod performance.
A rotarod apparatus (MED-Associates) was used to measure fore- and hindlimb motor coordination and balance. The mice were given a training session at 9 weeks of age (four trials per day for 3 consecutive days) to acclimate them to the rotarod apparatus. During the test period, each mouse was placed on the rotarod with increasing speed, from 4 rpm to 40 rpm in 300 s. The latency to fall off the rotarod within this time period was recorded. Mice were tested on the rotarod once a week. Each mouse received two consecutive trials and the mean latency to fall was used in the analysis.
Immunoprecipitation
Mouse brains were bisected mid-sagittally. One half was used in immunoprecipitation (IP), CaM kinase II activity, and TGase assays. The other half was used for immunohistochemistry assays. To prepare striatal homogenates for IP, a half brain was cut into 300 μm coronal sections with a cryostat (Leica). Striatum was punched out from five consecutive sections and homogenized in lysate buffer (10 mm Tris-HCl, pH 7.5, 0.14 m NaCl, 1 mm EDTA, and 1:1000 protease inhibitor mixture) in a Tissue Tek homogenizer. Protein concentration was determined using the BCA Protein Assay kit (Pierce Chemical) and tissue homogenates containing 150 μg of protein were centrifuge at 12,000 × g for 5 min at 4°C to separate the insoluble fraction. After removal of the supernatant, the insoluble fraction was resuspended in 60 μl of 95% formic acid and incubated in a shaking water bath at 37°C for 40 min. The formic acid was then evaporated using a CentriVap Speed Vacuum (Labconco) for 1.5 h at 45°C. The pellets were resuspended in 150 μl of IP wash buffer (10 mm Tris-HCl, pH 7.5, 0.14 m NaCl, and 0.1% Tween 20) and sonicated on ice at 10 × 5 s pulse. Immunopurification of proteins containing TGase-catalyzed bonds was performed using 81D4 monoclonal antibody prebound to Sepharose beads (CovalAb) using a protocol developed by CovalAb and as described previously (Norlund et al., 1999; Zainelli et al., 2005).
Immunoblot
Immunoaffinity-purified proteins were separated on 10% SDS-polyacrylamide gels and then electrophoretically transferred to nitrocellulose membranes. Membranes were then incubated in blocking buffer (5% nonfat dry milk, 0.1% Tween 20, and 1× TBS) for 1 h at room temperature. Membranes were incubated overnight at 4°C with primary antibody on a shaker. Primary antibody (Millipore Bioscience Research Reagents: anti-Huntingtin aa 1-82, mouse IgG, 1:500) was diluted in antibody buffer (1% nonfat dry milk, 0.1% Tween 20, and 1× TBS). The next day, membranes were washed with TBS/0.1% Tween 20, and then they were incubated with goat anti-mouse secondary antibody conjugated to horseradish peroxidase (Jackson ImmunoResearch Laboratories) diluted in antibody buffer. Membranes were washed, and signal was detected using enhanced chemiluminescence Western blotting detection reagents (GE Healthcare). Using Scion Image for Windows, immunoblots were quantified by calculating the integrated optical density (IOD) of each protein band on the film.
Evaluation of htt aggregates by immunofluorescence
Sections of frozen mouse brain tissue (20 μm thick) were mounted on glass slides, and then fixed in 4% paraformaldehyde. After fixation, the sections were washed in PBS, pH 7.4, and then nonspecific binding was blocked with 5% normal goat serum (NGS). Sections were incubated overnight with primary antibody: MAB5374 (mouse IgG, 1:100, directed against human huntingtin amino acids 1–256) in 1% NGS + 1× PBS. Next, sections were washed and incubated for 1 h with secondary antibody: goat anti-mouse IgG, Fcγ fragment specific, conjugated to DyLight 649 (1:400) (Jackson ImmunoResearch). Next, coverslips were mounted on tissue sections with Prolong Gold antifade reagent with DAPI (Invitrogen). As a control for nonspecific labeling with secondary antibody, omission of the primary antibody was also performed on a slide from each case. Htt-aggregate positive nuclei and size of htt-aggregates were measured in each of ten 215 ×215 μm microscope fields by Olympus IX-81 microscope, in each of five rostrocaudally spaced sections in the striatum of 5 mice from the three groups of R6/2 mice (wild-type littermates did not show htt immunoreactivity). At least 450 nuclei per mouse were obtained and the percentage of nuclei containing htt-aggregates and area of htt-aggregates were calculated by CellProfiler software (Whitehead Institute for Biomedical Research, Cambridge, MA) and a mean value was obtained for each group.
Measurement of striatal volume by Nissl staining
Cryostat sections (20 μm thick) were postfixed in 4% paraformaldehyde, then dehydrated in graded alcohols. After being delipidated in 1:1 alcohol/chloroform for 1.5 h, sections were rehydrated through graded alcohols and stained with 0.2% aqueous solution of cresyl violet (Sigma) for 5 min, followed by a brief rinse with water and dehydration in graded alcohols. Sections were cleared in xylene (two changes, 5 min each) and coverslipped with Permount (Fisher Scientific). The volume of the striatum was measured according to the principle of Cavalieri (Cyr et al., 2005) (volume = s 1 d 1 + s 2 d 2…s n d n, where s = surface area and d = distance between two sections). We considered 15 coronal levels of the striatal sections (from bregma 1.7 mm, with an interval of 200 μm between the sections) for the volumetric measurement study. Image capturing was performed by using an inverted microscope (Olympus IX-81) coupled to a digital camera (Hamamatsu EMCCD). The montage image of a coronal brain section was constructed by Slidebook (Intelligent Imaging Innovations). The surface area of striatum in each section was measured using NIH ImageJ (http://rsbweb.nih.gov/ij/). Data were expressed as the average Cavalieri volume ±SEM (mm3) of 4–5 mice per group.
CaM kinase II activity assay
CaM kinase II enzyme activity was analyzed using the SignaTECT Calcium/Calmodulin-Dependent Protein Kinase Assay System (Promega), according to the manufacturer's protocol. Mouse striatum was homogenized in the extraction buffer [20 mm Tris-HCl, pH 8.0, 2 mm EDTA, 2 mm EGTA, 2 mm DTT, 1 mm PMSF and 1:1000 Protease Inhibitor Cocktail (Sigma)]. The homogenate was centrifuged at 350 × g for 5 min at 4°C and the supernatant was mixed with [γ-32P]ATP (at 3000Ci/mmol, 10mCi/ml), 50 μm biotinylated CaM kinase II substrate, reaction buffer containing 50 mm Tris-HCl, pH 7.5, 10 mm MgCl2 and 0.5 mm DTT, with or without control buffer containing 1 mm EGTA. After incubation at 30°C for 2 min, the reaction was terminated by adding 7.5 m guanidine hydrochloride and spotted to a streptavidin-impregnated membrane. Membranes were washed and the retained radioactive substrate of CaM kinase II was quantitated by liquid scintillation counting (Beckman). Radioactive counts were used as an index of endogenous CaM kinase II activity in the sample.
TGase II activity assay
TGase activity was measured by detecting the incorporation of a biotinylated TGase amine substrate, 5-(biotinamido)pentylamine (Pierce) into N,N′-dimethylcasein (Calbiochem) as previously described (Dudek et al., 2009). Ninety-six-well Immulon 4 HBX plates (Dynatech) were coated with 100 μl of N,N′-dimethylcasein (20 mg/ml) in 0.05 m sodium bicarbonate, and stored at 4°C overnight. After the unbound N,N′-dimethylcasein was discarded, the plate was washed with PBS and blocked with nonfat dry milk (2% milk in PBS) for 1 h at 37°C, followed by 3 washes with PBS. Striatum from the various mice were homogenized in 0.1 m Tris-HCl pH 8.3, 1 mm EDTA, 1 mm PMSF, and Protease Inhibitor Cocktail. Striatal homogenates (15 μg of protein/sample) were added to the plate with 0.1 m Tris-HCl pH 8.5, 0.15 m NaCl, 5 mm DTT, 0.5 mm biotin-labeled pentylamine, and 5 mm CaCl2. The mixture was incubated for 1 h at 37°C. The plate was washed with TBS and 0.001% Tween followed by incubation with streptavidin conjugated to HRP (Jackson ImmunoResearch) for 1 h at room temperature. After five washes, 1 × TMB substrate solution (eBioscience) was added and color was allowed to develop for 3 min. Then the reaction was stopped with 3N sulfuric acid and the absorbance was read at 450 nm. For GTP-inhibited TGase enzymatic activity, the assay was performed in the presence of 500 μm GTPγS (Sigma-Aldrich). Negative controls were run in the absence of pentylamine or dimethylcasein. Each assay also included a standard curve of varying amounts of guinea pig TGase (Sigma). Each sample was measured in triplicate. TGase activity was converted to a percentage control based on the mean value for the Vec-WT mice.
Statistics
All data are presented as mean ± SEM and analyzed by one- or two-way ANOVA with repeated measures. Post hoc tests were conducted using Bonferroni's multiple-comparison tests. There were one or two mice dead after week 13 in each HD group, so the last observed value was used for the subsequent missing values. Survival time was demonstrated by Kaplan–Meier survival curves and analyzed by a log-rank test. GB-STAT 10.0 (Dynamic Microsystems) and GraphPad Prism 5.0 were used for all statistical analyses. A probability level of p < 0.05 was considered to be statistically significant for all statistical tests.
Results
AAV-mediated delivery of CaM-fragment in R6/2 mice attenuated body weight loss
Body weight was monitored from 7 weeks of age onwards. All groups of mice were of similar initial body weight (22.9 ± 0.3 g, n = 10–14 mice in each group). Changes in body weight were expressed as a percentage of body weight measured at 7 weeks of age (Fig. 1 a). Body weight in all groups slowly increased or remained unchanged until week 10. Thereafter, mice in the two wild-type (WT) groups continued gaining body weight. The Vec-HD (R6/2 mice injected with empty vector AAV expressing GFP alone) and Scr-HD mice (R6/2 mice injected with AAV expressing a scrambled version of the CaM-fragment) had profound weight loss over the duration of the observation period, whereas CaM-HD mice maintained their body weight, or slightly increased their body weight (Fig. 1 a). One-way ANOVA (F (4,50) = 82.48; p < 0.0001) followed by Bonferroni's multiple-comparison test indicated that mice in CaM-HD group had less change in body weight than those in the Vec-HD group at 12–16 weeks of age (p < 0.05), and there was no significant difference in body weight change between Scr-HD and Vec-HD at each time point. Similarly, body weight change in the CaM-WT was not significantly different from the Vec-WT.
Figure 1.
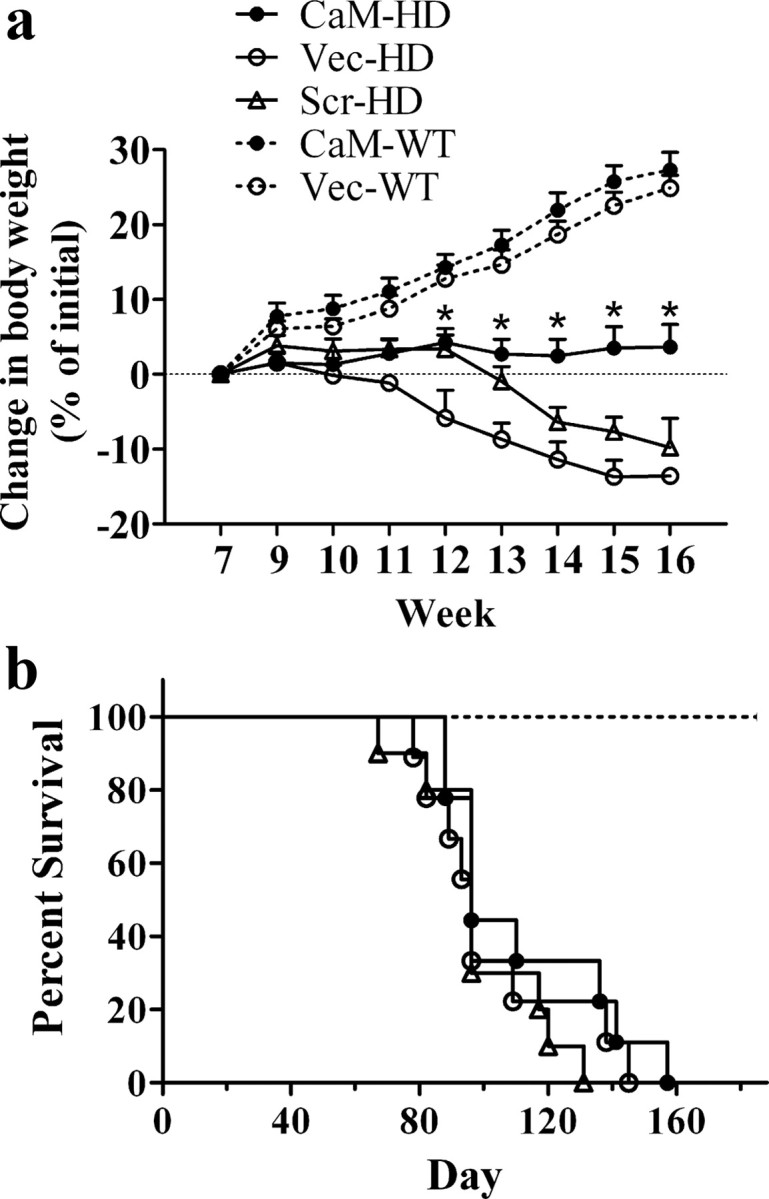
Effect of CaM-fragment on body weight and survival. a, Changes in body weight were expressed as a percentage of body weight measured at 7 weeks of age. One-way ANOVA and Bonferroni's test found that the change in body weight was significantly smaller in CaM-HD than in Vec-HD (p < 0.05) starting from week 12, and there is no significant difference between Scr-HD and Vec-HD. *p < 0.05 (CaM-HD vs Vec-HD). b, Kaplan–Meier survival curves showed the first death was at day 88, 78, and 67 in CaM-HD, Vec-HD, and Scr-HD mice, respectively. By log-rank comparison, three groups of HD mice did not differ from each other (p > 0.05) (n = 9–14 in each group).
CaM-fragment expression did not significantly increase R6/2 mice survival
Although the first death took place on days 88, 78, and 67 and mean survival time was 112, 102.9, and 99.7 d in CaM-HD, Vec-HD, and Scr-HD mice, respectively, suggesting a small (∼10%) increase in life span of CaM-HD mice, Kaplan–Meier survival curves demonstrated no statistically significant effect of CaM-fragment expression on the mortality in R6/2 mice (Fig. 1 b). By log-rank comparison, the three groups of HD mice did not significantly differ from each other (p > 0.05). None of the mice in WT groups died during the observation period (from 6 to 27 weeks of age).
CaM-fragment expression increases stride length and lowers stride frequency in R6/2 mice
Gait dynamics were characterized using the DigiGait Imaging System. Generally, there is a clear difference between WT mice and Vec-HD or Scr-HD mice starting from week 11 in all the parameters measured, whereas the difference between WT mice and CaM-HD mice is not significant in stride length and frequency. At a speed of 14 cm/s, WT mice maintained a regular alternating gait at a consistent stride length of 5.8 ± 0.3 cm and stride frequency of 2.5 ± 0.1 steps/s, whereas Vec-HD and Scr-HD mice walked at a gradually reduced stride length and higher stride frequency. For stride length (Fig. 2 a), the two-way ANOVA with repeated measures indicated a significant main effect of group (F (4,126) = 35.77; p < 0.0001) and week (F (3,126) = 12.12; p < 0.0001). There was also a significant interaction between group and week (F (12,126) = 5.71; p < 0.0001). The post hoc Bonferroni test indicated that stride length of CaM-HD mice is indistinguishable from the two HD control groups at 10 weeks of age, but CaM-HD mice displayed a longer stride length compared with Vec-HD or Scr-HD mice at weeks 11–13. Stride length of CaM-HD mice was not significantly shorter than WT mice until week 13. For stride frequency (Fig. 2 b), two-way ANOVA indicated significant differences among groups (F (4,126) = 11.99; p < 0.0001), but there is no significant effect of week (F (3,126) = 0.202; p = 0.895) or interaction between group and week (F (12,126) = 1.368; p = 0.190). The Bonferroni test indicated that CaM-HD mice had a significantly lower stride frequency than Vec-HD at weeks 11–13. Although Vec-HD and Scr-HD were statistically indistinguishable at any time point, there was a statistically significant difference between CaM-HD and Scr-HD at week 12. There was no significant difference between CaM-HD and WT mice in stride frequency. Analysis of some other gait dynamics revealed differences between genotypes. For example, the measure of stance width variation between steps was greater (Fig. 2 c) and paw area at peak stance was smaller (Fig. 2 d) in HD mice than in WT mice, but the CaM-fragment had no beneficial effects on these gait indices. For stance width variation, two-way ANOVA followed by Bonferroni test indicated significant differences between HD and WT groups (F (4,126) = 13.61; p < 0.0001), but no significant differences among HD groups or WT groups (p > 0.05). There is no significant effect of week (F (3,126) = 1.21; p = 0.31) although interaction between group and week is significant (F (12,126) = 1.97; p = 0.03). Similar results were obtained in the analysis of paw area at peak stance (Group factor: F (4,126) = 10.42; p < 0.0001), whereas week effect is significant (F (3,126) = 13.68; p < 0.0001) and interaction between group and week is not (F (12,126) = 1.55; p = 0.11).
Figure 2.

Video-based gait analysis on the treadmill. Mice were placed on a treadmill belt moving at a speed of 14 cm/s. Gait data were pooled from all four paws. CaM-HD exhibited a significantly greater stride length (a) and a lower stride frequency (b). There is no significant difference between CaM-HD and Vec-HD or Scr-HD in stance width variability (c) and paw area at peak stance (d). Two-way ANOVA with repeated measures followed by Bonferroni test. *p < 0.05 (CaM-HD vs Vec-HD), # p < 0.05 (CaM-HD vs Scr-HD). n = 6–14 in each group.
CaM-HD mice had fewer low mobility bouts and longer travel distance than Scr-HD or Vec-HD mice
Force-plate actometers were used to measure the locomotor activity of R6/2 and WT mice. The number of low mobility bouts (LMB, defined as remaining continuously in a virtual circle of 15 mm radius for 10 s) is presented in Figure 3 a. Two-way ANOVA with repeated measures indicated a significant effect of group (F (4,144) = 27.75, p < 0.001) and a significant effect of week (F (3,144) = 3.179, p < 0.05), but the interaction between group and week was not significant. The number of LMB gradually increased in HD mice without CaM-fragment expression, whereas CaM-HD mice, similar to WT mice, maintained LMB at a relatively lower level. At weeks 12 and 13, the Bonferroni post hoc test detected significantly fewer LMB in CaM-HD mice compared with either Scr-HD or Vec-HD mice. Total travel distance in a 30 min session provided another index of locomotor activity of HD mice (Fig. 3 b). Two-way ANOVA with repeated measures indicated a significant effect of group (F (4,138) = 3.40, p = 0.02), but the effect of week (F (3,138) = 0.17, p = 0.92) and interaction between group and week (F (12,138) = 1.12, p = 0.35) were not significant. Consistent with reduced LMB in HD mice treated with CaM-fragment, CaM-fragment expression also increased the locomotor activity in HD mice as indicated by a significantly greater travel distance in CaM-HD mice than in either Scr-HD (at weeks 12–13) or Vec-HD mice (at week 12). There was no significant difference between CaM-HD and WT mice in distance traveled in 30 min.
Figure 3.
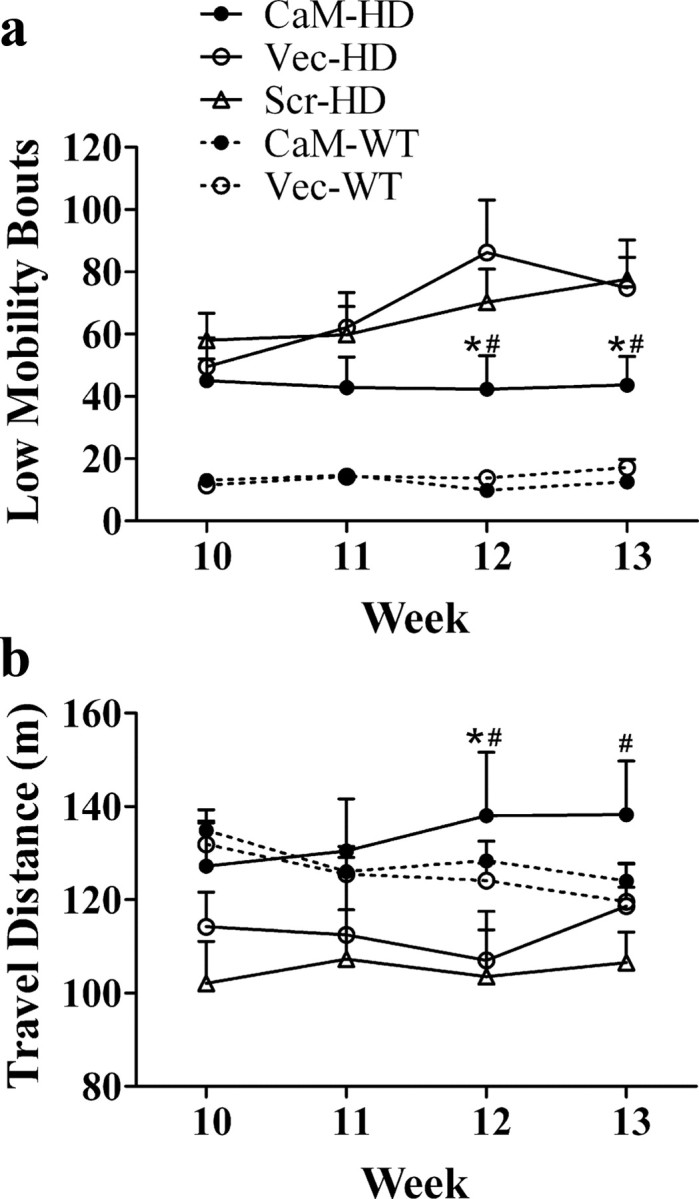
Expression of CaM-fragment enhanced the locomotor activity in R6/2 mice. Spontaneous locomotor activities of R6/2 and WT control mice were recorded in a force-plate actometer apparatus for 30 min. a, CaM-HD mice displayed a significantly smaller number of low mobility bouts than the two HD control groups at weeks 12–13. b, Distance traveled by CaM-HD mice was significantly longer than Scr-HD at weeks 12–13 and Vec-HD at week 12. Two-way ANOVA with repeated measures followed by Bonferroni test. *p < 0.05 (CaM-HD vs Vec-HD), # p < 0.05 (CaM-HD vs Scr-HD). n = 9–14 in each group.
CaM-fragment improved performance on the rotarod in R6/2 mice
Rotarod was used to measure fore- and hindlimb motor coordination and balance starting at 10 weeks of age (i.e., 3 weeks after the AAV injections). At 10 weeks of age, the two groups of WT mice were able to stay on the rod during the 300 s time period measured, whereas none of the three groups of HD mice could finish the test without falling in 300 s. Even at this early time point after injection of the AAV, the mean latency to fall was significantly differed among the HD groups, 245.9 ± 32.0, 150.2 ± 29.5, 98.1 ± 19.8 s in CaM-HD, Vec-HD, and Scr-HD mice, respectively. After 10 weeks of age, there was a progressive decline in performance of all HD mice, but the CaM-HD mice still achieved better performance than the other two HD groups (Fig. 4). Using analysis using two-way ANOVA with repeated measurements, we found a significant main effect of group (F (4,188) = 49.35, p < 0.0001), a significant main effect of week (F (4,188) = 18.86, p < 0.0001) and a significant interaction between group and week (F (16,188) = 4.714, p < 0.0001) on latency to fall off the rotarod. Bonferroni post hoc test indicated that CaM-HD mice had longer latency to fall compared with Scr-HD mice at 10–14 weeks of age and Vec-HD mice at 10–11 weeks of age (p < 0.05).
Figure 4.

CaM-fragment expression delayed the onset of the rotarod defects in R6/2 mice. Mice were placed on a rotating rod with increasing speed, from 4 rpm to 40 rpm in 300 s. The latency to fall off the rotarod within this time period was recorded. Two-way ANOVA with repeated measures followed by Bonferroni test found that CaM-HD mice had significantly longer latency to fall than Scr-HD starting from week 10, and than Vec-HD at weeks 10–11. *p < 0.05 (CaM-HD vs Vec-HD), # p < 0.05 (CaM-HD vs Scr-HD). n = 9–14 in each group.
CaM-fragment expression reduced TGase-modified htt in R6/2 mice striatum
Cell culture studies indicated that AAV-mediated CaM-fragment expression can significantly inhibit the formation of TGase-modified htt (Dudek et al., 2009). Here we used striatal homogenates from HD or WT mice to detect TGase-catalyzed cross-linking of htt. Immunopurification of proteins containing ε-(γ-glutamyl) lysine bonds was performed using 81D4 mAb prebound to Sepharose beads. TGase-modified htt was then detected on immunoblots as shown in the Figure 5 a. TGase-modified htt was only detected in HD but not WT mice. One-way ANOVA (F (2,15) = 18.44, p < 0.0001) followed by Bonferroni's comparison test found there was a significant reduction (by approximate 50%) in TGase-modified htt in the striatum of CaM-HD mice compared with Scr-HD or Vec-HD mice (Fig. 5 b).
Figure 5.

TGase-modified htt in R6/2 mice striatum was reduced by CaM-fragment expression. a, The insoluble fraction from mouse striatal homogenates was dissolved with formic acid. Immunopurification (IP) of proteins containing ε-(γ-glutamyl) lysine bonds was performed using 81D4 mAb prebound to Sepharose beads. Immunopurified proteins were then examined on immunoblots (IB) using an antibody against htt. b, Quantitation of immunoblots. Data shown are the mean IOD ± SEM, and they are normalized to CaM-HD mice. *p < 0.05 compared with CaM-HD (n = 5 in each group at 13–14 weeks of age).
CaM-fragment expression decreases the percentage of htt-positive nuclei and the size of htt aggregates in the striatum of R6/2 mice
Striatal intranuclear and neuropil inclusions containing mutant htt are prominent neuropathological hallmarks of HD and may play an important role in disease progression. In R6/2 mice, intranuclear aggregates are much larger than neuropil aggregates and amenable to quantification, so we examined the effect of CaM-fragment on intranuclear htt aggregates in the striatum of R6/2 mice. In CaM-HD mice, the percentage of htt-positive nuclei was lower and the size of intranuclear aggregates appeared smaller than in the control virus-treated HD mice (Fig. 6 a). These observations were confirmed by quantification of nuclear aggregates (Table 1). We found a significant reduction in the percentage of striatal nuclei containing htt-aggregates (∼18%) and the nuclear aggregate size (∼35%) in CaM-HD mice compared with either Vec- or Scr-HD mice. GFP fluorescence was observed in coronal brain sections from AAV-injected mice, indicating a widespread infection and expression of CaM-fragment in the striatum (Fig. 6 a). Both human HD and R6/2 mouse brains are characterized by atrophy of the striatum. To exam whether the CaM-fragment delivery decreases gross striatal atrophy, we used Nissl staining and the Cavalieri principle to estimate striatal volumes. There was no statistically significant difference in mean striatal volumes between CaM-HD and the two groups of control HD mice. However, all three groups of HD mice had significantly smaller striatal volumes compared with WT littermates (Fig. 6 b, Table 1). As expected, striatal neuronal atrophy was observed in HD mice in high-power images.
Figure 6.
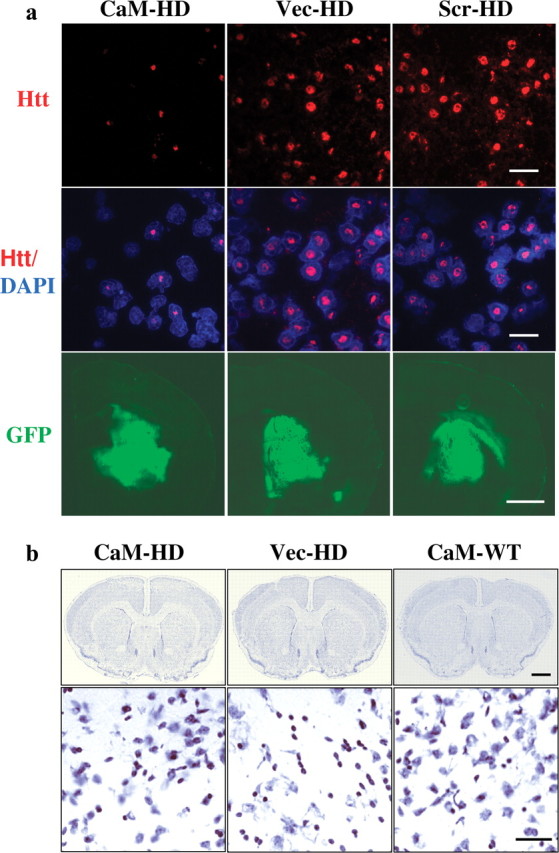
Histological evaluation of neuropathology. a, Top and middle, Immunofluorescent labeling of striatum in R6/2 mice at 14 weeks of age. Htt-aggregates were labeled with htt antibody MAB5374 (red), the nuclei were labeled with DAPI (blue). The composite images show that the percentage of htt-positive nuclei and the size of nuclear htt-aggregates were decreased in CaM-HD compared with the control-HD mice. Scale bar, 30 μm. Bottom, Photomicrographs of GFP distribution in the brain of representative AAV-injected animals indicated that the same vector-derived CaM-fragment was expressed in the striatum. Scale bar, 1 mm. b, Top, Montage images of Nissl-stained brain coronal sections from CaM-HD, Vec-HD, and CaM-WT mice at the level at which the corpus callosum starts to merge in the middle. Scale bar, 1 mm. Bottom, High magnification of micrograph of the dorsomedial aspect of the striatum from the sections above. There is marked neuronal atrophy with small angulated neurons in Vec-HD mouse, with relative preservation of neuronal size in CaM-HD mouse. Scale bar, 50 μm.
Table 1.
Quantitative analysis of neuropathology
| CaM-HD | Vec-HD | Scr-HD | CaM-WT | Vec-WT | |
|---|---|---|---|---|---|
| Percentage of nuclei containing htt aggregates (%) | 71.7 ± 5.6 | 85.1 ± 2.1* | 89.5 ± 0.8 a * | ||
| Size of intranuclear htt aggregates (μm2) | 248.4 ± 27.1 | 378.1 ± 31.5* | 391.0 ± 28.2 b * | ||
| Striatal volume (mm3) | 14.5 ± 0.3 | 13.8 ± 0.7 | 13.6 ± 0.3 | 17.0 ± 0.8 # | 17.2 ± 0.4 c # |
*p < 0.05 as compared to CaM-HD (one-way ANOVA, n = 5 in each group at 13–14 weeks of age).
# p < 0.05 as compared to CaM-HD (one-way ANOVA, n = 4–5 in each group at 14–20 weeks of age for HD mice, at 27–28 weeks of age for WT mice).
aF (2,12) = 6.83.
bF (2,12) = 7.38.
cF (4,17) = 11.77.
CaM kinase II activity was not affected by CaM-fragment expression
To test whether expression of the CaM-fragment would interfere with other CaM-dependent enzymes, we measured the activity of calmodulin-dependent protein kinase II (CaM kinase II) in mouse striatal homogenates. We found no significant differences in CaM kinase II activity among the various HD or WT mouse groups in either the presence or absence of EGTA, a calcium chelator (Fig. 7 a). However, EGTA caused a general decrease in CaM kinase II activity in all mouse groups. Two-way ANOVA indicated a significant main effect of EGTA (F (1,50) = 72.12; p < 0.0001), but there was no significant effect of mouse group (F (4,50) = 0.543; p = 0.705) or an interaction between EGTA and group (F (4,50) = 0.966; p = 0.435).
Figure 7.
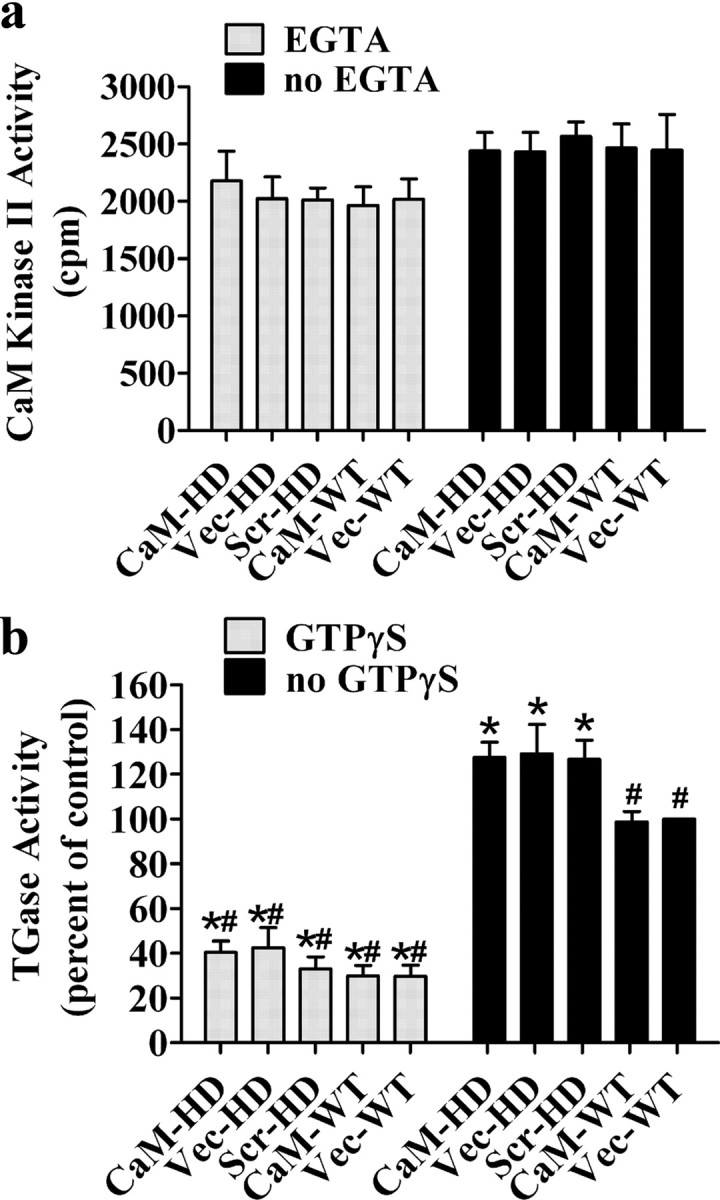
Expression of CaM-fragment in mouse striatum did not significantly affect the activity CaM kinase II or TGase. a, Mouse striatal homogenates were used to measure CaM kinase II activity. Two-way ANOVA indicates a significant main effect of EGTA. However, there was no significant main effect of mouse group. The interaction between EGTA and group was also not significant. Bonferroni post hoc test indicates no significant differences in CaM kinase II activity among the various groups in either the presence or absence of EGTA. b, The presence of the CaM-fragment did not significantly change the levels of TGase activity in either HD or WT mice. However, a significant elevation of TGase activity in three groups of HD mice was observed compared with WT mice. When 500 μm GTPγS was added, there is a dramatic reduction in all mouse groups. Two-way ANOVA indicates a significant main effect of GTPγS and mouse group, and there is a significant interaction between GTPγS and group. Bonferroni post hoc test. *p < 0.01 compared with Vec-WT; # p < 0.01 compared with CaM-HD in the absence of GTPγS. Each column represents the mean ± SEM (n = 5 in each group at 13–14 weeks of age).
CaM-fragment did not change total TGase activity
To explore whether the reduced TGase-modified htt in CaM-HD mice is due to a substrate-dependent inhibition or a general decrease in TGase enzymatic activity, we compared total TGase activity in the striatum among the different groups of mice. There is a significant increase in TGase activity in the three groups of HD mice compared with CaM-WT or Vec-WT mice (p < 0.01). Expression of the CaM-fragment did not have an effect on TGase activity in either HD or WT mice. In the presence of GTPγS, which specifically inhibits TGase activity, TGase activity is dramatically reduced (at least 60%) in all groups (Fig. 7 b). Two-way ANOVA indicated a significant main effect of GTPγS (F (1,50) = 428.149; p < 0.0001) and mouse group (F (4,50) = 5.535; p < 0.01), and there was a significant interaction between GTPγS and mouse group (F (4,50) = 5.032; p < 0.01). The activity of guinea pig liver TGase was measured in each assay in the range from 0.01 to 0.5 mU/ml and resulted in a linear correlation with an R-square value from 0.95 to 0.99. The activity of TGase in the striatal samples fell within this linear range for guinea pig liver TGase.
Discussion
Although there have been enormous strides in understanding the molecular and mechanistic pathways that mediate the progression of HD, an effective treatment to slow or prevent the disease-associated physical and mental decline has yet to be discovered. A number of reports in cultured cells have shown that interfering peptides can selectively inhibit pathological interactions between mutant htt and other proteins, and therefore are a potential therapeutic strategy for HD (Nagai et al., 2000; Dudek et al., 2008, 2009). However, only a few studies have been performed to verify the efficacy of these peptides in vivo (Kazantsev et al., 2002; Tang et al., 2009). We previously found that plasmid or AAV-mediated delivery of CaM-fragment significantly attenuated TGase-modified htt, htt-associated cytotoxicity and intracellular Ca2+ disturbances without interfering with the activity of other CaM-dependent enzymes in HD cell models (Dudek et al., 2008, 2009). We now report that expression of the CaM-fragment in the striatum ameliorated body weight loss, improved motor performance of R6/2 mice, reduced TGase-modified htt and decreased the number and size of nuclear htt-aggregates in the striatum.
Striatal delivery of CaM-fragment significantly delayed the onset of movement abnormalities and improved motor function as measured by analysis of gait, rotarod performance and locomotor behavior. It has been reported that gait deficiencies in R6/2 mice became apparent from 8 weeks of age. As visualized in the footprint analysis, R6/2 mice displayed a significantly shorter stride length, a staggering movement and a gait that lacked a uniform step pattern compared with WT littermates (Carter et al., 1999). Striatal injection of AAV-CaM-fragment conferred significant improvements in stride length and frequency measurements, and a mild, insignificant increase in paw area at peak stance in R6/2 mice. Decreased motor function on the rotarod is the most commonly reported measure among animal models of HD (Schilling et al., 1999; von Hörsten et al., 2003). Three weeks after CaM-fragment expression, R6/2 mice started to show significantly improved rotarod performance and this beneficial effect was maintained from 10 weeks of age onward. Although motor abnormalities such as bradykinesia (abnormally slow movements) and pronounced hypoactivity were frequently observed in HD animal models (Mangiarini et al., 1996; Keene et al., 2002), there have been relatively few quantitative studies on spontaneous locomotor activity of R6/2 mice. In the present experiments using a force-plate actometer, progressive locomotor deterioration (assessed by number of low mobility bouts and travel distance) was observed in R6/2 mice injected with control AAV, whereas delivery of AAV expressing CaM-fragment to R6/2 mice striatum significantly improved their locomotor deficits.
Compared with the TGase inhibitor cystamine, which prolonged the life span of R6/2 mice by 12–19% (Dedeoglu et al., 2002; Karpuj et al., 2002b), there was a nonsignificant increase in the mean survival time by ∼10% in the CaM-HD mice. Differences in other mechanisms of action could possibly account for the different outcomes. In addition to inhibiting TGase, cystamine increased transcription of neuroprotective genes, increased glutathione levels, and ameliorated apoptosis (Dedeoglu et al., 2002; Karpuj et al., 2002b). In contrast, that CaM-fragment decreased TGase-modified htt, normalized intracellular calcium release, and reduced mutant htt-associated cytotoxicity but had no effect on TGase activity (Dudek et al., 2008, 2009). Second, the different outcomes could be due to the different brain regions treated. The CaM-fragment was expressed in the striatum only while the cystamine-treatment was systemically delivered and could affect other brain regions that display neuropathology in HD such as the frontal cortex. Rather than nonspecific inhibition of TGase by cystamine, disrupting the CaM–htt interaction, as with the CaM-fragment, is a promising new approach for the treatment HD.
As a result of its expanded N-terminal polyglutamine region, mutant htt is processed and deposited as a component of insoluble protein aggregates that persist in neuronal nuclei, perikarya, and processes (DiFiglia et al., 1997). Although some evidence suggests that htt-associated toxicity is linked to soluble htt and its protein–protein interactions (Petersen et al., 1999), htt aggregates act as a phenotypic readout that can reflect pathologically relevant processes such as htt cleavage, misfolding, and sequestration (Chopra et al., 2007). A number of compounds which are neuroprotective in R6/2 mice, such as the TGase inhibitor cystamine (Dedeoglu et al., 2002), the antioxidant coenzyme Q10 (Ferrante et al., 2002) and the energy buffer creatine (Ferrante et al., 2000), significantly suppress htt aggregates. Our present results revealed that CaM-fragment significantly reduced TGase-modified htt in the insoluble fraction, the percentage of htt-positive nuclei and intranuclear htt-aggregate size in the striatum, which may contribute to the benefits of CaM-fragment. These findings support the hypothesis that CaM-regulated TGase modification of htt contributes to the formation and stabilization of htt-aggregates and may play a role in the pathogenesis of HD. However, it is difficult to speculate whether there would be an associated increase in soluble or oligomeric htt in the R6/2 mouse striatum with CaM-fragment expression. There is evidence that TGase modifications stabilize proteins (Tucholski et al., 1999), so inhibiting TGase modifications to htt may increase clearance of the htt protein.
R6/2 mice developed significant striatal atrophy compared with WT littermates, a pathological characteristic associated with the gradual neurodegeneration occurring in the mice. Although there is no significant difference in striatal volume between CaM-HD and control-HD mice by light microscopy, we could not exclude the possibility that expression of CaM-fragment would reduce the striatal atrophy if a more sensitive technology (MR scanning) was applied. Moreover, if striatal injections of AAV were performed at an earlier age, there might be more notable beneficial results including increases in survival and striatal volume.
Similar to increases in TGase protein level and enzymatic activity in human HD brain (Karpuj et al., 1999; Lesort et al., 1999), our results show that TGase activity is elevated in R6/2 mice compared with WT littermates. Interestingly, striatal delivery of CaM-fragment did not change the overall activity of TGase in either R6/2 mice or WT mice. These results are consistent with our previous studies in cell culture (Dudek et al., 2009), suggesting that CaM-fragment may specifically affect the CaM–mutant htt interaction and selectively alter TGase-catalyzed modifications of mutant htt without interfering with the activity of TGase on other substrate proteins. Furthermore, CaM-fragment expression did not affect CaM kinase II activity, indicating that CaM-fragment had no effect on the activity of other CaM-dependent enzymes. Most importantly, expression of CaM-fragment in WT mice had no effect on any of the behavioral tests nor striatal histopathology, suggesting there is minimal, if any, deleterious effect of long-term expression of CaM-fragment.
A prominent hypothesis on the pathogenesis of HD is that the aberrant interactions of the extended polyglutamine repeats (polyQs) of mutant htt, either with other proteins or with each other result in htt-aggregate formation (Wanker, 2000). Glutamine repeats may function as polar zippers and expansion of polyQs may increase the affinity of htt for its protein binding partners (Perutz et al., 1994). The principal proteins that are known to interact with htt are: htt associated protein 1 (Li et al., 1995), htt interacting protein (Kalchman et al., 1997), type 1 inositol 1,4,5-trisphosphate receptor (InsP3R1) (Tang et al., 2003) and calmodulin (Bao et al., 1996; Zainelli et al., 2004). As revealed by our previous in vitro assay (Dudek et al., 2009), the neuroprotective effects of CaM-fragment may depend on its ability to competitively bind to mutant htt thereby disrupting the CaM–mutant htt interaction. First, this disruption may result in the CaM and mutant htt no longer in close proximity to each other, thus the TGase activity in htt modification and aggregate formation may be reduced. Second, the association between mutant htt and CaM-fragment may prevent the further interaction of mutant htt with other proteins, such as InsP3R1, an intracellular Ca2+ release channel. Since mutant htt selectively associates with and activates InsP3R1(Tang et al., 2003), disrupting their interaction should stabilize neuronal Ca2+ homeostasis. This hypothesis is supported by in vitro experiments that indicated the CaM-fragment (Dudek et al., 2008) or InsP3R-fragment (Tang et al., 2009) attenuates mutant htt-associated intracellular Ca2+ disturbances.
Although it will be advantageous to confirm the results from the current study using a different HD animal model, special care should be taken to choose a different strain other than R6/2 mice. Instead of body weight loss, YAC128 mice have significantly (27%) increased body weight at 12 months of age (Van Raamsdonk et al., 2006, 2007), which in turn can cause bias in most behavioral tests. Obvious or diffuse nuclear htt accumulation is not detectable in the striatum or cortices of BAC HD mice even in 18-month-old mice (Gray et al., 2008). Intergenerational instability and somatic instability of the HD CAG repeat have been observed in HdhQ111 knock-in mice in all three background strains: C57BL/6, FVB/N and 129Sv (Lloret et al., 2006). The phenotype of N171-82Q mice is more variable than that of R6/2 mice, and therefore a much larger number of mice are necessary to provide adequate power (Hersch and Ferrante, 2004).
Many therapeutic targets for HD today are based on the pathogenesis hypotheses, from mitochondrial dysfunction to transcriptional perturbation to TGase hyperactivity (Beal and Ferrante, 2004). However, since clinical trials of mechanism-based therapies for HD have been limited by insufficient statistical power and insignificant improvement (Handley et al., 2006), current clinical treatments are symptomatic. The major neurological symptoms associated with HD include disordered voluntary movements: uncoordinated, arrhythmic, and slow fine motor movements; rigidity; and gait disturbances (Harper, 1991). Our in vivo results demonstrated that viral delivery of CaM-fragment to the striatum of R6/2 mice significantly improved motor coordination and balance, enhance locomotor activity and alleviated gait disturbances, suggesting the possibility of a better quality of life for HD patients if this new therapeutic strategy is successfully translated to the clinical trials.
The present studies provide, for the first time, in vivo evidence that CaM-fragment has significant efficacy in improving the behavioral deficits and neuropathological phenotype in the HD animal model. The positive effects of CaM-fragment in R6/2 mice provide further evidence that CaM-regulated TGase modification of htt may contribute to HD pathogenesis. More importantly, these studies have identified a novel therapeutic strategy for treating HD patients.
Footnotes
This work was supported by the High Q Foundation/CHDI, Inc. We thank Dr. David Moore and Heather Shinogle (Microscopy and Analytical Imaging Laboratory, University of Kansas) for their assistance with digital imaging and analysis.
References
- Bao J, Sharp AH, Wagster MV, Becher M, Schilling G, Ross CA, Dawson VL, Dawson TM. Expansion of polyglutamine repeat in huntingtin leads to abnormal protein interactions involving calmodulin. Proc Natl Acad Sci U S A. 1996;93:5037–5042. doi: 10.1073/pnas.93.10.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Ferrante RJ. Experimental therapeutics in transgenic mouse models of Huntington's disease. Nat Rev Neurosci. 2004;5:373–384. doi: 10.1038/nrn1386. [DOI] [PubMed] [Google Scholar]
- Billett HH, Puszkin EG. The red cell membrane contains calmodulin-regulated crosslinking and proteolytic activity. Hematol Pathol. 1991;5:185–193. [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung WY. Role of calmodulin in brain function. Prog Brain Res. 1982;56:237–253. doi: 10.1016/S0079-6123(08)63778-4. [DOI] [PubMed] [Google Scholar]
- Chopra V, Fox JH, Lieberman G, Dorsey K, Matson W, Waldmeier P, Housman DE, Kazantsev A, Young AB, Hersch S. A small-molecule therapeutic lead for Huntington's disease: preclinical pharmacology and efficacy of C2–8 in the R6/2 transgenic mouse. Proc Natl Acad Sci U S A. 2007;104:16685–16689. doi: 10.1073/pnas.0707842104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AJ, Sheu KF, Burke JR, Strittmatter WJ, Gentile V, Peluso G, Blass JP. Pathogenesis of inclusion bodies in (CAG)n/Qn-expansion diseases with special reference to the role of tissue transglutaminase and to selective vulnerability. J Neurochem. 1999;72:889–899. doi: 10.1046/j.1471-4159.1999.0720889.x. [DOI] [PubMed] [Google Scholar]
- Cyr M, Caron MG, Johnson GA, Laakso A. Magnetic resonance imaging at microscopic resolution reveals subtle morphological changes in a mouse model of dopaminergic hyperfunction. Neuroimage. 2005;26:83–90. doi: 10.1016/j.neuroimage.2005.01.039. [DOI] [PubMed] [Google Scholar]
- Dedeoglu A, Kubilus JK, Jeitner TM, Matson SA, Bogdanov M, Kowall NW, Matson WR, Cooper AJ, Ratan RR, Beal MF, Hersch SM, Ferrante RJ. Therapeutic effects of cystamine in a murine model of Huntington's disease. J Neurosci. 2002;22:8942–8950. doi: 10.1523/JNEUROSCI.22-20-08942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Dudek NL, Dai Y, Muma NA. Protective effects of interrupting the binding of calmodulin to mutant huntingtin. J Neuropathol Exp Neurol. 2008;67:355–365. doi: 10.1097/NEN.0b013e31816a9e60. [DOI] [PubMed] [Google Scholar]
- Dudek NL, Dai Y, Muma NA. Neuroprotective effects of calmodulin peptide 76-121aa: disruption of calmodulin binding to mutant huntingtin. Brain Pathol. 2009 doi: 10.1111/j.1750-3639.2008.00258.x. Advance online publication. Retrieved March 10, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Jenkins BG, Dedeoglu A, Kuemmerle S, Kubilus JK, Kaddurah-Daouk R, Hersch SM, Beal MF. Neuroprotective effects of creatine in a transgenic mouse model of Huntington's disease. J Neurosci. 2000;20:4389–4397. doi: 10.1523/JNEUROSCI.20-12-04389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. J Neurosci. 2002;22:1592–1599. doi: 10.1523/JNEUROSCI.22-05-01592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folk JE, Park MH, Chung SI, Schrode J, Lester EP, Cooper HL. Polyamines as physiological substrates for transglutaminases. J Biol Chem. 1980;255:3695–3700. [PubMed] [Google Scholar]
- Fowler SC, Birkestrand BR, Chen R, Moss SJ, Vorontsova E, Wang G, Zarcone TJ. A force-plate actometer for quantitating rodent behaviors: illustrative data on locomotion, rotation, spatial patterning, stereotypies, and tremor. J Neurosci Methods. 2001;107:107–124. doi: 10.1016/s0165-0270(01)00359-4. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego: Academic; 1997. [Google Scholar]
- Gentile V, Sepe C, Calvani M, Melone MA, Cotrufo R, Cooper AJ, Blass JP, Peluso G. Tissue transglutaminase-catalyzed formation of high-molecular-weight aggregates in vitro is favored with long polyglutamine domains: a possible mechanism contributing to CAG-triplet diseases. Arch Biochem Biophys. 1998;352:314–321. doi: 10.1006/abbi.1998.0592. [DOI] [PubMed] [Google Scholar]
- Gray M, Shirasaki DI, Cepeda C, André VM, Wilburn B, Lu XH, Tao J, Yamazaki I, Li SH, Sun YE, Li XJ, Levine MS, Yang XW. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28:6182–6195. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin M, Casadio R, Bergamini CM. Transglutaminases: nature's biological glues. Biochem J. 2002;368:377–396. doi: 10.1042/BJ20021234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton TG, Stasko MR, Kale A, Amende I, Costa AC. Gait dynamics in trisomic mice: quantitative neurological traits of Down syndrome. Physiol Behav. 2004;82:381–389. doi: 10.1016/j.physbeh.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Handley OJ, Naji JJ, Dunnett SB, Rosser AE. Pharmaceutical, cellular and genetic therapies for Huntington's disease. Clin Sci (Lond) 2006;110:73–88. doi: 10.1042/CS20050148. [DOI] [PubMed] [Google Scholar]
- Harper PS. Huntington's disease. London: Saunders; 1991. [Google Scholar]
- Hersch SM, Ferrante RJ. Translating therapies for Huntington's disease from genetic animal models to clinical trials. NeuroRx. 2004;1:298–306. doi: 10.1602/neurorx.1.3.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Kahlem P, Green H, Djian P. Transglutaminase action imitates Huntington's disease: selective polymerization of Huntingtin containing expanded polyglutamine. Mol Cell. 1998;1:595–601. doi: 10.1016/s1097-2765(00)80059-3. [DOI] [PubMed] [Google Scholar]
- Kalchman MA, Koide HB, McCutcheon K, Graham RK, Nichol K, Nishiyama K, Kazemi-Esfarjani P, Lynn FC, Wellington C, Metzler M, Goldberg YP, Kanazawa I, Gietz RD, Hayden MR. HIP1, a human homologue of S. cerevisiae Sla2p, interacts with membrane-associated huntingtin in the brain. Nat Genet. 1997;16:44–53. doi: 10.1038/ng0597-44. [DOI] [PubMed] [Google Scholar]
- Karpuj MV, Garren H, Slunt H, Price DL, Gusella J, Becher MW, Steinman L. Transglutaminase aggregates huntingtin into nonamyloidogenic polymers, and its enzymatic activity increases in Huntington's disease brain nuclei. Proc Natl Acad Sci U S A. 1999;96:7388–7393. doi: 10.1073/pnas.96.13.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpuj MV, Becher MW, Steinman L. Evidence for a role for transglutaminase in Huntington's disease and the potential therapeutic implications. Neurochem Int. 2002a;40:31–36. doi: 10.1016/s0197-0186(01)00060-2. [DOI] [PubMed] [Google Scholar]
- Karpuj MV, Becher MW, Springer JE, Chabas D, Youssef S, Pedotti R, Mitchell D, Steinman L. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med. 2002b;8:143–149. doi: 10.1038/nm0202-143. [DOI] [PubMed] [Google Scholar]
- Kazantsev A, Walker HA, Slepko N, Bear JE, Preisinger E, Steffan JS, Zhu YZ, Gertler FB, Housman DE, Marsh JL, Thompson LM. A bivalent Huntingtin binding peptide suppresses polyglutamine aggregation and pathogenesis in Drosophila . Nat Genet. 2002;30:367–376. doi: 10.1038/ng864. [DOI] [PubMed] [Google Scholar]
- Keene CD, Rodrigues CM, Eich T, Chhabra MS, Steer CJ, Low WC. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington's disease. Proc Natl Acad Sci U S A. 2002;99:10671–10676. doi: 10.1073/pnas.162362299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesort M, Chun W, Johnson GV, Ferrante RJ. Tissue transglutaminase is increased in Huntington's disease brain. J Neurochem. 1999;73:2018–2027. [PubMed] [Google Scholar]
- Li H, Li SH, Johnston H, Shelbourne PF, Li XJ. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet. 2000;25:385–389. doi: 10.1038/78054. [DOI] [PubMed] [Google Scholar]
- Li XJ, Li SH, Sharp AH, Nucifora FC, Jr, Schilling G, Lanahan A, Worley P, Snyder SH, Ross CA. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995;378:398–402. doi: 10.1038/378398a0. [DOI] [PubMed] [Google Scholar]
- Lloret A, Dragileva E, Teed A, Espinola J, Fossale E, Gillis T, Lopez E, Myers RH, MacDonald ME, Wheeler VC. Genetic background modifies nuclear mutant huntingtin accumulation and HD CAG repeat instability in Huntington's disease knock-in mice. Hum Mol Genet. 2006;15:2015–2024. doi: 10.1093/hmg/ddl125. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Mastroberardino PG, Iannicola C, Nardacci R, Bernassola F, De Laurenzi V, Melino G, Moreno S, Pavone F, Oliverio S, Fesus L, Piacentini M. ‘Tissue’ transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington's disease. Cell Death Differ. 2002;9:873–880. doi: 10.1038/sj.cdd.4401093. [DOI] [PubMed] [Google Scholar]
- Nagai Y, Tucker T, Ren H, Kenan DJ, Henderson BS, Keene JD, Strittmatter WJ, Burke JR. Inhibition of polyglutamine protein aggregation and cell death by novel peptides identified by phage display screening. J Biol Chem. 2000;275:10437–10442. doi: 10.1074/jbc.275.14.10437. [DOI] [PubMed] [Google Scholar]
- Norlund MA, Lee JM, Zainelli GM, Muma NA. Elevated transglutaminase-induced bonds in PHF tau in Alzheimer's disease. Brain Res. 1999;851:154–163. doi: 10.1016/s0006-8993(99)02179-4. [DOI] [PubMed] [Google Scholar]
- Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci U S A. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen A, Mani K, Brundin P. Recent advances on the pathogenesis of Huntington's disease. Exp Neurol. 1999;157:1–18. doi: 10.1006/exnr.1998.7006. [DOI] [PubMed] [Google Scholar]
- Puszkin EG, Raghuraman V. Catalytic properties of a calmodulin-regulated transglutaminase from human platelet and chicken gizzard. J Biol Chem. 1985;260:16012–16020. [PubMed] [Google Scholar]
- Ross CA, Margolis RL, Becher MW, Wood JD, Engelender S, Cooper JK, Sharp AH. Pathogenesis of neurodegenerative diseases associated with expanded glutamine repeats: new answers, new questions. Prog Brain Res. 1998;117:397–419. doi: 10.1016/s0079-6123(08)64029-7. [DOI] [PubMed] [Google Scholar]
- Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I. Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron. 2003;39:227–239. doi: 10.1016/s0896-6273(03)00366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Guo C, Wang H, Chen X, Bezprozvanny I. Neuroprotective effects of inositol 1,4,5-trisphosphate receptor C-terminal fragment in a Huntington's disease mouse model. J Neurosci. 2009;29:1257–1266. doi: 10.1523/JNEUROSCI.4411-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucholski J, Kuret J, Johnson GV. Tau is modified by tissue transglutaminase in situ: possible functional and metabolic effects of polyamination. J Neurochem. 1999;73:1871–1880. [PubMed] [Google Scholar]
- Van Raamsdonk JM, Gibson WT, Pearson J, Murphy Z, Lu G, Leavitt BR, Hayden MR. Body weight is modulated by levels of full-length huntingtin. Hum Mol Genet. 2006;15:1513–1523. doi: 10.1093/hmg/ddl072. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Metzler M, Slow E, Pearson J, Schwab C, Carroll J, Graham RK, Leavitt BR, Hayden MR. Phenotypic abnormalities in the YAC128 mouse model of Huntington disease are penetrant on multiple genetic backgrounds and modulated by strain. Neurobiol Dis. 2007;26:189–200. doi: 10.1016/j.nbd.2006.12.010. [DOI] [PubMed] [Google Scholar]
- von Hörsten S, Schmitt I, Nguyen HP, Holzmann C, Schmidt T, Walther T, Bader M, Pabst R, Kobbe P, Krotova J, Stiller D, Kask A, Vaarmann A, Rathke-Hartlieb S, Schulz JB, Grasshoff U, Bauer I, Vieira-Saecker AM, Paul M, Jones L, et al. Transgenic rat model of Huntington's disease. Hum Mol Genet. 2003;12:617–624. doi: 10.1093/hmg/ddg075. [DOI] [PubMed] [Google Scholar]
- Wanker EE. Protein aggregation and pathogenesis of Huntington's disease: mechanisms and correlations. Biol Chem. 2000;381:937–942. doi: 10.1515/BC.2000.114. [DOI] [PubMed] [Google Scholar]
- Zainelli GM, Ross CA, Troncoso JC, Muma NA. Transglutaminase cross-links in intranuclear inclusions in Huntington disease. J Neuropathol Exp Neurol. 2003;62:14–24. doi: 10.1093/jnen/62.1.14. [DOI] [PubMed] [Google Scholar]
- Zainelli GM, Ross CA, Troncoso JC, Fitzgerald JK, Muma NA. Calmodulin regulates transglutaminase 2 cross-linking of huntingtin. J Neurosci. 2004;24:1954–1961. doi: 10.1523/JNEUROSCI.4424-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zainelli GM, Dudek NL, Ross CA, Kim SY, Muma NA. Mutant huntingtin protein: a substrate for transglutaminase 1, 2, and 3. J Neuropathol Exp Neurol. 2005;64:58–65. doi: 10.1093/jnen/64.1.58. [DOI] [PubMed] [Google Scholar]