

Abstract
The molecular mechanisms that regulate survival of embryonic neural precursors are still relatively ill-defined. Here, we have asked whether the p53 family member p63 plays any role during this developmental window, focusing on the embryonic cerebral cortex. We show that genetic knockdown of p63 either in culture or in the embryonic telencephalon causes apoptosis of cortical precursors and newly born cortical neurons, and that this can be rescued by expression of ΔNp63, but not TAp63 isoforms. This cortical precursor apoptosis is the consequence of deregulated p53 activity, since both basal precursor apoptosis and that induced by loss of p63 are rescued by coincident genetic silencing of p53. Finally, we demonstrate that the third p53 family member, ΔNp73, does not regulate survival of cortical precursor cells, but that it collaborates with ΔNp63 to ensure the survival of newly born cortical neurons. Thus, the balance of ΔNp63 versus p53 determines the life versus death of embryonic cortical precursors, a role that these p53 family members may well play in other populations of developing and/or adult neural precursors.
Introduction
The cerebral cortex develops from a pool of precursor cells that sequentially generates neurons and then glial cells (Miller and Gauthier, 2007). Some of these precursors undergo cellular apoptosis from embryonic day 12 (E12) to E18 (Blaschke et al., 1996, 1998; Thomaidou et al., 1997) potentially as part of a cell death program that acts both to restrict the number of precursor cells to determine brain size, and as a correction mechanism to ensure the survival of only those cells that differentiate appropriately (Voyvodic, 1996; Haydar et al., 1999; Kuan et al., 2000; Buss et al., 2006). However, although it is clear that key enactors of apoptosis such as caspase 3 (Kuida et al., 1996) and caspase 9 (Hakem et al., 1998; Kuida et al., 1998) are required for death of developing CNS precursors, the mechanisms that regulate this cell death period are not well understood.
Potential insights into the underlying mechanisms come from the period of naturally occurring neuronal death, which has been extensively studied. In this regard, the p53 tumor suppressor and its related family members p63 and p73 have been shown to determine the life versus death of developing peripheral sympathetic neurons as they compete for limiting amounts of target-derived nerve growth factor (NGF) (Slack et al., 1996; Aloyz et al., 1998; Pozniak et al., 2000; Jacobs et al., 2005; Miller and Kaplan, 2007). p63 and p73 are expressed as full-length isoforms containing a transactivation (TA) domain (termed TAp63 and TAp73) or as truncated isoforms that lack the TA domain (termed ΔNp63 and ΔNp73) that function, at least in part, as naturally occurring dominant-inhibitory p53 family members (Jacobs et al., 2006). Both TAp63 and ΔNp63 are expressed in cortical precursors, cortical neurons and in vivo in the cortex of embryonic, postnatal, and adult brains (Jacobs et al., 2005). Importantly, this family has been implicated in regulation of the survival and maintenance of CNS neurons, with p53 functioning as a cell death protein, and a naturally occurring truncated form of p73, ΔNp73, as a major survival protein that prevents neuronal apoptosis and neurodegeneration (Jacobs et al., 2006; Wetzel et al., 2008). However, whether or not this family regulates neural precursor cell survival is still an open question. One clue that they might comes from p53−/− mice that display a partially penetrant embryonic lethality as a consequence of aberrant neural hypertrophy, a phenotype that might be explained by decreased precursor cell apoptosis (Armstrong et al., 1995; Sah et al., 1995). Concomitant ablation of p53 can also rescue neural apoptosis induced by the deletion of pRb (Macleod et al., 1996) possibly through E2F1 (Tsai et al., 1998), of E3 ubiquitin ligase Mdm4 (Migliorini et al., 2002), and of DNA repair proteins DNA Ligase IV (Frank et al., 2000) and XRCC4 (Gao et al., 2000). Finally, p53 is essential for neural precursor apoptosis in response to genotoxic stress (Akhtar et al., 2006a,b). However, whether p53 is essential for neural precursor apoptosis during normal development and/or how its activity might be regulated are still open questions.
Here, we have asked whether the p53 family plays a role in regulating neural precursor survival, and demonstrate that ΔNp63 is essential for the survival of embryonic cortical precursors and newly born cortical neurons, a role that it plays by antagonizing the proapoptotic actions of p53. Moreover, we demonstrate that ΔNp73 does not regulate survival of cortical precursor cells, but that it collaborates with ΔNp63 to ensure the survival of newly born postmitotic cortical neurons. Thus, the p53 family plays a key role in determining the life versus death of embryonic cortical precursors, a role that it might play throughout the developing CNS.
Materials and Methods
Animals and in utero electroporation.
p63fl/fl mice (Mills et al., 2002) were maintained through homozygous breeding pairs in a C57BL6 background. p73+/− mice were a gift from Dr. Frank McKeon (Harvard University, Cambridge, MA) (Yang et al., 2000). In utero electroporation was performed on E13–14 CD1 or p63fl/fl mice using a square electroporator CUY21 EDIT (TR Tech) delivering five 50 ms pulses of 50 V with 950 ms intervals per embryo (Paquin et al., 2005; Bartkowska et al., 2007; Gauthier et al., 2007). The embryos were injected with nuclear enhanced green fluorescent protein (EGFP) expression plasmid driven from the EF1α-promoter at a 1:3 ratio with nonsilencing shRNA, p63 shRNA, control plasmid, or Cre recombinase, for a total of 4 μg of DNA per embryo and 0.05% trypan blue as a tracer. When both p63 shRNA and p53 shRNA were coelectroporated, DNA was mixed at a ratio of 1 pEF-EGFP:3 p63 shRNA:3 p53 shRNA for a total of 7 μg per embryo. Embryonic brains were dissected in ice-cold HBSS, fixed in 4% PFA at 4°C for 24 h, cryoprotected, and cryosectioned coronally at 16 μm. This study was approved by The Hospital for Sick Children's Animal Care Committee, and use was in accordance with Canadian Council on Animal Care guidelines.
Cell cultures and transfections.
E12–E13 cortical precursors, from CD1 or p63fl/fl mice, were cultured in NeurobasalR (Invitrogen-21103) medium supplemented with 40 ng/ml FGF2 (Promega), 2% B27 supplement (Invitrogen), 1% penicillin-streptomycin (Invitrogen), and 500 μm l-glutamine on 4 well chamber slides (Nunc) coated with 2% laminin and 1% poly-d-lysine (BD Biosciences) as described previously (Barnabé-Heider and Miller, 2003; Barnabé-Heider et al., 2005) at a density of 150,000 cells/well for four-well chamber slides. Culture medium was not changed for the duration of the experiment. For transfections, 1–2 h after plating, 1 μg of DNA and 1.5 μl of Fugene 6.0 (Roche) mixed with 100 μl of Opti-MEM (Invitrogen) were incubated at room temperature for 45 min and added to the cultures. The sequence for p63 shRNA, p53 shRNA, and nonsilencing control are as described previously (Cam et al., 2006). The sequence used for ΔNp73 shRNA was 5′-AACTCTCAGTCCGCACGCC. The Cre recombinase (Cre) expression plasmid was from Dr. C. C. Hui (The Hospital for Sick Children, Toronto, ON, Canada) and the human ΔNp63α and TAp63α expression plasmids were from Dr. Meredith Irwin (The Hospital for Sick Children, Toronto, ON, Canada). The human and murine ΔNp63α and TAp63α proteins are 99% homologous (Yang et al., 1998).
Reverse transcriptase-PCR analysis.
Cortices were dissected from E12.5 embryos and Trizol (Invitrogen) was used to isolate RNA according to the manufacturer's protocol. RNA was treated with DNase (Fermentas) to avoid contamination with genomic DNA. A sample of RNA was subjected to PCR with GAPDH primers (Jacobs et al., 2005) to ensure the absence of genomic DNA. Reverse transcription was performed using RevertAid H Minus M-MuLV Reverse Transcriptase (Fermentas) primed with random hexamers, according to manufacturer's instructions. ΔNp73 expression pattern were obtained using primers specific to its N-terminal region (Walsh et al., 2004).
Quantitative reverse transcriptase-PCR.
HEK 293 cells were transfected with TAp73 or 5′UTR containing ΔNp73 cDNA and specific shRNA vectors against each isoform or a scrambled sequence using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Total RNA was extracted 48 h after transfection using Trizol reagent (Invitrogen) and RNeasy kit (QIAGEN). cDNA for quantitative reverse transcriptase (RT)-PCR was prepared using SuperScript III First-Strand Synthesis SuperMix (Invitrogen). Real-time PCR was performed according to the manufacturer's specifications using Chromo4 Real-Time PCR Detection System (Bio-Rad) and a Platinum Quantitative PCR Super-Mix-UDG (Invitrogen). Samples were analyzed in triplicate and were normalized to β-actin for each reaction. The primers used for TAp73 were 5′-AGTTCTCTAGAGCCAGACA and 5′-CAAATTGAACTGGGCCATG, for ΔNp73 were 5′-CTACCATGCTTTACGTCGG and 5′-CTGCCCATCTGGTCCAT. The primers for β-actin were as previously published (Keyes et al., 2005; Wetzel et al., 2008). All PCR products were single bands with predicted molecular weights.
Immunfluorescence, Western blots, and antibodies.
Immunocytochemistry and immunohistochemistry were performed as previously described (Barnabé-Heider et al., 2005; Gauthier et al., 2007). The primary antibodies used were mouse anti-GFP (1:1000; Invitrogen), rabbit anti-GFP (1:500; Millipore Bioscience Research Reagents), rabbit anti-cleaved caspase 3 (1:500; Cell Signaling Technology), mouse anti-Ki67 (1:200; PharMingen), mouse anti-HuD (1:200, Invitrogen), mouse anti-βIII-tubulin (1:800; Covance), rabbit anti-phospho-histone-H3 (1:1000; Upstate) and mouse 4A4 anti-p63 (1:200, Santa Cruz Biotechnology). The secondary antibodies (Jackson ImmunoResearch) used were indocarbocyanine (Cy3)-conjugated goat anti-mouse and anti-rabbit IgG (1:400), FITC conjugated anti-mouse and anti-rabbit IgG (1:200), dichlorotriazinyl amino fluorescein-conjugated streptavidin (1:1000), and Cy3-conjugated streptavidin (1:1000). To assess the efficacy of p63 shRNA to knockdown overexpressed p63, HEK293 cells were cotransfected with mouse ΔNp63α and either nonsilencing shRNA or p63 shRNA. Two days later, 30 μg of protein lysate was run on SDS-PAGE, and Western blots were performed as described previously (Barnabé-Heider et al., 2005). The primary antibodies (Santa Cruz Biotechnology) were mouse 4A4 anti-p63 (1:200), rabbit anti-GFP (1:1000; Millipore Bioscience Research Reagents), and rabbit anti-Erk (1:3000). Secondary antibodies were HRP-conjugated goat anti-mouse or anti-rabbit (1:10,000; Boehringer Mannheim).
Quantification and confocal analysis.
Microscopy of cultured cortical precursors was performed using a Zeiss Axioplan2 upright microscope equipped with fluorescence optics. Analysis of cultured cortical precursors was performed by scoring >200 transfected cells per condition per experiment in at least eight randomly selected fields spanning the culture well. Images were acquired with Northern Eclipse software (Empix) using a Sony XC-75CE CCD video camera. For all analyses, at least three independent culture experiments were performed at different times with comparable results. For quantification of immunocytochemistry on tissue sections, brains were chosen with a similar anatomical distribution of EGFP expression. Images of the electroporated dorsal telencephalon at the level of the developing hippocampus were taken and covered the ventricular zone (VZ), sub-VZ (SVZ), and cortical plate of each coronal section. Only those brains that were electroporated within this area were selected for analysis. A total of two to seven sections were analyzed per animal and three or more embryos were analyzed per condition. Images represented a mean of four scans taken with a 40× objective and were analyzed using a Zeiss Pascal confocal microscope and the manufacturer's software. The actual numbers of sections and cells counted are indicated in the figure legends. For definition of cellular location within electroporated cortices, the VZ, SVZ, and cortical plate were delineated using Hoechst 33258 staining to define cell density, and HuD and Ki67-immunostaining to define proliferative and neuronal layers, as described previously (Bartkowska et al., 2007; Gauthier et al., 2007). Image acquisition was performed using Northern Eclipse Software (Empix) with a Sony-XC-75CE CCD video camera. Error bars indicate SEM, and statistics were performed using either the Student's t test or one-way ANOVA, as appropriate.
Results
ΔNp63 is essential for survival of cultured cortical precursor cells and newly born neurons
To ask whether p63 is important for the development of neural precursor cells, we studied primary murine E12–13 cortical precursors, a system we have previously characterized (Toma et al., 2000; Ménard et al., 2002; Barnabé-Heider and Miller, 2003; Barnabé-Heider et al., 2005; Bartkowska et al., 2007; Gauthier et al., 2007). During plating in FGF2, these precursor cells are virtually all dividing, nestin-positive cells that sequentially generate neurons at 1 d in vitro (DIV), and glia at 5–6 DIV. The increase in differentiated cells is accompanied by a decrease in proliferating precursor cells.
We previously showed that p63 is expressed as both TA and ΔN variants in cortical precursors, in newly born cortical neurons, and in vivo in the embryonic cortex (Jacobs et al., 2005). To ask whether p63 plays a role in regulating cortical precursors, we knocked down p63 mRNA using a previously described shRNA that targets all of the full-length and truncated isoforms (Cam et al., 2006). We confirmed the efficacy and specificity of this shRNA by cotransfecting HEK293 cells with constructs encoding p63 shRNA and either murine ΔNp63α or GFP. Western blot analysis using a pan-p63 antibody showed that p63 levels, but not GFP levels, were reduced by p63 shRNA 48 h after transfection (Fig. 1A), in good agreement with previous data showing that this p63 shRNA specifically targets p63 but not p53 or p73 (Cam et al., 2006). We then cotransfected freshly plated E12–13 cortical precursor cells with EGFP and either p63 shRNA or nonsilencing shRNA and analyzed p63 levels immunocytochemically with an antibody to all isoforms of p63 at 2 DIV. This analysis revealed that the p63 shRNA caused an increase in the proportion of cells with no or low detectable p63 staining (73 ± 3% vs 42 ± 2% for p63 shRNA vs nonsilencing shRNA, p < 0.01) (Fig. 1B).
Figure 1.
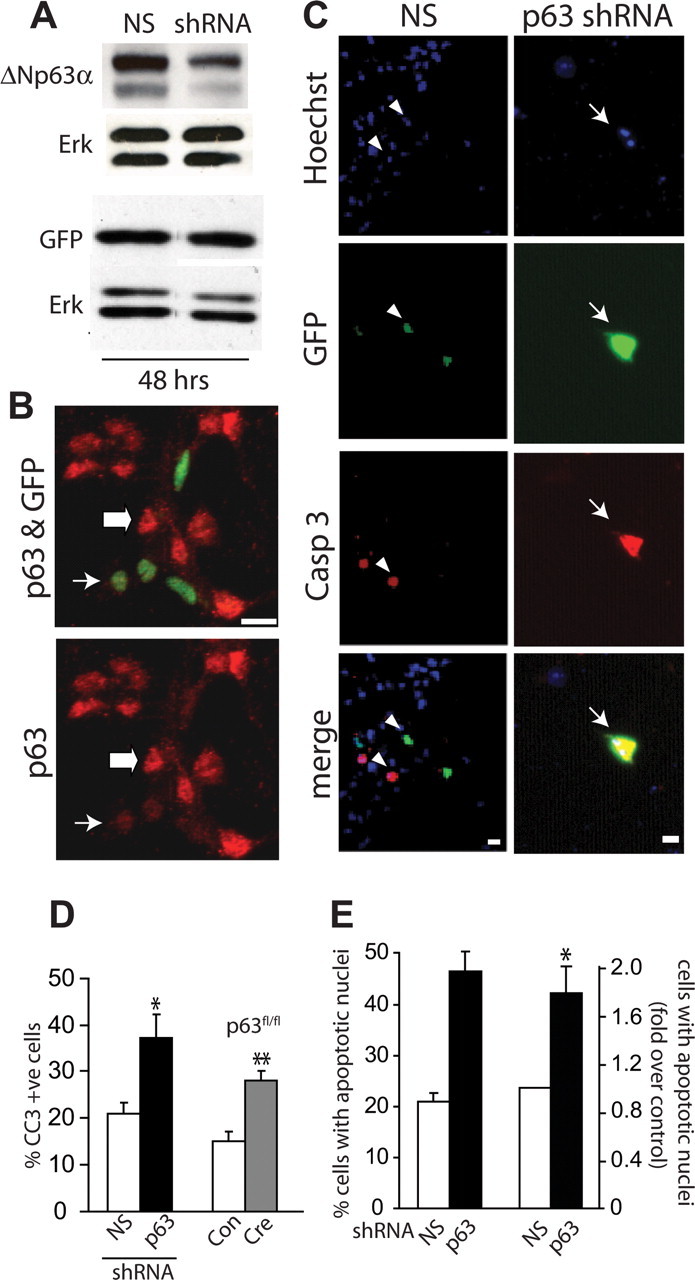
p63 is essential for the survival of cultured murine embryonic cortical precursor cells. A, Western blot analysis for ΔNp63α (top) and GFP (bottom) in lysates of HEK293 cells cotransfected with plasmids expressing ΔNp63α (top) or GFP (bottom) plus p63 shRNA, or a control, nonsilencing (NS) shRNA for 48 h. In the top panel, both bands are ΔNp63α. Blots were reprobed for Erk 1/2 to ensure equal protein loading. B, Immunocytochemistry for all isoforms of p63 (red) and EGFP (GFP, green) in cultured precursors cotransfected with plasmids encoding nuclear EGFP and p63 shRNA for 48 h, indicating no or low (thin arrow) or basal levels (thick arrow) of p63 expression. Note the lower levels of p63 in the transfected (green) cells. C, Fluorescence micrographs of cortical precursor cultures cotransfected with plasmids encoding EGFP and nonsilencing (NS) shRNA or p63 shRNA, and then analyzed at 2 d for EGFP (green, GFP), and cleaved caspase-3 (red, Casp 3). Cells were counterstained with Hoechst 33258 (blue) to show all nuclei. The bottom panel shows the merges. Arrows denote double-labeled and arrowheads single-labeled cells. Note the apoptotic morphology of the nucleus of the p63 shRNA-transfected cell shown in the right panels. D, Left bars, Quantification of the percentage of EGFP-positive cells positive for cleaved caspase-3 in cultures similar to those in C. Right bars, p63fl/fl cortical precursors were cotransfected with plasmids encoding EGFP and control vector (Con) or Cre recombinase (Cre) and analyzed and quantified as shown in C. Results in both cases are pooled from three independent experiments each. E, Quantification of the proportion of EGFP-positive cells displaying condensed fragmented nuclei as detected using Hoechst 33258 in experiments similar to those in C. The left pair shows the percentage of apoptotic cells in one representative experiment, whereas the right pair shows pooled data from three independent experiments expressed as fold-change relative to nonsilencing shRNA-transfected cells. Error bars indicate SEM. *p < 0.05; **p < 0.01, Student's t test. Scale bars, 10 μm.
We next asked whether this knockdown of p63 affected cell survival. Cortical precursors were cotransfected with plasmids encoding EGFP and either p63 shRNA or nonsilencing shRNA, and apoptosis was assessed 2 d later by immunostaining for EGFP and cleaved caspase-3 (Fig. 1C) (Bartkowska et al., 2007). Quantification revealed that p63 knockdown resulted in a twofold increase in the proportion of cells that were positive for cleaved caspase-3 (Fig. 1D). Similar results were obtained by counting the percentage of EGFP-positive cells with condensed, apoptotic nuclei as monitored by staining with Hoechst 33258 (Fig. 1E). To confirm these results, we took advantage of p63fl/fl mice (Mills et al., 2002), in which exons common to both TA and ΔN isoforms are flanked by lox P sites. Initially, we confirmed that Cre expression in p63wt/wt cortical precursors did not alter cell survival (data not shown). Then, we cotransfected p63fl/fl cortical precursor cells with plasmids expressing EGFP and Cre, and assessed survival 2 d later. As observed with p63 shRNA, acute p63 genetic ablation resulted in a twofold increase in cleaved caspase-3-positive cells (Fig. 1D), confirming the p63 shRNA data.
These findings indicate that p63 is an important survival protein for cortical precursor cells and/or for cortical neurons that have differentiated in these cultures over the first 2 d. To distinguish these two possibilities, we cotransfected cortical precursors with plasmids encoding EGFP and p63 shRNA or nonsilencing shRNA, and 3 d later immunostained them for the neuron-specific protein βIII-tubulin. We then counterstained these cultures with Hoechst 33258 to detect condensed, apoptotic nuclei, and quantified apoptotic neurons (βIII-tubulin-positive) versus apoptotic precursors (βIII-tubulin-negative). This analysis demonstrated that p63 knockdown resulted in an ∼2-fold increase in the death of both cortical precursor cells and newly born cortical neurons (Fig. 2A–C).
Figure 2.
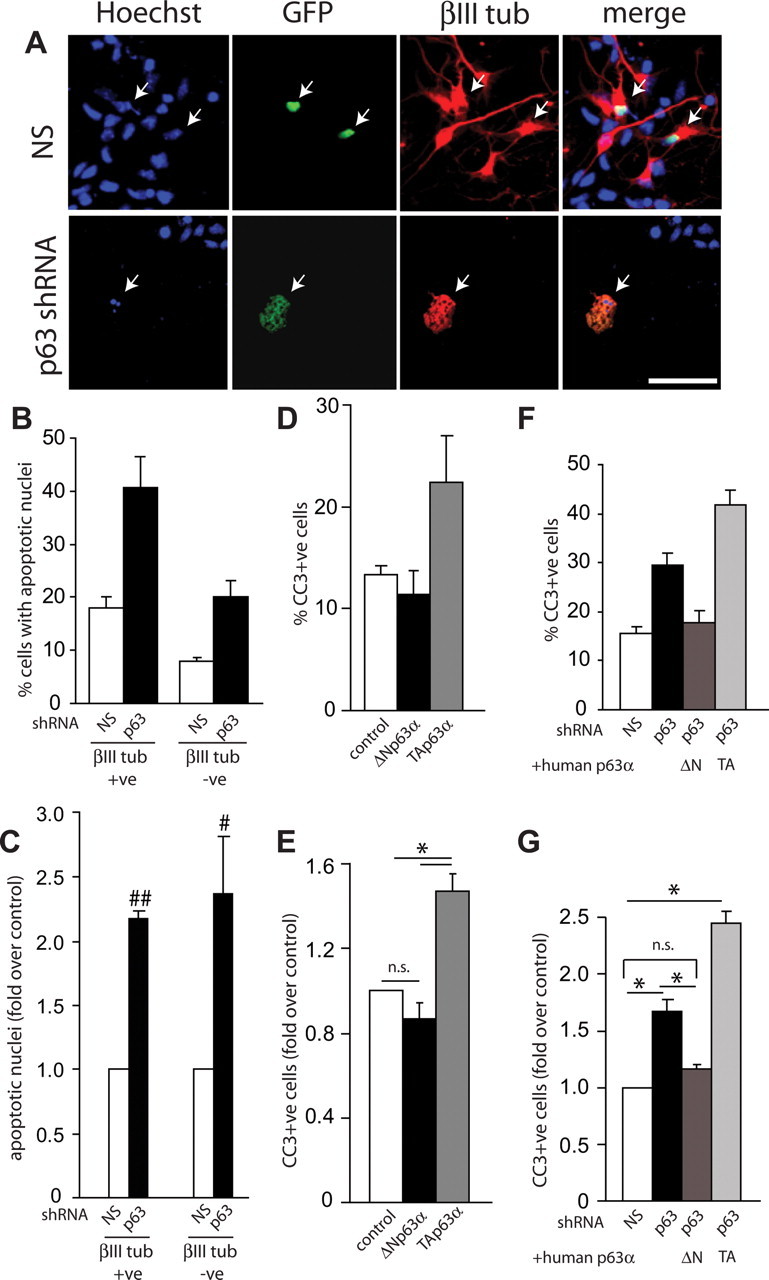
ΔNp63 is essential for survival of cortical precursors and newly born cortical neurons. A, Fluorescence micrographs of cortical precursor cultures cotransfected with plasmids encoding nuclear EGFP and nonsilencing (NS) shRNA or p63 shRNA and then analyzed at 3 d for EGFP (green, GFP), βIII-tubulin (βIII tub, red), and counterstained with Hoechst 33258 (blue) to show all nuclei. The right panels show the merges. Arrows denote double-labeled cells. Note that the βIII-tubulin-positive cell transfected with p63 shRNA is dying, as indicated by the apoptotic nuclear morphology. B, C, Quantification of EGFP-positive, βIII-tubulin-positive (neurons), and negative (precursors) cells with condensed, fragmented nuclei (as monitored by Hoechst staining) relative to nonsilencing (NS) shRNA-transfected cells in experiments similar to those shown in A. Graphs in B show percentage apoptotic cells from one representative experiment, whereas those in C show pooled data from three independent experiments expressed as fold-change relative to nonsilencing shRNA-transfected cells. D, E, Cortical precursor cells were cotransfected with plasmids encoding nuclear EGFP and control vector (control), murine ΔNp63α or TAp63α, and analyzed immunocytochemically at 2 d for EGFP and cleaved caspase 3 (CC3). Graphs in D show percentage cleaved caspase-3 (CC3)-positive-transfected cells from one representative experiment, whereas those in E show pooled data from three independent experiments expressed as fold-change relative to nonsilencing shRNA-transfected cells. F, G, Cortical precursor cells were cotransfected with plasmids encoding nuclear EGFP plus nonsilencing (NS) shRNA or with EGFP plus p63 shRNA and either human ΔNp63α (hΔNp63α) or human TAp63α (hTAp63α) and analyzed immunocytochemically at 2 d for EGFP and cleaved caspase 3. Graphs in F show percentage cleaved caspase-3 (CC3)-positive-transfected cells from one representative experiment, whereas those in G show pooled data from three independent experiments expressed as fold-change relative to nonsilencing shRNA-transfected cells. Error bars indicate SEM. #p < 0.05; ##p < 0.001, Student's t test; *p < 0.01, ANOVA; n.s., not significant. Scale bar, 50 μm.
We have previously shown that TAp63 isoforms are death proteins for postmitotic neurons (Jacobs et al., 2005), whereas others have shown that ΔNp63 isoforms are prosurvival proteins for non-neural cell types (Liefer et al., 2000; Cheng et al., 2006; Carroll et al., 2007; Borrelli et al., 2009). To ask which of these two isoforms promotes cortical precursor survival, we initially overexpressed murine ΔNp63α versus TAp63α in cortical precursors and measured cell survival 2 d later. Immunostaining for EGFP and cleaved caspase-3 revealed that, whereas ΔNp63α had no effect on the basal levels of survival, TAp63α increased the number of apoptotic cells, indicating that it is a death protein in cortical precursors (Fig. 2D,E), as it is in other cell types. We then asked which of these two isoforms would rescue the enhanced cell death seen with p63 shRNA. To do this, cortical precursor cells were transfected with EGFP, p63 shRNA, and with human ΔNp63α or human TAp63α (both of which would not be targeted by the murine shRNA). Immunostaining of these cultures 2 d later for cleaved caspase-3 demonstrated that human ΔNp63α rescued the increased apoptosis caused by murine p63 shRNA, whereas TAp63α enhanced the level of cell death (Fig. 2F,G) Together, these data argue that it is ΔNp63 isoforms that are essential for cortical precursor cell survival.
p63 is important for survival but not proliferation in the embryonic cortex in vivo
To ask whether p63 is important for survival in the embryonic cortex, as it is in culture, we performed in utero electroporation to genetically manipulate murine cortical precursors in vivo (Barnabé-Heider et al., 2005; Paquin et al., 2005; Bartkowska et al., 2007; Gauthier et al., 2007). We previously showed that 24 h after electroporation of E13–14 embryos, the majority of electroporated cells reside in the VZ/SVZ and that ∼85% of these cells are proliferating precursors (Paquin et al., 2005). Over the subsequent 4 d, many of the electroporated cells differentiate into neurons, at which point they migrate out of the VZ/SVZ into the cortical plate. Of those precursors that remain, some differentiate into astrocytes and oligodendrocytes during the early postnatal period.
We therefore performed in utero electroporation at E13–14 with plasmids encoding a nuclear EGFP and either p63 shRNA or nonsilencing shRNA and analyzed the cortex 1 and 3 d after electroporation. Immunocytochemistry for EGFP at 1 d revealed that, as previously reported (Paquin et al., 2005), virtually all of the electroporated cells were localized to the VZ/SVZ in both control and p63 shRNA-electroporated brains (Fig. 3A). However, by 3 d, many fewer EGFP-positive cells were present within the cortices that were electroporated with p63 shRNA (Fig. 3B). Quantification of cell numbers at these two time points revealed that similar numbers of EGFP-positive cells were present at 1 d (E14–15) in both conditions but that by 3 d at E16–17, there were threefold to fourfold fewer cells in cortices electroporated with the p63 shRNA (Fig. 3C). This decrease in cell numbers was observed both in the cortical mantle and in the VZ/SVZ (Fig. 3D), suggesting that, as seen in culture, p63 knockdown led to death of both cortical precursors and newly born cortical neurons. To confirm these results, we electroporated a plasmid expressing Cre recombinase into the lateral ventricles of E13/14 p63fl/fl embryos and counted the number of EGFP-positive cells 3 d later. As observed with p63 shRNA, acute Cre recombinase-mediated p63 ablation resulted in ∼3-fold fewer EGFP-positive cells (Fig. 3C).
Figure 3.
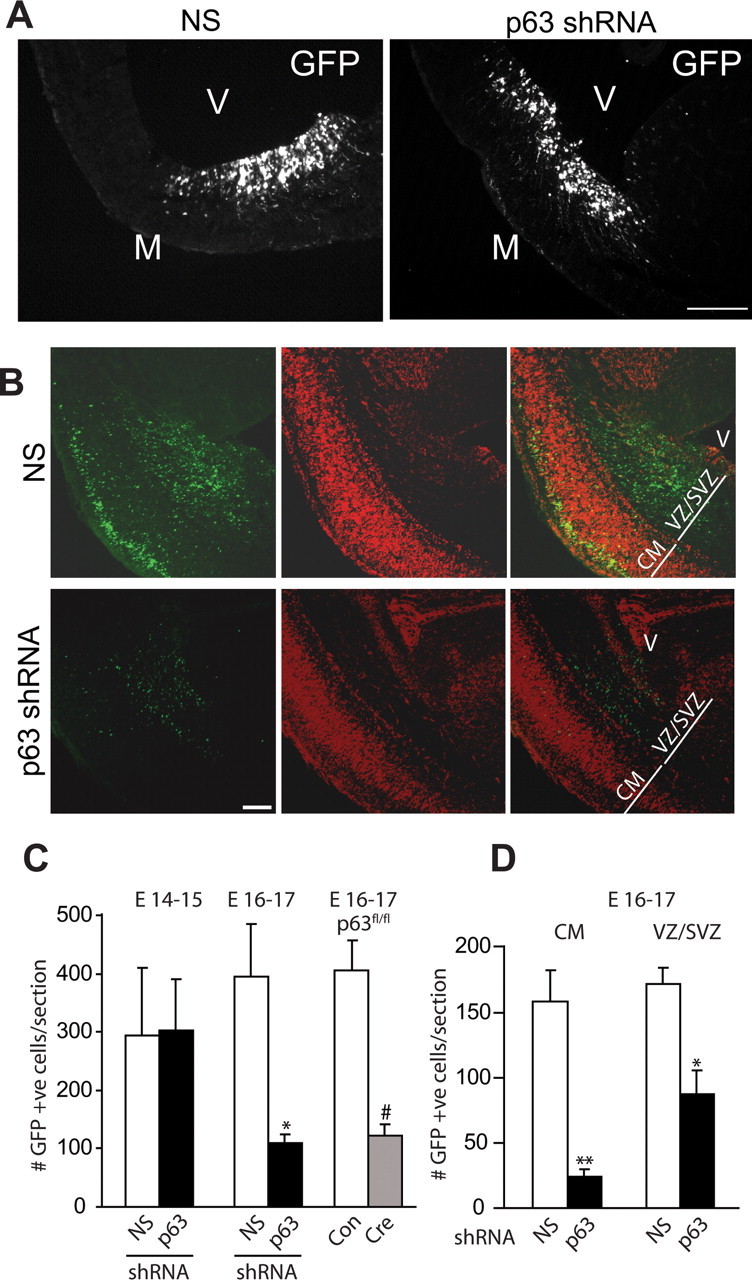
p63 is necessary for survival of neural precursors and newly born neurons within the embryonic cortex. Murine cortices were electroporated at E13/14 and analyzed 1–3 d later. A, Fluorescence micrographs of coronal sections through cortices cotransfected with EGFP and nonsilencing shRNA (NS; left) or p63 shRNA (right) at 1 d after electroporation. Sections were immunostained for EGFP (GFP). Note that the vast majority of transfected cells are localized to the VZ/SVZ region adjacent to the ventricles. M, Meninges; V, ventricle. B, Fluorescence micrographs as in A but at 3 d after electroporation. Note that in the control, nonsilencing shRNA-electroporated cortex, many transfected cells are located within both the cortical mantle and the VZ/SVZ at this time point. V, Ventricle; CM, cortical mantle. C, Wild-type cortices were electroporated with plasmids encoding nuclear EGFP and nonsilencing shRNA (NS) or p63 shRNA, or p63fl/fl cortices were electroporated with EGFP plus control empty vector (Con) or Cre recombinase (Cre), and the number of EGFP-positive cells within the entire cortex was quantified either 1 d later at E14–15, or 3 d later at E16–17. In all cases, results were pooled from three independent electroporations. For the left pair of bars, n = at least 6 animals per condition, 3–5 sections/embryo, for a total of 29 sections (7131 cells) and 24 sections (7125 cells) for nonsilencing and p63 shRNAs, respectively. For the middle pair of bars, n = 7 and 5 embryos with 2–7 sections/embryo, for a total of 32 sections (13,294 cells) and 18 sections (2103 cells) for nonsilencing and p63 shRNAs, respectively. For the right pair of bars, n = 4 and 3 embryos with 2–4 sections/embryo for a total of 12 sections (4875 cells) and 10 sections (1217 cells) for control and Cre-electroporated cortices, respectively. D, Quantification of sections similar to those in B for the number of EGFP-positive-transfected cells in the CM (left pair) and VZ/SVZ (right pair) at 3 d after electroporation. For the left pair, n = 7 and 5 embryos with 2–7 sections/embryo, for a total of 32 sections (5189 cells) and 18 sections (494 cells) for nonsilencing and p63 shRNAs, respectively. Results were pooled from three independent electroporations. For the right pair, n = 5 and 4 embryos with 2–7 sections/embryo for a total of 18 sections (2963 cells) and 15 sections (1357 cells) for nonsilencing and p63 shRNAs, respectively. Results are pooled from two independent electroporations. Error bars indicate SEM. *p < 0.05; **p < 0.01; #p < 0.0001, Student's t test; n.s., not significant. Scale bar, 100 μm.
Although these findings suggest that p63 silencing causes enhanced cell death in vivo as it does in culture, an alternative explanation is that it decreases cell number by decreasing precursor proliferation. To address this possibility, cultured cortical precursors were transfected with p63 shRNA, and cells were immunostained 2 d later for the proliferation marker Ki67 (Fig. 4A). This analysis demonstrated that p63 knockdown had no effect on precursor proliferation (Fig. 4B). Similar results were obtained when cultured p63fl/fl cortical precursors were transfected with Cre recombinase for 2 d (Fig. 4B). To confirm that p63 silencing also did not affect precursor proliferation in vivo, we performed immunocytochemistry for Ki67 on sections of cortices that were electroporated either 1 or 3 d earlier with p63 versus nonsilencing shRNAs. Confocal microscopy demonstrated that the percentage of Ki67-positive cells was similar in cortical precursors expressing p63 shRNA versus nonsilencing shRNA at both time points (Fig. 4C–E). Similar results were obtained when electroporated cortices were immunostained for a second marker of cell division, phosphohistone H3, at 3 d after electroporation (4 ± 1% vs 5 ± 1% for nonsilencing vs p63 shRNA, respectively; p > 0.05). Previously, we used this marker to show reduced proliferation in embryos electroporated with shRNA against TrkB (Bartkowska et al., 2007). Thus, p63 is essential for survival of cortical precursors and newly born neurons in vivo as it is in culture.
Figure 4.
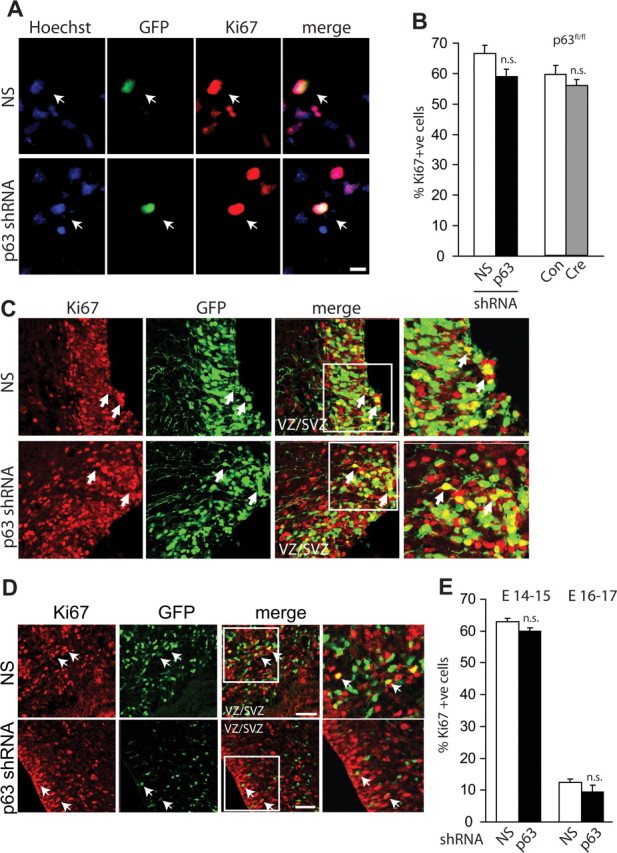
p63 is dispensable for proliferation of cortical precursor cells in vitro and in vivo. A, Cortical precursors were cotransfected with plasmids encoding nuclear EGFP and nonsilencing (NS) shRNA or p63 shRNA, cultured for 2 d, and then immunostained for EGFP (green, GFP) and Ki67 (red). Cells were counterstained with Hoechst 33258 (blue). The right panels show the merges. Arrows denote double-labeled cells. Scale bar, 10 μm. B, Wild-type cortical precursors were cotransfected with plasmids encoding nuclear EGFP and nonsilencing (NS) or p63 shRNA for 2 d before immunostaining as in A and quantitative analysis (left bars). Alternatively, p63fl/fl precursors were cotransfected with EGFP and Cre recombinase (Cre) or control vector (Con) and analyzed in the same way (right bars). In both cases, results indicate the percentage of Ki67-positive, transfected cells, pooled from three independent experiments each. C–E, E13/14 cortices were electroporated with plasmids encoding nuclear EGFP and nonsilencing shRNA (NS) or p63 shRNA and analyzed 1–3 d later. C, Confocal micrographs of coronal sections through cortices immunostained for EGFP (GFP, green) and Ki67 (Ki67, red) at 1 d. The center right panels show the merges. Arrows indicate transfected, Ki67-positive cells. Areas denoted by the boxes are shown at higher magnification in the right panels. D, Confocal micrographs as in C but at 3 d after electroporation. E, Quantification of the percentage of transfected, Ki67-positive cells in sections at 1 d (E14–15) or 3 d (E16–17) after electroporation as shown in C, D. In both cases, results are pooled from three independent electroporations. For the left bars, n = 6 embryos per condition, 3–5 sections per embryo for a total of 29 sections (3525 cells) and 24 sections (3796 cells) for nonsilencing and p63 shRNA-electroporated cortices, respectively. For the right bars, n = 5 and 6 embryos with 2–5 sections/embryo for a total of 32 sections (6164 cells) and 18 sections (1115 cells) for nonsilencing and p63 shRNA-electroporated cortices, respectively. Error bars indicate SEM. n.s., Not significant. Scale bar, 50 μm.
Coincident silencing of p53 rescues precursor cell death resulting from p63 knockdown
One way that the ΔNp63 and p73 isoforms support cell survival is by antagonizing the proapoptotic effects of p53 (Liefer et al., 2000; Lee et al., 2004; Jacobs et al., 2006; Pietsch et al., 2008). We therefore determined whether this was the case for p63 and cortical precursors. Initially, we asked whether p53 knockdown would affect basal levels of cortical precursor survival. Cultured precursors were cotransfected with plasmids encoding EGFP and a previously described p53 shRNA (Cam et al., 2006), and the percentage of EGFP-positive, cleaved caspase-3-positive cells was determined 2 d later. This analysis demonstrated that p53 knockdown had no effect on the low level of cell death present in these cultures (16 ± 2% vs 15 ± 2% in nonsilencing vs p53 shRNA transfected cells, respectively; p > 0.05). We then asked whether p53 knockdown would rescue the increased apoptosis seen when p63 was silenced. Precursors were cotransfected with EGFP, p63 shRNA, and either p53 shRNA or nonsilencing shRNA. Immunocytochemistry for cleaved caspase-3 2 d later demonstrated that p53 knockdown completely rescued the apoptosis induced by p63 shRNA (Fig. 5A–C), suggesting that ΔNp63 regulates survival of cortical precursor cells by antagonizing a p53-dependent pathway.
Figure 5.
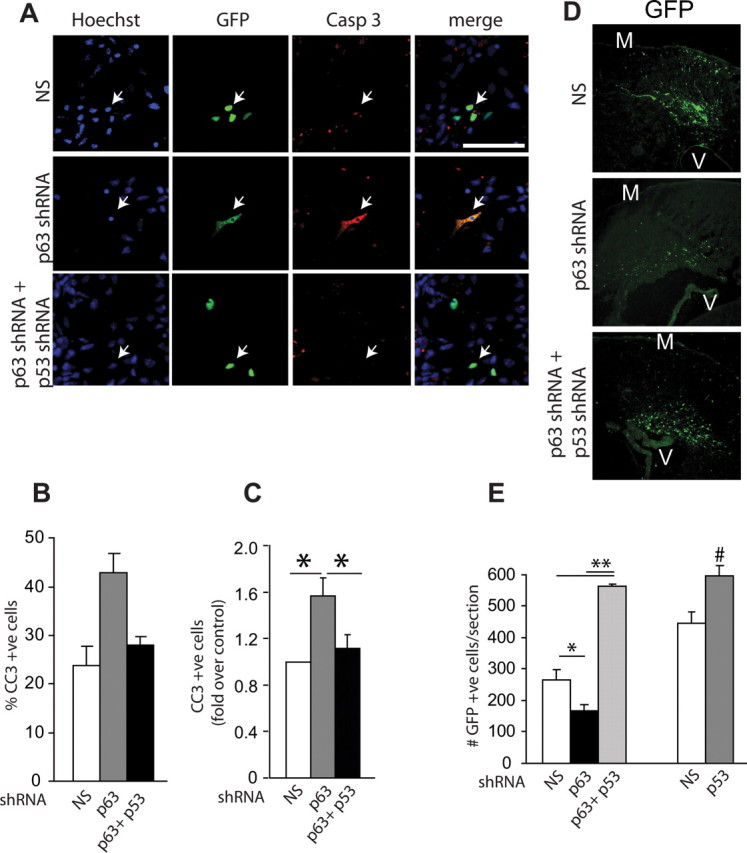
p63 antagonizes a p53-dependent pathway to regulate cortical precursor survival. A, Fluorescence micrographs of cortical precursor cultures cotransfected with plasmids encoding nuclear EGFP and nonsilencing shRNA, p63 shRNA, or both p63 shRNA and p53 shRNA and immunostained 2 d later for EGFP (green, GFP) and cleaved caspase-3 (Casp 3, red). Cells were counterstained with Hoechst 33258 (blue). The right panels show the merges. Arrows denote double-labeled cells. Scale bar, 100 μm. B, C, Quantification of the percentage of transfected, cleaved caspase-3-positive cells in experiments similar to those shown in A. The graph in B is from one representative experiment, whereas that in C shows pooled data from three independent experiments expressed as fold-change relative to nonsilencing shRNA-transfected cells. D, E, Left bars, E13/14 cortices were electroporated with plasmids encoding nuclear EGFP and nonsilencing shRNA (NS) or p63 shRNA plus or minus p53 shRNA and analyzed 3 d later. D, Confocal micrographs of coronal sections through cortices immunostained for EGFP (GFP, green) at 3 d. M, Meninges; V, ventricle. Scale bar, 100 μm. E, Left bars, Quantification of the number of transfected, EGFP-positive cells in sections similar to those shown in D. n = at least 3 animals per condition, 3–6 sections/embryo, for a total of 18 sections (4805 cells), 13 sections (2162 cells), and 11 sections (6166 cells) for cortices electroporated with nonsilencing shRNA, p63 shRNA, and p63 plus p53 shRNAs, respectively. Results are pooled from four independent electroporations. E, Right bars, E13/14 cortices were electroporated with plasmids encoding nuclear EGFP and nonsilencing shRNA (NS) or p53 shRNA and immunostained 3 d later for EGFP. The number of transfected, GFP-positive cells was quantified. n = at least 3 animals per condition, 2–5 sections/embryo for a total of 9 sections (4136 cells) and 33 sections (19450 cells) for nonsilencing and p53 shRNAs, respectively. Results are pooled from three independent electroporations. Error bars indicate SEM. *p < 0.05; **p < 0.01 based on ANOVA, #p < 0.05, Student's t test.
We then asked whether this was also the case in vivo; cortices were coelectroporated with plasmids encoding EGFP, p63 shRNA, and either nonsilencing shRNA or p53 shRNA, and the number of EGFP-positive cells were counted 3 d later. As predicted, p63 shRNA reduced the total number of EGFP-positive cells relative to nonsilencing shRNA and silencing of p53 rescued the loss of cells seen with p63 shRNA (Fig. 5D,E). However, silencing of p53 also apparently increased transfected cell survival over the normal basal levels (Fig. 5E). These data suggest that p53 regulates precursor cell survival in vivo, even when p63 levels are normal. To ask whether this was the case, we coelectroporated cortices with EGFP and either nonsilencing shRNA or p53 shRNA. Quantification of the number of EGFP-positive cells 3 d later confirmed that silencing of p53 increased the number of cortical precursor cells (Fig. 5E). Thus, precursors normally die via p53, and p63 promotes precursor survival by antagonizing this p53-dependent cell death.
ΔNp73 and p63 collaborate to promote survival of newly born neurons but not cortical precursor cells
Our data indicate that ΔNp63 is necessary for the survival of at least a subpopulation of cortical precursors and newly born cortical neurons, and we have previously shown that ΔNp73 is essential for the survival and maintenance of postnatal cortical neurons (Pozniak et al., 2002). To clarify the precise stages at which these two family members act, we silenced one versus the other in cultured cortical precursors. ΔNp73 mRNA is robustly expressed in newly born neurons of the cortical plate (Yang et al., 2000), but lower levels of ΔNp73 are also expressed in the cortex at E12.5, a time point when it is almost completely composed of neural precursors (Fig. 6A). We therefore first designed an shRNA that would selectively knockdown ΔNp73, but not TAp73 isoforms. This shRNA was cotransfected with a ΔNp73 expression construct that included the ΔNp73 5′UTR (which is targeted by the shRNA) into HEK293 cells, and the levels of ΔNp73 mRNA were quantified. Quantitative RT-PCR demonstrated that 48 h after transfection, the ΔNp73-specific shRNA was able to knockdown ΔNp73 mRNA (1 ± 0.3 vs 0.5 ± 0.2 in nonsilencing vs ΔNp73 shRNA transfected cells, respectively; p < 0.05). We then cotransfected cortical precursors with EGFP and ΔNp73 shRNA or p63 shRNA, and immunostained these cultures for EGFP and cleaved caspase-3 2 d later. This analysis demonstrated that ΔNp73 knockdown had no effect on precursor cell survival, whereas p63 knockdown decreased survival, as predicted (Fig. 6B,C). We next asked whether ΔNp73 knockdown had any effect on survival of newly born neurons; cultures were cotransfected with EGFP and ΔNp73 shRNA, and 3 d later, after substantial numbers of neurons were generated, were immunostained for EGFP and the neuronal marker βIII-tubulin. This analysis confirmed that ΔNp73 knockdown had no effect on the survival of cortical precursors, but that it did increase the number of apoptotic neurons by ∼2-fold (Fig. 6D–F).
Figure 6.
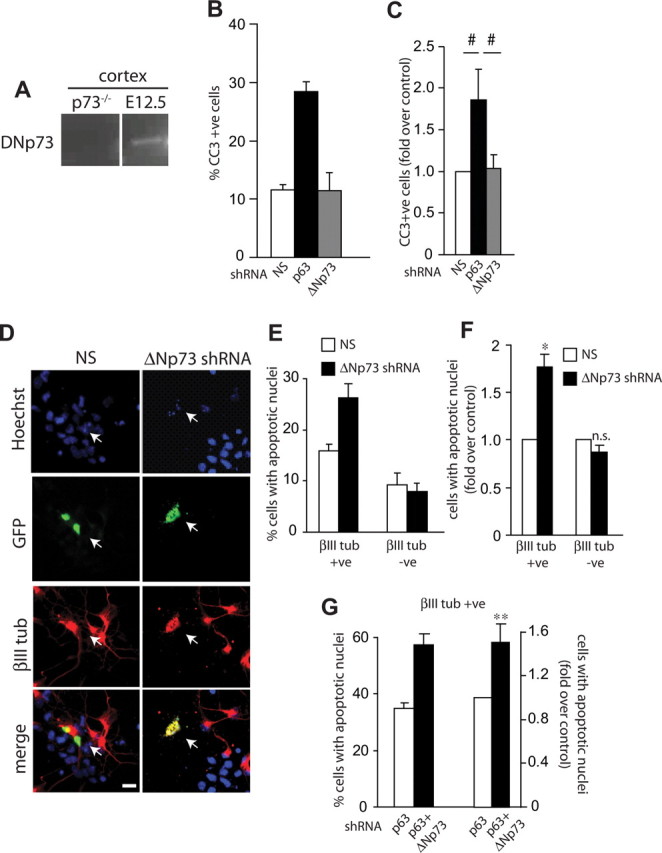
ΔNp73 collaborates with p63 to regulate survival of newly born neurons. A, RT-PCR analysis for ΔNp73 mRNA in E12.5 murine cortex from wild-type or p73−/− embryos. B, C, Cortical precursor cells were cotransfected with plasmids encoding nuclear EGFP and nonsilencing shRNA (NS), p63 shRNA, or ΔNp73 shRNA and immunostained 2 d later for cleaved caspase 3 (CC3)-positive cells. The graph in B is from one representative experiment, whereas that in C shows pooled data from three independent experiments expressed as fold-change relative to nonsilencing shRNA-transfected cells. D, Fluorescence micrographs of cortical precursor cultures cotransfected with plasmids encoding nuclear EGFP and nonsilencing (NS) shRNA or ΔNp73 shRNA and analyzed at 3 d for EGFP (green, GFP), βIII-tubulin (red, βIII tub), and counterstained with Hoechst 33258 (blue) to show all nuclei. The bottom panel shows the merges. Arrows denote double-labeled cells. Note that the ΔNp73 shRNA-transfected, βIII-tubulin-positive neuron is dying, as indicated by the apoptotic nuclear morphology. E, F, Quantification of the percentage of EGFP-positive cells that are βIII-tubulin-positive (neurons) or -negative (precursors) with condensed fragmented nuclei in experiments as shown in D. The graphs in E are from one representative experiment, whereas those in F show pooled data from three independent experiments expressed as fold-change relative to nonsilencing shRNA-transfected cells. G, Cortical precursors were cotransfected with plasmids encoding EGFP and p63 shRNA with or without ΔNp73 shRNA and immunostained 3 d later for EGFP and βIII tubulin. Cells were counterstained with Hoechst 33258 to quantitate apoptotic nuclear morphology. The percentage of EGFP-positive, βIII-tubulin-positive neurons with condensed fragmented nuclei was quantified. The left bars represent data from one representative experiment, whereas the right bars show pooled data from three independent experiments expressed as fold-change relative to nonsilencing shRNA-transfected cells. Error bars indicate SEM. *p < 0.05, **p < 0.01 Student's t test; #p < 0.05, one-way ANOVA; n.s., not significant. Scale bar, 10 μm.
These data indicate that p63, but not p73, is important for cortical precursor survival, but that both ΔNp63 and ΔNp73 support the survival of newly born cortical neurons. To ask whether the two collaborate to support neuronal survival, we cotransfected cultured cortical precursor cells with shRNAs for p63 and ΔNp73, and immunostained them 3 d later for βIII-tubulin and cleaved caspase-3. Quantification of these experiments demonstrated that coincident silencing of ΔNp73 and p63 increased neuronal apoptosis relative to silencing of p63 alone (Fig. 6G). Thus, ΔNp63 and ΔNp73 collaborate together to promote the survival of newly born neurons.
Discussion
The results from this study support three major conclusions. First, our experiments genetically manipulating p63 levels indicate that ΔNp63 promotes the survival of cortical precursor cells and newly born neurons in vitro and in vivo, an unexpected role for p63 in the embryonic neuroepithelium. These manipulations do not, however, affect precursor proliferation, indicating that the observed increase in apoptosis is a primary consequence of decreased p63 levels. Second, the acute genetic knockdown of p53 indicates that, surprisingly, p53 normally functions in the developmental apoptosis of cortical precursors, and that ΔNp63 promotes precursor survival by antagonizing p53. Finally, our experiments demonstrate that ΔNp73 is not essential for cortical precursor survival, but that, as we have shown previously (Pozniak et al., 2002; Wetzel et al., 2008), it promotes the survival of postmitotic cortical neurons, a function that it subserves during embryogenesis by collaborating with ΔNp63. Together, these data indicate that the balance of ΔNp63 versus p53 determines the life versus death of embryonic cortical precursors, and suggests that these p53 family members may well play a similar role in other populations of developing and/or adult neural precursors.
The number of precursor cells in the developing cortex is determined by the rate and extent of precursor cell proliferation, and by programmed cell death, both of which are required to generate the correct cell types and appropriate brain size. Precursor cell survival during embryogenesis is regulated by secreted factors that support cell survival and by proapoptotic mechanisms that eliminate cells with genomic instability or with errors in differentiation (Voyvodic, 1996; Haydar et al., 1999; Kuan et al., 2000; Buss et al., 2006). With regard to extracellular factors, CPG15 (Putz et al., 2005), erythropoietin (Yu et al., 2002), BDNF (Barnabé-Heider and Miller, 2003), and ephrins (Depaepe et al., 2005) have all been shown to promote the survival of cortical precursor cells. These environmental cues ultimately regulate a number of well characterized proapoptotic proteins, including caspase-3 (Kuida et al., 1996), caspase-9 (Kuida et al., 1996), Apaf-1 (Cecconi et al., 1998; Yoshida et al., 1998), bax, and bak (Lindsten et al., 2000, 2003), all of which play important prodeath roles in the embryonic brain. The importance of these apoptotic mechanisms is exemplified by mouse models of Down syndrome, where enhanced, deregulated apoptosis of precursor cells and neurons leads to microcephaly (Bambrick and Krueger, 1999; Haydar et al., 2000; Seidl et al., 2001; Kadota et al., 2002).
Despite this previous work, the signaling pathways that govern survival versus death decisions in neural precursors remain unclear. How does a precursor integrate various survival and apoptotic cues, and do these cues converge on a central signaling pathway? One such central, integrative pathway in both central and peripheral neurons involves the p53 family (for review, see Jacobs et al., 2006; Miller and Kaplan, 2007). In particular, ΔNp73 functions as a major neuronal survival and maintenance protein (Pozniak et al., 2000, 2002; Walsh et al., 2004), a function it fulfills at least partially by antagonizing full-length isoforms of p53 (Aloyz et al., 1998; Pozniak et al., 2000; Lee et al., 2004) and p63 (Jacobs et al., 2005). The importance of ΔNp73 has recently been emphasized by the finding that p73 haploinsufficiency leads to neuronal degeneration and accumulation of aberrantly phosphorylated tau in the brains of aging and Alzheimer's disease model mice (Wetzel et al., 2008), phenotypes that are at least partially caused by dysregulation of JNK activity (Lee et al., 2004; Wetzel et al., 2008). In contrast, the full-length TAp73 isoform has recently been shown to be critical not for cell survival, but for appropriate genesis of the hippocampus (Tomasini et al., 2008). Thus, positive cues converge on to the prosurvival protein ΔNp73, whereas stress signals converge on to the proapoptotic p53 family members, with the balance between the two ultimately determining neuronal life versus death. However, ΔNp73 is predominantly expressed in postmitotic neurons in the embryonic brain (Yang et al., 2000), indicating that it likely does not play a role in neural precursors, a conclusion supported by our findings here. Instead, we provide evidence that the relevant prosurvival family member for CNS neural precursors is ΔNp63. Interestingly, loss of ΔNp63 has previously been shown to result in apoptosis of epithelial stem cells, but this phenotype was thought to be secondary to defective proliferation (Senoo et al., 2007) or cell adhesion (Carroll et al., 2006). Our results raise the intriguing possibility that ΔNp63 is a key prosurvival protein in multiple stem cell populations, and that the dramatic epithelial cell phenotypes seen in p63−/− embryos (Mills et al., 1999; Yang et al., 1999) are at least partially directly attributable to the loss of this survival activity. Indeed, this has been proposed to be the case in the embryonic bladder epithelium (Cheng et al., 2006).
Our results indicate that p53 itself plays an important role in embryonic neural development, functioning as a proapoptotic protein for neural precursors during normal embryonic development. In this regard, previous work has shown that when either cell cycle regulation or DNA integrity are perturbed, p53 becomes the major proapoptotic protein in neural precursors, since deletion of p53 is sufficient to rescue the greatly increased apoptosis seen in mice lacking pRb (Macleod et al., 1996), DNA Ligase IV (Frank et al., 2000) or XRCC4 (Gao et al., 2000). Moreover, p53 is clearly important for neural precursor apoptosis induced by genotoxic stress (Akhtar et al., 2006a,b). However, a role for p53 in neural precursor apoptosis during normal development has not been previously demonstrated, although it was suggested by the observation that p53−/− mice exhibit a partially penetrant phenotype of exencephaly (Armstrong et al., 1995; Sah et al., 1995), a phenotype also observed after deletion of other key proapoptotic genes such as caspase 3 and caspase 9 (Kuida et al., 1996, 1998; Hakem et al., 1998). In fact, it is likely that p53 mediates precursor apoptosis via caspase 3 and caspase 9, as well as other previously defined p53 targets such as PUMA, Noxa, and bak/bax, since all of these proteins have been shown to be downstream of p53 during genotoxic stress of precursors (Akhtar et al., 2006a,b). In this regard, our data showing that ΔNp63 promotes survival of cortical precursors by antagonizing p53 infers that ΔNp63 promotes survival by inhibiting the intrinsic cell death pathways that involve these downstream proteins. Interestingly, the finding that adult SVZ stem cells from p53−/− mice display enhanced proliferation and reduced apoptosis (Meletis et al., 2006) suggests that this may be a role that ΔNp63 and p53 continue to play in neural precursors throughout their lifetime.
Our data showing that ΔNp73 is important for the survival of newly born cortical neurons, but not for cortical precursors, confirms previous work emphasizing the importance of this protein for neuronal survival (Pozniak et al., 2000, 2002; Walsh et al., 2004; Wetzel et al., 2008). In this regard, although our data indicate that ΔNp63 collaborates with ΔNp73 to promote survival of newly born cortical neurons, it is still unclear whether ΔNp63 is important throughout a neuron's lifetime, as is ΔNp73, or whether it only supports survival during the transition from a precursor to a postmitotic neuron. One clue that it might be important in mature neurons comes from p63+/− mice; these animals display premature aging, including aberrant circling behavior and reduced general fitness (Keyes et al., 2005). The possibility that ΔNp63 collaborates with ΔNp73 to maintain the longevity of mature CNS neurons becomes increasingly important in light of the recent demonstration that p73+/− mice display neurodegeneration and formation of phospho-tau filaments reminiscent of the tangles seen in Alzheimer's disease (Wetzel et al., 2008).
The signaling mechanisms that regulate the survival of neural precursors are poorly understood. Here, we have identified roles for the p53 family members in embryonic cortical precursors, and suggest that the balance between the different prosurvival and proapoptotic family members determines embryonic neural precursor survival. Whether or not ΔNp63 plays a similar role in survival of adult neural stem cells and/or in other populations of stem cells outside the nervous system is a key question for the future.
Footnotes
This work was supported by Canadian Institutes of Health Research (CIHR) Grants MOP 13958 and MGP 38021 to F.D.M. and D.R.K. F.D.M. is a Howard Hughes Medical Institute International Research Scholar and a Canada Research Chair, and D.R.K. is a Canada Research Chair. C.B.D. was supported by Natural Sciences and Engineering Research Council of Canada and a CIHR MD/PhD Scholarship, A.P. by scholarships from the Ontario Government and the Hospital for Sick Children, and M.F. by a fellowship from the Japan Society for the Promotion of Science. We are grateful to Dr. Thorsten Stiewe (Philipps-University Marburg, Germany) for the generous gifts of shRNAs against p63, p53, and p73. We thank Alea Mills for the kind gift of the floxed p63 mice, Meredith Irwin for her advice, and members of the Kaplan–Miller laboratories, in particular Dennis Aquino, for their input and assistance.
References
- Akhtar RS, Geng Y, Klocke BJ, Latham CB, Villunger A, Michalak EM, Strasser A, Carroll SL, Roth KA. BH3-only proapoptotic Bcl-2 family members Noxa and Puma mediate neural precursor cell death. J Neurosci. 2006a;26:7257–7264. doi: 10.1523/JNEUROSCI.0196-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar RS, Geng Y, Klocke BJ, Roth KA. Neural precursor cells possess multiple p53-dependent apoptotic pathways. Cell Death Differ. 2006b;13:1727–1739. doi: 10.1038/sj.cdd.4401879. [DOI] [PubMed] [Google Scholar]
- Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, Miller FD. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol. 1998;143:1691–1703. doi: 10.1083/jcb.143.6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong JF, Kaufman MH, Harrison DJ, Clarke AR. High-frequency developmental abnormalities in p53-deficient mice. Curr Biol. 1995;5:931–936. doi: 10.1016/s0960-9822(95)00183-7. [DOI] [PubMed] [Google Scholar]
- Bambrick LL, Krueger BK. Neuronal apoptosis in mouse trisomy 16: mediation by caspases. J Neurochem. 1999;72:1769–1772. doi: 10.1046/j.1471-4159.1999.721769.x. [DOI] [PubMed] [Google Scholar]
- Barnabé-Heider F, Miller FD. Endogenously produced neurotrophins regulate survival and differentiation of cortical progenitors via distinct signaling pathways. J Neurosci. 2003;23:5149–5160. doi: 10.1523/JNEUROSCI.23-12-05149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnabé-Heider F, Wasylnka JA, Fernandes KJ, Porsche C, Sendtner M, Kaplan DR, Miller FD. Evidence that embryonic neurons regulate the onset of cortical gliogenesis via cardiotrophin-1. Neuron. 2005;48:253–265. doi: 10.1016/j.neuron.2005.08.037. [DOI] [PubMed] [Google Scholar]
- Bartkowska K, Paquin A, Gauthier AS, Kaplan DR, Miller FD. Trk signaling regulates neural precursor cell proliferation and differentiation during cortical development. Development. 2007;134:4369–4380. doi: 10.1242/dev.008227. [DOI] [PubMed] [Google Scholar]
- Blaschke AJ, Staley K, Chun J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development. 1996;122:1165–1174. doi: 10.1242/dev.122.4.1165. [DOI] [PubMed] [Google Scholar]
- Blaschke AJ, Weiner JA, Chun J. Programmed cell death is a universal feature of embryonic and postnatal neuroproliferative regions throughout the central nervous system. J Comp Neurol. 1998;396:39–50. doi: 10.1002/(sici)1096-9861(19980622)396:1<39::aid-cne4>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Borrelli S, Candi E, Alotto D, Castagnoli C, Melino G, Viganò MA, Mantovani R. p63 regulates the caspase-8-FLIP apoptotic pathway in epidermis. Cell Death Differ. 2009;16:253–263. doi: 10.1038/cdd.2008.147. [DOI] [PubMed] [Google Scholar]
- Buss RR, Sun W, Oppenheim RW. Adaptive roles of programmed cell death during nervous system development. Annu Rev Neurosci. 2006;29:1–35. doi: 10.1146/annurev.neuro.29.051605.112800. [DOI] [PubMed] [Google Scholar]
- Cam H, Griesmann H, Beitzinger M, Hofmann L, Beinoraviciute-Kellner R, Sauer M, Hüttinger-Kirchhof N, Oswald C, Friedl P, Gattenlöhner S, Burek C, Rosenwald A, Stiewe T. p53 family members in myogenic differentiation and rhabdomyosarcoma development. Cancer Cell. 2006;10:281–293. doi: 10.1016/j.ccr.2006.08.024. [DOI] [PubMed] [Google Scholar]
- Carroll DK, Carroll JS, Leong CO, Cheng F, Brown M, Mills AA, Brugge JS, Ellisen LW. p63 regulates an adhesion programme and cell survival in epithelial cells. Nat Cell Biol. 2006;8:551–561. doi: 10.1038/ncb1420. [DOI] [PubMed] [Google Scholar]
- Carroll DK, Brugge JS, Attardi LD. p63, cell adhesion and survival. Cell Cycle. 2007;6:255–261. doi: 10.4161/cc.6.3.3799. [DOI] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- Cheng W, Jacobs WB, Zhang JJ, Moro A, Park JH, Kushida M, Qiu W, Mills AA, Kim PC. DeltaNp63 plays an anti-apoptotic role in ventral bladder development. Development. 2006;133:4783–4792. doi: 10.1242/dev.02621. [DOI] [PubMed] [Google Scholar]
- Depaepe V, Suarez-Gonzalez N, Dufour A, Passante L, Gorski JA, Jones KR, Ledent C, Vanderhaeghen P. Ephrin signalling controls brain size by regulating apoptosis of neural progenitors. Nature. 2005;435:1244–1250. doi: 10.1038/nature03651. [DOI] [PubMed] [Google Scholar]
- Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, Manis JP, Horner J, DePinho RA, Alt FW. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000;5:993–1002. doi: 10.1016/s1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- Gao Y, Ferguson DO, Xie W, Manis JP, Sekiguchi J, Frank KM, Chaudhuri J, Horner J, DePinho RA, Alt FW. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. 2000;404:897–900. doi: 10.1038/35009138. [DOI] [PubMed] [Google Scholar]
- Gauthier AS, Furstoss O, Araki T, Chan R, Neel BG, Kaplan DR, Miller FD. Control of CNS cell-fate decisions by SHP-2 and its dysregulation in Noonan syndrome. Neuron. 2007;54:245–262. doi: 10.1016/j.neuron.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- Haydar TF, Kuan CY, Flavell RA, Rakic P. The role of cell death in regulating the size and shape of the mammalian forebrain. Cereb Cortex. 1999;9:621–626. doi: 10.1093/cercor/9.6.621. [DOI] [PubMed] [Google Scholar]
- Haydar TF, Nowakowski RS, Yarowsky PJ, Krueger BK. Role of founder cell deficit and delayed neuronogenesis in microencephaly of the trisomy 16 mouse. J Neurosci. 2000;20:4156–4164. doi: 10.1523/JNEUROSCI.20-11-04156.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs WB, Govoni G, Ho D, Atwal JK, Barnabé-Heider F, Keyes WM, Mills AA, Miller FD, Kaplan DR. p63 is an essential proapoptotic protein during neural development. Neuron. 2005;48:743–756. doi: 10.1016/j.neuron.2005.10.027. [DOI] [PubMed] [Google Scholar]
- Jacobs WB, Kaplan DR, Miller FD. The p53 family in nervous system development and disease. J Neurochem. 2006;97:1571–1584. doi: 10.1111/j.1471-4159.2006.03980.x. [DOI] [PubMed] [Google Scholar]
- Kadota M, Shirayoshi Y, Oshimura M. Elevated apoptosis in pre-mature neurons differentiated from mouse ES cells containing a single human chromosome 21. Biochem Biophys Res Commun. 2002;299:599–605. doi: 10.1016/s0006-291x(02)02686-4. [DOI] [PubMed] [Google Scholar]
- Keyes WM, Wu Y, Vogel H, Guo X, Lowe SW, Mills AA. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev. 2005;19:1986–1999. doi: 10.1101/gad.342305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan CY, Roth KA, Flavell RA, Rakic P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 2000;23:291–297. doi: 10.1016/s0166-2236(00)01581-2. [DOI] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Lee AF, Ho DK, Zanassi P, Walsh GS, Kaplan DR, Miller FD. Evidence that DeltaNp73 promotes neuronal survival by p53-dependent and p53-independent mechanisms. J Neurosci. 2004;24:9174–9184. doi: 10.1523/JNEUROSCI.1588-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liefer KM, Koster MI, Wang XJ, Yang A, McKeon F, Roop DR. Down-regulation of p63 is required for epidermal UV-B-induced apoptosis. Cancer Res. 2000;60:4016–4020. [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten T, Golden JA, Zong WX, Minarcik J, Harris MH, Thompson CB. The proapoptotic activities of Bax and Bak limit the size of the neural stem cell pool. J Neurosci. 2003;23:11112–11119. doi: 10.1523/JNEUROSCI.23-35-11112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod KF, Hu Y, Jacks T. Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J. 1996;15:6178–6188. [PMC free article] [PubMed] [Google Scholar]
- Meletis K, Wirta V, Hede SM, Nistér M, Lundeberg J, Frisén J. p53 suppresses the self-renewal of adult neural stem cells. Development. 2006;133:363–369. doi: 10.1242/dev.02208. [DOI] [PubMed] [Google Scholar]
- Ménard C, Hein P, Paquin A, Savelson A, Yang XM, Lederfein D, Barnabé-Heider F, Mir AA, Sterneck E, Peterson AC, Johnson PF, Vinson C, Miller FD. An essential role for a MEK-C/EBP pathway during growth factor-regulated cortical neurogenesis. Neuron. 2002;36:597–610. doi: 10.1016/s0896-6273(02)01026-7. [DOI] [PubMed] [Google Scholar]
- Migliorini D, Lazzerini Denchi E, Danovi D, Jochemsen A, Capillo M, Gobbi A, Helin K, Pelicci PG, Marine JC. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol. 2002;22:5527–5538. doi: 10.1128/MCB.22.15.5527-5538.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FD, Gauthier AS. Timing is everything: making neurons versus glia in the developing cortex. Neuron. 2007;54:357–369. doi: 10.1016/j.neuron.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR. To die or not to die: neurons and p63. Cell Cycle. 2007;6:312–317. doi: 10.4161/cc.6.3.3795. [DOI] [PubMed] [Google Scholar]
- Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398:708–713. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- Mills AA, Qi Y, Bradley A. Conditional inactivation of p63 by Cre-mediated excision. Genesis. 2002;32:138–141. doi: 10.1002/gene.10067. [DOI] [PubMed] [Google Scholar]
- Paquin A, Barnabé-Heider F, Kageyama R, Miller FD. CCAAT/enhancer-binding protein phosphorylation biases cortical precursors to generate neurons rather than astrocytes in vivo. J Neurosci. 2005;25:10747–10758. doi: 10.1523/JNEUROSCI.2662-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietsch EC, Sykes SM, McMahon SB, Murphy ME. The p53 family and programmed cell death. Oncogene. 2008;27:6507–6521. doi: 10.1038/onc.2008.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science. 2000;289:304–306. doi: 10.1126/science.289.5477.304. [DOI] [PubMed] [Google Scholar]
- Pozniak CD, Barnabé-Heider F, Rymar VV, Lee AF, Sadikot AF, Miller FD. p73 is required for survival and maintenance of CNS neurons. J Neurosci. 2002;22:9800–9809. doi: 10.1523/JNEUROSCI.22-22-09800.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putz U, Harwell C, Nedivi E. Soluble CPG15 expressed during early development rescues cortical progenitors from apoptosis. Nat Neurosci. 2005;8:322–331. doi: 10.1038/nn1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T. A subset of p53-deficient embryos exhibit exencephaly. Nat Genet. 1995;10:175–180. doi: 10.1038/ng0695-175. [DOI] [PubMed] [Google Scholar]
- Seidl R, Cairns N, Lubec G. The brain in Down syndrome. J Neural Transm Suppl. 2001;61:247–261. doi: 10.1007/978-3-7091-6262-0_20. [DOI] [PubMed] [Google Scholar]
- Senoo M, Pinto F, Crum CP, McKeon F. p63 is essential for the proliferative potential of stem cells in stratified epithelia. Cell. 2007;129:523–536. doi: 10.1016/j.cell.2007.02.045. [DOI] [PubMed] [Google Scholar]
- Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Lochmüller H, Aloyz R, Haghighi A, Lach B, Seth P, Cooper E, Miller FD. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J Cell Biol. 1996;135:1085–1096. doi: 10.1083/jcb.135.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomaidou D, Mione MC, Cavanagh JF, Parnavelas JG. Apoptosis and its relation to the cell cycle in the developing cerebral cortex. J Neurosci. 1997;17:1075–1085. doi: 10.1523/JNEUROSCI.17-03-01075.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toma JG, El-Bizri H, Barnabé-Heider F, Aloyz R, Miller FD. Evidence that helix-loop-helix proteins collaborate with retinoblastoma tumor suppressor protein to regulate cortical neurogenesis. J Neurosci. 2000;20:7648–7656. doi: 10.1523/JNEUROSCI.20-20-07648.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC, Khan F, Itie-Youten A, Wakeham A, Tsao MS, Iovanna JL, Squire J, Jurisica I, Kaplan D, Melino G, Jurisicova A, Mak TW. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008;22:2677–2691. doi: 10.1101/gad.1695308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai KY, Hu Y, Macleod KF, Crowley D, Yamasaki L, Jacks T. Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol Cell. 1998;2:293–304. doi: 10.1016/s1097-2765(00)80274-9. [DOI] [PubMed] [Google Scholar]
- Voyvodic JT. Cell death in cortical development: How much? Why? So what? Neuron. 1996;16:693–696. doi: 10.1016/s0896-6273(00)80089-6. [DOI] [PubMed] [Google Scholar]
- Walsh GS, Orike N, Kaplan DR, Miller FD. The invulnerability of adult neurons: a critical role for p73. J Neurosci. 2004;24:9638–9647. doi: 10.1523/JNEUROSCI.1299-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel MK, Naska S, Laliberté CL, Rymar VV, Fujitani M, Biernaskie JA, Cole CJ, Lerch JP, Spring S, Wang SH, Frankland PW, Henkelman RM, Josselyn SA, Sadikot AF, Miller FD, Kaplan DR. p73 regulates neurodegeneration and phospho-tau accumulation during aging and Alzheimer's disease. Neuron. 2008;59:708–721. doi: 10.1016/j.neuron.2008.07.021. [DOI] [PubMed] [Google Scholar]
- Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dötsch V, Andrews NC, Caput D, McKeon F. p63, a p53 homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell. 1998;2:305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, McKeon F. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, Vagner C, Bonnet H, Dikkes P, Sharpe A, McKeon F, Caput D. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404:99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/s0092-8674(00)81733-x. [DOI] [PubMed] [Google Scholar]
- Yu X, Shacka JJ, Eells JB, Suarez-Quian C, Przygodzki RM, Beleslin-Cokic B, Lin CS, Nikodem VM, Hempstead B, Flanders KC, Costantini F, Noguchi CT. Erythropoietin receptor signalling is required for normal brain development. Development. 2002;129:505–516. doi: 10.1242/dev.129.2.505. [DOI] [PubMed] [Google Scholar]