

Abstract
Hippocampal interneurons synchronize the activity of large neuronal ensembles during memory consolidation. Although the latter process is manifested as increases in synaptic efficacy which require new protein synthesis in pyramidal neurons, it is unknown whether such enduring plasticity occurs in interneurons. Here, we uncover a long-term potentiation (LTP) of transmission at individual interneuron excitatory synapses which persists for at least 24 h, after repetitive activation of type-1 metabotropic glutamate receptors [mGluR1-mediated chemical late LTP (cL-LTPmGluR1)]. cL-LTPmGluR1 involves presynaptic and postsynaptic expression mechanisms and requires both transcription and translation via phosphoinositide 3-kinase/mammalian target of rapamycin and MAP kinase kinase-extracellular signal-regulated protein kinase signaling pathways. Moreover, cL-LTPmGluR1 involves translational control at the level of initiation as it is prevented by hippuristanol, an inhibitor of eIF4A, and facilitated in mice lacking the cap-dependent translational repressor, 4E-BP. Our results reveal novel mechanisms of long-term synaptic plasticity that are transcription and translation-dependent in inhibitory interneurons, indicating that persistent synaptic modifications in interneuron circuits may contribute to hippocampal-dependent cognitive processes.
Introduction
Long-term memory consolidation requires both gene expression and the synthesis of new proteins (Kandel, 2001). In the hippocampus, the strengthening or weakening of synaptic connectivity, long-term potentiation (LTP) and depression (LTD), respectively, are considered to be cellular models for memory storage (Bliss and Collingridge, 1993; Malenka and Bear, 2004). The late phase of LTP (L-LTP) is distinguished from its early phase (E-LTP) by its dependence on new protein synthesis (Kandel, 2001; Kelleher et al., 2004a). L-LTP is generally induced by a variety of repetitive training or stimulation protocols both in vivo, lasting from days to weeks (Bliss and Gardner-Medwin, 1973; Barnes and McNaughton, 1985), and in vitro, lasting up to several hours, respectively (Frey et al., 1988; Nguyen et al., 1994; Kelleher et al., 2004b). The newly synthesized proteins in response to L-LTP presumably serve to strengthen connectivity at the level of single synapses within hippocampal circuits, allowing the capture of memories (Kandel, 2001).
Local hippocampal circuits consist of networks of principal excitatory projection cells and local inhibitory interneurons. Although long-term synaptic modifications of hippocampal interneuronal circuits occur in pathological conditions such as seizure (Ben-Ari, 2006), the study of synaptic plasticity in interneurons has mainly focused on early forms of plasticity. Indeed, excitatory synapses onto interneurons have been shown to undergo LTP and LTD (Kullmann and Lamsa, 2007). Moreover, inhibitory synapses from interneurons onto pyramidal cells also display LTP and LTD (Nugent and Kauer, 2008). Such bidirectional modifications in excitation or inhibition have been implicated in the regulation of temporal fidelity of firing and synchronization of extensive hippocampal circuits (Traub et al., 2004; Kullmann and Lamsa, 2007; Pelletier and Lacaille, 2008). However, whether such changes in synaptic efficacy in interneuron circuits persist beyond several hours remains unknown. In the present study, we used an mGluR1-mediated chemical induction protocol in slice cultures and whole-cell voltage clamp recordings to uncover a persistent long-term potentiation of excitatory synaptic transmission in individual CA1 oriens-alveus interneurons (OA-INs) which lasts for at least 24 h. Using pharmacological and genetic approaches, we show that this long-term synaptic plasticity is both transcription and translation-dependent. In addition, we provide genetic and pharmacological evidence that the phosphatidylinositol 3-kinase (PI3K), extracellular signal-regulated protein kinase (ERK), and the mammalian target of rapamycin (mTOR)/4E-BPs signaling pathways regulate this process.
Materials and Methods
Hippocampal slice cultures.
Experiments were performed in accordance with the Canadian animal care guidelines at Université de Montréal. Unless otherwise stated, hippocampal slice cultures were obtained from Sprague Dawley rats (7- to 10-d-old), as described previously (Muller et al., 2001; Bourdeau et al., 2007). In brief, the brain was removed and dissected in HBSS (Invitrogen)-based medium. Cortico-hippocampal slices (400 μm thick) were then obtained using a McIlwain tissue chopper (The Mickle Laboratory Engineering Co. Ltd). After dissection, slices were placed on Millicell culture plate inserts (Millipore) and allowed to recover in OptiMEM (Invitrogen) kept at 37°C in a humidified atmosphere (95% air, 5% CO2) for 24 h. They were then maintained in a Neurobasal medium (Invitrogen) supplemented with B27 and Glutamax I (Invitrogen) for 3–7 d. In electrophysiology experiments with transgenic mice (see Fig. 6), slice cultures were obtained in the same manner as described above but from C57BL/6 wild-type (Charles River) and eIF4E-BP1/2−/− (Le Bacquer et al., 2007) 4- to 5-d-old mice. In immunofluorescence experiments with S6 phosphorylation assays, slices were obtained from heterozygous GAD67-GFP mice (see below).
Figure 6.

Facilitation of cL-LTPmGluR1 in 4E-BP1/2 knock-out mice. A, Example of EPSCs obtained from OA-INs in slices of wild-type (left) and 4E-BP1/2−/− mice (right) showing that repeated, but not single, mGluR1 stimulation increased EPSCs in wild-type mice, but single and repeated mGluR1 stimulation increased EPSCs in 4E-BP1/2−/− mice. B, Summary bar graphs of effect of single versus repeated mGluR1 stimulation on EPSC potency and paired-pulse ratio. C, Representative mEPSCs (top) and cumulative distribution plots for all cells (bottom) from slices of wild-type (left) and 4E-BP1/2−/− mice (right) showing that repeated, but not single, mGluR1 stimulation increased mEPSC amplitude and frequency in wild-type mice, and both single and repeated mGluR1 stimulation increased mEPSC amplitude and frequency in 4E-BP1/2−/− mice. D, Summary bar graphs showing effects of single and repeated mGluR1 stimulation on mEPSC amplitude and frequency in wild-type (open) and 4E-BP1/2−/− (filled) mice. Values expressed as mean ± SEM; * corresponds to p < 0.05. Number above bar indicates cells analyzed.
Induction of cL-LTPmGluR1.
The chemical induction protocol consisted of three applications (10 min duration each at 30 min intervals) of the mGluR1/5 agonist (S)-3,5-dihydroxyphenylglycine (DHPG, 5 μm; Tocris Biosciences) in the presence of the mGluR5 antagonist 2-methyl-6-(phenylethynyl)-pyridine (MPEP, 25 μm; Ascent Scientific). For concomitant application of inhibitors, drugs were applied from 30 min before to 30 min after DHPG/MPEP treatment. For delayed application of inhibitors, drugs were applied for the same period starting at 3 h after DHPG/MPEP treatment. LY367,385, anisomycin, actinomycin D, rapamycin, wortmannin, U0126, and U0124 were purchased from Calbiochem. Hippuristanol was provided by J. Pelletier (McGill University, Montreal, QC, Canada) (Bordeleau et al., 2006). After treatments, slices were allowed to recover for 24 h before recordings. Experimenters were blind to all treatment groups and mice genotype.
Whole-cell recordings.
Slices were transferred in oxygenated artificial CSF (ACSF) at room temperature containing (in mm): 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 4 MgSO4, 4 CaCl2, 26 NaHCO3, and 10 d-(+)-glucose (pH 7.35–7.45; 295–305 mOsmol). Slices were allowed to recuperate for at least 45 min before electrophysiological recordings. Thereafter, the CA3 and CA1 were disconnected by a surgical cut and slices were transferred in a submerged recording chamber perfused (2–3 ml/min) with oxygenated ACSF at 31 ± 0.5°C. OA-INs were visualized and identified based on their soma shape and position in stratum oriens using an upright microscope (Nikon Eclipse E600FN) equipped with a long-range water-immersion objective (x40, Nomarski Optics) and an infrared camera (70 Series; Dage-MTI). Whole-cell recordings were obtained from OA-INs using borosilicate pipettes (3–6 MΩ). Whole cell recording solution contained (in mm): 135 CsMeSO3, 10 phosphocreatine, 5 NaCl, 10 HEPES, 1 MgCl2, 0.1 spermine, 2 QX-314, 2 ATP–Tris, 0.4 GTP–Tris and 0.1% biocytin (pH 7.2–7.3; 280–290 mOsmol). Recordings were performed in voltage-clamp mode using a Multiclamp 700A amplifier (Molecular Devices). The signal was low-pass-filtered at 2 kHz, digitized at 20 kHz and stored on a PC. Acquisition and off-line analyses were performed using 1322A Digidata acquisition board, and pClamp 9.0–9.2 (Molecular Devices) and Origin 7.5 (OriginLab) software. Cells were maintained at a holding potential of −60 mV and series resistance was monitored regularly. Data were discarded if the holding current was unstable or if series resistance varied >25% of initial value.
Evoked and miniature EPSCs.
EPSCs mediated by AMPA/kainate receptors were recorded in the presence of dl-2-amino-5-phosphonovaleric acid (50 μm; Tocris Biosciences) and GABAzine (5 μm; Tocris Biosciences) to block NMDA and GABAA receptors respectively. Pairs of EPSCs were evoked at 0.5 Hz by constant current pulses (50 μs duration, 50 ms interpulse interval) using a bipolar θ-glass electrode filled with ACSF and positioned in the stratum oriens. Putative single-fiber EPSCs were evoked using minimal stimulation as previously described (Perez et al., 2001; Lapointe et al., 2004). Briefly, at the beginning of each experiment, the stimulus strength was adjusted to a value that yielded ∼ 50% successes (range 40 – 60%). Amplitude of average EPSC (including failures), failure rate (number of failures as percentage of total number of stimulations) and EPSC potency (amplitude of EPSCs excluding failures) were calculated from 5-min bins over a 10–20 min period. Because the failure rate was an adjusted parameter, we used EPSC potency to characterize amplitude changes in evoked transmission. In a subset of experiments on Hebbian LTP induced by a pairing protocol involving electrical synaptic stimulation, we used average EPSC amplitude and failure rate in addition to potency, comparing them to the baseline period before induction (described in supplemental material, available at www.jneurosci.org). Miniature EPSCs (mEPSCs) were recorded after further addition of tetrodotoxin (1 μm, Alomone Labs). For analysis, mEPSCs were detected with a running template (average of 20 events) which had a well defined baseline, using pClamp 9 software. Detection threshold was set at 3 pA and 200–250 events were sampled per neuron over a 15–25 min period.
GAD67-GFP mice.
Heterozygous GAD67-GFP mice were genotyped as described originally (Tamamaki et al., 2003). Briefly, DNA from tails was extracted with DNeasy tissue kit (Qiagen) according to manufacturer's instructions. DNA was amplified 30 cycles (68°C-3 min) by PCR on a ICycler (Bio-Rad Laboratories) using forward primer 5′-GGCACAGCTCTCCCTTCTGTTTGC-3′ and reverse primer 5′-CTGCTTGTCGGCCATGATATAGACG-3′ (Alpha DNA). PCR products were visualized on 1.5% agarose gel containing 2 μg/ml ethidium bromide.
Immunofluorescence.
GAD67-GFP mice (17- to 22-d-old) were deeply anesthetized with sodium pentobarbital (MTC Pharmaceuticals, Cambridge, Ontario, Canada) and perfused transcardially with ice-cold ACSF containing (in mm): 110 choline-chloride, 2.5 KCl, 7 MgCl2, 26 NaHCO3, 7 dextrose, 1.3 ascorbic acid and 0.5 CaCl2 and saturated with 95% O2 and 5% CO2. Coronal hippocampal slices (300 μm thick) were obtained using a vibratome (Leica VT 10005) and transferred to normal oxygenated ACSF containing (in mm): 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, 26 NaHCO3, 10 d-(+)-glucose, 1.3 ascorbic acid maintained at 31−33°C. After a 1 h recovery period, slices were treated as described. Slices were fixed immediately after treatment and resectioned as described (Perez et al., 2001). Sections were permeabilized with 0.3% Triton X-100 in PBS (15 min) and unspecific binding was blocked with 10% normal goat serum in 0.1% Triton X-100/PBS (1 h). Rabbit polyclonal phospho-S6 ribosomal protein (S235/S236) antibody (1:200; Cell Signaling Technology, catalog # 2211) was incubated overnight at 4°C. Sections were subsequently incubated at room temperature with Texas-Red-conjugated goat anti-rabbit IgG (1:400; 90 min; Jackson Immunoresearch Laboratories). Images were acquired using a confocal microscope (LSM510; Zeiss) at excitation wavelengths 488 and 543 nm. Images from different treatment/groups were acquired using the exact same parameters. Cell fluorescence was quantified using ImageJ software (National Institutes of Health; freely available) by comparing integrated density in cells corrected for background. In preliminary experiments, we found that basal levels of phospho-S6 were highly variable in interneurons in slice cultures but more constant in acute slices. Therefore, we performed the phospho-S6 immunofluorescence experiments on acute slices.
Statistical analysis.
Unless otherwise stated, between groups comparisons were performed using ANOVA followed by post hoc Tukey's test. Kolmogorov–Smirnov test was used to compare distributions of mEPSC parameters between groups (200 events per neuron and 6 neurons per treatment/group, except for wild-type and 4E-BP1/2−/− mice with 250 events per neuron and 5 neurons per treatment/group), as well as immunofluorescence intensity data. Values were expressed as mean ± SEM and a P value <0.05 was considered significant.
Results
Repeated mGluR1 stimulation induces a persistent long-term potentiation at OA-IN excitatory synapses
At OA-IN excitatory synapses, an early form of Hebbian LTP lasting 30 min is induced by pairing theta-burst stimulation with postsynaptic depolarization (Perez et al., 2001; Lapointe et al., 2004) or chemically by a single application of mGluR1 agonist (Le Vasseur et al., 2008). Because repeated electrical stimulation at hippocampal Schaffer collateral-pyramidal cell synapses elicits long lasting changes in synaptic strength (Frey et al., 1988; Nguyen et al., 1994) and mGluR1 is highly expressed in OA-INs in vivo (Baude et al., 1993; Lapointe et al., 2004) and slice cultures (Fig. S1A, available at www.jneurosci.org as supplemental material), we predicted that repeated agonist stimulation of mGluR1 would elicit a persistent LTP in OA-INs in hippocampal slice cultures. To explore this possibility, we used a protocol which consisted of three applications (10 min duration, 30 min interval) of the mGluR1/5 agonist (S)-3,5-dihydroxyphenylglycine (DHPG) in the presence of the mGluR5 antagonist 2-methyl-6-(phenylethynyl)pyridine (MPEP) (Fig. 1A) in slice cultures. After a 24 h recovery period, EPSCs evoked by minimal stimulation and subsequently spontaneous mEPSCs (in 1 μm TTX) (Fig. 1) were recorded in individual identified OA-INs (Fig. S1B, available at www.jneurosci.org as supplemental material). An increase in both evoked and spontaneous transmission was observed at 24 h postinduction in slices treated with repetitive activation of mGluR1 (3×DHPG/MPEP) relative to sham-treated (Fig. 1). For EPSCs evoked by minimal stimulation, potency was increased by 99% and this was accompanied by a 40% decrease in paired-pulse ratio (PPR) (Fig. 1B–D). Thus, both presynaptic and postsynaptic mechanisms may underlie the persistent changes in evoked transmission. Similarly, in OA-INs from slices treated with repeated mGluR1 stimulation, spontaneous mEPSCs increased in both amplitude (157%) and frequency (113%) relative to values from sham-treated slices (Fig. 1E,F), indicating long-term presynaptic and postsynaptic changes in synaptic efficacy. Importantly, there was no difference in input resistance between 3× DHPG/MPEP- and sham-treated groups (256 ± 19 MΩ vs 251 ± 16 MΩ, respectively), ruling out nonspecific membrane effects of agonist stimulation. Unlike repeated stimulations of mGluR1, a single agonist application did not elicit persistent potentiation (Fig. 1B–F). The activation of mGluR1 was necessary for induction of persistent changes since repeated agonist stimulation (3 × DHPG/MPEP) in the presence of the mGluR1 antagonist LY367,385 (100 μm) blocked the potentiation of evoked EPSCs and spontaneous mEPSCs at 24 h poststimulation (Fig. 1C,D,F). Taken together, these data demonstrate that repeated mGluR1 stimulation induces a persistent long-term potentiation at OA-IN excitatory synapses (mGluR1-mediated chemical late LTP, cL-LTPmGluR1).
Figure 1.
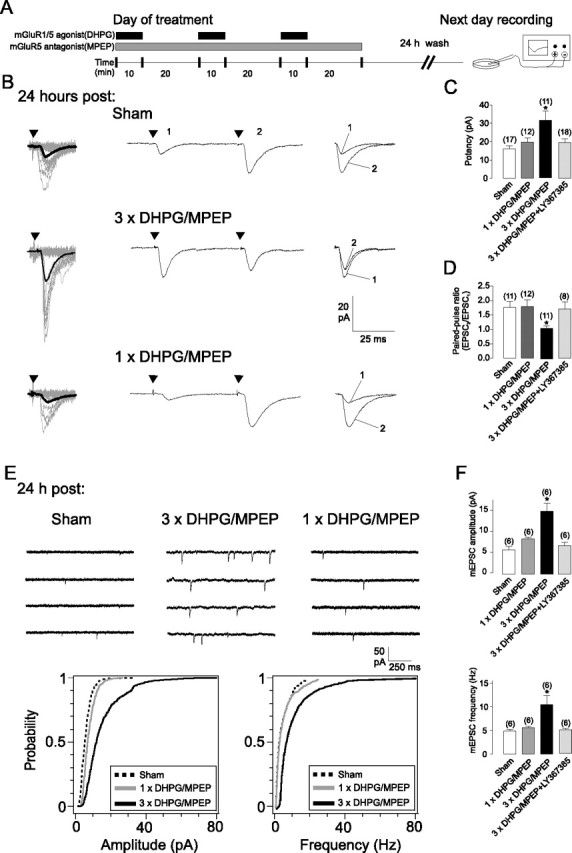
Persistent long-term potentiation of synaptic currents in hippocampal OA-INs at 24 h after repetitive mGluR1 activation (cL-LTPmGluR1). A, Schematics of induction and recording protocol. Cultured hippocampal slices were treated with repetitive (3×) or single (1×) application of mGluR1/5 agonist (DHPG, 5 μm, black bars) in the presence of an mGluR5 antagonist (MPEP, 25 μm, gray bar). On the next day, after a 24 h wash-out, whole cell recordings were obtained from visually identified CA1 OA-INs in agonist- or sham-treated slices. B, Representative EPSCs evoked by minimal stimulation at 24 h after sham-treatment (top), repetitive (3×; middle) and single (1×; bottom) mGluR1 agonist-stimulation, showing larger responses after repetitive treatment. Left, Superimposed 20 successive events (EPSCs + failures; gray) with average EPSC (including failures; solid black line) of 100 events. Middle, Average of EPSC pairs (100 events) evoked by paired-pulse stimulation (50 ms interstimulus interval), showing loss of paired-pulse facilitation after repetitive treatment. Right, Superimposed first and second EPSCs of average pair. Black triangles indicate time of stimulation. C, D, Summary bar graphs of effects on EPSC potency and paired-pulse ratio for all cells, showing potentiation of EPSC potency and reduced paired-pulse ratio 24 h after repetitive mGluR1 activation, relative to sham-treated, but no effect after single agonist application, or repetitive activation in the presence of the mGluR1 antagonist (LY367,385). E, Representative examples of spontaneous mEPSCs (in 1 μm TTX) at 24 h after treatment. Top, Continuous records of mEPSCs after sham treatment (right), repeated (middle) or single (right) mGluR1 agonist application, showing more frequent and larger mEPSCs after repeated treatment. Bottom, Cumulative distribution plots for all cells illustrating increase in mEPSC amplitude and frequency after repetitive mGluR1 stimulation. F, Summary bar graphs for all cells, showing increase in mean mEPSC amplitude and frequency 24 h after repetitive mGluR1 activation, relative to sham-treated, but no effect after single agonist application or in presence of LY367,385. Values are expressed as mean ± SEM and * corresponds to p < 0.05. Number of cells are indicated above bar.
In CA1 pyramidal neurons, the early phase of LTP typically decays and is converted to the late phase within a 1–3 h period (Kandel, 2001; Kelleher et al., 2004a). Because OA-INs display an early phase chemical LTP in response to a single stimulation of mGluR1 (Le Vasseur et al., 2008), we examined the magnitude of cL-LTPmGluR1 at earlier time points (1–3 h) after repeated mGluR1 stimulation. We found a 101% increase in potency and a 33% decrease in PPR of evoked EPSCs at 1–3 h after induction (Fig. S2, available at www.jneurosci.org as supplemental material). Although the magnitude of changes in evoked EPSC parameters tended to be larger at 1 h, they were not significantly different from those at 2–3, or 24 h postinduction (Fig. S2C, available at www.jneurosci.org as supplemental material). Hence, the conversion of early to late phase during cL-LTPmGluR1 occurs without apparent changes in synaptic properties.
We next examined whether cL-LTPmGluR1 involves similar mechanisms as LTP induced by a Hebbian protocol of pairing theta-burst stimulation of afferents (TBS) and postsynaptic depolarization at OA-IN synapses (Perez et al., 2001). If so, we should expect occlusion of Hebbian LTP by prior establishment of cL-LTPmGluR1. Effectively, we found that in OA-INs from sham-treated slices, pairing TBS and postsynaptic depolarization induced LTP, which was reflected as increases (relative to baseline) in mean EPSC amplitude (189%) and potency (206%), and a decrease (50%) in failure rate at 25–30 min postpairing. However, in OA-INs from slices treated 24 h previously with repeated mGluR1 stimulation, LTP was not induced by the Hebbian pairing protocol (Fig. S3, available at www.jneurosci.org as supplemental material). Occlusion of Hebbian LTP by cL-LTPmGluR1 suggests that both phenomena involve similar mechanisms. Thus, long-term plasticity induced by repeated mGluR1 stimulation is closely related to LTP elicited by physiological stimulation of afferents.
cL-LTPmGluR1 is transcription and translation dependent
In many forms of persistent long-term synaptic plasticity, gene expression is required (Nguyen et al., 1994; Martin et al., 1997). To determine whether cL-LTPmGluR1 requires transcription, we used the transcription inhibitor, actinomycin D (actinoD). Treatment of slices with actinoD during stimulation prevented the increase in evoked EPSCs and mEPSCs induced by repeated mGluR1 stimulation (Fig. 2), but not when applied 3 h after repeated mGluR1 stimulation (200% increase in potency, 29% decrease in PPR, 130% increase in mEPSC amplitude and 146% increase in mEPSC frequency relative to sham) (Fig. 2B,C,E,F). These data indicate that the critical time window for transcription in cL-LTPmGluR1 occurs during or shortly after (<3 h) induction. The effect of actinoD was not attributable to nonspecific inhibition of transcription since evoked and miniature synaptic activity in OA-INs were unaffected in slices treated with actinoD alone (Fig. 2) as in sham treated slices (Fig. S4, available at www.jneurosci.org as supplemental material).
Figure 2.
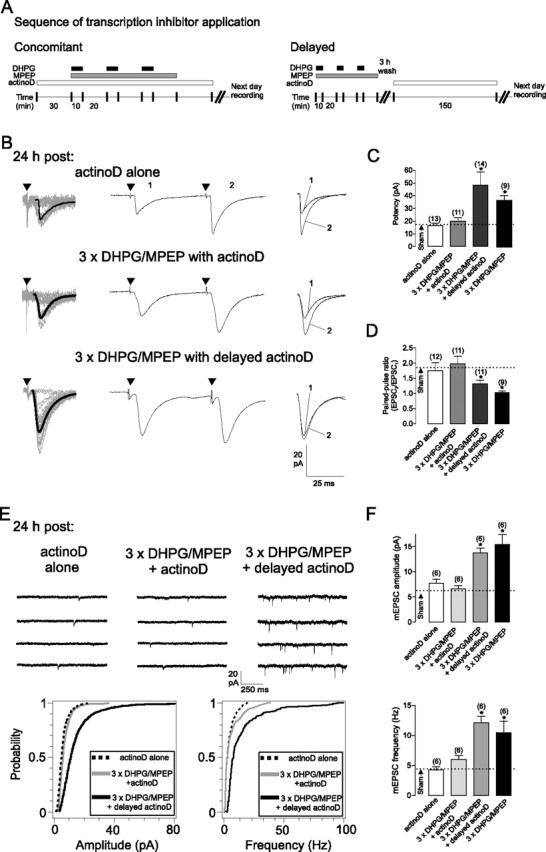
cL-LTPmGluR1 is dependent on transcription. A, Treatment protocols for transcription inhibitor actinomycin D. For concomitant application (left), slices were treated with actinomycin D (actinoD) from 30 min before to 30 min after repetitive mGluR1 stimulation (3× DHPG/MPEP). For delayed application (right), actinoD was applied for a similar period (150 min) starting 3 h after repeated mGluR1 stimulation. Slices were washed and recordings were made on the following day. B, Concomitant (middle), but not delayed (bottom), inhibition of transcription by actinoD prevented increase in EPSC amplitude and decrease in paired-pulse ratio induced by repetitive mGluR1 activation. Treatment with actinoD alone (top) did not affect EPSCs. C, D, Summary of effects of inhibition of transcription by actinoD on changes in EPSC potency and paired-pulse ratio induced by repeated mGluR1 stimulation. Dotted line corresponds to sham-treatment values (see also Fig. S4, available at www.jneurosci.org as supplemental material). E, Concomitant, but not delayed, inhibition of transcription by actinoD prevented increases in mEPSC amplitude and frequency induced by repeated mGluR1 stimulation. Top, Representative mEPSCs traces at 24 h after treatment. Bottom,: Cumulative distribution plots for mEPSC amplitude and frequency for all cells. F, Summary of actinoD treatment effects on mEPSCs. Values expressed as mean ± SEM; * corresponds to p < 0.05. Number of cells is indicated above bars.
De novo protein synthesis is required for L-LTP at CA1 Schaffer collateral synapses (Frey et al., 1988; Kelleher et al., 2004b). Moreover, mGluR1/5-induced late LTD (L-LTD) at hippocampal synapses is translation-dependent but transcription-independent (Snyder et al., 2001). Therefore, we examined whether cL-LTPmGluR1 in OA-INs required translation using the protein synthesis inhibitor anisomycin (aniso). Application of aniso prevented the increases in evoked EPSCs and mEPSCs induced by repeated mGluR1 stimulation (Fig. 3). The effects of aniso were not attributable to nonspecific protein synthesis inhibition since treatment with aniso alone was without effect on evoked and spontaneous transmission (Fig. 3) and not different from sham treatment (Fig. S4, available at www.jneurosci.org as supplemental material). As was the case with actinoD, a delayed application of aniso starting at 3 h after repeated mGluR1 stimulation did not prevent cL-LTPmGluR1 of evoked EPSCs (82% increase in potency and 31% decrease in PPR relative to sham) and mEPSCs (168% increase in mEPSC amplitude and 197% increase in mEPSC frequency relative to sham) (Fig. 3). In addition, we found that application of hippuristanol, a recently identified inhibitor of the eukaryotic translation initiation factor 4A (eIF4A) (Bordeleau et al., 2006), prevented cL-LTPmGluR1 expression (Fig. 3B,C,E). Thus, persistent long-term potentiation induced by repeated mGluR1 stimulation requires de novo protein synthesis which takes place during or immediately subsequent to induction.
Figure 3.
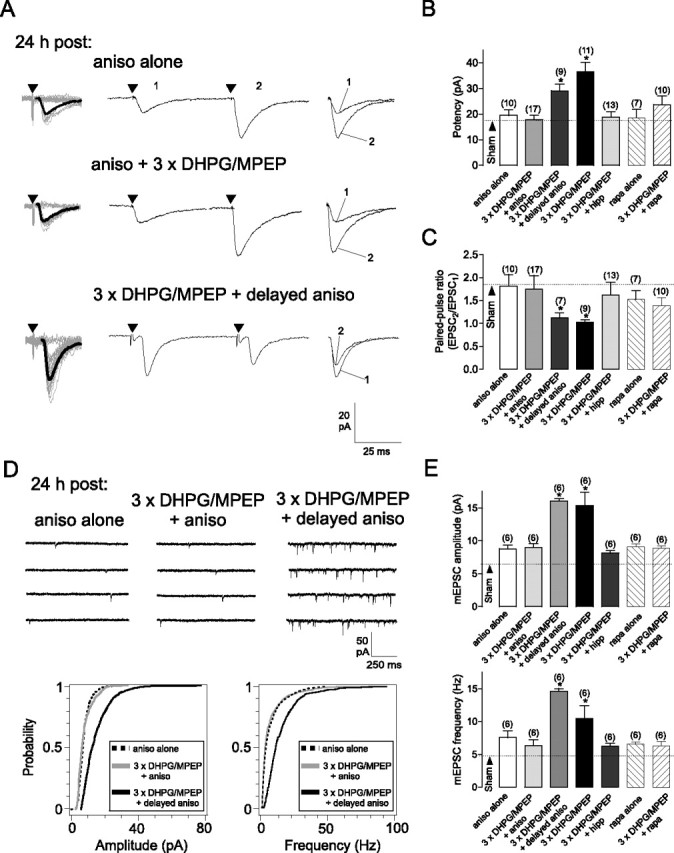
cL-LTPmGluR1 is dependent on translation. A, Concomitant (middle), but not delayed (bottom), inhibition of protein synthesis by anisomycin prevented the increase in EPSC amplitude and decrease in paired-pulse ratio induced by repetitive mGluR1 activation. Treatment with anisomycin alone (top) did not affect EPSCs. B, C, Summary of effects of protein synthesis inhibitors on changes in EPSC potency and paired-pulse ratio induced by repeated mGluR1 stimulation. Dotted line corresponds to sham-treatment values (see also Fig. S4, available at www.jneurosci.org as supplemental material). D, Concomitant, but not delayed, inhibition of protein synthesis prevented increases in mEPSC amplitude and frequency induced by repeated mGluR1 stimulation. Top, Representative mEPSCs traces at 24 h after treatment. Bottom, Cumulative distribution plots for mEPSC amplitude and frequency in all cells. E, Summary bar graphs of effects of protein synthesis inhibitor treatment on mEPSCs. Values expressed as mean ± SEM; * corresponds to p < 0.05. Number above bar indicates cells analyzed.
Inhibition of the PI3K/mTOR and ERK signaling pathways blocks cL-LTPmGluR1
The mammalian target of rapamycin (mTOR) is a key regulator of translation in persistent forms of synaptic plasticity (Hay and Sonenberg, 2004; Banko and Klann, 2008). Hence, we determined its role in cL-LTPmGluR1. Indeed, application of the mTOR inhibitor, rapamycin, prevented the increases in evoked EPSCs (Fig. 3B,C) and mEPSCs (Fig. 3E) induced by repeated mGluR1 stimulation. Application of rapamycin alone was without effect (Fig. S4, available at www.jneurosci.org as supplemental material). Rapamycin sensitivity suggested that mTOR-dependent mechanisms were triggered during or shortly after cL-LTPmGluR1 induction.
Because of the brief time window for protein synthesis, we next used an mTOR read-out assay to identify the signaling pathways activated by repeated mGluR1 stimulation directly in OA-INs. The PI3K, a key signaling pathway upstream of mTOR, has been implicated in the induction of L-LTP at CA1 Schaffer collateral synapses (Tang et al., 2002; Banko et al., 2005; Tsokas et al., 2007). Thus, we sought also to examine the role of PI3K/mTOR signaling in eliciting cL-LTPmGluR1 at excitatory OA-IN synapses. The PI3K and its downstream target the kinase mTOR regulate protein synthesis via multiple effectors, including via phosphorylation of the translational regulatory protein ribosomal S6 by activation of S6 kinase (Hay and Sonenberg, 2004; Banko and Klann, 2008). Therefore, changes in S6 phosphorylation, which provide a bona fide readout of PI3K/mTOR activity, were examined in GFP-labeled OA-INs from GAD67-GFP knock-in mice (Tamamaki et al., 2003) (Fig. 4). First, we confirmed the presence of mTOR in GFP-labeled OA-INs (Fig. S5A, available at www.jneurosci.org as supplemental material). Subsequently, we established that these signaling pathways were activated in OA-INs in nonculture conditions and signaling experiments were performed in acute hippocampal slices. Consistent with our prediction, repeated mGluR1 stimulation increased S6 phosphorylation in GFP-labeled OA-INs. Importantly, the increase in S6 phosphorylation in response to repeated mGluR1 stimulation was prevented by the respective PI3K and mTOR inhibitors wortmannin and rapamycin (Fig. 4). Application of each inhibitor alone did not affect basal S6 phosphorylation level (Fig. 4). Thus, the PI3K/mTOR signaling pathway appears to be activated in OA-INs by repeated mGluR1 stimulation.
Figure 4.

Repeated mGluR1 stimulation increases ribosomal S6 phosphorylation in OA-INs. A, Immunostaining of phosphorylated S6 (p-S6; serine 235/236, middle panel) in GAD67-GFP labeled OA-INs in acute hippocampal slices (left panel). Arrows point to OA-INs positive for p-S6. Staining is increased immediately after repeated mGluR1 stimulation (3× DHPG/MPEP) relative to sham-treatment, which is prevented in the presence of inhibitors rapamycin (1 μm; +rapa), U0126 (20 μm; +U0126), or wortmannin (100 nm; +wort). B, Summary of effects on S6 phosphorylation for all cells. Top, Cumulative distribution plots showing increase in fluorescence after repeated mGluR1 stimulation that is prevented by inhibitors. Fluorescence is expressed in integrated density (i.d.). Bottom, Summary bar graph of phospho-S6 fluorescence values (i.d.). Open bars show values obtained with each inhibitor alone. Results are expressed as mean ± SEM and are representative of three independent experiments (2 animals). Three of six representative sections from each animal were quantified. * corresponds to p < 0.05 using Kolmogorov–Smirnov test (for cumulative distribution) or Student's t test (for the mean). Scale bar, 20 μm.
The mitogen-activated protein kinase kinase (MEK) and ERK is another signaling pathway upstream of mTOR implicated in translation-dependent forms of long-term synaptic plasticity (Hay and Sonenberg, 2004; Banko and Klann, 2008). Hence, we examined whether MEK-ERK signaling contributed to the increases in mTOR activity induced by repeated mGluR1 stimulation in OA-INs. We found that application of U0126, an inhibitor of MEK, prevented the increase in S6 phosphorylation induced in OA-INs by repeated mGluR1 stimulation (Fig. 4). Application of U0126 alone did not affect basal S6 phosphorylation level (Fig. 4). Thus, the MEK-ERK signaling pathway is implicated in mTOR activation by cL-LTPmGluR1 induction protocol in OA-INs.
Since the PI3K/mTOR and MEK-ERK signaling pathways were recruited in OA-INs shortly after repeated mGluR1 stimulation, we next determined their contribution in cL-LTPmGluR1. In the presence of the PI3K inhibitor, wortmannin, the increase in EPSC amplitude and potency and the decrease in PPR induced by repeated mGluR1 stimulation were prevented (Fig. 5A–C). Similarly, the potentiation of mEPSC amplitude and frequency induced by repeated mGluR1 stimulation were impaired by application of wortmannin during induction (Fig. 5D,E). Application of the PI3K inhibitor alone was without effects (Fig. 5; Fig. S4, available at www.jneurosci.org as supplemental material). Thus, cL-LTPmGluR1 induction requires PI3K signaling. We next established the involvement of MEK-ERK signaling in cL-LTPmGluR1 expression, using the MEK inhibitor U0126 and its inactive analog U0124. The potentiation of evoked EPSCs and mEPSCs induced by repeated mGluR1 stimulation were blocked by induction in the presence of the inhibitor U0126 but not by the inactive analog U0124 (237% increase in potency, 46% decrease in PPR, 169% increase in mEPSC amplitude and 59% increase in frequency) (Fig. 5F–J). Application of U0126 alone had no effect on EPSCs and mEPSCs (Fig. 5; Fig. S4, available at www.jneurosci.org as supplemental material). Hence, MEK-ERK signaling is necessary for the induction of cL-LTPmGluR1. Collectively, these results indicate that cL-LTPmGluR1 requires new protein synthesis via activation of PI3K/mTOR and MEK-ERK signaling pathways during or shortly after its induction, which implicates convergence onto their downstream target, the translational repressor of initiation, 4E-BP.
Figure 5.

Contributions of PI3K and ERK signaling pathways to cL-LTPmGluR1. A, Application of the PI3K inhibitor wortmannin (wort, 100 nm, 150 min) prevented the increase in EPSC amplitude and decrease in paired-pulse ratio induced by repetitive mGluR1 activation (middle vs bottom). Treatment with wortmannin alone did not affect EPSCs (top). B, C, Summary bar graphs of effects of wortmannin on changes in EPSC potency and paired-pulse ratio induced by repeated mGluR1 stimulation. Dotted line corresponds to sham-treatment values (see also Fig. S4, available at www.jneurosci.org as supplemental material). D, Representative mEPSCs (top) and cumulative distribution plots for all cells (bottom) showing that wortmannin prevented increases in mEPSC amplitude and frequency induced by repeated mGluR1 stimulation. E, Summary bar graphs of effects of PI3K inhibitor on mEPSCs. F, The increase in EPSC amplitude and decrease in paired-pulse ratio induced by repetitive mGluR1 activation was prevented by the MEK/ERK inhibitor U0126 (20 μm, 150 min) (middle) but not by the inactive MEK/ERK inhibitor U0124 (bottom). Treatment with U0126 alone did not affect EPSCs (top). G, H, Summary bar graphs of effects of MEK/ERK inhibitor U0126 and inactive analog U0124 on changes in EPSC potency and paired-pulse ratio induced by repeated mGluR1. I, Representative mEPSCs (top) and cumulative distribution plots for all cells (bottom) showing that U0126 prevented increases in mEPSC amplitude and frequency induced by repeated mGluR1 stimulation, but not U0124. J, Summary bar graphs of effects of MEK/ERK inhibitor on mEPSCs. Values expressed as mean ± SEM; * corresponds to p < 0.05. Number above bar indicates cells analyzed.
cL-LTPmGluR1 induction is facilitated in 4E-BP1/2−/− mice
PI3K/mTOR and MEK-ERK pathways have both been implicated in regulating the phosphorylation of the translational repressor 4E-BP (Hay and Sonenberg, 2004; Banko and Klann, 2008). Furthermore, electrically induced L-LTP (Banko et al., 2005) or L-LTD induced chemically by the mGluR1/5 agonist DHPG (Banko et al., 2006) are both facilitated in mice lacking 4E-BP2, the major isoform of 4E-BPs in rodent forebrain (Hay and Sonenberg, 2004). Given the above, we used a transgenic knock-out mice approach to examine the role of 4E-BP-dependent regulatory mechanisms in cL-LTPmGluR1 at OA-IN synapses.
Using immunofluorescence, we first established that the two isoforms of 4E-BPs present in the brain, 4E-BP1 and 4E-BP2, were present in GAD67-GFP OA-INs (Fig. S5, available at www.jneurosci.org as supplemental material). Mice with double knock-out of 4E-BP1 and 4E-BP2 (4E-BP1/2−/−) are viable, and exhibit a phenotype with low threshold for obesity, insulin resistance, and increased S6K1 activity leading to abnormal Akt signaling in muscle, liver, and adipose tissue (Le Bacquer et al., 2007). Next, we prepared hippocampal slice cultures from mice with 4E-BP1/2−/− double knock-out to examine whether the threshold for cL-LTPmGluR1 induction was altered in OA-INs. Consistent with the expectation that removal of translational repressors 4E-BP1/2 enhances protein synthesis (Banko et al., 2005, 2006), we found a facilitation of cL-LTPmGluR1 in OA-INs of 4E-BP1/2−/− mice (Fig. 6). A single mGluR1 stimulation, which did not alter evoked EPSCs in slices from wild-type mice, produced a long-term increase in EPSC potency (73%) and decrease in PPR (51%) relative to sham in slices from 4E-BP1/2−/− mice (Fig. 6A,B). Similarly, single mGluR1 stimulation induced a long-term increase in mEPSC amplitude (108%) and frequency (237%) in slices of 4E-BP1/2−/− mice but not of wild type mice (Fig. 6C,D). Repeated mGluR1 stimulation induced similar long-term increases in evoked EPSCs and mEPSCs in OA-INs of both 4E-BP1/2−/− and wild-type (Fig. 6). Thus, genetic removal of the cap-dependent translation repressors 4E-BP1/2 lowered threshold and did not impair cL-LTPmGluR1 induction, providing further support that mTOR- and protein synthesis-dependent plasticity in OA-INs is regulated by repression of cap-dependent translation initiation.
Discussion
cL-LTPmGluR1: a 24 h persistent synaptic plasticity in interneurons
Persistent strengthening of pyramidal neuron excitatory synapses is critical for hippocampal memory formation (Bliss and Collingridge, 1993; Kandel, 2001). Remarkably, it is still unknown whether interneuron circuits undergo such persistent synaptic modifications, despite their essential role in hippocampal information-processing (Buzsáki, 2005; Diba and Buzsáki, 2007; Ego-Stengel and Wilson, 2007). Here, we identified a persistent form of interneuron plasticity (cL-LTPmGluR1), which may contribute to the above process. Maintained for at least 24 h after repetitive mGluR1 stimulation, cL-LTPmGluR1 was observed as increases in transmission using two independent measures of unitary excitatory synaptic responses in OA-INs (Figs. 1, 7). The persistent potentiation was expressed as: (1) decreased paired-pulse ratio and increased potency of putative single-fiber evoked EPSCs, and (2) increased amplitude and frequency of mEPSCs. At other central glutamate synapses, such changes may result from higher release probability (Stevens and Wang, 1994; Lisman et al., 2007), addition of new release sites (Bolshakov et al., 1997; Bozdagi et al., 2000), or postsynaptic receptor insertion (Liao et al., 1995; Bolshakov et al., 1997; Bozdagi et al., 2000)). Our findings on evoked and spontaneous unitary transmission in OA-INs suggest that analogous presynaptic and postsynaptic mechanisms underlie the persistent expression of cL-LTPmGluR1.
Figure 7.

Expression and induction mechanisms of persistent long-term potentiation at OA-IN excitatory synapses (cL-LTPmGluR1). A, Expression mechanisms. At OA-IN excitatory synapses, presynaptic action potential firing releases glutamate which activates Ca2+-permeable AMPA receptors (CP-AMPARs) to generate an EPSC. At 24 h post 3× mGluR1 stimulation, newly synthesized gene products and proteins lead to potentiation, likely via presynaptic and postsynaptic expression mechanisms: presynaptically, by increasing transmitter release (new release sites, or modulation); postsynaptically, by increasing responsiveness (new receptor insertion or increased channel conductance via kinase modulation). B, Induction mechanisms. Summary diagram of signaling cascade based on (1) immunocytochemical detection of 4E-BPs, phospho-S6, and mTOR in OA-INs, (2) observed pharmacological effects of inhibitors of PI3K, MEK, mTOR, eIF4A, transcription and translation on S6 phosphorylation and/or cL-LTPmGluR1, and (3) effects of transgenic knock-out of 4E-BP1/2 on cL-LTPmGluR1. Repeated mGluR1 stimulation (chemically, or presumably by repeated presynaptic firing) triggers PI3K, and ERK signaling pathways to activate mTOR. mTOR leads to (1) ribosomal S6 protein phosphorylation and (2) direct phosphorylation of 4E-BP1/2 repressor proteins which releases eIF4E. Subsequently, eIF4E associates with eIF4A and eIF4G to form the cap-binding complex, eIF4F, which initiates translation. Repeated mGluR1 stimulation also activates transcription by not yet established mechanisms (which may involve ERK).
Although L-LTP is mostly documented using field recordings, which reflect transmission at multiple synapses of large neuronal populations (Frey et al., 1988; Nguyen et al., 1994), we studied cL-LTPmGluR1 at individual synapses using whole-cell patch-clamp recordings. Remarkably, the effects were sufficiently robust to detect lasting changes at putative single synapses at 24 h after induction. A similar slice culture approach was used to monitor L-LTP using field recordings for periods of weeks (Tominaga-Yoshino et al., 2008). Thus, cL-LTPmGluR1 maintenance mechanisms could be monitored for longer periods (> 24 h) and could possibly even be monitored with paired recordings from CA1 pyramidal neurons and OA-INs to examine the nature of long-term changes at identified unitary synapses. Moreover, cL-LTPmGluR1 in slice cultures could be combined with functional knock-down approaches to study underlying molecular mechanisms. Indeed, such approach would be highly complementary to address the selectivity issue with pharmacological inhibitors as used in the present study to dissect the signaling cascade underlying long-term plasticity in OA-INs. Finally, if coupled to in vivo models, cL-LTPmGluR1 can be used to examine the contribution of persistent interneuron plasticity mechanisms to hippocampal-dependent memory tasks.
cL-LTPmGluR1 induction
We used a combination of biochemical, pharmacological and genetic approaches to uncover induction mechanisms of cL-LTPmGluR1. These induction mechanisms were generally consistent with but not identical to those underlying persistent plasticity at CA1 Schaffer collateral synapses (Fig. 7). cL-LTPmGluR1 occluded the early Hebbian LTP at OA-IN synapses induced by physiological stimulation paired with postsynaptic depolarization (Fig. S3, available at www.jneurosci.org as supplemental material). These results indicate that both cL-LTPmGluR1 and Hebbian LTP occur at the same OA-IN synapses and may involve similar mechanisms. This finding is consistent with previous evidence that Hebbian LTP at OA-IN synapses involves presynaptic and postsynaptic mechanisms (Perez et al., 2001). Occlusion was also found at Schaffer collateral synapses (Huang et al., 1994). Thus, cL-LTPmGluR1 may entail expression mechanisms related to those activated by physiological stimulation of OA-IN afferents during early Hebbian LTP.
We found that cL-LTPmGluR1 depends on transcription and translation within a brief time window (Figs. 2, 3). Likewise, translation (Frey et al., 1988; Kelleher et al., 2004a; Costa-Mattioli et al., 2005) and transcription (Nguyen et al., 1994; Costa-Mattioli et al., 2007) take place within a short period (< 1 h) of L-LTP induction at CA1 Schaffer collateral synapses. Interestingly, transcription that is triggered by MEK-ERK signaling in chemically induced L-LTP, results in rapid mRNA synthesis and translocation to dendrites (Ying et al., 2002). Similarly, cL-LTPmGluR1 was also MEK-ERK dependent. Given ERK role in regulating transcription, it will be important to identify the gene products and regulatory mechanisms involved in cL-LTPmGluR1. Yet, cL-LTPmGluR1 differs from L-LTD induced by mGluR1/5 stimulation (mGluR-L-LTD) at CA1 Schaffer collateral synapses since the latter involves a translation-dependent rapid internalization of glutamate receptors but does not require transcription (Snyder et al., 2001). Conceivably, cL-LTPmGluR1 and L-LTP may engage similar mechanisms, distinct from those of mGluR-L-LTD (Waung et al., 2008), that are limited by mRNA turnover, and hence, require a fast onset of transcription.
Using the S6 phosphorylation assay in OA-INs from GAD67-GFP knock-in mice (Fig. 4), we showed that repeated mGluR1 stimulation leads to mTOR activation in OA-INs via PI3K-ERK signaling pathways. Moreover, using specific inhibitors (Fig. 5), we demonstrated that cL-LTPmGluR1 required both PI3K/mTOR and MEK-ERK signaling for induction (Fig. 7). Evidence using pharmacological (Banko and Klann, 2008) or genetic (Kelleher et al., 2004b) approaches also implicated MEK-ERK signaling pathway in triggering translation during plasticity at CA1 Schaffer collateral synapses. Remarkably, the respective downstream effectors of PI3K/mTOR and MEK-ERK signaling pathways, mTOR or ERK, interact directly with the repressor of translation initiation, 4E-BP, during plasticity at CA1 Schaffer collateral synapses (Banko and Klann, 2008). Hence, pathway convergence suggests that cL-LTPmGluR1 induction may require concomitant activation of distinct signaling mechanisms.
Finally, our findings indicate that the induction threshold for c-LTPmGluR1 was facilitated in absence of repressors of cap-dependent translation, 4E-BP1 and 4E-BP2 (Fig. 6). Importantly, we found that, in 4E-BP1/2−/− mice, basal transmission was not facilitated in sham-treated conditions, similar to observations at Schaffer collateral synapses in 4E-BP2−/− mice (Banko et al., 2005, 2006), thus ruling out constitutive effect of de-repression of translation (Fig. 6). In addition, we found that cL-LTPmGluR1 induced by repeated mGluR1 stimulation was not impaired in 4E-BP1/2−/− mice. This differs from observations in 4E-BP2−/− mice in which L-LTP induced by repeated electrical stimulation was impaired whereas that induced by single train was facilitated (Banko et al., 2005). The apparent disparity may be attributed to multiple factors such as greater selectivity of chemical (mGluR1) than electrical stimulation, or different genotypes (single 4E-BP2−/− vs double 4E-BP1/2−/− knock-out). Nonetheless, the same translational control mechanism appears implicated in opposing forms of plasticity, cL-LTPmGluR1 and mGluR-LTD, although they involve different receptor and cell-type (mGluR1 in OA-INs; mGluR5 in CA1 pyramidal cells) (Banko et al., 2006). Interestingly, cL-LTPmGluR1 was also prevented by hippuristanol (Fig. 3, 7), implicating an additional requirement for eIF4A, in forming the eIF4F cap-binding translation initiation complex (Hay and Sonenberg, 2004). The contribution of distinct components downstream of mTOR, 4E-BP and eIF4A to cL-LTPmGluR1 suggest that de-repression of eIF4E alone is insufficient to trigger translation initiation (Hay and Sonenberg, 2004). Furthermore, these results are consistent with the notion that translational control via repression may be rate-limiting (Hay and Sonenberg, 2004). Our findings that hippocampal inhibitory interneurons and pyramidal cells use similar translational regulatory mechanisms during persistent forms of plasticity provide further evidence that such mechanisms are highly preserved across cell types (Hay and Sonenberg, 2004; Banko and Klann, 2008) and species (Martin et al., 1997; Costa-Mattioli et al., 2007), thus suggesting a possible role in other cortical circuit plasticity.
Implication for hippocampal function
There are several indications that interneuron plasticity contributes to exploratory behavior during formation of hippocampal memories (Maurer et al., 2006; Diba and Buzsáki, 2007; Ego-Stengel and Wilson, 2007). During spatial exploration, pyramidal neurons adapt preferred firing-rates to specific locations in the environment (place-fields) and sequentially follow the predominant theta rhythm of the entire hippocampal network (phase precession (Maurer et al., 2006)). The precise contribution of interneurons to such behavior is unknown, however, interneurons synchronize large populations of pyramidal neurons at theta rhythm (4–8 Hz) under anesthesia (Klausberger et al., 2003) or during spatial exploration (Buzsáki, 2005). Moreover, interneurons that are strongly coupled to pyramidal neurons form place-field maps (Maurer et al., 2006). Hence, interneurons are able to store spatial information (place-field or phase precession) via synchronization of pyramidal neurons or by inheriting the spatial selectivity (place-field map) of pyramidal neurons to which they are coupled.
Considering the above, the persistent increases in interneuron excitation underlying cL-LTPmGluR1 can potentially facilitate the functional coupling of OA-INs to pyramidal neurons during exploration (Buzsáki, 2005). This raises two inter-related questions of whether cL-LTPmGluR1-dependent increases in OA-IN excitation occur in vivo, and if so, whether such synaptic modifications play a role during memory formation? First, since cL-LTPmGluR1 (Fig. S3, available at www.jneurosci.org as supplemental material) occludes Hebbian LTP in OA-INs, it is conceivable that physiological activation of CA1 collaterals during training would elicit cL-LTPmGluR1 in OA-INs in vivo. Second, a number of observations implicate mGluR1s in hippocampal-dependent memory. Notably, mGluR1s are highly expressed in OA-INs (Baude et al., 1993; Lapointe et al., 2004). Moreover, mGluR1s are implicated in hippocampal memory consolidation. CA1 Schaffer collateral LTP and context-specific associative learning are reduced in mGluR1 null mice, suggesting that mGluR1s modulate the plasticity process (Aiba et al., 1994). Similarly, mGluR1 antagonism in CA1 region severely reduces avoidance learning (Simonyi et al., 2007). Thus, mGluR1s are critical for CA1 Schaffer collateral LTP and associated memory. Therefore, if cL-LTPmGluR1 occurred at OA-IN excitatory synapses during exploration (Geisler et al., 2007), the persistent synaptic plasticity mechanisms uncovered here would strengthen the coupling of pyramidal-interneuron circuits—a critical step in acquisition of spatial memory (Buzsáki, 2005).
Footnotes
This work was supported by the Canadian Institutes of Health Research (operating grant MOP-10848 to J.C.L.) and Fonds de la Recherche en Santé du Québec (Groupe de Recherche sur le système nerveux central). J.C.L. is a recipient of the Canada Research Chair in Cellular and Molecular Neurophysiology. GAD67-GFP mice were kindly provided by Y. Yanagawa and L. Mei. We thank Drs. W. Sossin, J. G. Pelletier, and A. Panatier for excellent comments.
References
- Aiba A, Chen C, Herrup K, Rosenmund C, Stevens CF, Tonegawa S. Reduced hippocampal long-term potentiation and context-specific deficit in associative learning in mGluR1 mutant mice. Cell. 1994;79:365–375. doi: 10.1016/0092-8674(94)90204-6. [DOI] [PubMed] [Google Scholar]
- Banko JL, Klann E. Cap-dependent translation initiation and memory. Prog Brain Res. 2008;169:59–80. doi: 10.1016/S0079-6123(07)00004-0. [DOI] [PubMed] [Google Scholar]
- Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005;25:9581–9590. doi: 10.1523/JNEUROSCI.2423-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko JL, Hou L, Poulin F, Sonenberg N, Klann E. Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2006;26:2167–2173. doi: 10.1523/JNEUROSCI.5196-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CA, McNaughton BL. An age comparison of the rates of acquisition and forgetting of spatial information in relation to long-term enhancement of hippocampal synapses. Behav Neurosci. 1985;99:1040–1048. doi: 10.1037//0735-7044.99.6.1040. [DOI] [PubMed] [Google Scholar]
- Baude A, Nusser Z, Roberts JD, Mulvihill E, McIlhinney RA, Somogyi P. The metabotropic glutamate receptor (mGluR1 alpha) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron. 1993;11:771–787. doi: 10.1016/0896-6273(93)90086-7. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Seizures beget seizures: the quest for GABA as a key player. Crit Rev Neurobiol. 2006;18:135–144. doi: 10.1615/critrevneurobiol.v18.i1-2.140. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Gardner-Medwin AR. Long-lasting potentiation of synaptic transmission in the dentate area of the unanaestetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:357–374. doi: 10.1113/jphysiol.1973.sp010274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolshakov VY, Golan H, Kandel ER, Siegelbaum SA. Recruitment of new sites of synaptic transmission during the cAMP-dependent late phase of LTP at CA3-CA1 synapses in the hippocampus. Neuron. 1997;19:635–651. doi: 10.1016/s0896-6273(00)80377-3. [DOI] [PubMed] [Google Scholar]
- Bordeleau ME, Mori A, Oberer M, Lindqvist L, Chard LS, Higa T, Belsham GJ, Wagner G, Tanaka J, Pelletier J. Functional characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat Chem Biol. 2006;2:213–220. doi: 10.1038/nchembio776. [DOI] [PubMed] [Google Scholar]
- Bourdeau ML, Morin F, Laurent CE, Azzi M, Lacaille JC. Kv4.3-mediated A-type K+ currents underlie rhythmic activity in hippocampal interneurons. J Neurosci. 2007;27:1942–1953. doi: 10.1523/JNEUROSCI.3208-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW. Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron. 2000;28:245–259. doi: 10.1016/s0896-6273(00)00100-8. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. Theta rhythm of navigation: link between path integration and landmark navigation, episodic and semantic memory. Hippocampus. 2005;15:827–840. doi: 10.1002/hipo.20113. [DOI] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M, Bidinosti M, Ben Mamou C, Marcinkiewicz E, Yoshida M, Imataka H, Cuello AC, Seidah N, Sossin W, Lacaille JC, Ron D, Nader K, Sonenberg N. Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature. 2005;436:1166–1173. doi: 10.1038/nature03897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, Sossin W, Kaufman R, Pelletier J, Rosenblum K, Krnjeviæ K, Lacaille JC, Nader K, Sonenberg N. eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell. 2007;129:195–206. doi: 10.1016/j.cell.2007.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diba K, Buzsáki G. Forward and reverse hippocampal place-cell sequences during ripples. Nat Neurosci. 2007;10:1241–1242. doi: 10.1038/nn1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ego-Stengel V, Wilson MA. Spatial selectivity and theta phase precession in CA1 interneurons. Hippocampus. 2007;17:161–174. doi: 10.1002/hipo.20253. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res. 1988;452:57–65. doi: 10.1016/0006-8993(88)90008-x. [DOI] [PubMed] [Google Scholar]
- Geisler C, Robbe D, Zugaro M, Sirota A, Buzsáki G. Hippocampal place cell assemblies are speed-controlled oscillators. Proc Natl Acad Sci U S A. 2007;104:8149–8154. doi: 10.1073/pnas.0610121104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Huang YY, Li XC, Kandel ER. cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and macromolecular synthesis-dependent late phase. Cell. 1994;79:69–79. doi: 10.1016/0092-8674(94)90401-4. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Tonegawa S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron. 2004a;44:59–73. doi: 10.1016/j.neuron.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004b;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Magill PJ, Márton LF, Roberts JD, Cobden PM, Buzsáki G, Somogyi P. Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature. 2003;421:844–848. doi: 10.1038/nature01374. [DOI] [PubMed] [Google Scholar]
- Kullmann DM, Lamsa KP. Long-term synaptic plasticity in hippocampal interneurons. Nat Rev Neurosci. 2007;8:687–699. doi: 10.1038/nrn2207. [DOI] [PubMed] [Google Scholar]
- Lapointe V, Morin F, Ratté S, Croce A, Conquet F, Lacaille JC. Synapse-specific mGluR1-dependent long-term potentiation in interneurones regulates mouse hippocampal inhibition. J Physiol. 2004;555:125–135. doi: 10.1113/jphysiol.2003.053603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bacquer O, Petroulakis E, Paglialunga S, Poulin F, Richard D, Cianflone K, Sonenberg N. Elevated sensitivity to diet-induced obesity and insulin resistance in mice lacking 4E-BP1 and 4E-BP2. J Clin Invest. 2007;117:387–396. doi: 10.1172/JCI29528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Vasseur M, Ran I, Lacaille JC. Selective induction of metabotropic glutamate receptor 1- and metabotropic glutamate receptor 5-dependent chemical long-term potentiation at oriens/alveus interneuron synapses of mouse hippocampus. Neuroscience. 2008;151:28–42. doi: 10.1016/j.neuroscience.2007.09.071. [DOI] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Raghavachari S, Tsien RW. The sequence of events that underlie quantal transmission at central glutamatergic synapses. Nat Rev Neurosci. 2007;8:597–609. doi: 10.1038/nrn2191. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Martin KC, Casadio A, Zhu H, Yaping E, Rose JC, Chen M, Bailey CH, Kandel ER. Synapse-specific, long-term facilitation of Aplysia sensory to motor synapses: a function for local protein synthesis in memory storage. Cell. 1997;91:927–938. doi: 10.1016/s0092-8674(00)80484-5. [DOI] [PubMed] [Google Scholar]
- Maurer AP, Cowen SL, Burke SN, Barnes CA, McNaughton BL. Phase precession in hippocampal interneurons showing strong functional coupling to individual pyramidal cells. J Neurosci. 2006;26:13485–13492. doi: 10.1523/JNEUROSCI.2882-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D, Toni N, Buchs P-A, Parisi L, Stoppini L. Interface organotypic hippocampal slice cultures. In: Fedoroff S, Richardson A, editors. Protocols for neural cell cultures. Totowa, NJ: Humana; 2001. [Google Scholar]
- Nguyen PV, Abel T, Kandel ER. Requirement of a critical period of transcription for induction of a late phase of LTP. Science. 1994;265:1104–1107. doi: 10.1126/science.8066450. [DOI] [PubMed] [Google Scholar]
- Nugent FS, Kauer JA. LTP of GABAergic synapses in the ventral tegmental area and beyond. J Physiol. 2008;586:1487–1493. doi: 10.1113/jphysiol.2007.148098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier JG, Lacaille JC. Long-term synaptic plasticity in hippocampal feedback inhibitory networks. Prog Brain Res. 2008;169:241–250. doi: 10.1016/S0079-6123(07)00014-3. [DOI] [PubMed] [Google Scholar]
- Perez Y, Morin F, Lacaille JC. A hebbian form of long-term potentiation dependent on mGluR1a in hippocampal inhibitory interneurons. Proc Natl Acad Sci U S A. 2001;98:9401–9406. doi: 10.1073/pnas.161493498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonyi A, Serfozo P, Shelat PB, Dopheide MM, Coulibaly AP, Schachtman TR. Differential roles of hippocampal metabotropic glutamate receptors 1 and 5 in inhibitory avoidance learning. Neurobiol Learn Mem. 2007;88:305–311. doi: 10.1016/j.nlm.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4:1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Wang Y. Changes in reliability of synaptic function as a mechanism for plasticity. Nature. 1994;371:704–707. doi: 10.1038/371704a0. [DOI] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol. 2003;467:60–79. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga-Yoshino K, Urakubo T, Okada M, Matsuda H, Ogura A. Repetitive induction of late-phase LTP produces long-lasting synaptic enhancement accompanied by synaptogenesis in cultured hippocampal slices. Hippocampus. 2008;18:281–293. doi: 10.1002/hipo.20391. [DOI] [PubMed] [Google Scholar]
- Traub RD, Bibbig A, LeBeau FE, Buhl EH, Whittington MA. Cellular mechanisms of neuronal population oscillations in the hippocampus in vitro. Annu Rev Neurosci. 2004;27:247–278. doi: 10.1146/annurev.neuro.27.070203.144303. [DOI] [PubMed] [Google Scholar]
- Tsokas P, Ma T, Iyengar R, Landau EM, Blitzer RD. Mitogen-activated protein kinase upregulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J Neurosci. 2007;27:5885–5894. doi: 10.1523/JNEUROSCI.4548-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron. 2008;59:84–97. doi: 10.1016/j.neuron.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR. Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci. 2002;22:1532–1540. doi: 10.1523/JNEUROSCI.22-05-01532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]