

Abstract
GABA, the main inhibitory neurotransmitter in the adult brain, has recently emerged as an important signal in network development. Most of the trophic functions of GABA have been attributed to depolarization of the embryonic and neonatal neurons via the activation of ionotropic GABAA receptors. Here we demonstrate a novel mechanism by which endogenous GABA selectively regulates the development of GABAergic synapses in the developing brain. Using whole-cell patch-clamp recordings on newborn mouse hippocampi lacking functional GABAB receptors (GABAB-Rs) and time-lapse fluorescence imaging on cultured hippocampal neurons expressing GFP-tagged brain-derived neurotrophic factor (BDNF), we found that activation of metabotropic GABAB receptors (GABAB-Rs) triggers secretion of BDNF and promotes the development of perisomatic GABAergic synapses in the newborn mouse hippocampus. Because activation of GABAB-Rs occurs during the characteristic ongoing physiological network-driven synaptic activity present in the developing hippocampus, our results reveal a new mechanism by which synaptic activity can modulate the development of local GABAergic synaptic connections in the developing brain.
Introduction
The proper development of highly organized structures in the CNS is a complex process that determines appropriate connectivity in the adult. Neurotransmitters released during spontaneous and experience-driven synaptic activity play a crucial role in the formation of neuronal networks (Katz and Shatz, 1996; Zhang and Poo, 2001). The most well documented example is glutamate, the major excitatory transmitter in the vertebrate CNS, which regulates nearly all aspects of neuronal network formation from migration to synaptogenesis (Zhang and Poo, 2001; Manent and Represa, 2007). Recent advances have shown that GABA acts beyond its classical inhibitory role and also functions as an important developmental signal by regulating proliferation, migration, growth, and synapse formation (Ben-Ari et al., 2007). With the observation that the activation of chloride-permeable GABAA receptors (GABAA-Rs) depolarizes developing neurons (Cherubini et al., 1991), it was proposed that part of the trophic action of GABA relies on membrane depolarization and subsequent activation of voltage-dependent calcium channels (Ben-Ari et al., 2007). Consistent with this hypothesis, early conversion of GABA-induced depolarization into hyperpolarization impaired synapse formation and dendritic development of the target neurons both in vitro (Chudotvorova et al., 2005) and in vivo (Ge et al., 2006; Cancedda et al., 2007; Wang and Kriegstein, 2008; Reynolds et al., 2008).
In addition to ionotropic GABAA-Rs, GABA also binds to metabotropic GABAB-Rs. These receptors are ubiquitously expressed at early stages of development, even before synapses are formed (Fritschy et al., 1999; Behar et al., 2001; López-Bendito et al., 2002, 2004), and are activated by endogenous GABA released during early network-driven synaptic activity (McLean et al., 1996; Obrietan and Van den Pol, 1999; Catsicas and Mobbs, 2001; López-Bendito et al., 2003). Although GABAB-R activity has been reported to modulate cortical neuronal migration (Behar et al., 1996, 2001; López-Bendito et al., 2003), little attention has been paid to the possible contribution of GABAB-Rs in the functional development of neuronal networks.
In the present study, we tested the ability of CA3 hippocampal pyramidal cells to develop functional synaptic connections in the absence of functional GABAB-Rs. We found that activation of GABAB-Rs selectively promotes the development of hippocampal GABAergic synapses via the induction of brain-derived neurotrophic factor (BDNF) secretion. We further show that this process occurs during physiological patterns of synaptic activity. This study reveals a novel role of GABAB-Rs in regulating the self-refinement of GABAergic synaptic connections in the developing brain.
Materials and Methods
All animal experiments were carried out according to the guidelines laid down by the Inserm animal welfare committee and the European Communities Council Directive of 24 November 1986 (86/609/EEC).
Intact hippocampal formation and slice preparation.
Experiments were performed on intact hippocampal formation (IHF) and hippocampal slices obtained from newborn [postnatal day 1 (P1) to P24] GABAB1 receptor subunit knock-out (GABAB1-KO) and wild-type (GABAB1-WT) mice (Prosser et al., 2001) and BDNF-KO and BDNF-WT mice (The Jackson Laboratory). The procedure for the preparation of the intact IHFs was similar to that previously described (Khalilov et al., 1997). Brains were removed from anesthetized (350 mg/kg chloral hydrate, i.p.) animals and immersed into ice-cold (2–4°C) artificial CSF (ACSF) of the following composition (in mm): 126 NaCl, 3.5 KCl, 2 CaCl2, 1.3 MgCl2, 1.2 NaH2PO4, 25 NaHCO3, and 11 glucose, pH 7.4, when equilibrated with 95% O2 and 5% CO2. The hippocampi were then incubated for 12–16 h at 32°C in ACSF (oxygenated with 95% O2 and 5% CO2) alone or supplemented with different drugs. After the incubation, hippocampal slices (600 μm thick) were cut with a McIlwain tissue chopper and kept in ACSF at 25°C for 60 min before use. Slices were then transferred to a submerged recording chamber and perfused with ACSF (3 ml/min) at 34°C. For each experiment, half of the dissected hippocampi were incubated in control conditions, and the other half incubated in treated conditions. Electrophysiological recordings on slices obtained from control and treated hippocampi were performed on the same day.
Electrophysiological recordings.
Whole-cell patch-clamp recordings of CA3 pyramidal neurons were performed with an Axopatch 200B amplifier (Molecular Devices). To record miniature activity, borosilicate microelectrodes (4–8 MΩ) were filled with the following solution (in mm): 110 CsCl, 30 potassium gluconate, 10 HEPES, 1.1 EGTA, 0.1 CaCl2, 4 MgATP, 0.3 NaGTP, 5-(and-6)-tetramethylrhodamine biocytin (rhodamine, 0.5–1%), pH = 7.25, osmolarity = 275 mosmol/L. Criteria for accepting a recording included a resting potential of <−55 mV, an Ri of >400 MΩ, and an Rs of <25 MΩ. Capacitance, input, and series resistances were measured online with Acquis Software (Biologic).
Miniature GABAA receptor-mediated postsynaptic currents (mGABAA-PSCs) were isolated in the presence of the ionotropic glutamatergic receptor antagonists [10 μm 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX), 40 μm D-2-amino-5-phosphovaleric acid (D-APV)] and tetrodotoxin (1 μm TTX) and recorded at a holding potential of −70 mV. Miniature glutamatergic postsynaptic currents were recorded in the presence of TTX (1 μm) and the GABAA receptor antagonist bicuculline (10 μm). Neurons were clamped at −70 mV. The currents were stored on an Axoscope 8.1 (Molecular Devices) and analyzed off-line with the Mini Analysis program (Synaptosoft 5.1). The fact that no false events would be identified was confirmed by visual inspection for each experiment. To generate the average PSCs, multiple overlapping events were discarded, and the remaining events were aligned on their rising phase. In the figures, the histograms were constructed using miniature PSCs recorded for 10–30 min. To determine the probability of presence of giant depolarizing potentials (GDPs), an average of 13 ± 6 cells (ranging from n = 3 at P24 to n = 23 at P1) were recorded in GABAB1-WT slices and 14 ± 7 cells (ranging from n = 3 at P24 to n = 27 at P2) in GABAB1-KO slices.
Peak-scaled analysis of mGABAA-PSCs was performed as described by Traynelis et al. (1993) using Mini Analysis program (Synaptosoft 5.1). For each recording, we verified the absence of correlation between the decay time course and peak amplitude of mGABAA-PSCs. For each recording, we used between 50 and 200 events and eliminated all events with a decay time distorted by multiple peaks or anomalous noise. Each individual event was scaled to the peak of the mean waveform of the averaged event and subtracted. The mean variance was plotted against mean current. The plot was well fit by a parabolic function that yields the single-channel current i0 and the number of channels contributing to mGABAA-PSCs, N0. From i0, the single-channel conductance γ can be calculated.
Paired-pulse relationship (PPR) of GABAergic synapses impinging onto CA3 pyramidal neurons was measured using pairs of identical stimuli at a 100 ms interval, delivered at a frequency of 0.01 Hz with a bipolar tungsten electrode placed in the CA3 stratum radiatum. The resulting pairs of postsynaptic GABAA currents (GABAA-PSCs) were isolated in the presence of 10 μm CNQX, 40 μm D-APV, and 5 μm CGP55845. PPR was measured as the following amplitude ratio: second GABAA-PSCs/first GABAA-PSCs. Average PPR values were calculated from 20–30 paired stimulations.
Neuron reconstruction and morphometric analysis.
Biocytin filling was done using the whole-cell patch-clamp technique. Briefly, after whole-cell access, biocytin (1% in internal pipette solution) filling was done for 15 min. The slices were then fixed in PFA–PBS at 4°C overnight, and the biocytin-filled neurons were visualized using the avidin–biotin method. For 3-D reconstruction, a dendritic tree was digitized directly from the sections by use of a 20× objective on a Nikon microscope, equipped with a motorized stage and coupled to a computer running Neurolucida software (Microbrightfield), thereby allowing for x–z coordinates of digitized points to be stored and analyzed.
Immunohistochemistry.
Brains from P7 mice were fixed in 4% paraformaldehyde (overnight). Cryostat-cut hippocampal sections (30 μm) were preincubated (1 h) in PBS–Triton X-100 (0.3%)–goat serum (3%) and then coincubated overnight at 4°C with antibodies to glutamic acid decarboxylase (Chemicon MAB351) and synaptophysin (Chemicon AB9272). Slices were washed with PBS, and Alexa Fluor 488 donkey antibody to mouse IgG (FluoProbes) and Cy3 donkey antibody to rabbit IgG (Chemicon) were applied (2 h). Sections were visualized with confocal microscopy (LSM 510, Zeiss). Five areas were sampled per animal, in the stratum radiatum and the stratum pyramidale. After the recording sessions, the optical sections were displayed in the form of digital images of 1024 × 1024 pixels and processed using the ImageJ software (NIH, Bethesda, MD; http://rsb.info.nih.gov/ij/). All the pictures were reviewed to set a threshold to optimize the representation of puncta, and then the same threshold was applied to all images. The GAD65-labeled area fraction, defined as the percentage of GAD65-immunopositive pixels per field (1024 × 1024 pixels), and the size of GAD65 puncta were measured. The proportion of colocalized immunoreactivity (IR) was expressed as the ratio: synaptophysin and GAD65 colocalized area/synaptophysin IR area or GAD65 IR area. For each section, counts were performed blindly in slices taken from three WT and three KO animals.
Cell cultures and transfections.
Neurons from postnatal day 0 rat hippocampus were dissociated using trypsin and plated on coverslips coated with polyethylenimine as previously described (Kuczewski et al., 2008b). Eleven days after plating, neurons were transfected with cDNAs coding for BDNF-GFP (gift from Dr. V. Lessmann, Institute of Physiology, Otto-von-Guericke University Magdeburg, Magdeburg, Germany) according to the OZ Biosciences protocol (Buerli et al., 2007). After transfection, the cultures were incubated at 30°C in 5% CO2. Immunostaining confirmed that BDNF-GFP is targeted to the dendrite and packed into secretory granules of the regulated pathway of secretion (Kuczewski et al., 2008b).
Time-lapse imaging.
Real-time imaging was performed as previously described (Kuczewski et al., 2008b). Fluorescence intensity was measured from dendritic regions containing clusters of BDNF-GFP with ImageJ software after subtraction of background fluorescence. Clusters in which the fluorescence intensity varied during the 5 min control period by >5% were discarded from the analysis. Fluorescence decreases caused by photobleaching and constitutive release were corrected by subtracting the extrapolation of the fluorescence decrease in the first 5 min over the whole recording time. Values are plotted as the percentage of the fluorescence intensity of the last frame before stimulation. Percentage variation in the text and statistical analysis were calculated by comparing the relative fluorescence of the interval −100 to 0 s (control) with that of 400–500 s (after stimuli).
Surface GFP immunofluorescence staining.
The procedure for surface BDNF-GFP immunostaining was similar to that previously described (Kuczewski et al., 2008b). Briefly, after being washed in ACSF, the living cultures were incubated at 4°C for 1 h in the presence of an anti-GFP antibody (10 μg/ml; Molecular Probes). Cultures were then washed with 0.1 m PBS (4°C, pH 7.4) and fixed for 15 min with 4% paraformaldehyde–4% sucrose. After fixation, the neurons were exposed to a saturating concentration (10 μg/ml) of either anti-rabbit secondary antibody coupled to Cy3 (FluoProbes) for 1.5 h under a nonpermeabilized condition. Quantifications were performed with ImageJ. Surface-bound BDNF-GFP on BDNF-GFP-expressing neurons was expressed as the following ratio: Cy3 and BDNF-GFP colocalized area/BDNF-GFP area.
Phospho-CREB activation and immunocytochemistry.
The procedure for phospho-CREB activation and immunocytochemistry was similar to that previously described (Kuczewski et al., 2008b). Briefly, 1 d before stimulation (at 13 DIV) one-half of the culture medium was changed to MEM with 2% B27 supplement. To reduce the basal level of CREB phosphorylation, cultures were incubated for 30 min in TTX (1 μm). Five minutes before stimulation, NBQX (10 μm), D-APV (40 μm), and bicuculline (20 μm) were added to medium. The cultures were then stimulated with baclofen (50 μm) for 10 min in the absence or presence of TrkB-IgG (2 μg/ml) or CGP55845 (10 μm). Five to 10 min after stimulation, neurons were fixed for 15 min (4% paraformaldehyde) at 4°C and rinsed several times. Coverslips were then preincubated in PBS–Triton-100 (0.1%)–goat serum (3%) for 1 h at room temperature and incubated overnight with mouse anti-CREB (1:1000) and rabbit anti-phospho-CREB (pCREB, 1:1000) antibodies (Cell Signaling Technology). Immunoreactivities for pCREB and CREB were detected with an Alexa 488-coupled (A488) rabbit secondary antibody (1:500; FluoProbes) and a Cy3-coupled mouse secondary antibody (1:500; Jackson ImmunoResearch Laboratories), respectively. All procedures were performed in phosphate-free solution containing 140 mm NaCl, 5 mm KCl, and 10 mm HEPES-Na, pH 7.4. Images were acquired with an LSM 510 laser scanning confocal microscope (Zeiss). The acquisition of A488 (pCREB) and then Cy3 (CREB) was sequential to avoid overlap of excitation and emission of fluorescence. The optical sections were digitized (1024 × 1024 pixels) and processed using ImageJ software. The pCREB-to-CREB intensity ratio was expressed as means value ratio of the pCREB-A488 staining intensity versus the CREB-Cy3 staining intensity.
ELISA.
Brains of wild-type, GABAB1-KO, and BDNF heterozygote mice were rapidly removed from their skulls at P6. Hippocampi were rapidly dissected out, weighed, and snap frozen in liquid nitrogen and stored at −80°C. BDNF was extracted from hippocampi by mechanical homogenization in a buffer containing 100 mm Tris–HCl, pH 7.0, containing 1 m NaCl, 4 mm EDTA, 2% Triton X-100, and the protease inhibitors 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 17 μg/ml PMSF. Homogenates were centrifuged at 14,000 × g for 20 min. Supernatants were collected and analyzed with a commercial two-antibody sandwich ELISA (BDNF Emax immunoassay system; Promega) according to the protocol of the manufacturer. The total protein content of each supernatant was measured with a Bradford protein assay. The BDNF level was expressed as the ratio of BDNF to the total soluble protein concentration. There was no significant difference in the weight of GABAB1-WT and GABAB1-KO hippocampi (9.87 ± 0.31 mg/hippocampi vs 9.12 ± 0.56 mg/hippocampi, respectively).
Drugs.
NBQX, D-APV, bicuculline, and CGP55845 were purchased from Tocris Cookson. Tetrodotoxin and baclofen were purchased from Sigma. k252a was from Calbiochem. TrkB-IgG and TrkC-IgG were from R&D Systems.
Results
Miniature GABAergic synaptic activity is altered in mice lacking functional GABAB receptors
To address the contribution of GABAB-Rs to the development of the hippocampal circuit, we recorded miniature GABAA and glutamatergic-receptor-mediated postsynaptic currents (mGABAA-PSCs and mGlu-PSCs, respectively) from hippocampal CA3 pyramidal cells from P6 to P8 GABAB1-KO mice, which display a complete loss of GABAB receptor function (Prosser et al., 2001). The frequency of mGABAA-PSCs was significantly reduced in GABAB1-KO (0.73 ± 0.12 Hz in GABAB1-KO, n = 13) when compared with their wild-type littermates (1.61 ± 0.26 Hz, n = 10, p = 0.03) (Fig. 1A,B). This decrease in frequency was observed at all developmental stages studied (i.e., from P1 to P10) (Fig. 1C; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The mean amplitude, coefficient of variation of amplitude (CVa), and kinetic properties of mGABAA-PSCs were unchanged (Fig. 1B; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In contrast to mGABAA-PSCs, the average frequency of mGlu-PSCs was not different between GABAB1-WT (0.11 ± 0.017 Hz, n = 10) and GABAB1-KO (0.11 ± 0.016 Hz, n = 12) P6–P8 neurons. The other parameters of mGlu-PSCs were also unchanged (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The membrane capacitance of the recorded cells, an indicator of neuronal size (Colin-Le Brun et al., 2004) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material), was not different between GABAB1-WT and GABAB1-KO (Fig. 1B; supplemental Fig. 1, available at www.jneurosci.org as supplemental material), suggesting that the morphological development of the target CA3 pyramidal cells was not affected. This observation was confirmed by quantitative morphometric analysis of intracellularly biocytin-loaded CA3 pyramidal neurons (Fig. 1D–G). Therefore, the development of GABAergic synapses is selectively impaired in mice lacking functional GABAB-Rs.
Figure 1.

Miniature GABAergic activity is impaired in GABAB1-KO hippocampal slice. A, Representative traces of mGABAA-PSCs. Averaged mGABAA-PSCs (n = 30) are shown at an expanded time scale. B, Summary plot of the mGABAA-PSC parameters and cell capacitance in GABAB1-KO mice at P6–P8 expressed as percentage of GABAB1-WT values. C, Summary plot of the mGABAA-PSC frequency in recordings from GABAB1-WT (open bars) and GABAB1-KO (filled bars) mice at different postnatal developmental stages. D, Two-dimensional projection of three-dimensional reconstruction of biocytin-filled P6–P7 CA3 pyramidal cells. Scale bar, 100 μm. E, F, Summary plot of the total apical and basal dendritic length (E) and dendritic branching point (F) of the biocytin-filled CA3 pyramidal cells of GABAB1-KO and GABAB1-WT mice. The average membrane capacitance was 35.9 ± 3.8 pF for the GABAB1-WT pyramidal cells and 38.2 ± 3.1pF for the GABAB1-KO pyramidal cells. n = 14 for each genotype. G, Sholl analysis of biocytin-filled CA3 pyramidal neurons obtained from GABAB1-KO and GABAB1-WT mice. The number of intersections within each concentric ring (10 μm beginning from the soma) is plotted versus the distance from the soma. n = 14 for each genotype. H, Left, Summary plot of the mGABAA-PSC frequency in recordings from GABAB1-WT intact hippocampi incubated in vitro for 12–16 h in control condition (ACSF) or with CGP55845 (CGP; 5 μm), and from GABAB1-KO intact hippocampi incubated in vitro for 12–16 h in control condition (ACSF). Right, Logarithmic plot of the mGABAA-PSC frequency in recordings from GABAB1-WT and GABAB1-KO intact hippocampi incubated in the corresponding conditions. Each symbol represents the result of one single cell. The dashed line represents the mean value obtained from GABAB1-WT incubated in ACSF. In this and following figures, *p < 0.05, and the number of cells recorded is shown within brackets.
Blockade of GABAB-Rs in vitro mimics the deficit in GABAergic transmission observed in GABAB1-KO mice
In a previous study, we showed that the factors required for the functional maturation of GABAergic synapses in vivo are preserved in the IHFs in vitro (Colin-Le Brun et al., 2004). We therefore used this preparation to overcome the potential pitfalls of genetic manipulations often observed in KO animals. IHFs obtained from P1 GABAB1-WT mice were incubated at 32°C for 12–16 h in oxygenated control ACSF or with the GABAB-R antagonist CGP55845 (5 μm). After the incubation period, hippocampal slices were prepared to record mGABAA-PSCs. In slices obtained from CGP55845-treated IHFs, the frequency of mGABAA-PSCs was significantly lower compared with control IHFs [0.107 ± 0.024 Hz (n = 11) vs 0.247 ± 0.049 Hz (n = 10), p = 0.031] (Fig. 1H). The other mGABAA-PSC parameters were unchanged (supplemental Fig. 4a, available at www.jneurosci.org as supplemental material). P1 IHFs obtained from GABAB1-KO mice were also incubated in control ACSF. The mean frequency of mGABAA-PSCs (0.117 ± 0.026 Hz, n = 8) was significantly lower compared with control GABAB1-WT IHFs (p = 0.046) but similar to the value obtained in CGP55845-treated GABAB1-WT IHFs (Fig. 1H). The other mGABAA-PSC parameters were unchanged (supplemental Fig. 4a, available at www.jneurosci.org as supplemental material). Therefore, the pharmacological blockade of the GABAB-Rs in vitro reproduces the deficit in GABAergic transmission observed in GABAB1-KO mice, showing that it reflects the absence of GABAB-R activation by endogenous GABA.
Reduced perisomatic GABAergic synapses in mice lacking GABAB receptors
We next asked whether the deficit in GABAergic synaptic transmission resides in the presynaptic or the postsynaptic site. We first performed a peak-scaled variance analysis of mGABAA-PSCs which allows an estimate of the mean unitary conductance and the number of channels open at the peak of the synaptic current (Traynelis et al., 1993) and found no difference between GABAB1-KO (n = 6) and GABAB1-WT (n = 11) neurons (Fig. 2A–C). To detect possible changes in the probability of GABA release, we then measured the paired-pulse ratio and CVa of evoked GABAA-PSCs. We found no difference between GABAB1-WT and GABAB1-KO mice (n = 5 for both) (Fig. 2D–F). Finally, to investigate possible changes in the density of GABAergic terminals, we performed immunolabeling against GAD65, the synthetic enzyme for GABA. Double immunostaining against GAD65 and synaptophysin confirmed that GAD65 staining corresponds to GABAergic terminals (supplemental Fig. 5a–c, available at www.jneurosci.org as supplemental material). Quantitative analysis shows that, compared with the GABAB1-WT hippocampi, the labeled area fraction was reduced by ∼20% in the stratum pyramidale of GABAB1-KO (p = 0.001), while the average size of GAD-positive puncta was unchanged (n = 5 pairs of mice, 5 sections per mouse) (Fig. 2G–I). There was, however, no difference in the labeled area fraction and average size of GAD65-positive puncta in the stratum radiatum of GABAB1-KO and GABAB1-WT hippocampi (Fig. 2G–I). Similar results were obtained with synaptophysin, a presynaptic marker (supplemental Fig. 5d,e, available at www.jneurosci.org as supplemental material). Although functional presynaptic modifications cannot be completely excluded, together these data suggest that the deficit in GABAergic synaptic activity observed in GABAB1-KO mice results at least in part from a decrease in the density of proximal GABAergic terminals.
Figure 2.

Presynaptic reduction in the number of perisomatic GABAergic terminals in GABAB1-KO hippocampi. A, Example of mGABAA-PSCs superimposed to the scaled mean waveform and associated relationship between mean mGABAA-PSCs amplitude and variance recorded from a representative GABAB1-KO neuron. B, C, Summary plot of the unitary conductance (B) and the number of GABAA channels at the peak of the mGABAA-PSCs (C) in GABAB1-WT (open bars) and GABAB1-KO (filled bars) mice. D, Representative averaged (n = 20) evoked GABAA-PSCs from GABAB1-WT and KO mice (interstimulus = 100 ms). E, F, Summary plot of the paired-pulse ratio (E) and the coefficient of variation (F) of evoked GABAA-PSCs in GABAB1-WT (open bars) and GABAB1-KO (filled bars) mice. Both GABAB-R-dependent and GABAB-R independent PPRs have been reported for eGABAA-PSCs in hippocampus. The PPR experiment were therefore performed in the presence of the GABAB-R antagonist CGP55845 (5 μm) in both GABAB1-KO mice and GABAB1-WT. G, Microscopic confocal images showing GAD65-positive puncta in the strata pyramidale and radiatum of GABAB1-WT and GABAB1-KO hippocampi at postnatal day 7. Scale bars, 10 μm. H, I, Summary plot of the fraction area of GAD65 fluorescence (H) and average size of GAD65 fluorescent puncta (I) in GABAB1-WT (open bars) and GABAB1-KO (filled bars). S., Stratum.
GABAB-Rs are primarily activated by spontaneous network-driven synaptic activity
We next asked whether and when GABAB-Rs are activated during ongoing synaptic activity. To address this point, we investigated the effect of the GABAB-R antagonist CGP55845 on GABAB1-WT slices. We found that the duration of GDPs, the characteristic primitive network-driven pattern of synaptic activity (Ben-Ari et al., 1989), was significantly increased in the presence of CGP55845 (5 μm) (from 0.757 ± 0.146 s to 1.738 ± 0.617 s, n = 8, p = 0.0002) (Fig. 3A). However, when applied in the presence of NBQX (10 μm) and D-APV (40 μm), to isolate spontaneous GABAA-PSCs, CGP55845 had no significant effect on the frequency, amplitude and charge transfer of sGABAA-PSCs in GABAB1-WT neurons (n = 9) (Fig. 3B,C). In agreement with previous findings that GABAB-Rs are activated by concomitant release of GABA from several interneurons (Scanziani, 2000), these data show that the activation of GABAB-Rs by endogenous GABA required the presence of spontaneous GDPs.
Figure 3.

GABAB-Rs are primarily activated during spontaneous network-driven synaptic activity. A, Representative current traces of spontaneous GDPs recorded in GABAB1-WT pyramidal neurons in control conditions and with the GABAB-R antagonist, CGP55845 (5 μm). B, Representative traces of spontaneous GABAA receptor-mediated postsynaptic currents (sGABAA-PSCs) recorded in the presence of NBQX (10 μm) and D-APV (40 μm) with or without CGP55845 (5 μm). C, Summary plot of the effect of CGP55845 on sGABAA-PSC frequency, amplitude, and charge transfer in GABAB1-WT neurons expressed as percentages relative to control conditions (ACSF).
The lack of GABAB-R activation is responsible for the deficit in GABAergic transmission
Network construction is modulated by the level and pattern of spontaneous synaptic activity generated in the developing nervous system. Thus, the deficit in GABAergic synaptic transmission observed in the GABAB1-KO could be accounted for by alterations in spontaneous synaptic activity resulting from the lack of functional GABAB-Rs. We therefore characterized the spontaneous synaptic activity generated in the GABAB1-KO. We found that GDPs were present in both GABAB1-WT and GABAB1-KO hippocampal slices from P1 to P10 and progressively disappeared by the end of the second postnatal week (Fig. 4A,B). No difference in timing of the disappearance of GDPs was observed between the two groups (Fig. 4B). There was also no difference in the frequency or amplitude of GDPs between GABAB1-KO (n = 33) and GABAB1-WT (n = 40) mice (Fig. 4C,D). However, GDP duration was two- to threefold longer in GABAB1-KO slices (1630 ± 381 ms, n = 40) compared with WT (740 ± 97 ms, n = 33, p = 0.02) (Fig. 4A,E). The longer GDPs in GABAB1-KO slices are network-driven synaptic events because they were recorded with field electrodes and abolished by TTX (1 μm) (data not shown). As expected, application of CGP55845 (5 μm) had no effect on GDP duration in GABAB1-KO slices (1555 ± 72 ms in CGP55845, n = 4). These data therefore show that GDPs are longer in GABAB1-KO mice and that the lengthening of GDPs results from the lack of functional GABAB-R-mediated inhibition.
Figure 4.

Characterization of synaptic activity in the developing mouse hippocampus. A, Representative current traces of spontaneous GDPs recorded in GABAB1-WT and KO CA3 pyramidal neurons. B, Plot of the percentage of cells showing GDPs at different postnatal stages of development in GABAB1-WT (open symbols) and GABAB1-KO (filled symbols). An average of 13 ± 6 cells (ranging from n = 3 at P24 to n = 23 at P1) were recorded in GABAB1-WT slices, and 14 ± 7 cells (ranging from n = 3 at P24 to n = 27 at P2) in GABAB1-KO slices were also recorded to construct this graph. The Boltzmann fit shows that there is no significant difference in the disappearance of GDPs between GABAB1-WT (dashed line) and GABAB1-KO (dark line). C–E, Summary plot of GDP frequency (C), amplitude (D), and duration (E) in GABAB1-WT (open bars) and GABAB1-KO (filled bars). F, Left, Summary plot of the mGABAA-PSC frequency in recordings from GABAB1-WT intact hippocampi incubated in vitro for 12–16 h in control condition (ACSF), in the presence of TTX (1 μm) alone, TTX and baclofen (5 μm), and baclofen alone. Right, Logarithmic plot of the mGABAA-PSC frequency in recordings from GABAB1-WT and GABAB1-KO intact hippocampi incubated in the corresponding conditions. Each symbol represents the result of one single cell. The dashed line represents the mean value obtained from GABAB1-WT incubated in ACSF. baclo, Baclofen.
We next asked whether the deficit in GABAergic transmission observed in hippocampi deficient in GABAB-Rs is due to the lack of GABAB-R activity per se or is an indirect consequence of the lack of GABAB-R activity, i.e., the lengthening of GDP duration. To address this point, P1 WT IHFs were incubated for 12–16 h in the presence of TTX (1 μm) to block action potential-dependent activity and GDPs. Since GABAB receptors are activated during GDPs, we hypothesized that blockade of GDPs with TTX treatment would reduce activation of GABAB-R and lead to a GABAergic deficit similar to that observed in GABAB-R-deficient hippocampi. We found that mGABAA-PSC frequency was indeed significantly reduced in TTX-treated IHFs (0.158 ± 0.037 Hz, n = 15) compared with control IHFs (0.270 ± 0.036 Hz, n = 15, p = 0.045) (Fig. 4F). The other mGABAA-PSC parameters were unchanged (supplemental Fig. 4b, available at www.jneurosci.org as supplemental material). We next attempted to rescue the deficit induced by TTX with the specific GABAB-R agonist baclofen. Baclofen (5 μm for 12–16 h) completely rescued the TTX-induced deficit [from 0.158 ± 0.037 Hz (n = 15) to 0.383 ± 0.076 Hz (n = 11), respectively, p = 0.023] (Fig. 4F; supplemental Fig. 4b, available at www.jneurosci.org as supplemental material). Finally, we investigated the consequences of treatment with baclofen alone, which also blocked GDPs (Tosetti et al., 2004) while activating GABAB-Rs. We found that incubation with baclofen (5 μm) had no significant effect on mGABAA-PSC frequency (0.390 ± 0.082 Hz, n = 11) when compared with control WT IHFs (Fig. 4F; supplemental Fig. 4b, available at www.jneurosci.org as supplemental material). Together, these data show that the deficit in GABAergic synaptic transmission observed in GABAB-R-deficient IHFs is not a consequence of abnormal synaptic activity but rather results from the lack of GABAB-R activation during ongoing synaptic activity.
GABAB-Rs and BDNF signaling interact to regulate the formation of functional GABAergic synapses
We next asked how activity-dependent activation of GABAB-Rs translates into functional development of GABAergic synapses. Considering that BDNF is a powerful modulator of GABAergic synapse development (Lessmann et al., 2003), the recent observation that GABAB-R activity increases BDNF expression in cultured hippocampal neurons (Ghorbel et al., 2005) prompted us to test the possible contribution of BDNF-tropomyosin-related kinase (TrkB) receptor signaling to this synaptic development. P1 WT IHFs were incubated for 12–16 h with k252a (200 nm), a membrane-permeable inhibitor of protein tyrosine kinase. In k252a-treated IHFs, the frequency of mGABAA-PSCs was reduced compared with control IHFs [0.270 ± 0.036 Hz (n = 15) vs 0.105 ± 0.028 Hz (n = 14), p = 0.002] (Fig. 5A). The other mGABAA-PSC parameters were unchanged (supplemental Fig. 4c, available at www.jneurosci.org as supplemental material). The deficit induced by k252a was not rescued by baclofen [0.116 ± 0.026 Hz k252a-treated IHFs (n = 10) and 0.105 ± 0.028 Hz (n = 14) in k252a/baclofen-treated IHFs] (Fig. 5A; supplemental Fig. 4c, available at www.jneurosci.org as supplemental material). Moreover, when incubated with k252a, the GABAB-R antagonist had no further effect on mGABAA-PSCs [0.088 ± 0.024 Hz (n = 11) in CGP55845/k252a-treated IHFs vs 0.105 ± 0.028 Hz (n = 14) in k252a-treated IHFs] (Fig. 5A; supplemental Fig. 4c, available at www.jneurosci.org as supplemental material). These data show that GABAB-Rs promote the functional maturation of GABAergic synapses by means of a cascade involving tyrosine kinase signaling.
Figure 5.

GABAB and BDNF-TrkB signaling interact to promote the formation of functional GABAergic synapses. A, Left, Summary plot of the mGABAA-PSC frequency in recordings from GABAB1-WT intact hippocampi incubated in vitro for 12–16 h in control conditions (ACSF) or with K252a alone (200 nm), K252a plus CGP55845 (5 μm), or K252a plus baclofen (baclo; 5 μm). B, Left, Summary plot of the mGABAA-PSC frequency in recordings from GABAB1-WT intact hippocampi incubated in vitro for 12–16 h in the presence of TrkC-IgG (1 μg/ml) alone, TrkC-IgG plus CGP55845 (CGP, 5 μm), TrkB-IgG (1 μg/ml) alone, or TrkB-IgG plus CGP55845 (5 μm). C, Left, Summary plot of the mGABAA-PSC frequency in recordings from BDNF-WT and BDNF-KO intact hippocampi incubated in vitro for 12–16 h in control conditions (ACSF) or with CGP55845 (5 μm). D, Left, Summary plot of the mGABAA-PSC frequency in recordings from GABAB1-WT intact hippocampi incubated in vitro for 12–16 h in control conditions (ACSF) or with BDNF (50 ng/ml), CGP55845 (CGP; 5 μm), or BDNF (50 ng/ml) plus CGP55845 (5 μm). The graphs on the right in A–D represent the logarithmic plot of the mGABAA-PSC frequency in recordings from the intact hippocampi incubated in the corresponding conditions. Each symbol represents the result of one single cell. The dashed line represents the mean value obtained from GABAB1-WT incubated in ACSF.
P1 WT IHFs were then incubated with the BDNF scavenger TrkB-IgG (1 μg/ml) or with the NT3 scavenger TrkC-IgG (1 μg/ml). The frequency of mGABAA-PSCs was significantly reduced in TrkB-IgG-treated IHFs (0.105 ± 0.033 Hz, n = 8) compared with TrkC-IgG-treated IHFs (0.481 ± 0.087 Hz, n = 14, p = 0.001) (Fig. 5B). The other mGABAA-PSC parameters were not affected (supplemental Fig. 4d, available at www.jneurosci.org as supplemental material). The GABAB-R antagonist had no further effect on mGABAA-PSC frequency when incubated with TrkB-IgG (0.117 ± 0.015 Hz, n = 12) (Fig. 5B; supplemental Fig. 4d, available at www.jneurosci.org as supplemental material) but significantly decreased the frequency of mGABAA-PSCs when incubated with TrkC-IgG (0.481 ± 0.087 Hz, n = 14, p = 0.006 compared TrkC-IgG-treated IHF) (Fig. 5B; supplemental Fig. 4d, available at www.jneurosci.org as supplemental material). These data show that endogenous GABA and BDNF, acting on GABAB-Rs and TrkB-Rs, respectively, interact to promote the functional maturation of hippocampal GABAergic synapses.
To confirm the requirement of GABAB-R and BDNF interaction for the functional maturation of GABAergic synapses, P1 IHFs obtained from BDNF-KO and WT littermates were incubated for 12–16 h in control ACSF or with CGP55845 (5 μm). Incubation with the GABAB-R antagonist had no effect on the frequency of mGABAA-PSCs recorded from BDNF-KO IHFs [0.074 ± 0.019 Hz in control IHF (n = 8) vs 0.082 ± 0.019 Hz in CGP55845-treated IHFs (n = 8)] but significantly decreased the frequency of mGABAA-PSCs recorded from BDNF-WT IHFs [0.248 ± 0.052 Hz in control IHF (n = 12) vs 0.108 ± 0.018 Hz in CGP55845-treated IHFs (n = 9), p = 0.03] (Fig. 5C; supplemental Fig. 4f, available at www.jneurosci.org as supplemental material). Moreover, the frequency of mGABAA-PSCs from IHFs incubated in control ACSF was significantly reduced in BDNF-KO when compared with their wild-type littermates (p = 0.01) (Fig. 5C), showing that BDNF is required for the functional maturation of hippocampal GABAergic synapses. Altogether, these data strengthen the conclusion that GABAB-Rs and BDNF interact to promote the maturation of GABAergic synapses in the developing mouse hippocampus.
To better determine the interplay between GABAB-Rs and BDNF-TrkB signaling, we attempted to rescue the deficit induced by CGP55845 with exogenous BDNF. P1 WT IHFs were incubated with CGP55845 (5 μm) alone or with CGP55845 (5 μm) and BDNF (50 ng/ml) for 12–16 h. BDNF rescued the deficit induced by CGP55845. The mGABAA-PSC frequency was 0.165 ± 0.042 Hz in CGP55845-treated IHFs (n = 21) and 0.331 ± 0.069 Hz in CGP55845 and BDNF-treated IHFs (n = 17, p = 0.014) (Fig. 5D; supplemental Fig. 4e, available at www.jneurosci.org as supplemental material). Incubation with BDNF (50 ng/ml) alone, however, had no significant effect on mGABAA-PSC frequency when compared with control WT IHFs [0.36 ± 0.088 Hz in control IHFs (n = 17) vs 0.407 ± 0.038 Hz in BDNF-treated IHFs (n = 14)] (Fig. 5D; supplemental Fig. 4e, available at www.jneurosci.org as supplemental material). With the observation that baclofen failed to rescue the deficit in GABAergic synaptic activity induced by k252a (Fig. 5A), these data show that GABAB-Rs promote the maturation of GABAergic synapses by controlling the amount of available extracellular BDNF.
Activation of GABAB-Rs triggers BDNF release
We next asked how GABAB-Rs control extracellular BDNF availability. GABAB-Rs could modulate the production and/or secretion of BDNF. To determine whether GABAB-Rs modulate BDNF production, we measured the level of BDNF in hippocampi of GABAB1-KO and GABAB1-WT by use of an ELISA and found no significant difference (110 ± 7 pg/ml total protein vs 109 ± 10 pg/ml total protein, respectively, n = 5 for both).
In an attempt to determine whether GABAB-Rs could modulate the secretion of BDNF, we sought to measure the amount of BDNF released from mouse hippocampi in vitro upon GABAB-R activation using ELISA. However, levels of BDNF in these samples were below the threshold for detection of the assay. We therefore examined whether activation of GABAB-Rs triggers BDNF release at the cellular level. With this aim, we transfected cultured hippocampal neurons with GFP-tagged BDNF and monitored dendritic BDNF-GFP secretion with time-lapse fluorescent imaging in living neurons. Using this approach, BDNF-GFP secretion was detectable as a decrease in intracellular GFP fluorescence intensity (Kuczewski et al., 2008b). To exclude indirect effects of synaptic activity, GABAergic and glutamatergic ionotropic receptor antagonists were present. Baclofen (10 μm) led to a significant decrease of dendritic BDNF-GFP fluorescence intensity [−2.6 ± 0.63% change 5 min after baclofen application, n = 62 regions of interest (ROIs) from 14 cells, p = 0.0008 compared with control period, p = 0.01 compared with nonstimulated neurons, n = 107 ROIs from 23 cells] (Fig. 6A–C). The decrease in fluorescence intensity induced by baclofen was prevented by CGP55845 (10 μm, −0.74 ± 0.53% change 5 min after baclofen application, n = 42 ROIs from 11 cells, p = 0.03 compared with baclofen alone) (Fig. 6D) and by nominally Ca2+-free extracellular solution (0.005 ± 0.005% change after 5 min of baclofen application, n = 31 ROIs from 7 cells, p = 0.0006 compared with baclofen alone) (Fig. 6E). The baclofen-induced secretion of BDNF-GFP was confirmed by surface-bound BDNF-GFP immunostaining (Kuczewski et al., 2008b). Bath-applied baclofen (10 μm, 10 min) increases GFP staining surrounding the BDNF-GFP-expressing cell (Fig. 6F). Quantification of surface-bound BDNF-GFP on transfected neurons shows that baclofen significantly increased BDNF-GFP release (n = 10 cells for both, 3 cultures, p = 0.04 compared with control) (Fig. 6G). This effect was prevented by CGP55845 (10 μm, n = 11 cells, 3 cultures) (Fig. 6G), which when applied alone had no effect on surface-bound BDNF-GFP in control conditions (n = 5 cells, 3 cultures) (Fig. 6G).
Figure 6.
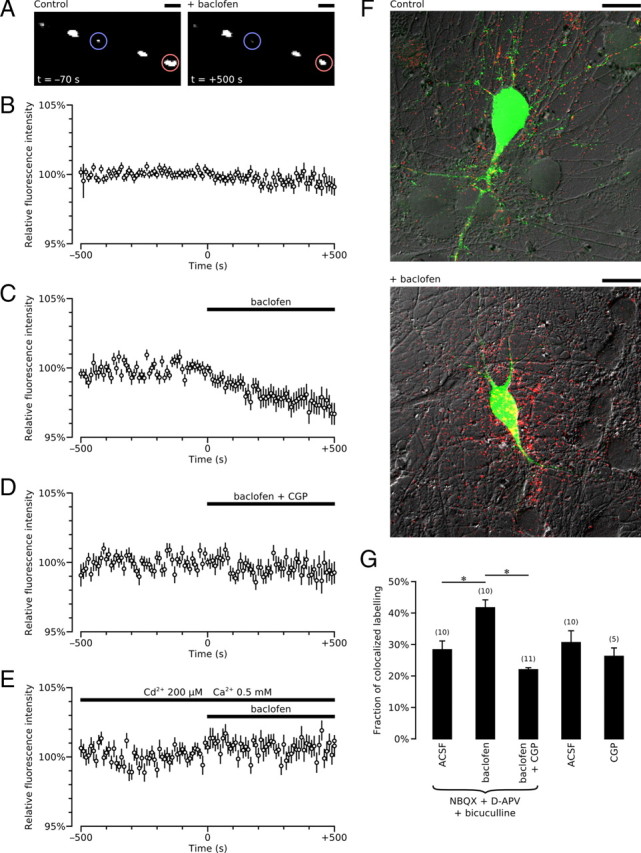
GABAB-R activation triggers BDNF-GFP secretion. A, Examples of dendritic BDNF-GFP granules are shown before and after baclofen application. Blue and red circles highlight examples of the BDNF-GFP clusters of fluorescence analyzed. The fluorescence was enhanced to near-saturation levels to make fluorescence variation visible. Scale bar, 5 μm. B–E, Average time course of dendritic fluorescence variation in control, nonstimulated neurons (B) (n = 107 ROIs from 23 cells), and in baclofen-stimulated neurons in the absence (C) (n = 62 ROIs from 14 cells) or presence (D) (10 μm, n = 42 ROIs from 11 cells) of CGP35845, or in nominally Ca2+-free external solution (E) (0.5 mm Ca2+, 200 μm Cd2+, n = 31 ROIs from 7 cells). All experiments were performed in the presence of bicuculline (20 μm), NBQX (10 μm), and D-APV (40 μm). F, Surface staining confirmed the BDNF-GFP secretion produced by baclofen. Overlapped images showing intracellular BDNF-GFP fluorescence (green) and secreted BDNF-GFP detected using anti-GFP antibody (red) under nonpermeabilized conditions in control [bicuculline (20 μm), NBQX (10 μm), and D-APV (40 μm)] and baclofen-treated sister cultures. Released BDNF-GFP that bound to the extracellular membrane of BDNF-GFP-expressing neurons appears as a yellow signal. Scale bar, 20 μm. G, Quantitative analysis of surface-bound BDNF-GFP on BDNF-GFP-expressing neurons (yellow signal/green signal) in control (n = 10 cells), baclofen-treated (n = 10 cells), and baclofen (10 μm) and CGP55845 (10 μm)-treated cultures (n = 11 cells) in the presence of bicuculline (20 μm), NBQX (10 μm), and D-APV (40 μm). Surface-bound BDNF-GFP on BDNF-GFP-expressing neurons was also quantified in nontreated (n = 10 cells) and CGP55845 (CGP; 10 μm)-treated cultures (n = 5 cells). Three different sister cultures were used in each condition.
These data show that GABAB-R activation can trigger secretion of BDNF-GFP from hippocampal neurons in culture. However, protein overexpression could have modified the release properties of BDNF. To determine whether GABAB-R activation can trigger secretion of endogenous BDNF, we used the phosphorylated form of the cAMP response element-binding protein CREB (pCREB) as a sensor of endogenous BDNF release (Kuczewski et al., 2008b). Once released, BDNF interacts with TrkB receptors to activate downstream signaling pathways. One of the most common is the ERK (extracellular signal-regulated kinase) pathway, which leads to the phosphorylation of CREB (Ghosh et al., 1994). We therefore tested whether baclofen can induce a BDNF-dependent phosphorylation of CREB. Hippocampal neuronal cultures were stimulated with baclofen (3 and 50 μm) for 10 min in the presence of NBQX (10 μm), D-APV (50 μm), and bicuculline (20 μm). To rule out a possible effect of baclofen treatment on CREB synthesis and to normalize the results obtained from different cultures, the pCREB/CREB ratio was quantified (Fig. 7A,B). At 3 μm, baclofen induced a significant increase of the pCREB/CREB ratio [30 ± 11% in control (n = 74 cells, 3 cultures) versus 43 ± 7% in baclofen-treated (n = 64 cells, 3 cultures) cultures, p = 0.001]. At 50 μm, baclofen induced a threefold increase of the pCREB/CREB ratio [from 27 ± 5% in control (n = 42 cells, 3 cultures) versus 77 ± 13% in baclofen-treated (n = 54 cells, 3 cultures) cultures, p = 0.00008] (Fig. 7B). This increase was prevented by CGP55845 (10 μm, n = 43 cells; 3 cultures, p = 0.0003 compared with baclofen) (Fig. 7B), showing that GABAB-R activation is required. No difference in the pCREB/CREB ratio was observed when the cultures were stimulated by baclofen in the presence of the BDNF scavenger TrkB-IgG (2 μg/ml, n = 21 cells, 3 cultures, p = 0.0002 compared with baclofen) (Fig. 7B). As expected, BDNF (20 ng/ml) triggered a phosphorylation of CREB and an increase in the pCREB/CREB ratio (n = 40 cells, 3 cultures, p = 0.0001 compared with control) (Fig. 7B). Thus, GABAB-R activation triggers secretion of endogenous BDNF.
Figure 7.

GABAB-R activation induces a BDNF-dependent phosphorylation of CREB in hippocampal neuronal cultures. A, Immunofluorescence signal of CREB (red, left panels) and phosphorylated CREB (pCREB, green, middle panels) in hippocampal neuronal cultures in control conditions (top panels) or treated with baclofen in the absence (middle panels) or presence (bottom panels) of TrkB-IgG. Merged images are shown on the right. Scale bar, 10 μm. B, Average pCREB/CREB ratio in the different conditions. For each condition, three different sister cultures were used, and quantification was based on 7–18 cells per culture. baclo, Baclofen; CGP, CGP55845.
Discussion
GABA, the main inhibitory transmitter in the adult vertebrate brain, has recently emerged as an important signal for neuronal development. Besides its classical role in regulating synaptic activity, GABA modulates nearly all key steps of network construction from neuronal migration to experience-dependent refinement of local connections (Ben-Ari et al., 2007). Most of these effects have been attributed to the depolarizing action of GABA, which leads to a postsynaptic rise in intracellular Ca2+ concentration in developing neurons via the activation of chloride-permeable GABAA-Rs (Ben-Ari et al., 2007). Here, we reveal a novel mechanism by which endogenous GABA selectively regulates the development of GABAergic synapses. We found that activation of metabotropic GABAB-Rs triggers secretion of BDNF and promotes the development of GABAergic synapses in the hippocampus of newborn mice. Moreover, we show that this process occurs during ongoing physiological patterns of activity.
Our study shows that GABAB-R signaling plays a role in self-regulating inhibitory synapse development. We have identified a selective deficit in miniature GABAergic synaptic activity in GABAB1-KO hippocampal pyramidal cells that can be reproduced in vitro in wild-type hippocampi in which GABAB-Rs were pharmacologically blocked. Miniature GABAergic activity was also reduced in wild-type hippocampi incubated with TTX, to block action potential-dependent synaptic activity and subsequent activation of GABAB-Rs. The TTX-induced deficit was rescued by the selective GABAB receptor agonist, baclofen, showing that GABAB-R activation is required for the functional maturation of GABAergic synapses. Although we cannot completely rule out functional presynaptic modifications, we provide morphological data suggesting that the reduced GABAergic activity in GABAB1-KO mice is at least in part due to a decrease in the number of perisomatic GABAergic terminals. This result is consistent with previous findings that synaptic activity (Chattopadhyaya et al., 2004) and endogenous GABA levels (Chattopadhyaya et al., 2007) regulate cortical basket cell axon branching and perisomatic synapse formation through the activation of GABAA and GABAB receptors. Since miniature activity has been proposed to originate from proximal GABAergic synapses (Soltesz et al., 1995), a decrease in the density of those synapses will significantly affect the frequency of mGABAA-PSCs in GABAB1-KO pyramidal cells.
The timing of disappearance of GDPs, whose generation critically depends on depolarizing action of GABA (Ben-Ari et al., 2007), was not affected between GABAB1-KO and WT mice. This result suggests that the depolarizing-to-hyperpolarizing developmental shift in GABAA receptor-mediated responses is not affected in GABAB1-KO mice. Accordingly, we found that bath-applied isoguvacine, a selective GABAA-R agonist, increased the spiking activity of CA3 pyramidal neurons during the first postnatal week of life in both GABAB1-KO and WT mice (unpublished observations). Several studies have shown that the depolarizing and excitatory action of GABAA receptors is an important signal for neuronal network development (Barbin et al., 1993; Manent et al., 2005; Cancedda et al., 2007; Wang and Kriegstein, 2008); this might explain the lack of structural alterations or modifications in the morphology of CA3 pyramidal neurons in GABAB1-KO mice.
We show that the mechanism by which GABAB-Rs promote the formation of GABAergic synapses in vivo likely involves BDNF secretion and subsequent activation of the TrkB signaling pathway. k252a or TrkB-IgG mimicked and occluded the deleterious effect of GABAB-R blockade, the GABAB-R antagonist had no effect on BDNF-KO mice, and GABAB-R activation triggered dendritic Ca2+-dependent secretion of BDNF in hippocampal cultures. The fact that BDNF is necessary for the maturation of GABAergic synapses has been clearly established, but the source and mechanism of its activity-dependent release have remained unexplored. So far, at least three distinct signals regulating dendritic BDNF secretion have been directly identified in neuronal cultures (Kuczewski et al., 2009): (1) tetanic stimulation of presynaptic glutamatergic fibers (Hartmann et al., 2001), (2) action potentials that propagate backwards into the dendrites (Kuczewski et al., 2008b), and (3) prolonged depolarization of the postsynaptic neuron (Magby et al., 2006). Our study provides a novel and unexpected mechanism by which synaptic activity can trigger a Ca2+-dependent dendritic release of BDNF.
Important questions remain about the cell type and the location of GABAB-Rs involved in vivo. Because GABAergic interneurons and glial cells do not produce neurotrophins themselves (Ernfors et al., 1990), BDNF is likely provided by CA3 pyramidal neurons (Brigadski et al., 2005) or the mossy fibers arising from dentate gyrus granular cells (Danzer and McNamara, 2004). GABAB-R activation can increase the intracellular Ca2+ concentration in neurons (Shen and Slaughter, 1999; Hirono et al., 2001; New et al., 2006). A recent study has shown that the phosphorylation of α-CaMKII, a critical step in BDNF secretion (Kolarow et al., 2007), is enhanced by GABAB-R activation in the developing rat hippocampus (Xu et al., 2008). Thus, postsynaptic Ca2+ elevation and α-CaMKII phosphorylation might underlie the GABAB-R-induced secretion of BDNF and development of GABAergic synapses that we found in the present study. Alternatively, GABAB-Rs can increase Ca2+ concentration in glial cells (Kang et al., 1998; Meier et al., 2008) and indirectly trigger neuronal secretion of BDNF (Elmariah et al., 2005). Further experiments will be required to address this point.
Regulated activity-dependent release of BDNF is crucial for many different aspects of GABAergic and glutamatergic synapse development (Lu et al., 2005). However, because BDNF diffusion (Horch and Katz, 2002) and Ca2+ rise are rather restricted events, spatial proximity is required between the signal that triggers the secretion of BDNF and the target. Synaptic activation of ionotropic glutamatergic receptors has been previously reported to trigger a localized dendritic release of BDNF (Hartmann et al., 2001) and a potentiation of GABAergic synaptic activity in the developing rat hippocampus (Kuczewski et al., 2008a). However, glutamatergic synapses are restricted to the dendrites. Thus, activation of somatic GABAB-Rs might trigger the local and specific secretion of BDNF needed for the development of perisomatic GABAergic synapses. Such specificity in the control of BDNF secretion might explain why perisomatic GABAergic synapses, but not glutamatergic or dendritic GABAergic synapses, are impaired in GABAB1-KO hippocampi. Thus multiple triggers of BDNF secretion might coexist in the hippocampus. Depending on the pattern of activity generated by the neuronal network, BDNF could exert a selective control on the development of different subpopulations of GABAergic synapses. The reason why GABAB-R-mediated trophic action is selective for perisomatic GABAergic synapses remains to be clarified. One likely possibility is that the effectors linking GABAB-R activation to BDNF secretion are exclusively localized at the perisomatic level.
GABAB-Rs are located at perisynaptic or extrasynaptic sites, and thus GABA spillover is needed to activate them. GABA spillover becomes substantial during high-frequency stimulation (Isaacson et al., 1993; Xu et al., 2008) or concomitant activation of several interneurons or when the GABA uptake is blocked (Scanziani, 2000). In the developing hippocampus, most of the ongoing synaptic activity is provided by a primitive network-driven pattern, termed GDPs, present both in vitro (Ben-Ari et al., 1989) and in vivo (Leinekugel et al., 2002). GABAergic interneurons fire synchronously during GDPs (Khazipov et al., 1997). GDPs therefore fulfil the criteria for GABAB-R activation (McLean et al., 1996). Accordingly, we found that the lack of GABAB-R function in GABAB1-KO neurons resulted in lengthening of GDPs. In contrast, when GDPs were blocked in GABAB1-WT neurons, the GABAB-R antagonist had no significant effect on spontaneous GABAergic activity. Thus, the activation of GABAB-Rs required the presence of GDPs. We therefore propose that GDPs play an instructive role in the development of the hippocampal GABAergic circuit, providing the critical amount of GABA needed to activate GABAB-Rs and trigger a subsequent secretion of BDNF. Accordingly, a deficit in GABAergic synaptic activity was observed when GDPs were blocked with TTX, but not when GDPs were blocked with the GABAB-R agonist baclofen. Thus, baclofen substitutes for GDPs to activate GABAB-Rs showing that the activation of these receptors is sufficient to support a proper development of at least perisomatic GABAergic synapses. This conclusion is strengthened by the observation that the TTX-induced deficit was rescued by baclofen. Since early spontaneous network-driven activity similar to GDPs and subsequent activation of GABAB-Rs appear to occur in virtually every developing circuit (Obrietan and Van den Pol, 1999; Catsicas and Mobbs, 2001), it is possible that this mechanism is important for synaptic maturation throughout the nervous system.
Footnotes
This work was supported by Inserm, Centre National de la Recherche Scientifique, and Agence Nationale pour la Recherche (ANR). H.F. was a recipient of a Ministère de la Recherche et de l'Education fellowship. N.K. was a recipient of Fondation pour la Recherche Médicale and ANR fellowships. We wish to thank Drs. I. Medina, R. Khazipov, and M. Phillips for critical reading of this manuscript, Dr. V. Lessmann for the generous gift of BDNF-GFP cDNA, and S. Corby and C. Pellegrino for technical help.
References
- Barbin G, Pollard H, Gaïarsa JL, Ben-Ari Y. Involvement of GABAA receptors in the outgrowth of cultured hippocampal neurons. Neurosci Lett. 1993;152:150–154. doi: 10.1016/0304-3940(93)90505-f. [DOI] [PubMed] [Google Scholar]
- Behar TN, Li YX, Tran HT, Ma W, Dunlap V, Scott C, Barker JL. GABA stimulates chemotaxis and chemokinesis of embryonic cortical neurons via calcium-dependent mechanisms. J Neurosci. 1996;16:1808–1818. doi: 10.1523/JNEUROSCI.16-05-01808.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar TN, Smith SV, Kennedy RT, McKenzie JM, Maric I, Barker JL. GABA(B) receptors mediate motility signals for migrating embryonic cortical cells. Cereb Cortex. 2001;11:744–753. doi: 10.1093/cercor/11.8.744. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaïarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol. 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Brigadski T, Hartmann M, Lessmann V. Differential vesicular targeting and time course of synaptic secretion of the mammalian neurotrophins. J Neurosci. 2005;25:7601–7614. doi: 10.1523/JNEUROSCI.1776-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerli T, Pellegrino C, Baer K, Lardi-Studler B, Chudotvorova I, Fritschy JM, Medina I, Fuhrer C. Efficient transfection of DNA or shRNA vectors into neurons using magnetofection. Nat Protoc. 2007;2:3090–3101. doi: 10.1038/nprot.2007.445. [DOI] [PubMed] [Google Scholar]
- Cancedda L, Fiumelli H, Chen K, Poo MM. Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J Neurosci. 2007;27:5224–5235. doi: 10.1523/JNEUROSCI.5169-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catsicas M, Mobbs P. GABAB receptors regulate chick retinal calcium waves. J Neurosci. 2001;21:897–910. doi: 10.1523/JNEUROSCI.21-03-00897.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ. Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci. 2004;24:9598–9611. doi: 10.1523/JNEUROSCI.1851-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Wu CZ, Knott G, Kuhlman S, Fu Y, Palmiter RD, Huang ZJ. GAD67-mediated GABA synthesis and signaling regulate inhibitory synaptic innervation in the visual cortex. Neuron. 2007;54:889–903. doi: 10.1016/j.neuron.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini E, Gaiarsa JL, Ben-Ari Y. GABA: an excitatory transmitter in early postnatal life. Trends Neurosci. 1991;14:515–519. doi: 10.1016/0166-2236(91)90003-d. [DOI] [PubMed] [Google Scholar]
- Chudotvorova I, Ivanov A, Rama S, Hübner CA, Pellegrino C, Ben-Ari Y, Medina I. Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J Physiol. 2005;566:671–679. doi: 10.1113/jphysiol.2005.089821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colin-Le Brun I, Ferrand N, Caillard O, Tosetti P, Ben-Ari Y, Gaïarsa JL. Spontaneous synaptic activity is required for the formation of functional GABAergic synapses in the developing rat hippocampus. J Physiol. 2004;559:129–139. doi: 10.1113/jphysiol.2004.065060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer SC, McNamara JO. Localization of brain-derived neurotrophic factor to distinct terminals of mossy fiber axons implies regulation of both excitation and feedforward inhibition of CA3 pyramidal cells. J Neurosci. 2004;24:11346–11355. doi: 10.1523/JNEUROSCI.3846-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmariah SB, Oh EJ, Hughes EG, Balice-Gordon RJ. Astrocytes regulate inhibitory synapse formation via Trk-mediated modulation of postsynaptic GABAA receptors. J Neurosci. 2005;25:3638–3650. doi: 10.1523/JNEUROSCI.3980-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernfors P, Wetmore C, Olson L, Persson H. Identification of cells in rat brain and peripheral tissues expressing mRNA for members of the nerve growth factor family. Neuron. 1990;5:511–526. doi: 10.1016/0896-6273(90)90090-3. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Meskenaite V, Weinmann O, Honer M, Benke D, Mohler H. GABAB-receptor splice variants GB1a and GB1b in rat brain: developmental regulation, cellular distribution and extrasynaptic localization. Eur J Neurosci. 1999;11:761–768. doi: 10.1046/j.1460-9568.1999.00481.x. [DOI] [PubMed] [Google Scholar]
- Ge S, Goh EL, Sailor KA, Kitabatake Y, Ming GL, Song H. GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature. 2006;439:589–593. doi: 10.1038/nature04404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghorbel MT, Becker KG, Henley JM. Profile of changes in gene expression in cultured hippocampal neurones evoked by the GABAB receptor agonist baclofen. Physiol Genomics. 2005;22:93–98. doi: 10.1152/physiolgenomics.00202.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J. 2001;20:5887–5897. doi: 10.1093/emboj/20.21.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirono M, Yoshioka T, Konishi S. GABA(B) receptor activation enhances mGluR-mediated responses at cerebellar excitatory synapses. Nat Neurosci. 2001;4:1207–1216. doi: 10.1038/nn764. [DOI] [PubMed] [Google Scholar]
- Horch HW, Katz LC. BDNF release from single cells elicits local dendritic growth in nearby neurons. Nat Neurosci. 2002;5:1177–1184. doi: 10.1038/nn927. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Solís JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Esclapez M, Medina I, Aggoun D, Lamsa K, Leinekugel X, Khazipov R, Ben-Ari Y. A novel in vitro preparation: the intact hippocampal formation. Neuron. 1997;19:743–749. doi: 10.1016/s0896-6273(00)80956-3. [DOI] [PubMed] [Google Scholar]
- Khazipov R, Leinekugel X, Khalilov I, Gaïarsa J-L, Ben-Ari Y. Synchronization of GABAergic interneuronal network in CA3 subfield of neonatal rat hippocampal slices. J Physiol. 1997;498:763–772. doi: 10.1113/jphysiol.1997.sp021900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolarow R, Brigadski T, Lessmann V. Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase II signaling and proceeds via delayed fusion pore opening. J Neurosci. 2007;27:10350–10364. doi: 10.1523/JNEUROSCI.0692-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczewski N, Langlois A, Fiorentino H, Bonnet S, Marissal T, Diabira D, Ferrand N, Porcher C, Gaiarsa JL. Spontaneous glutamatergic activity induces a BDNF-dependent potentiation of GABAergic synapses in the newborn rat hippocampus. J Physiol. 2008a;586:5119–5128. doi: 10.1113/jphysiol.2008.158550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczewski N, Porcher C, Ferrand N, Fiorentino H, Pellegrino C, Kolarow R, Lessmann V, Medina I, Gaiarsa JL. Backpropagating action potentials trigger dendritic release of BDNF during spontaneous network activity. J Neurosci. 2008b;28:7013–7023. doi: 10.1523/JNEUROSCI.1673-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczewski N, Porcher C, Lessmann V, Medina I, Gaiarsa JL. Activity-dependent dendritic release of BDNF and biological consequences. Mol Neurobiol. 2009;39:37–49. doi: 10.1007/s12035-009-8050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinekugel X, Khazipov R, Cannon R, Hirase H, Ben-Ari Y, Buzsáki G. Correlated burst of activity in the neonatal rat hippocampus in vivo. Science. 2002;296:2049–2052. doi: 10.1126/science.1071111. [DOI] [PubMed] [Google Scholar]
- Lessmann V, Gottmann K, Malcangio M. Neurotrophin secretion: current facts and future prospects. Prog Neurobiol. 2003;69:341–374. doi: 10.1016/s0301-0082(03)00019-4. [DOI] [PubMed] [Google Scholar]
- López-Bendito G, Shigemoto R, Kulik A, Paulsen O, Fairén A, Luján R. Expression and distribution of metabotropic GABA receptor subtypes GABABR1 and GABABR2 during rat neocortical development. Eur J Neurosci. 2002;15:1766–1778. doi: 10.1046/j.1460-9568.2002.02032.x. [DOI] [PubMed] [Google Scholar]
- López-Bendito G, Luján R, Shigemoto R, Ganter P, Paulsen O, Molnár Z. Blockade of GABA(B) receptors alters the tangential migration of cortical neurons. Cereb Cortex. 2003;13:932–942. doi: 10.1093/cercor/13.9.932. [DOI] [PubMed] [Google Scholar]
- López-Bendito G, Shigemoto R, Kulik A, Vida I, Fairén A, Luján R. Distribution of metabotropic GABA receptor subunits GABAB1a/b and GABAB2 in the rat hippocampus during prenatal and postnatal development. Hippocampus. 2004;14:836–848. doi: 10.1002/hipo.10221. [DOI] [PubMed] [Google Scholar]
- Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci. 2005;6:603–614. doi: 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- Magby JP, Bi C, Chen ZY, Lee FS, Plummer MR. Single-cell characterization of retrograde signaling by brain-derived neurotrophic factor. J Neurosci. 2006;26:13531–13536. doi: 10.1523/JNEUROSCI.4576-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manent JB, Represa A. Neurotransmitters and brain maturation: early paracrine actions of GABA and glutamate modulate neuronal migration. Neuroscientist. 2007;13:268–279. doi: 10.1177/1073858406298918. [DOI] [PubMed] [Google Scholar]
- Manent JB, Demarque M, Jorquera I, Pellegrino C, Ben-Ari Y, Aniksztejn L, Represa A. A noncanonical release of GABA and glutamate modulates neuronal migration. J Neurosci. 2005;25:4755–4765. doi: 10.1523/JNEUROSCI.0553-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean HA, Caillard O, Khazipov R, Ben-Ari Y, Gaiarsa JL. Spontaneous release of GABA activates GABAB receptors and controls network activity in the neonatal rat hippocampus. J Neurophysiol. 1996;76:1036–1046. doi: 10.1152/jn.1996.76.2.1036. [DOI] [PubMed] [Google Scholar]
- Meier SD, Kafitz KW, Rose CR. Developmental profile and mechanisms of GABA-induced calcium signaling in hippocampal astrocytes. Glia. 2008;56:1127–1137. doi: 10.1002/glia.20684. [DOI] [PubMed] [Google Scholar]
- New DC, An H, Ip NY, Wong YH. GABAB heterodimeric receptors promote Ca2+ influx via store-operated channels in rat cortical neurons and transfected Chinese hamster ovary cells. Neuroscience. 2006;137:1347–1358. doi: 10.1016/j.neuroscience.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Van den Pol AN. GABAB receptor-mediated regulation of glutamate-activated calcium transients in hypothalamic and cortical neuron development. J Neurophysiol. 1999;81:94–102. doi: 10.1152/jn.1999.82.1.94. [DOI] [PubMed] [Google Scholar]
- Prosser HM, Gill CH, Hirst WD, Grau E, Robbins M, Calver A, Soffin EM, Farmer CE, Lanneau C, Gray J, Schenck E, Warmerdam BS, Clapham C, Reavill C, Rogers DC, Stean T, Upton N, Humphreys K, Randall A, Geppert M, Davies CH, Pangalos MN. Epileptogenesis and enhanced prepulse inhibition in GABA(B1)-deficient mice. Mol Cell Neurosci. 2001;17:1059–1070. doi: 10.1006/mcne.2001.0995. [DOI] [PubMed] [Google Scholar]
- Reynolds A, Brustein E, Liao M, Mercado A, Babilonia E, Mount DB, Drapeau P. Neurogenic role of the depolarizing chloride gradient revealed by global overexpression of KCC2 from the onset of development. J Neurosci. 2008;28:1588–1597. doi: 10.1523/JNEUROSCI.3791-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M. GABA spillover activates postsynaptic GABA(B) receptors to control rhythmic hippocampal activity. Neuron. 2000;25:673–681. doi: 10.1016/s0896-6273(00)81069-7. [DOI] [PubMed] [Google Scholar]
- Shen W, Slaughter MM. Metabotropic GABA receptors facilitate L-type and inhibit N-type calcium channels in single salamander retinal neurons. J Physiol. 1999;516:711–718. doi: 10.1111/j.1469-7793.1999.0711u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltesz I, Smetters DK, Mody I. Tonic inhibition originates from synapses close to the soma. Neuron. 1995;14:1273–1283. doi: 10.1016/0896-6273(95)90274-0. [DOI] [PubMed] [Google Scholar]
- Tosetti P, Bakels R, Colin-Le Brun I, Ferrand N, Gaiarsa JL, Caillard O. Acute desensitization of presynaptic GABAB-mediated inhibition and induction of epileptiform discharges in the neonatal rat hippocampus. Eur J Neurosci. 2004;19:3227–3234. doi: 10.1111/j.0953-816X.2004.03413.x. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Silver RA, Cull-Candy SG. Estimated conductance of glutamate receptor channels activated during EPSCs at the cerebellar mossy fiber-granule cell synapse. Neuron. 1993;11:279–289. doi: 10.1016/0896-6273(93)90184-s. [DOI] [PubMed] [Google Scholar]
- Wang DD, Kriegstein AR. GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J Neurosci. 2008;28:5547–5558. doi: 10.1523/JNEUROSCI.5599-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Zhao MX, Poo MM, Zhang XH. GABA(B) receptor activation mediates frequency-dependent plasticity of developing GABAergic synapses. Nat Neurosci. 2008;11:1410–1418. doi: 10.1038/nn.2215. [DOI] [PubMed] [Google Scholar]
- Zhang LI, Poo MM. Electrical activity and development of neural circuits. Nat Neurosci. 2001;4(Suppl):1207–1214. doi: 10.1038/nn753. [DOI] [PubMed] [Google Scholar]