

Abstract
Alzheimer's disease (AD) is characterized by memory impairment, neurochemically by accumulation of β-amyloid peptide (namely Aβ1-42) and morphologically by an initial loss of nerve terminals. Caffeine consumption prevents memory dysfunction in different models, which is mimicked by antagonists of adenosine A2A receptors (A2ARs), which are located in synapses. Thus, we now tested whether A2AR blockade prevents the early Aβ1-42-induced synaptotoxicity and memory dysfunction and what are the underlying signaling pathways. The intracerebral administration of soluble Aβ1-42 (2 nmol) in rats or mice caused, 2 weeks later, memory impairment (decreased performance in the Y-maze and object recognition tests) and a loss of nerve terminal markers (synaptophysin, SNAP-25) without overt neuronal loss, astrogliosis, or microgliosis. These were prevented by pharmacological blockade [5-amino-7-(2-phenylethyl)-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine (SCH58261); 0.05 mg · kg−1 · d−1, i.p.; for 15 d] in rats, and genetic inactivation of A2ARs in mice. Moreover, these were synaptic events since purified nerve terminals acutely exposed to Aβ1-42 (500 nm) displayed mitochondrial dysfunction, which was prevented by A2AR blockade. SCH58261 (50 nm) also prevented the initial synaptotoxicity (loss of MAP-2, synaptophysin, and SNAP-25 immunoreactivity) and subsequent loss of viability of cultured hippocampal neurons exposed to Aβ1-42 (500 nm). This A2AR-mediated control of neurotoxicity involved the control of Aβ1-42-induced p38 phosphorylation and was independent from cAMP/PKA (protein kinase A) pathway. Together, these results show that A2ARs play a crucial role in the development of Aβ-induced synaptotoxicity leading to memory dysfunction through a p38 MAPK (mitogen-activated protein kinase)-dependent pathway and provide a molecular basis for the benefits of caffeine consumption in AD.
Introduction
Alzheimer's disease (AD) is the most common chronic neurodegenerative disease and is clinically characterized by a progressive impairment of cognitive functions such as learning and memory. Although the traditional neuropathologic hallmarks of AD are the presence of neurofibrillary tangles and the accumulation of the senile plaques resulting from β-amyloid peptide (Aβ) aggregation, the neurochemical parameter best correlated with memory dysfunction in AD is the levels of soluble Aβ, mainly Aβ1-42 (Selkoe, 2001). Also, the earliest morphological trait and the best correlated with initial memory impairment in AD is the loss of synapses in the limbic cortex, namely in the hippocampus (Coleman et al., 2004). In fact, synapses seem to be the primordial target of toxic Aβ oligomers, with the resulting synaptic failure underlying memory impairment in AD (Hardy and Selkoe, 2002; Klein et al., 2004). Thus, the early Aβ1-42-induced synaptotoxicity and associated mechanisms constitute major targets in the development of novel therapeutic strategies for AD.
Adenosine modulates synaptic transmission through inhibitory A1 or facilitatory A2A receptors (A2ARs), both of which are predominantly located in synapses, namely in the limbic and neocortex (Fredholm et al., 2005). Given the ability of A1Rs to inhibit calcium entry into neurons, glutamate release, and NMDA receptor activation, A1Rs have been considered promising candidate targets to prevent neuronal damage. However, their rapid downregulation and functional desensitization after insults limits their neuroprotective potential (de Mendonça et al., 2000). More recently, major interest has been devoted to A2ARs since their blockade affords neuroprotection against chronic insults in the adult brain (Cunha, 2005; Chen et al., 2007), which also trigger major increases in the extracellular levels of adenosine (de Mendonça et al., 2000). This is in notable agreement with the ability of caffeine (a nonselective adenosine receptor antagonist) to protect against cognitive impairment in different animal models, an effect that mainly seems to involve A2ARs (for review, see Cunha, 2008b; Takahashi et al., 2008). Likewise, caffeine consumption inversely correlates with the incidence of AD (Maia and de Mendonça, 2002) and prevents memory impairment in animal models of AD (Arendash et al., 2006; Dall'Igna et al., 2007), an effect mimicked by selective A2AR antagonists (Dall'Igna et al., 2007). Interestingly, A2AR blockade selectively prevented Aβ-induced, but not scopolamine- or dizocilpine maleate (MK801)-induced, memory impairment (Cunha et al., 2008). Notably, memory impairment by Aβ (but not scopolamine or MK801) involves synaptotoxicity. This suggests that A2AR blockade prevents memory impairment by selectively controlling synaptotoxicity, which would provide a molecular basis to support a neuroprotective action of A2ARs.
The present study tested the ability of A2ARs to prevent Aβ1-42-induced synaptotoxicity and memory impairment and investigated the underlying mechanisms. Results show that A2AR blockade (pharmacologic or genetic) prevents Aβ1-42-induced synaptotoxicity and subsequent memory dysfunction by a mechanism involving the control of the p38 mitogen-activated protein kinase (MAPK) pathway.
Materials and Methods
Animals.
Wistar rats (8–10 week males) were from Charles River. C57BL/6 mice (8–10 week males), both wild-type (WT) and A2AR knock-out (KO), were generously provided by Jiang-Fan Chen (University of Boston, Boston, MA). Animals were maintained under controlled environment (23 ± 2°C; 12 h light/dark cycle; ad libitum access to food and water) and handled according to European Union guidelines (86/609/EEC). Behavioral experiments were conducted between 10:00 A.M. and 4:00 P.M.
Analysis of β-amyloid peptides and in vivo administration procedures.
The β-amyloid (1-42) peptide fragment (Aβ1-42) or the nonamyloidogenic reverse peptide Aβ42-1 (Aβ42-1) was dissolved in water at a concentration of 2.25 mg/ml and 2 nmol in 4 μl was administered intracerebroventricularly, as previously described (Dall'Igna et al., 2007). Control animals were intracerebroventricularly infused with a similar volume of water. Behavioral analysis was performed 2 or 15 d after Aβ1-42 or Aβ42-1 administration. The selective A2AR antagonist 5-amino-7-(2-phenylethyl)-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo [1,5-c]pyrimidine (SCH58261) (generously provided by Scott Weiss, Vernalis, Wokingham, UK) was injected intraperitoneally at an efficacious dose (0.05 mg/kg of SCH58261) (Cunha et al., 2006, 2008; Dall'Igna et al., 2007), in saline (0.9% sodium chloride) with 10% dimethylsulfoxide, applied daily starting 30 min before Aβ1-42 administration. Control animals were injected intraperitoneally with saline with 10% dimethylsulfoxide.
The qualitative analysis of the oligomerization status of the Aβ peptide solution was evaluated by Western blot analysis using the 6E10 antibody that recognizes different human Aβ homomeric forms, as previously described (Evans et al., 2008). Briefly, 10 μl of the different batches of Aβ solutions was mixed with sample buffer (40% glycerol, 2% SDS, 0.2 m Tris-HCl, pH 6.8, and 0.005% Coomassie blue) and analyzed by electrophoresis (40 mA for 3 h and 30 min) using a tricine running buffer (Gibson et al., 2004). The blots were revealed with Coomassie blue (using a Coomassie blue R-250 solution made of 40% methanol, 10% acetic acid, and 0.1% Coomassie blue R-250 for 30 min, followed by destaining with 40% methanol and 10% acetic acid) or with 6E10 antibody (1:1000 dilution; Covance), as described below (see Western blot analysis).
The Aβ1-42 levels in the hippocampus were quantified using two ELISA kits (Invitrogen), one detecting Aβ1-42 (and isoforms with lower length) and the other Aβ1-40, as previously described (Cao et al., 2009). Briefly, one hippocampus was homogenized in RIPA buffer (100 mm Tris, pH 8.0, 150 mm NaCl, 0.5% deoxycholate, 1% IGEPAL, 0.2% SDS, and protease inhibitor mixture containing leupeptin, pepstatin A, chymostatin, and aprotinin, all 1 mg/ml from Sigma-Aldrich). The mixture was centrifuged (30 min at 27,000 × g) and the supernatant was stored at −80°C until ELISA quantifications, which were performed following the manufacturer's instructions. Aβ1-42 levels were estimated by subtracting the estimated amount of Aβ1-40 from those of Aβ1-42 and were normalized by tissue weight and/or amount of protein, determined with the bicinchoninic acid (BCA) method (Pierce Biotechnology).
The detection of Aβ aggregates in the hippocampus was performed using Congo Red (Puchtler et al., 1985) or Thioflavin-S histochemical analysis (Reyes et al., 2004) of hippocampal sections (see below), as previously described (Melo et al., 2009).
Behavioral analysis.
Locomotor activity was monitored in an open-field arena (50 × 50 cm, divided in four squares of 25 cm for rats, and 30 × 30 cm, divided in nine squares for mice, respectively), and the exploratory behavior of the animals was evaluated by counting the total number of line crossings and the number of rearings over a 5 min period. Hippocampal-dependent memory performance was assessed by measuring spontaneous alternation performance during 8 min in the Y-maze test, which allows evaluating cognitive searching behavior, although it does not allow isolating memory performance (for review, see Hughes, 2004). The series of arm entries was recorded visually and an alternation was defined as entries in all three arms on consecutive occasions. The percentage of alternation was calculated as follows: total of alternations/(total arm entries − 2), as previously described (Dall'Igna et al., 2007). Memory performance was also evaluated using the object recognition test consisting of two 3 min sessions (24 h after habituation): the first with two identical objects (training session) and the second (test session, 30 min after) with two dissimilar objects (a familiar and a novel one); recognition object index was calculated by the ratio of the time spent exploring novel object over the total exploration time of both objects, as previously described (Costa et al., 2008b). The experimenter conducting behavioral analysis was blinded to treatment conditions.
Histochemistry and immunohistochemistry.
Brain fixation was performed through transcardiac perfusion with 4% paraformaldehyde (in 0.9% sodium chloride and 4% sucrose), as previously described (Cunha et al., 2006). Frozen brain were sectioned (20 μm coronal slices) with a Leica CM1850 cryostat (Leica Microsystems), mounted on slides coated with 2% gelatin with 0.08% chromalin (chromium and potassium sulfate), allowed to dry at room temperature, and stored at −20°C until use.
Neuronal morphology in hippocampal sections was evaluated by cresyl violet staining of Nissl bodies, as previously described (Lopes et al., 2003). Briefly, sections were incubated for 10 min with cresyl violet (Sigma-Aldrich) solution (0.5% in acetate buffer). Sections were then washed twice with acetate buffer, twice in 100% ethanol, cleared with xylene, and mounted with Vector medium (Vector Laboratories). Degenerating neurons were detected using Fluoro-Jade C, which fluorescently labels them independently of the mechanism of cell death (Schmued et al., 2005). We used a 0.0001% solution of Fluoro-Jade C (Histo-Chem), as previously described (Cunha et al., 2006).
Detection of nerve terminals was performed as previously described (Cunha et al., 2006), using immunohistochemical detection of synaptophysin, a protein located in synaptic vesicles (Masliah and Terry, 1993). Immunohistochemistry detection of CD11b (a marker of microglia) (Jensen et al., 1997) and of glial fibrillary acidic protein (GFAP) (a marker of astrocytes) (Pekny and Nilsson, 2005) was performed to evaluated microgliosis and astrogliosis, respectively. The sections were first rinsed for 5 min with PBS (140 mm NaCl, 3 mm KCl, 20 mm Na2HPO4, 1.5 mm KH2PO4) and then three times for 5 min with Trizma base solution (TBS) (0.05 m containing 150 mm NaCl, pH 7.2) at room temperature. Sections were then permeabilized and blocked with TBS containing 0.2% Triton X-100 and 10% goat serum during 45 min, incubated in the presence of the mouse anti-synaptophysin antibody (1:500) or rat anti-CD11b (1:600; Serotec) or anti-GFAP-Cy3 (1:500; Sigma-Aldrich) for 72 h at 4°C, rinsed three times for 10 min in TBS, and subsequently incubated with goat anti-mouse or goat anti-rat secondary antibody conjugated with a fluorophore (Alexa Fluor 488; Invitrogen) (1:100) for 2 h at room temperature. After rinsing twice for 10 min in TBS and once for 10 min in distilled water, the sections were dehydrated and passed through xylene before mounting on slides, using Vectashield mounting medium (Vector Laboratories).
All sections were examined under a transmission and fluorescence Zeiss Axiovert 200 microscope, with AxioVision software 4.6 (PG-HITEC).
Assays in hippocampal synaptosomes.
Synaptosomes (i.e., enriched nerve terminals) were prepared from the hippocampus using a sucrose/Percoll-based series of centrifugations, as previously described (Rebola et al., 2005). Briefly, the two hippocampi from one animal were homogenized at 4°C in sucrose solution (0.32 m) containing 1 mm EDTA, 10 mm HEPES, 1 mg/ml bovine serum albumin (BSA), and 1 mm dithiothreitol (DTT), pH 7.6, centrifuged at 3000 × g for 10 min at 4°C, the supernatants were collected and centrifuged at 14,000 × g for 12 min at 4°C, and the pellet was resuspended in 1 ml of a 45% (v/v) Percoll solution in Krebs' buffer (140 mm NaCl, 5 mm KCl, 25 mm HEPES, 1 mm EDTA, 10 mm glucose, pH 7.4). After centrifugation at 14,000 × g for 2 min at 4°C, the top layer was removed (synaptosomal fraction) and washed in 1 ml of Krebs' buffer. Protein determination was performed with the BCA method.
The redox status of synaptosomes, known to be affected by exposure to β-amyloid peptides (Mattson et al., 1998), was measured by a colorimetric assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich), as previously described (Silva et al., 2007). Synaptosomes were incubated for 2 h at 37°C in Krebs' buffer in the absence or presence of Aβ1-42 (500 nm) and/or SCH58261 (50 nm). MTT (0.5 mg/ml) was then added and incubated for 1 h at 37°C in the dark. As MTT is converted to a water-insoluble blue product (formazan) by viable terminals, the precipitated dye can be spectrophotometrically (570 nm) quantified after exposing synaptosomes to isopropanol containing 0.04 m HCl. Values were expressed as the percentage of optical density of control synaptosomes, in the absence of added drugs.
The mitochondrial membrane potential of synaptosomes was measured by a fluorimetric assay adapted and optimized for synaptosomes from a fluorimetric protocol used in isolated brain mitochondria (Oliveira et al., 2007). Synaptosomes were incubated for 2 h at 37°C in Krebs' buffer in the absence or presence of Aβ1-42 (500 nm) and/or SCH58261 (50 nm), followed by 1 h incubation with 2 nm tetramethyl rhodamine methyl ester (TMRM+) (Invitrogen) and a short-spin centrifugation. The pellet was resuspended in 150 μl of Krebs–HEPES with 2 nm TMRM+. The functional assay was performed in a fluorescence spectrometer (Spectra Max Gemini EM; Molecular Devices), using 540 nm excitation and 590 nm emission, with a cutoff of 570 nm, and analyzed with SoftMax Pro V5 (Molecular Devices). The experiment is initiated by measuring a baseline (370 ± 8 fluorescent arbitrary units; n = 8) for 10 min, followed by the simultaneous addition of carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP) (2 μm) and oligomycin (1 μg/ml) and sequential measurement during 10 min to establish the new baseline yielding a change of relative fluorescence of 607 ± 28 fluorescent arbitrary units (control, n = 8). The effect of tested drugs was measured as changes in this difference between final and initial baseline and are expressed as the percentage of the difference observed in control conditions.
Primary cultures of neurons.
Hippocampal neurons were cultured from 17- to 19-d-old Wistar rat embryos, as previously described (Silva et al., 2007), and plated on poly-d-lysine-coated 16-mm-diameter coverslips or six-well plates at densities of 5 × 104/coverslip (viability and immunocytochemistry assays) or 1 × 106/well (Western blot analysis). Neurons were grown at 37°C in a 5% CO2 humidified atmosphere in Neurobasal medium with B-27 supplement, glutamate (25 μm), glutamine (0.5 mm), and gentamicin (0.12 mg/ml).
Drug treatments and evaluation of cell death.
Aβ1-42-induced neuronal damage was evaluated after culturing the neurons for 5–7 d. After 1 week, the culture matures and forms functional synaptic connections, and most of the regions exhibit spontaneous synaptic transmission (Rui et al., 2006). Either Aβ1-42 (500 nm) or Aβ42-1 (500 nm) were directly added to the medium and incubated for 12–48 h. To test the ability of any drug [SCH58261, 8-Br-cAMP, or N-[2-((o-bromocinamyl)amino)ethyl]-5-isoquinolinesulfonamide (H-89) from Sigma-Aldrich or 4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole (SB202190) from Tocris] to modify the effects of Aβ1-42, these drugs were added 15 min before addition of Aβ1-42 onward.
Viability assays were performed by double labeling (3 min incubation) with the fluorescent probes Syto-13 (4 μm) and propidium iodide (PI) (4 μg/ml; Invitrogen) followed by fluorescence microscopy cell counting. As previously described (Silva et al., 2007), viable neurons present nuclei homogenously labeled with Syto-13 (green fluorescent nuclei), whereas apoptotic neurons show condensed and fragmented nuclei labeled with Syto-13 (primary apoptosis) or with Syto-13 plus PI (secondary apoptosis) and necrotic neurons present intact nuclei labeled with PI (red fluorescent nuclei). Each experiment was repeated using different cell cultures in duplicate, and cell counting was performed in at least six fields per coverslip, with a total of ∼300 cells. Results are mean ± SEM and statistical significance (p < 0.05) was evaluated by one-way ANOVA followed by Newman–Keuls multiple-comparison test.
Immunocytochemical evaluation of synaptotoxicity.
After fixation with 4% paraformaldehyde, cells were permeabilized with PBS with 0.2% Triton X-100 for 2 min and incubated with 3% of BSA in PBS for 30 min for the simultaneous immunocytochemical analysis of a presynaptic marker [synaptophysin or 25 kDa synaptosomal-associated protein (SNAP-25)] and a dendritic marker [microtubule-associated protein-2 (MAP-2)] (Silva et al., 2007). Cells were incubated with rabbit anti-MAP-2 (1:400; Santa Cruz Biotechnology) and mouse anti-synaptophysin (1:200; Sigma-Aldrich) or mouse anti-SNAP-25 (1:200; Sigma-Aldrich) for 1 h. After three washes with PBS, cells were incubated with anti-mouse or anti-rabbit secondary antibody conjugated with a fluorophore (Alexa Fluor 488 and Alexa Fluor 594, respectively; 1:200; Invitrogen). The cells were visualized by confocal microscopy (MRC 600).
Western blot analysis.
Cultured hippocampal neurons were washed twice with PBS and gently scraped with ice-cold lysis buffer composed of 25 mm HEPES-Na, 2 mm MgCl2, 1 mm EDTA, 1 mm EGTA, and supplemented with 2 mm DTT, 100 μm phenylmethanesulfonyl fluoride (PMSF), 2 mm orthovanadate, 50 mm sodium fluoride, and a protease inhibitor mixture containing leupeptin, pepstatin A, chymostatin, and aprotinin (1 mg/ml; all from Sigma-Aldrich). The synaptosomal extract from rat or mice was solubilized in 5% SDS supplemented with 2 mm DTT and 100 μm PMSF and rapidly sonicated. After determining the amount of protein using the BCA method, a 1/6 vol of 6× SDS-PAGE sample buffer was added before storage at −20°C. Electrophoresis was performed using a 10 or 7.5% SDS-PAGE gel after loading of different amounts of each sample. Proteins were then transferred to PVDF (polyvinylidene difluoride) membranes (GE Healthcare). Membranes were blocked for 1 h at room temperature with 5% low-fat milk in Tris-buffered saline or 3% bovine serum albumin (depending on the antibodies used), pH 7.6, and containing 0.1% Tween 20 (TBS-T). Membranes were then incubated overnight at 4°C with primary antibodies, namely mouse anti-synaptophysin (1:5000–20,000), mouse anti-SNAP-25 (1:5000–20,000), mouse anti-phospho-c-Jun N-terminal kinase (JNK) (1:1000; Cell Signaling), or mouse anti-phospho-p38 MAPK (1:1000; Cell Signaling). After washing with TBS-T, membranes were incubated either with anti-mouse or anti-rabbit IgG secondary antibodies (1:10,000–20,000 in TBS-T; Invitrogen). After washing, membranes were revealed using an ECF kit (GE Healthcare) and visualized in a VersaDoc 3000 (Bio-Rad). The membranes were then reprobed and tested for α-tubulin immunoreactivity using a mouse anti-α-tubulin antibody (1:10,000–20,000; Zymed), as previously described (Rebola et al., 2005). To determine phosphorylation ratio of p38 and JNK, the membranes were reprobed with rabbit anti-total JNK/SAPK (stress-activated protein kinase) or rabbit anti-p38 MAPK total (both 1:1000; Cell Signaling).
HPLC quantification of adenosine levels in the incubation medium.
After addition at time 0 of Aβ1-42 (500 nm), cultured neurons were maintained at 37°C in a 5% CO2 humidified atmosphere with 1.2 ml of medium, and samples (125 μl) were collected from the incubation medium after 0, 3, 12, 24, and 48 h. Each sample was filtered through 0.22 μm filters (Millex-GV from Millipore; Interface) and stored at −20°C until analysis by reverse-phase HPLC, as previously described (Cunha and Sebastião, 1993). The quantification of adenosine was achieved by calculating the peak area and then converting to concentration values (correcting the change of incubation volume over time) by calibration with known standards (0.03–3 μm).
Statistical analysis.
Results are presented as mean ± SEM. Data were analyzed with one-way ANOVA and Newman–Keuls multiple-comparison test (unless otherwise stated), using a significance level of 0.05.
Results
Characterization of Aβ1-42-induced memory impairment and morphological modifications
Western blot analysis of the Aβ1-42 solutions used in this study showed that they were mainly constituted by monomers (4 kDa) and oligomers constituted by up to four monomers (Fig. 1A). The intracerebroventricular administration of Aβ1-42 (2 nmol) led to an accumulation of Aβ1-42 in the hippocampus (91.1 ± 25.1 pg/mg of protein; n = 4; p < 0.05), indicating that 11.3 ± 2.9% of the total amount of administered Aβ1-42 accumulated in hippocampal tissue after 2 d. The hippocampal Aβ1-42 levels decreased over time (Fig. 1B), since only 26.6 ± 8.9 pg/mg of protein (n = 4) was detected after 15 d (p < 0.05; F = 12.39 compared with Aβ1-42 levels at 2 d).
Figure 1.

Intracerebroventricular administration of soluble β-amyloid peptides leads to an accumulation of soluble but not aggregated forms of Aβ in the hippocampus, causing delayed memory impairment without evident acute effects. The Coomassie R-250 staining and 6E10 antibody-based Western blot analysis of the two different batches of Aβ1-42 used showed that they were mainly constituted by monomers and oligomer containing up to four monomers (A). Rats were treated with Aβ1-42 (2 nmol, i.c.v.) or water (control), which accumulated in the hippocampus after 2 and 15 d (B), as measured by ELISA (n = 4 rats treated with water and n = 6 treated with Aβ1-42). Congo Red and Thioflavin S staining (C) failed to reveal the presence of Aβ aggregates in hippocampal sections collected 15 d after Aβ1-42 administration (images representative of 3 animals). D, Spontaneous alternation in the Y-maze test of control and Aβ1-42-treated rats after 2 or 15 d (n = 6 animals treated with water and n = 9 treated with Aβ1-42). E, Object recognition index in the object recognition test of control and Aβ1-42-treated rats after 2 or 15 d (n = 4 animals treated with water and n = 6–7 animals treated with Aβ1-42). Data in bar graphs are mean ± SEM; *p < 0.05.
The intracerebroventricular administration of Aβ1-42 (2 nmol) caused a time-delayed (within 2 weeks) memory impairment, in agreement with previous reports (Dall'Igna et al., 2007; Cunha et al., 2008), whereas it failed to affect memory performance within 2 d, as evaluated both in the Y-maze (Fig. 1D) or the object recognition test (Fig. 1E), without changes in locomotor activity 2 or 15 d after Aβ1-42 administration (data not shown). The control peptide (Aβ42-1; 2 nmol) changed neither Y-maze behavior nor locomotor activity (n = 4) (data not shown). This indicates that Aβ1-42 might trigger a cascade of events leading to a delayed rather than acute perturbation of memory performance, which likely results from the action of soluble forms of Aβ1-42 since we only found soluble Aβ1-42 and no evidence of the presence of Aβ aggregates 15 d after the intracerebroventricular administration of Aβ1-42 (Fig. 1C).
Histological analysis of hippocampal sections, 2 weeks after the injection of Aβ1-42, revealed a preservation of cresyl violet staining of Nissl bodies (Fig. 2A, showing CA3, which is identical with CA1) and absence of neuronal loss evaluated by Fluoro-Jade C, which is indistinguishable from control rats (Fig. 2B). Furthermore, there was no evidence of microgliosis (evaluated by CD11b immunoreactivity) or astrogliosis (evaluated by GFAP immunoreactivity), neither after 15 d (data not shown) nor after 2 d (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) of Aβ1-42 administration. Further excluding acute toxic effects of Aβ1-42 administration, there was no difference of cresyl violet or Fluoro-Jade C staining 2 d after the injection of Aβ1-42 or vehicle (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). However, immunohistochemical analysis revealed a decrease in the synaptic marker synaptophysin in hippocampal sections obtained from rats 15 d after Aβ1-42 injection (Fig. 2C), which was confirmed by quantitative Western blot analysis. As illustrated in Figure 2D, synaptophysin immunoreactivity was lower (−25.7 ± 4.3%; n = 7; p < 0.001) in hippocampal membranes collected from rats 15 d after Aβ1-42 administration when compared with controls. In contrast, the nonamyloidogenic Aβ42-1 peptide failed to modify synaptophysin immunoreactivity (data not shown).
Figure 2.

β-Amyloid administration causes a selective synaptotoxicity and memory dysfunction, which is prevented by blockade of adenosine A2A receptors. Rats were treated with Aβ1-42 (2 nmol, i.c.v.) or water (control). The A2AR antagonist SCH58261 (0.05 mg/kg, i.p.) was administered daily starting 30 min before Aβ, and rats were behaviorally analyzed after 15 d. A, B, Cresyl violet staining of Nissl bodies (A) and Fluoro-Jade C staining of neuronal death (B) in hippocampal sections from control and Aβ1-42-injected rats. C, D, Immunohistochemical labeling with anti-synaptophysin in hippocampal sections from rats injected with water (control), Aβ1-42 (Aβ), SCH58261 (SCH), and Aβ plus SCH (images representative of 5 experiments) (C) and quantification by Western blot analysis (D) of synaptophysin immunoreactivity in hippocampal membranes from these different experimental groups (data are mean ± SEM from 7 experiments; *p < 0.05). E, Spontaneous alternation in the Y-maze test of the same groups of rats, as well as rats injected with the nonamyloidogenic scrambled Aβ1-42 peptide (scAβ) (data are mean ± SEM from 9 rats; *p < 0.001).
Pharmacological blockade of adenosine A2A receptor protects from Aβ1-42-induced synaptotoxicity and memory impairment
We then tested whether the blockade of A2ARs prevented the loss of synaptic markers and memory impairment observed 2 weeks after the intracerebroventricular administration of Aβ1-42. For that purpose, we used a selective A2AR antagonist (SCH58261) in a dose (0.05 mg/kg, i.p.) that has previously been shown to preserve memory performance without peripheral or locomotor effects (Dall'Igna et al., 2007; Cunha et al., 2008). As illustrated in Figure 2C, SCH58261 (0.05 mg/kg) completely prevented the decrease of synaptophysin immunoreactivity caused by Aβ1-42. In fact, synaptophysin immunoreactivity in hippocampal sections was indistinguishable in control conditions and in Aβ1-42-injected rats that were treated daily with SCH58261 (Fig. 2C). Accordingly, Western blot analysis confirmed that the decrease in synaptophysin density on Aβ1-42 injection was prevented by SCH58261 (p < 0.001) (Fig. 2D). In parallel, SCH58261 was also able to significantly (p < 0.001) prevent the decreased Y-maze spontaneous alternation on Aβ1-42 injection. In contrast, SCH58261 did not modify synaptophysin immunoreactivity (Fig. 2D) or spontaneous alternation in control rats (Fig. 2E) nor did it affect locomotion in control or Aβ1-42-treated rats (data not shown).
Genetic inactivation of A2A receptor abolishes Aβ1-42-induced synaptotoxicity and memory deficits
The memory impairment and loss of synaptic markers observed in rats could also be reproduced on Aβ1-42 administration in wild-type (C57BL/6) mice. In fact, 2 weeks after the intracerebroventricular administration of Aβ1-42 (2 nmol), WT mice displayed a decreased memory performance, measured as a decreased (−23.0 ± 1.7%; n = 7; p < 0.001) spontaneous alternation in the Y-maze (Fig. 3A), without modification of locomotor activity (Fig. 3B), and a decreased density of two synaptic markers, synaptophysin (−26.7 ± 3.7%; n = 4; p < 0.001) and SNAP-25 (−25.8 ± 2.3%; n = 4; p < 0.001) (Fig. 3C), when compared with vehicle-injected (i.e., control) mice. Furthermore, the histological analysis of hippocampal sections of Aβ1-42-treated WT mice showed the absence of the following: neuronal loss evaluated by Fluoro-Jade C, microgliosis evaluated by CD11b immunoreactivity, and astrogliosis evaluated by GFAP immunoreactivity (Fig. 3D, showing CA1 area, with similar results obtained for CA3 area) (data not shown).
Figure 3.
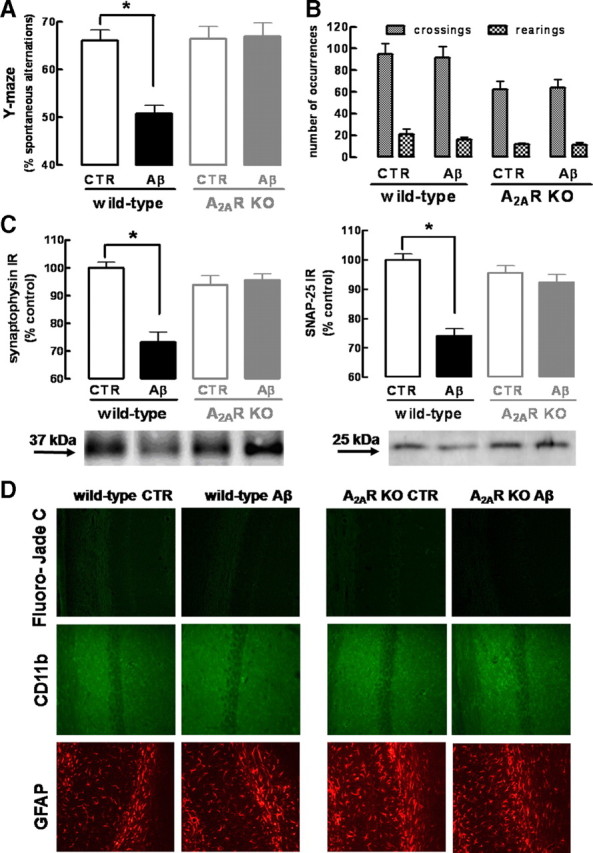
Genetic inactivation of adenosine A2A receptors prevents β-amyloid-induced synaptotoxicity and memory impairment. Wild-type C57BL/6 or A2AR KO mice were treated with Aβ1-42 (2 nmol, i.c.v.) or water [control (CTR)] and analyzed after 15 d. A, B, Spontaneous alternation in the Y-maze test (A) and spontaneous locomotion evaluated in an open-field arena (B) (data are mean ± SEM of n = 7 mice per experimental group; *p < 0.001). C, Western blot comparing synaptophysin and SNAP-25 immunoreactivity in hippocampal membranes obtained from wild-type or A2AR KO mice injected with water (CTR) or Aβ1-42 (data are mean ± SEM of n = 4 mice per experimental group; *p < 0.001). D, Fluoro-Jade C staining of neuronal death, CD11-b immunohistochemistry evaluating microgliosis, and GFAP immunohistochemistry evaluating astrogliosis in hippocampal sections from wild-type or A2AR KO mice injected with water (CTR) or Aβ1-42 (images representative of n = 4 mice per experimental group).
To confirm the key role of A2ARs in controlling Aβ1-42-induced loss of synaptic markers and memory impairment, we tested the effects of Aβ1-42 in A2AR KO mice. The A2AR genetic inactivation in KO mice led to a decrease in the number of crossings (30 ± 7; n = 14; p < 0.05) and rearings (7 ± 2; n = 14; p < 0.05) when compared with WT mice; however, this does not affect the Y-maze alternation, on comparison of saline-injected WT and KO mice (Fig. 3B). As shown in Figure 3, Aβ1-42 administration induced neither loss of synaptic markers nor memory impairment in A2AR KO mice. Indeed, Aβ1-42-injected A2AR KO mice did not display a decrease of spontaneous alteration in the Y-maze (Fig. 3A) or a decrease in the density of the synaptic markers, synaptophysin or SNAP-25 (Fig. 3C). Furthermore, vehicle- or Aβ1-42-injected A2AR KO mice did not display cell death, microgliosis, or astrogliosis (Fig. 3D).
Blockade of A2A receptors prevents Aβ1-42-induced dysfunction of purified nerve terminals
The observations that Aβ1-42 triggered an A2AR-sensitive selective loss of synaptic markers prompted the hypothesis that this A2AR-sensitive Aβ1-42-induced toxicity could be replicated in enriched nerve terminals (synaptosomes). Previous studies have already reported that exposure of synaptosomes to β-amyloid peptides triggers mitochondrial dysfunction (Mattson et al., 1998), which has been argued to be a key feature of Alzheimer's disease (Moreira et al., 2006). Accordingly, synaptosomes exposed for 2 h to 500 nm Aβ1-42 display a decrease (−8.3 ± 3.6% compared with control; n = 4; p < 0.001) in MTT reduction (Fig. 4A), which measures the redox status of synaptosomes, indicative of synaptosomal viability (Mattson et al., 1998; Silva et al., 2007). Furthermore, a decrease in TMRM+ accumulation, indicative of decreased mitochondrial membrane potential (−11.5 ± 2.5%; n = 8; p < 0.05) in Aβ1-42-treated synaptosomes was also observed (Fig. 4B).
Figure 4.

Exposure to Aβ1-42 directly decreases the function of rat hippocampal synaptosomes, which is prevented by blockade of adenosine A2A receptors. Synaptosomes were incubated for 2 h with 500 nm Aβ1-42 or Krebs' buffer, in the absence or presence of the A2AR antagonist, SCH58261 (50 nm), added 15 min before. A, Synaptosomal viability was measured using the MTT assay (data are mean ± SEM of n = 4; *p < 0.05). B, Measurement of mitochondrial membrane potential Δ (difference between the final and initial baseline) using the TMRM+ indicator after adding FCCP and oligomycin (data are mean ± SEM of n = 8; *p < 0.05).
On blockade of A2ARs with SCH58261 (50 nm), there was a prevention of the Aβ1-42-induced disruption of the functionality (Fig. 4A) and mitochondrial membrane potential of synaptosomes (Fig. 4B), whereas SCH58261 was devoid of effects in control synaptosomes (i.e., not treated with Aβ1-42) or treated with the nonamyloidogenic Aβ42-1 peptide (data not shown).
Blockade of A2A receptor protects hippocampal neurons from Aβ1-42-induced toxicity
To investigate the mechanism involved in the A2AR-mediated control of Aβ1-42-induced neurotoxicity, we used a cell culture model, namely, primary cultures of hippocampal neurons. Cultured hippocampal neurons were exposed for 12, 24, and 48 h to 500 nm Aβ1-42, and neuronal death was analyzed by double labeling with Syto-13 and PI (Fig. 5A,B). After 12 h of exposure to Aβ1-42, hippocampal neurons did not present any significant decrease (−1.0 ± 1.0%; n = 5; p > 0.05) of either cell viability (Fig. 5A) or number of apoptotic-like neurons (Fig. 5B) when compared with control neurons (either not exposed to Aβ1-42 or exposed to the nonamyloidogenic Aβ42-1 peptide). In fact, a decrease of cell viability (−9.0 ± 2.0%; n = 5; p < 0.001) was only observed 24 h after Aβ1-42 exposure (Fig. 5A), which was accompanied by an increased number of apoptotic-like neurons (5 ± 1%; n = 5; p < 0.001) (Fig. 5B). This Aβ1-42-induced neuronal death was larger after 48 h of exposure to Aβ1-42, as evaluated by the decreased number of viable neurons (−12.3 ± 3.7%; n = 5; p < 0.001) (Fig. 5A) and the increased number of apoptotic-like neurons (9 ± 2%; n = 5; p < 0.001) (Fig. 5B), indicating a time-dependent evolving profile of Aβ1-42-induced neurodegeneration. As occurred in vivo and in native brain preparations, this Aβ1-42-induced neurotoxicity was prevented by the A2AR antagonist, SCH58261 (50 nm), which did not affect neuronal viability in control neurons (Fig. 5C,D).
Figure 5.

Temporal analysis of neuronal death caused by Aβ1-42 and neuroprotection by blockade of adenosine A2A receptors. Hippocampal neurons were preincubated with the A2AR antagonist SCH58261 (50 nm) 15 min before addition of 500 nm Aβ1-42. Neurons were double labeled with Syto-13 and PI probes. Viable neurons presented green nuclei stained with Syto-13, whereas apoptotic neurons presented shrinkage nuclei stained with PI and Syto-13. A, B, Aβ-induced neuronal death is time dependent. C, D, Blockade of A2AR with SCH58261 prevents neuronal death on 48 h of incubation with Aβ. A total of ∼300 cells per coverslip was counted. Results are means ± SEM of duplicate coverslips from five independent hippocampal cultures; *p < 0.05.
We next investigated whether the exposure of cultured neurons to Aβ1-42 caused an initial synaptotoxicity preceding neuronal death. Since we observed that neurons incubated for 12 h with Aβ1-42 did not display loss of viability or damage, we evaluated whether Aβ1-42-induced synaptotoxicity would be present after 12 h of exposure to Aβ1-42, by evaluating the double staining of MAP-2 and synaptophysin or SNAP-25. As shown in Figure 6(and in supplemental Fig. 2, available at www.jneurosci.org as supplemental material), there was a retraction of MAP-2-labeled segments and a decrease in the number of synaptophysin-immunoreactive spots after 12 h of exposure to Aβ1-42 (i.e., at the time when neuronal damage is not yet present) (Fig. 5). To quantify this Aβ1-42-induced synaptotoxicity, we used Western blotting analysis, which showed a decrease in the density of synaptophysin (−30.3 ± 7.5%; n = 6; p < 0.05) and SNAP-25 (−37.0 ± 6.6%; n = 6; p < 0.05) on exposure to Aβ1-42. As occurred in vivo, this initial and evolving Aβ1-42-induced synaptotoxicity in neuronal cultures was also prevented by A2AR blockade with the selective A2AR antagonist, SCH58261 (50 nm) (Fig. 6A; supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 6.
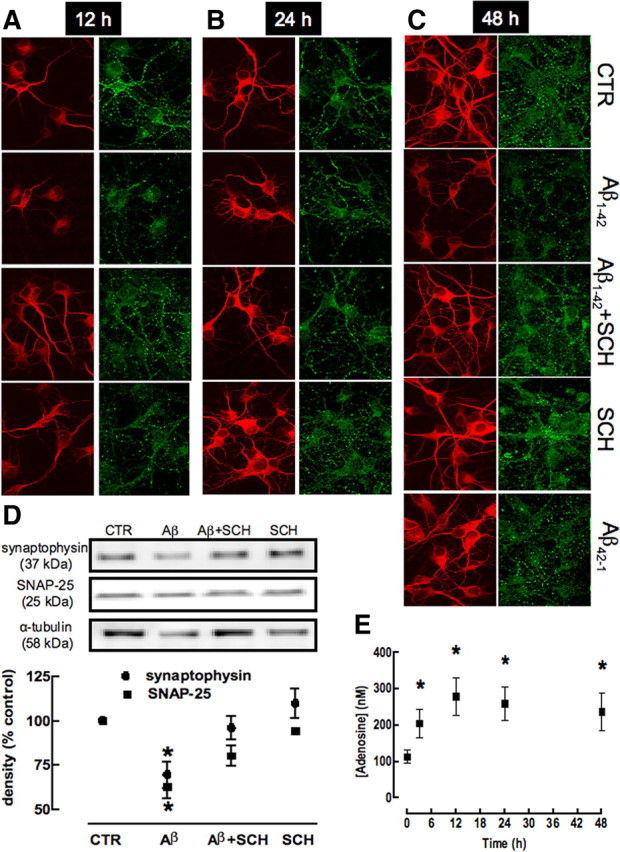
Temporal analysis of synaptotoxicity caused by Aβ1-42 and neuroprotection by blockade of adenosine A2A receptors. Hippocampal neurons were preincubated with the A2AR antagonist SCH58261 (50 nm) 15 min before addition of 500 nm Aβ1-42. Hippocampal neurons were double-labeled for MAP-2 (red) and synaptophysin (green) after 12 h (A), 24 h (B), and 48 h (C) of incubation and analyzed by confocal microscopy. Magnification, 400×. Aβ1-42 causes a decrease of MAP-2 and synaptophysin immunoreactivities at all type points, which is prevented by SCH58261 and is not mimicked by the nonamyloidogenic scrambled peptide Aβ42-1. D, Western blot analysis (15 μg of protein loaded in each lane) quantifying the loss of synaptophysin and SNAP-25 immunoreactivities in cultures treated with Aβ, which is prevented by SCH58261 (data are mean ± SEM of 6 independent cultures; *p < 0.05). E, Time course analysis of the extracellular levels of adenosine (quantified by HPLC) in hippocampal neurons incubated with Aβ (data are mean ± SEM of 5 independent cultures; *p < 0.05).
This observation that SCH58261 prevents Aβ1-42-induced neurotoxicity but is devoid of effects in controls suggests that the levels of extracellular adenosine might be increased on exposure to Aβ1-42, which is in accordance with the general concept that noxious stimuli are expected to increase the extracellular levels of adenosine (Fredholm et al., 2005). As predicted, incubation of hippocampal neurons with Aβ1-42 (500 nm) caused a >100% increase of the extracellular concentration of adenosine (104.7 ± 38.8 nm; n = 5; p < 0.05) after 3 h that is persistent until 48 h of incubation (Fig. 6C).
Signaling pathways involved in the neuroprotection afforded by A2A receptor blockade against Aβ1-42-induced neurotoxicity
Since one of main transducing systems operated by A2ARs involves cAMP/protein kinase A (PKA) pathway (Fredholm et al., 2005), we investigated whether the neuroprotective effects afforded by SCH58261 involved this pathway. As observed in the Figure 7A, the manipulation of the cAMP/PKA pathway influences Aβ1-42-induced neurotoxicity, as described by others (Parvathenani et al., 2000; Gong et al., 2004; Shrestha et al., 2006). In fact, the activation of PKA with the cell-permeable cAMP analog 8-Br-cAMP (200 μm) attenuated Aβ1-42-induced neurotoxicity, an effect prevented by the PKA inhibitor H-89 (1 μm) (Fig. 7A). However, the neuroprotection by SCH58261 persisted even in the presence of H-89, ruling out the participation of cAMP/PKA pathway in the neuroprotection resulting from the blockade of A2ARs (Fig. 7A).
Figure 7.

The neuroprotection afforded by blockade of adenosine A2A receptors against Aβ1-42-induced neurotoxicity involves the p38 MAPK rather than the cAMP/protein kinase A signaling pathway. Hippocampal neurons were preincubated with the A2AR antagonist SCH58261 (50 nm) or with the cAMP analog 8-Br-cAMP (200 μm) 15 min before addition of 500 nm Aβ1-42. All inhibitors tested were added 30 min Aβ1-42. A, Neuroprotection by SCH58261 does not involve the cAMP/PKA signaling pathway since the PKA inhibitor H-89 (1 μm) prevents the neuroprotection afforded by 8-Br-cAMP, but fails to modify the neuroprotection afforded by SCH58261, as evaluated after 24 h of exposure to Aβ1-42 (*p < 0.05 vs control #p < 0.05 vs Aβ1-42; &p < 0.05 vs Aβ1-42 + 8-Br-cAMP). B, C, Aβ1-42 triggered the activation of JNK (B) and p38 MAPK (C), evaluated by their degree of phosphorylation after 2 h, and SCH58261 enhanced JNK phosphorylation, whereas it blocked p38 MAPK phosphorylation (data are mean ± SEM from 6 independent cultures; *p < 0.05 vs control; **p < 0.05 vs effect of Aβ). D, The p38 MAPK inhibitor SB202190 prevents neuronal death induced by Aβ1-42 (data are mean ± SEM from 5 independent cultures; *p < 0.05 vs control).
It is also suggested that deregulation of the MAPK pathways, namely of JNK and p38 MAPK family of proteins, might play a role in the intracellular mechanisms of neurodegeneration, in particular in Aβ1-42-induced neurotoxicity (Troy et al., 2001; Minogue et al., 2003; Wang et al., 2004b; Zhu et al., 2005; Muñoz et al., 2007), and A2ARs can also signal through the MAPK pathway (for review, see Fredholm et al., 2005). To test the involvement of JNK and p38 MAPK in the A2AR-mediated protection against Aβ1-42-induced neurotoxicity, we first investigated the time course of Aβ1-42-induced activation of p38 MAPK and JNK (evaluated as their degree of phosphorylation) to determine the time points at which this process occurs (data not shown). It was found that, after 2 h of incubation with Aβ1-42 (500 nm), there was an increase of JNK (69 ± 21%; n = 6; p < 0.01) (Fig. 7B) and p38 MAPK (41 ± 15%; n = 7; p < 0.05) phosphorylation (Fig. 7C). At this time point, A2AR blockade with SCH58261 (50 nm) increased the Aβ1-42-induced JNK phosphorylation (210 ± 74%; n = 6; p < 0.01), whereas it abolished the Aβ1-42-induced p38 MAPK phosphorylation (Fig. 7B,C). Confirming the key role of p38 MAPK in the Aβ1-42-induced neurotoxicity (Zhu et al., 2005; Muñoz et al., 2007; Origlia et al., 2008), we found that the p38 MAPK inhibitor SB202190 (200 nm) prevented the Aβ1-42-induced loss of neuronal viability and increased number of apoptotic-like neurons (Fig. 7D).
Discussion
The present results provide the first demonstration that blockade of a membrane receptor enriched in hippocampal synapses, namely, A2ARs, abolishes the loss of nerve terminal markers (i.e., synaptotoxicity) triggered by Aβ to culminate in memory dysfunction, the two cardinal features of early phases of AD. These results are relevant for the following three different reasons: (1) they provide evidence that control of a presynaptic modulation system that prevents synaptotoxicity also prevents memory dysfunction, strengthening the hypothesis that synaptic dysfunction is a precocious core modification of AD; (2) they provide additional evidence that A2ARs, the density of which is increased in AD (Albasanz et al., 2008), are a novel promising target to control AD; (3) they provide a clear demonstration that neuroprotection afforded by A2AR blockade is independent of cAMP/PKA transducing system and results suggest that it is instead mediated by p38 MAPK.
We now observed that intracerebroventricular administration of soluble forms of Aβ1-42 (Resende et al., 2008) caused a delayed loss of memory performance only after 15 d that was selectively associated with loss of synaptic markers. In fact, the only morphological change found in the hippocampus of Aβ-injected rodents displaying memory deficits was the loss of synaptic markers, whereas neither overt neuronal damage nor astrogliosis nor microgliosis were observed, neither 15 nor 2 d after Aβ1-42 administration. Accordingly, in cultured neurons (in which peripheral, vascular, glial, or immune influences are absent), we also found that exposure to Aβ caused first a synaptotoxicity (Roselli et al., 2005; Calabrese et al., 2007; Shankar et al., 2007; Evans et al., 2008), which is only later followed by overt neuronal damage. Further strengthening that Aβ causes direct effects on nerve terminals, we showed that Aβ indeed directly impairs synaptosomal function, as observed by others (Mattson et al., 1998; Arias et al., 2002). Together, these observations indicate that Aβ, which can bind to synaptic proteins (Lacor et al., 2007) and accumulates synaptically in AD patients (Takahashi et al., 2002; Gylys et al., 2004; Fein et al., 2008), causes a primordial synaptotoxicity that precedes overt neuronal damage, as occurs in different transgenic animal models of AD (Hsia et al., 1999; Mucke et al., 2000; Oddo et al., 2003; Wu et al., 2004; Jacobsen et al., 2006) and in frontal cortical and hippocampal regions early in AD (Scheff et al., 2006, 2007). It should be stressed that we only obtained evidence that Aβ1-42 caused loss of synaptic markers, modification of the viability of nerve terminals (synaptosomes), and degeneration of synapses, which we collectively called synaptotoxicity; however, it remains to be determined to what extent this synaptotoxicity relates to the known Aβ-induced functional impairment of hippocampal synapses (Venkitaramani et al., 2007).
This tight relationship between synaptotoxicity and memory dysfunction is further strengthened by the key observation of the present study [i.e., that blockade of A2ARs (pharmacological or genetic inactivation) simultaneously prevents synaptotoxicity and memory impairment caused by Aβ administration]. Furthermore, the initial synaptotoxicity that precedes overt neuronal damage on exposure of cultured neurons to Aβ was also prevented by A2AR blockade. Finally, the direct Aβ-induced impairment of nerve terminal function was also prevented by A2AR blockade. All these observations are in agreement with the predominant synaptic localization of A2ARs in cortical regions (Rebola et al., 2005). These synaptic A2ARs play a key role controlling NMDA-dependent synaptic plasticity (Rebola et al., 2008), which is severely hampered early in AD (Roselli et al., 2005; Shankar et al., 2007; Venkitaramani et al., 2007). Thus, synaptic A2ARs normalize the function of these glutamatergic synapses (for review, see Cunha, 2008a), which are dysfunctional in AD (Bell et al., 2007), and their blockade prevents synaptotoxicity caused by different stimuli (Cunha et al., 2006; Silva et al., 2007) that leads to subsequent overt neurodegeneration on stressful conditions (Silva et al., 2007). This implies that the ability of A2ARs to control memory impairment should be particularly evident when synaptotoxicity is involved. Accordingly, we have previously shown that A2AR blockade can prevent memory impairment caused by Aβ, which we now show to involve synaptotoxicity, but are ineffective in controlling acute memory dysfunction caused by pharmacological manipulation of the cholinergic or glutamatergic systems (Cunha et al., 2008), which is reversible and does not involve synaptotoxicity. Overall, this supports the notion that prevention of synaptic impairment on A2AR blockade may underlie the ability of A2AR antagonists to prevent Aβ-induced memory impairment, which illustrates that control of synaptic dysfunction may be a relevant strategy to alleviate memory dysfunction associated with neurodegenerative conditions (Coleman et al., 2004; Wishart et al., 2006).
This putative relevance of targeting A2ARs to control memory impairment associated with neurodegenerative conditions is strongly supported by the ability of caffeine to counteract the development of neurodegenerative conditions and, in particular, the development of cognitive deficits (for review, see Cunha, 2008b; Takahashi et al., 2008). In fact, although it is doubtful that caffeine is a cognitive enhancer, its long-term consumption is clearly associated with decreased memory impairment caused by different perturbing conditions (Cunha, 2008b; Takahashi et al., 2008) such as on aging (Ritchie et al., 2007; Costa et al., 2008a) or Alzheimer's disease (Maia and de Mendonça, 2002; Eskelinen et al., 2009). The only known mechanisms of action of nontoxic doses of caffeine are the antagonism of adenosine receptors (Fredholm et al., 1999). Animal studies indicate that the ability of chronic caffeine consumption to prevent memory deterioration caused by different insults is mimicked by antagonists of A2ARs rather than A1Rs (for review, see Cunha, 2008b; Takahashi et al., 2008). Accordingly, we have previously shown that the beneficial effects of caffeine on Aβ-induced neurotoxicity and memory impairment are mimicked by antagonists of A2ARs but not of A1Rs (Dall'Igna et al., 2003, 2007). Thus, it is tempting to propose that the promising beneficial effects of caffeine consumption as a strategy to prevent the burden of AD might be related to the synaptoprotective effect afforded by A2AR blockade. This proposal does not exclude other possible concurring mechanisms by which caffeine may afford protection in AD, such as control of Aβ production (Arendash et al., 2006), control of the disruption of the blood–brain barrier (Chen et al., 2008), or control of neuroinflammation (Angulo et al., 2003). Thus, although the present data combining the use of fractionated nerve terminals, cultured neurons, and in vivo models strongly argue for the predominant importance of synaptic A2ARs in controlling Aβ-induced neurotoxicity, it does not exclude the possibility that other mechanisms may also contribute for neuroprotection against Aβ-induced neurotoxicity and memory impairment.
Finally, this study demonstrates that neuroprotection resulting from A2AR blockade does not involve the cAMP/protein kinase A transducing system but instead depends on control of p38 MAPK. In fact, the mechanisms by which A2ARs impact on neurodegeneration are still unresolved (for discussion, see Cunha, 2005; Chen et al., 2007). For historical reasons, there is a general consensus that A2ARs signal through activation of the adenylate cyclase/cAMP/PKA pathway (Fredholm et al., 2005). However, this is unlikely to be the relevant transducing system related to A2AR control of neurodegeneration since enhanced cAMP levels afford neuroprotection against Aβ-induced neurotoxicity (Parvathenani et al., 2000; Gong et al., 2004; Shrestha et al., 2006), whereas it is A2AR blockade (expected to decrease cAMP levels) that affords neuroprotection. Accordingly, neuroprotection afforded by A2AR blockade against Aβ-induced neurotoxicity was insensitive to the PKA inhibitor H-89, which prevented neuroprotection afforded by enhanced cAMP levels. Other transducing pathways have been documented to control degeneration in AD models, namely, the MAPK pathways (Zhu et al., 2005; Muñoz et al., 2007; Origlia et al., 2008), and, accordingly, we confirmed that Aβ triggered activation of both JNK and p38 MAPKs. Interestingly, we observed that A2AR blockade prevented the Aβ-induced activation of p38, whereas it enhanced JNK phosphorylation, an aspect that needs attention in view of the association between JNK activation and neurodegeneration (Wang et al., 2004a). Given that inhibition of p38 activation is sufficient to prevent Aβ-induced neurotoxicity, as also observed by others (Zhu et al., 2005; Muñoz et al., 2007; Origlia et al., 2008), this indicates that A2ARs signal through p38 MAPK to control neurodegeneration. Indeed, previous studies have documented the ability of A2ARs to control MAPK pathways in a cAMP-independent manner (Schulte and Fredholm, 2003; Fredholm et al., 2005; Gsandtner et al., 2005), and it has previously been suggested that the control by A2ARs of the ischemia-induced brain damage was related to the ability of A2AR antagonists to blunt the ischemia-induced accumulation of phosphorylated forms of p38 (Melani et al., 2006). Thus, the present results indicate that A2ARs control Aβ-induced neurotoxicity through control of p38 MAPK phosphorylation. However, this conclusion derives solely from in vitro studies and remains to be confirmed in vivo.
In summary, the present observations that blockade of A2ARs prevents the early synaptotoxicity in both in vitro and in vivo models pertinent to AD, strengthen the interest of exploring the prophylactic and therapeutic potential of A2AR antagonists, which are about to be introduced into clinical practice as novel antiparkinsonian drugs (Schwarzschild et al., 2006).
Footnotes
This work was supported by Fundação para a Ciência e para a Tecnologia Grant POCTI/44740/2002 and by a Pfizer award from the Portuguese Society of Neuroscience. L.O.P. was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico–Brazil. We thank Jiang Fan Chen for generously providing A2A receptor knock-out mice, Gary Arendash and Chuanhai Cao for their generous help in the assays measuring Aβ levels, Rosa Resende for her assistance in the native gel analysis, and Rogério Candeias for his efforts in performing some initial experiments in cortical neurons.
References
- Albasanz JL, Perez S, Barrachina M, Ferrer I, Martín M. Up-regulation of adenosine receptors in the frontal cortex in Alzheimer's disease. Brain Pathol. 2008;18:211–219. doi: 10.1111/j.1750-3639.2007.00112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo E, Casadó V, Mallol J, Canela EI, Viñals F, Ferrer I, Lluis C, Franco R. A1 adenosine receptors accumulate in neurodegenerative structures in Alzheimer disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathol. 2003;13:440–451. doi: 10.1111/j.1750-3639.2003.tb00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendash GW, Schleif W, Rezai-Zadeh K, Jackson EK, Zacharia LC, Cracchiolo JR, Shippy D, Tan J. Caffeine protects Alzheimer's mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience. 2006;142:941–952. doi: 10.1016/j.neuroscience.2006.07.021. [DOI] [PubMed] [Google Scholar]
- Arias C, Montiel T, Quiroz-Báez R, Massieu L. β-Amyloid neurotoxicity is exacerbated during glycolysis inhibition and mitochondrial impairment in the rat hippocampus in vivo and in isolated nerve terminals: implications for Alzheimer's disease. Exp Neurol. 2002;176:163–174. doi: 10.1006/exnr.2002.7912. [DOI] [PubMed] [Google Scholar]
- Bell KF, Bennett DA, Cuello AC. Paradoxical upregulation of glutamatergic presynaptic boutons during mild cognitive impairment. J Neurosci. 2007;27:10810–10817. doi: 10.1523/JNEUROSCI.3269-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese B, Shaked GM, Tabarean IV, Braga J, Koo EH, Halpain S. Rapid, concurrent alterations in pre- and postsynaptic structure induced by naturally-secreted amyloid-beta protein. Mol Cell Neurosci. 2007;35:183–193. doi: 10.1016/j.mcn.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C, Arendash GW, Dickson A, Mamcarz MB, Lin X, Ethell DW. Aβ-specific Th2 cells provide cognitive and pathological benefits to Alzheimer's mice without infiltrating the CNS. Neurobiol Dis. 2009;34:63–70. doi: 10.1016/j.nbd.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Sonsalla PK, Pedata F, Melani A, Domenici MR, Popoli P, Geiger J, Lopes LV, de Mendonça A. Adenosine A2A receptors and brain injury: broad spectrum of neuroprotection, multifaceted actions and “fine tuning” modulation. Prog Neurobiol. 2007;83:310–331. doi: 10.1016/j.pneurobio.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Chen X, Gawryluk JW, Wagener JF, Ghribi O, Geiger JD. Caffeine blocks disruption of blood brain barrier in a rabbit model of Alzheimer's disease. J Neuroinflammation. 2008;5:12. doi: 10.1186/1742-2094-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman P, Federoff H, Kurlan R. A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology. 2004;63:1155–1162. doi: 10.1212/01.wnl.0000140626.48118.0a. [DOI] [PubMed] [Google Scholar]
- Costa MS, Botton PH, Mioranzza S, Souza DO, Porciúncula LO. Caffeine prevents age-associated recognition memory decline and changes brain-derived neurotrophic factor and tirosine kinase receptor (TrkB) content in mice. Neuroscience. 2008a;153:1071–1078. doi: 10.1016/j.neuroscience.2008.03.038. [DOI] [PubMed] [Google Scholar]
- Costa MS, Botton PH, Mioranzza S, Ardais AP, Moreira JD, Souza DO, Porciúncula LO. Caffeine improves adult mice performance in the object recognition task and increases BDNF and TrkB independent on phospho-CREB immunocontent in the hippocampus. Neurochem Int. 2008b;53:89–94. doi: 10.1016/j.neuint.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Cunha GM, Canas PM, Oliveira CR, Cunha RA. Increased density and synapto-protective effect of adenosine A2A receptors on sub-chronic restraint stress. Neuroscience. 2006;141:1775–1781. doi: 10.1016/j.neuroscience.2006.05.024. [DOI] [PubMed] [Google Scholar]
- Cunha GM, Canas PM, Melo CS, Hockemeyer J, Müller CE, Oliveira CR, Cunha RA. Adenosine A2A receptor blockade prevents memory dysfunction caused by beta-amyloid peptides but not by scopolamine or MK-801. Exp Neurol. 2008;210:776–781. doi: 10.1016/j.expneurol.2007.11.013. [DOI] [PubMed] [Google Scholar]
- Cunha RA. Neuroprotection by adenosine in the brain: from A1 receptor activation to A2A receptor blockade. Purinergic Signal. 2005;1:111–134. doi: 10.1007/s11302-005-0649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA. Different cellular sources and different roles of adenosine: A1 receptor-mediated inhibition through astrocytic-driven volume transmission and synapse-restricted A2A receptor-mediated facilitation of plasticity. Neurochem Int. 2008a;52:65–72. doi: 10.1016/j.neuint.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Cunha RA. Caffeine, adenosine receptors, memory and Alzheimer disease. Med Clin. 2008b;131:790–795. doi: 10.1016/s0025-7753(08)75506-4. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Sebastião AM. Adenosine and adenine nucleotides are independently released from both the nerve terminals and the muscle fibres on electrical stimulation of the innervated skeletal muscle of the frog. Pflugers Arch. 1993;424:503–510. doi: 10.1007/BF00374914. [DOI] [PubMed] [Google Scholar]
- Dall'Igna OP, Porciúncula LO, Souza DO, Cunha RA, Lara DR. Neuroprotection by caffeine and adenosine A2A receptor blockade of β-amyloid neurotoxicity. Br J Pharmacol. 2003;138:1207–1209. doi: 10.1038/sj.bjp.0705185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall'Igna OP, Fett P, Gomes MW, Souza DO, Cunha RA, Lara DR. Caffeine and adenosine A2a receptor antagonists prevent beta-amyloid (25–35)-induced cognitive deficits in mice. Exp Neurol. 2007;203:241–245. doi: 10.1016/j.expneurol.2006.08.008. [DOI] [PubMed] [Google Scholar]
- de Mendonça A, Sebastião AM, Ribeiro JA. Adenosine: does it have a neuroprotective role after all? Brain Res Brain Res Rev. 2000;33:258–274. doi: 10.1016/s0165-0173(00)00033-3. [DOI] [PubMed] [Google Scholar]
- Eskelinen MH, Ngandu T, Tuomilehto J, Soininen H, Kivipelto M. Midlife coffee and tea drinking and the risk of late-life dementia: a population-based CAIDE study. J Alzheimers Dis. 2009;16:85–91. doi: 10.3233/JAD-2009-0920. [DOI] [PubMed] [Google Scholar]
- Evans NA, Facci L, Owen DE, Soden PE, Burbidge SA, Prinjha RK, Richardson JC, Skaper SD. Aβ1-42 reduces synapse number and inhibits neurite outgrowth in primary cortical and hippocampal neurons: a quantitative analysis. J Neurosci Methods. 2008;175:96–103. doi: 10.1016/j.jneumeth.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Fein JA, Sokolow S, Miller CA, Vinters HV, Yang F, Cole GM, Gylys KH. Co-localization of amyloid beta and tau pathology in Alzheimer's disease synaptosomes. Am J Pathol. 2008;172:1683–1692. doi: 10.2353/ajpath.2008.070829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Bättig K, Holmén J, Nehlig A, Zvartau EE. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev. 1999;51:83–133. [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. Int Rev Neurobiol. 2005;63:191–270. doi: 10.1016/S0074-7742(05)63007-3. [DOI] [PubMed] [Google Scholar]
- Gibson G, Gunasekera N, Lee M, Lelyveld V, El-Agnaf OM, Wright A, Austen B. Oligomerization and neurotoxicity of the amyloid ADan peptide implicated in familial Danish dementia. J Neurochem. 2004;88:281–290. doi: 10.1046/j.1471-4159.2003.02134.x. [DOI] [PubMed] [Google Scholar]
- Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest. 2004;114:1624–1634. doi: 10.1172/JCI22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gsandtner I, Charalambous C, Stefan E, Ogris E, Freissmuth M, Zezula J. Heterotrimeric G protein-independent signaling of a G protein-coupled receptor. Direct binding of ARNO/cytohesin-2 to the carboxyl terminus of the A2A adenosine receptor is necessary for sustained activation of the ERK/MAP kinase pathway. J Biol Chem. 2005;280:31898–31905. doi: 10.1074/jbc.M506515200. [DOI] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer's disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165:1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RN. The value of spontaneous alternation behavior (SAB) as a test of retention in pharmacological investigations of memory. Neurosci Biobehav Rev. 2004;28:497–505. doi: 10.1016/j.neubiorev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MB, Finsen B, Zimmer J. Morphological and immunophenotypic microglial changes in denervated fascia dentate of adult rats: correlation with blood brain barrier damage and astroglial reactions. Exp Neurol. 1997;143:103–116. doi: 10.1006/exnr.1996.6337. [DOI] [PubMed] [Google Scholar]
- Klein WL, Stine WB, Jr, Teplow DB. Small assemblies of unmodified amyloid β-protein are the proximate neurotoxin in Alzheimer's disease. Neurobiol Aging. 2004;25:569–580. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes LV, Rebola N, Pinheiro PC, Richardson PJ, Oliveira CR, Cunha RA. Adenosine A3 receptors are located in neurons of the rat hippocampus. Neuroreport. 2003;14:1645–1648. doi: 10.1097/00001756-200308260-00021. [DOI] [PubMed] [Google Scholar]
- Maia L, de Mendonça A. Does caffeine intake protect from Alzheimer's disease? Eur J Neurol. 2002;9:377–382. doi: 10.1046/j.1468-1331.2002.00421.x. [DOI] [PubMed] [Google Scholar]
- Masliah E, Terry R. The role of synaptic proteins in the pathogenesis of disorders of the central nervous system. Brain Pathol. 1993;3:77–85. doi: 10.1111/j.1750-3639.1993.tb00728.x. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Partin J, Begley JG. Amyloid beta-peptide induces apoptosis-related events in synapses and dendrites. Brain Res. 1998;807:167–176. doi: 10.1016/s0006-8993(98)00763-x. [DOI] [PubMed] [Google Scholar]
- Melani A, Gianfriddo M, Vannucchi MG, Cipriani S, Baraldi PG, Giovannini MG, Pedata F. The selective A2A receptor antagonist SCH 58261 protects from neurological deficit, brain damage and activation of p38 MAPK in rat focal cerebral ischemia. Brain Res. 2006;1073–1074:470–480. doi: 10.1016/j.brainres.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Melo JB, Sousa C, Garção P, Oliveira CR, Agostinho P. Galantamine protects against oxidative stress induced by amyloid β peptide in cortical neurons. Eur J Neurosci. 2009;29:455–464. doi: 10.1111/j.1460-9568.2009.06612.x. [DOI] [PubMed] [Google Scholar]
- Minogue AM, Schmid AW, Fogarty MP, Moore AC, Campbell VA, Herron CE, Lynch MA. Activation of the c-Jun N-terminal kinase signaling cascade mediates the effect of amyloid-beta on long term potentiation and cell death in hippocampus: a role for interleukin-1beta? J Biol Chem. 2003;278:27971–27980. doi: 10.1074/jbc.M302530200. [DOI] [PubMed] [Google Scholar]
- Moreira PI, Cardoso SM, Santos MS, Oliveira CR. The key role of mitochondria in Alzheimer's disease. J Alzheimers Dis. 2006;9:101–110. doi: 10.3233/jad-2006-9202. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz L, Ranaivo HR, Roy SM, Hu W, Craft JM, McNamara LK, Chico LW, Van Eldik LJ, Watterson DM. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer's disease mouse model. J Neuroinflammation. 2007;4:21. doi: 10.1186/1742-2094-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oliveira JM, Jekabsons MB, Chen S, Lin A, Rego AC, Gonçalves J, Ellerby LM, Nicholls DG. Mitochondrial dysfunction in Huntington's disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. J Neurochem. 2007;101:241–249. doi: 10.1111/j.1471-4159.2006.04361.x. [DOI] [PubMed] [Google Scholar]
- Origlia N, Righi M, Capsoni S, Cattaneo A, Fang F, Stern DM, Chen JX, Schmidt AM, Arancio O, Yan SD, Domenici L. Receptor for advanced glycation end product-dependent activation of p38 mitogen-activated protein kinase contributes to amyloid-β-mediated cortical synaptic dysfunction. J Neurosci. 2008;28:3521–3530. doi: 10.1523/JNEUROSCI.0204-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvathenani LK, Calandra V, Roberts SB, Posmantur R. cAMP delays beta-amyloid (25–35) induced cell death in rat cortical neurons. Neuroreport. 2000;11:2293–2297. doi: 10.1097/00001756-200007140-00045. [DOI] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Puchtler H, Waldrop FS, Meloan SN. A review of light, polarization and fluorescence microscopic methods for amyloid. Appl Pathol. 1985;3:5–17. [PubMed] [Google Scholar]
- Rebola N, Canas PM, Oliveira CR, Cunha RA. Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and striatum of the rat. Neuroscience. 2005;132:893–903. doi: 10.1016/j.neuroscience.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Rebola N, Lujan R, Cunha RA, Mulle C. Long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses: an essential role for adenosine A2A receptors. Neuron. 2008;57:121–134. doi: 10.1016/j.neuron.2007.11.023. [DOI] [PubMed] [Google Scholar]
- Resende R, Ferreiro E, Pereira C, Resende de Oliveira C. Neurotoxic effect of oligomeric and fibrillar species of amyloid-beta peptide 1-42: involvement of endoplasmic reticulum calcium release in oligomer-induced cell death. Neuroscience. 2008;155:725–737. doi: 10.1016/j.neuroscience.2008.06.036. [DOI] [PubMed] [Google Scholar]
- Reyes AE, Chacón MA, Dinamarca MC, Cerpa W, Morgan C, Inestrosa NC. Acetylcholinesterase-Aβ complexes are more toxic than Aβ fibrils in rat hippocampus: effect on rat β-amyloid aggregation, laminin expression, reactive astrocytosis, and neuronal cell loss. Am J Pathol. 2004;164:2163–2174. doi: 10.1016/s0002-9440(10)63774-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie K, Carrière I, de Mendonça A, Portet F, Dartigues JF, Rouaud O, Barberger-Gateau P, Ancelin ML. The neuroprotective effects of caffeine: a prospective population study (the Three City Study) Neurology. 2007;69:536–545. [Google Scholar]
- Roselli F, Tirard M, Lu J, Hutzler P, Lamberti P, Livrea P, Morabito M, Almeida OF. Soluble β-amyloid 1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J Neurosci. 2005;25:11061–11070. doi: 10.1523/JNEUROSCI.3034-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui Y, Li R, Liu Y, Zhu S, Yu X, Sheng Z, Xie Z. Acute effect of β amyloid on synchronized spontaneous Ca2+ oscillations in culture hippocampal networks. Cell Biol Int. 2006;30:733–740. doi: 10.1016/j.cellbi.2006.05.005. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Stowers CC, Scallet AC, Xu L. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 2005;1035:24–31. doi: 10.1016/j.brainres.2004.11.054. [DOI] [PubMed] [Google Scholar]
- Schulte G, Fredholm BB. Signalling from adenosine receptors to mitogen-activated protein kinases. Cell Signal. 2003;15:813–827. doi: 10.1016/s0898-6568(03)00058-5. [DOI] [PubMed] [Google Scholar]
- Schwarzschild MA, Agnati L, Fuxe K, Chen JF, Morelli M. Targeting adenosine A2A receptors in Parkinson's disease. Trends Neurosci. 2006;29:647–654. doi: 10.1016/j.tins.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha BR, Vitolo OV, Joshi P, Lordkipanidze T, Shelanski M, Dunaevsky A. Amyloid beta peptide adversely affects spine number and motility in hippocampal neurons. Mol Cell Neurosci. 2006;33:274–282. doi: 10.1016/j.mcn.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Silva CG, Porciúncula LO, Canas PM, Oliveira CR, Cunha RA. Blockade of adenosine A2A receptors prevents staurosporine-induced apoptosis of rat hippocampal neurons. Neurobiol Dis. 2007;27:182–189. doi: 10.1016/j.nbd.2007.04.018. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RN, Pamplona FA, Prediger RD. Adenosine receptor antagonists for cognitive dysfunction: a review of animal studies. Front Biosci. 2008;13:2614–2632. doi: 10.2741/2870. [DOI] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, Greene LA. beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem. 2001;77:157–164. doi: 10.1046/j.1471-4159.2001.t01-1-00218.x. [DOI] [PubMed] [Google Scholar]
- Venkitaramani DV, Chin J, Netzer WJ, Gouras GK, Lesne S, Malinow R, Lombroso PJ. β-Amyloid modulation of synaptic transmission and plasticity. J Neurosci. 2007;27:11832–11837. doi: 10.1523/JNEUROSCI.3478-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LH, Besirli CG, Johnson EM., Jr Mixed-lineage kinases: a target for the prevention of neurodegeneration. Annu Rev Pharmacol Toxicol. 2004a;44:451–474. doi: 10.1146/annurev.pharmtox.44.101802.121840. [DOI] [PubMed] [Google Scholar]
- Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid β-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004b;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart TM, Parson SH, Gillingwater TH. Synaptic vulnerability in neurodegenerative disease. J Neuropathol Exp Neurol. 2006;65:733–739. doi: 10.1097/01.jnen.0000228202.35163.c4. [DOI] [PubMed] [Google Scholar]
- Wu CC, Chawla F, Games D, Rydel RE, Freedman S, Schenk D, Young WG, Morrison JH, Bloom FE. Selective vulnerability of dentate granule cells prior to amyloid deposition in PDAPP mice: digital morphometric analyses. Proc Natl Acad Sci U S A. 2004;101:7141–7146. doi: 10.1073/pnas.0402147101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Mei M, Lee HG, Wang Y, Han J, Perry G, Smith MA. P38 activation mediates amyloid-beta cytotoxicity. Neurochem Res. 2005;30:791–796. doi: 10.1007/s11064-005-6872-x. [DOI] [PubMed] [Google Scholar]