

Abstract
The dopaminergic system plays an important role in the etiology of schizophrenia, and most antipsychotic drugs exert their functions by blocking dopamine D2 receptors (D2Rs). Since the signaling strength mediated by D2Rs is regulated by internalization and degradation processes, it is crucial to identify molecules that modulate D2R localization at the cell surface. Here, we show that the neural cell adhesion molecule (NCAM) promotes D2R internalization/desensitization and subsequent degradation via direct interaction with a short peptide in the third intracellular loop of the D2R. NCAM deficiency in mice leads to increased numbers of D2Rs at the cell surface and augmented D2R signaling as a result of impaired D2R internalization. Furthermore, NCAM-deficient mice show higher sensitivity to the psychostimulant apomorphine and exaggerated activity of dopamine-related locomotor behavior. These results demonstrate that, in addition to its classical function in cell adhesion, NCAM is involved in regulating the trafficking of the neurotransmitter receptor D2R as well as receptor-mediated signaling and behavior, thus implicating NCAM as modulator of the dopaminergic system and a potential pharmacological target for dopamine-related neurological and psychiatric disorders.
Introduction
The dopaminergic system is involved in the regulation of locomotion, cognition, emotional behavior, and endocrine secretion. Aberrant dopaminergic signaling is implicated in several neurological and psychiatric disorders, such as Parkinson's disease, depression, schizophrenia, and drug abuse (Zhou and Palmiter, 1995; Carlsson, 2001; Greengard, 2001; Nestler, 2001). Dopamine (DA) exerts its effects through two classes of dopamine receptors, D1-like receptors (D1R and D5R) and D2-like receptors (D2R, D3R, and D4R) (Seeman and Van Tol, 1994; Missale et al., 1998; Beaulieu et al., 2005). Increased activity of D2R signaling is believed to play an important role in the pathogenesis of schizophrenia. Schizophrenic patients show increased baseline occupancy of D2Rs by dopamine, and the number of D2Rs is elevated in the striatum of schizophrenic brains (Wong et al., 1986; Abi-Dargham et al., 2000). Major antipsychotic drugs exert their functions by blocking D2Rs, and the dopamine-releasing drugs worsen emotional symptoms (Creese et al., 1976; Gray and Roth, 2007).
D2R-mediated signaling is extensively regulated by multiple processes, and endocytosis is a major mechanism of D2R signal attenuation. D2R internalization occurs through clathrin-mediated endocytosis (Paspalas et al., 2006), a common mechanism for protein internalization from the plasma membrane (Mousavi et al., 2004). After endocytosis, internalized D2Rs are targeted to lysosomes for degradation; thus, D2R responses fail to resensitize after agonist treatment (Bartlett et al., 2005). D2R internalization is finely tuned, and impaired internalization has been implicated in schizophrenia (Iizuka et al., 2007). However, apart from the essential role of phosphorylation in D2R internalization (Ito et al., 1999; Kabbani et al., 2002; Namkung and Sibley, 2004), the specific molecular mechanisms that modulate D2R endocytosis have remained poorly understood.
Neural cell adhesion molecule (NCAM) is a glycoprotein highly expressed and first discovered in the nervous system (Edelman, 1985). Three major isoforms are generated by alternative splicing: NCAM180 and NCAM140 are transmembrane proteins, whereas NCAM120 is attached to the plasma membrane via a glycophosphatidyl inositol linkage (Maness and Schachner, 2007). NCAM is widely expressed in midbrain dopaminergic neurons and exerts a regulatory role on the development and survival of dopaminergic neurons via mediating the signaling of the neurotrophic factors, glial cell line-derived neurotrophic factor (GDNF) and brain-derived neurotrophic factor (Hyman et al., 1991; Lin et al., 1993; Levivier et al., 1995; Muller et al., 2000; Chao et al., 2003; Paratcha et al., 2003). Increasing evidence indicates that NCAM is related to psychiatric and neurodegenerative disorders, such as schizophrenia and bipolar disorders (Brennaman and Maness, 2008). NCAM180-deficient mice show impaired prepulse inhibition of startle (PPI), which is one characteristic of schizophrenic patients (Wood et al., 1998). Transgenic mice expressing the extracellular domain of NCAM show higher basal locomotor activity and enhanced responses to amphetamine, an indirect dopamine agonist, and a deficit in PPI (Pillai-Nair et al., 2005). In addition, schizophrenic patients show reduced polysialylated NCAM levels in hippocampus (Barbeau et al., 1995) and increased soluble NCAM fragments in CSF or in hippocampus and cortex (Lyons et al., 1988; van Kammen et al., 1998; Vawter et al., 1998).
Since both D2R and NCAM are associated with schizophrenia, we became interested in a potential functional relationship between D2R and NCAM. Here, we show that NCAM plays an important role in the regulation of D2R-dependent locomotor activity by modulating D2R internalization. NCAM deficiency in mice leads to hyperactivity of dopamine-related locomotion because of a disrupted D2R internalization process, which in turn results in augmented D2R signaling.
Materials and Methods
Experimental animals
C57BL/6J mice bred and maintained at the Universitätsklinikum Hamburg-Eppendorf were used for all experiments. NCAM-deficient (NCAM−/−) mice (Cremer et al., 1994) kindly provided by H. Cremer (Developmental Biology Institute of Marseille Luminy, Centre National de la Recherche Scientifique/Université de Méditerranée, Marseille, France) have been backcrossed onto the C57BL/6J background for more than eight generations and their age-matched wild-type (NCAM+/+) mice were used as controls. In all experiments, 2- to 3-month-old mice were used with the exception of hippocampal cultures, which used 1- to 2-d-old mice. Animals were housed at 25°C on a 12 h light/dark cycle with ad libitum access to food and water.
Antibodies
NCAM monoclonal D3 antibody (Schlosshauer, 1989) reacting with an epitope encoded by the NCAM180-specific exon 18, monoclonal 5B8 antibody (Dodd et al., 1988) recognizing the intracellular domains (ICDs) of NCAM140 and NCAM180, and polyclonal NCAM antibody 1β2 recognizing the extracellular domain of mouse NCAM have been described previously (Niethammer et al., 2002). Monoclonal antibody against D2R and myc were obtained from Santa Cruz Biotechnology. Dopamine and cAMP-regulated phosphoprotein with molecular weight 32 kDa (DARPP32) antibody was purchased from Cell Signaling Technology. Tyrosine hydroxylase (TH) and D1R antibodies were from Millipore. Antibodies against phospho-TH Ser40 or phospho-DARPP32 Thr34 were obtained from AbD Serotec. Mouse antibody against penta-His was from QIAGEN. Goat antibody against glutathione S-transferase (GST) was from GE Healthcare. All horseradish peroxidase (HRP)-coupled secondary antibodies and Cy2, Cy3, and Cy5-coupled secondary antibodies were obtained from Jackson ImmunoResearch Laboratories. Neutravidin-HRP was from Sigma-Aldrich.
DNA constructs
Rat pcDNA3-NCAM140 and rat pcDNA3-NCAM180 were kind gifts from P. Maness (University of North Carolina, Chapel Hill, NC). Exon 18, NCAM140-ICD, N-terminal truncated NCAM140-ICD (140-ICDΔN, lacking amino acids 730-772), and NCAM180-ICD in pQE30 were used to generate various His-tagged NCAM fragments. The full-length rat D2R cDNA cloned in pcDNA3 was kindly provided by D. R. Sibley (National Institute of Neurological Disorders and Stroke–National Institutes of Health, Bethesda, MD). IC3-D2R was subcloned into pGEX-4T-2 to generate the GST-tagged protein. Mutation of IC3-D2R (S311C), which is associated with schizophrenia (Itokawa et al., 1993), was generated by using QuickChange II XL-Site Directed Mutagenesis kit (Stratagene). To create the IC3-D2R (S311C) mutant, the sense primer 5′-CTC ACT CTC CCT GAT CCA TGC CAC CAC GGC CTA CAT AGC-3′ and the antisense primer 5′-GCT ATG TAG GCC GTG GTG GCA TGG ATC AGG GAG AGT GAG-3′ were used (sequence differences to wild-type D2R are given in bold letters). All DNA constructs were generated by PCR and sequences were verified.
Cell culture and transfection
Cultures of hippocampal neurons were prepared from 1- to 2-d-old C57BL/6J mice. Hippocampi were isolated, digested in trypsin (Sigma-Aldrich) plus DNase (Sigma-Aldrich), and triturated in DNase with fire-polished glass pipettes. Cells were then centrifuged, resuspended, and plated in Neurobasal A medium (Invitrogen) supplemented with B-27 (Invitrogen), 5 μg/ml gentamycin, 1 mm l-glutamine, 5 μm cytosine β-d-arabinofuranoside, and 12.5 ng/ml fibroblast growth factor (PeproTech) on coverslips coated with 100 μg/ml poly-l-lysine (Sigma-Aldrich). One-half of the medium was replaced with fresh medium every 2 d.
Stably myc-D2R-expressing HEK293 cells (Liu et al., 2007), kindly provided by K. A. Neve (Department of Behavioral Neuroscience, Oregon Health & Science University, Portland, OR), were maintained in DMEM supplemented with 10% fetal calf serum and 2 μg/ml puromycin (Sigma-Aldrich). HEK293 cells were transiently transfected with pcDNA3-NCAM180 by FuGENE 6 transfection reagent (Roche Diagnostics) according to the protocol of the manufacturer.
For analysis of D2R degradation, HEK293-D2R cells transfected with NCAM were stimulated with 10 μm dopamine (Sigma-Aldrich) at 37°C during different time periods in the presence of 10 μg/ml protein synthesis inhibitor cycloheximide (Tocris Bioscience). Cells were then lysed with RIPA buffer (50 mm Tris-HCl, 150 mm NaCl, 1 mm Na4P2O7, 1 mm NaF, 1 mm EDTA, 2 mm Na3VO4, 1 mm PMSF, 1% NP-40 plus complete EDTA-free protease inhibitor mixture, pH 7.5) for 30 min at 4°C and subjected to SDS-PAGE and Western blot analysis.
Phage display
A phage library (New England Biolabs) displaying 108–1010 random 12-mer peptides at the pili of M13-like phage particles in fusion with the N terminus of the pVIII major coat protein was used. All in vitro selection steps were performed according to the instruction manual, version 2.0, of Ph.D.-12 Phage Display Peptide Library kit (New England Biolabs).
Western blot analysis
For analysis of phosphorylated proteins, brains from 2- to 3-month-old mice were homogenized in 1% SDS and immediately boiled for 10 min. Protein extracts were separated by 10% SDS-PAGE and transferred onto nitrocellulose membrane (Protran; Whatman Schleicher and Schuell). For immunoblotting, membranes were blocked with 5% nonfat dry milk powder in PBS, pH 7.4, and incubated overnight at 4°C with primary antibodies. After washes with PBST (PBS with 0.1% Tween 20), membranes were incubated with appropriate HRP-conjugated secondary antibodies for 1 h at room temperature. For detection of biotinylated proteins, membranes were incubated with streptavidin coupled to HRP. After extensive washes, immunoreactive or streptavidin-reactive bands were visualized using either the enhanced chemiluminescent substrate (ECL) or the chemiluminescent substrate with extended duration (Pierce) on x-ray films (Kodak Biomax-ML; Sigma-Aldrich). Band intensities were densitometrically quantified using the image software TINA 2.09.
GST pull-down assay
GST-tagged IC3-D2R (GST-IC3-D2R) was expressed in Escherichia coli and purified using glutathione agarose beads (Sigma-Aldrich) according to the manufacturer's instructions. NCAM-ICDs and various fragments of NCAM140-ICD or NCAM180-ICD were expressed in E. coli and captured by Ni-nitrilotriacetic acid (NTA) agarose beads. Equal amounts of GST proteins were incubated with His-tagged NCAM-ICDs in PBS containing 1% BSA at 4°C overnight with gentle rotation. Afterward, glutathione agarose beads were added and incubated at 4°C for 8 h under constant agitation, followed by extensive washes with PBS containing 1% NP-40. The pulled down proteins were subjected to SDS-PAGE and Western blot analysis.
Immunoprecipitation
Brains from 2- to 3-month-old C57BL/6J mice were homogenized in 50 mm Tris-HCl, pH 7.5, 1 mm CaCl2, 1 mm MgCl2 and 1 mm NaHCO3 plus complete EDTA-free protease inhibitor mixture (Roche Diagnostics). Brain protein extracts were generated by lysing brain homogenates with RIPA buffer for 1 h at 4°C. Protein extracts of HEK293 cells were generated from stably myc-D2R-expressing HEK293 cells transiently transfected with pcDNA3-NCAM. Two days later, cells were rinsed and lysed in RIPA buffer, followed by additional disruption with repeated aspiration through a 25 gauge needle. After centrifugation at 700 × g for 10 min at 4°C, the resulting supernatant was taken as cell lysate for immunoprecipitation.
Protein extracts were cleared with protein A/G-agarose beads (Santa Cruz Biotechnology) for 3 h at 4°C, and then incubated with anti-D2R antibody or control IgG overnight at 4°C with gentle rotation. Protein A/G-agarose was added to capture immunocomplexes for 6 h at 4°C under constant agitation. After extensive washes with RIPA buffer, immunoprecipitated proteins were eluted from agarose beads by 2× SDS sample buffer (125 mm Tris-HCl, 4% SDS, 30% glycerol, 10% β-mercaptoethanol, and 0.00625% bromophenol blue, pH 6.8).
Biochemical cross-linking
ICDs of NCAM180, NCAM140, or close homolog of L1 (CHL1) were coupled to the trifunctional cross-linker Sulfo-SBED (Pierce) by incubation for 1 h at room temperature in the dark. Unbound cross-linker was removed by overnight dialysis against PBS at 4°C. Brains from 2- to 3-month-old C57BL/6J mice were homogenized at 4°C in lysis buffer P+, which contains PBS plus 1 mm MgCl2, 1 mm MnCl2, 1 mm EDTA, 1 mm NaF, 0.5 mm Na3VO4, 0.5 mm H2O2, 1 μm okadaic acid, and complete EDTA-free protease inhibitor mixture, or lysis buffer P−, which contains PBS plus protein kinase C inhibitor peptide (Sigma-Aldrich) and complete EDTA-free protease inhibitor mixture. Brain homogenates were added to the ICD-cross-linker baits and incubated for 1 h at room temperature, followed by photoactivation under ultraviolet (UV) light (365 nm). Potential interaction partners bound to ICD-cross-linker baits were isolated by Ni-NTA beads (QIAGEN), followed by streptavidin beads (Pierce). After extensive washes, precipitated proteins were separated by SDS-PAGE and subjected to Western blot analysis.
Preparation of subcellular fractions
Mouse brains were homogenized in HOMO buffer (5 mm Tris-HCl, 0.32 m sucrose, 1 mm MgCl2, 1 mm CaCl2, 1 mm NaHCO3 plus complete EDTA-free protease inhibitor mixture, pH 7.5) and centrifuged at 1000 × g for 10 min at 4°C to pellet nuclei and mitochondria. The resulting supernatant was centrifuged at 17,000 × g for 20 min at 4°C.
Isolation of synaptosomes.
The 17,000 × g pellet was resuspended in HOMO buffer and loaded onto a discontinuous sucrose gradient, which consisted of 0.8, 1.0, and 1.2 m sucrose from the top to the bottom. After centrifugation at 100,000 × g for 1 h at 4°C, the interphase materials between 1.0 and 1.2 m sucrose, which contains synaptosomes, were collected and pelleted by centrifugation.
Isolation of plasma membranes.
The 17,000 × g pellet was subjected to a hypotonic shock by adding 9 vol of ice-cold H2O plus complete EDTA-free protease inhibitor mixture, and rapidly adjusted to 5 mm Tris-HCl by adding 1 m Tris-HCl, pH 7.5. After centrifugation at 25,000 × g for 20 min at 4°C, the pellet fraction containing lysed membrane was resuspended in HOMO buffer and loaded onto the discontinuous sucrose gradient, which consisted of 0.8, 1.0, and 1.2 m sucrose from the top to the bottom. After centrifugation at 150,000 × g for 2 h at 4°C, the interphase materials between 1.0 and 1.2 m sucrose, which contains plasma membranes, were collected and pelleted by centrifugation.
Isolation of endosomes.
The 17,000 × g supernatant was collected and centrifuged at 100,000 × g for 1 h at 4°C. The pellet was resuspended in 2 m sucrose gradient solution, and a sucrose gradient containing 0.25, 0.8, 1.15, 1.3 m of sucrose was loaded on the top of 2 m samples. After centrifugation at 100,000 × g for 2 h at 4°C, the interphase fraction between 0.8 and 1.15 m sucrose, which contains endosomes, was collected, and proteins were precipitated by methanol–chloroform.
Isolation of lysosomes.
Lysosomes were prepared by a discontinuous OptiPrep gradient using the lysosome isolation kit (Sigma-Aldrich). Fractions were collected at the interphases of the OptiPrep gradient and assayed for the lysosomal marker protein, Lamp (lysosome-associated membrane protein). The fraction at the interphase between 8 and 12% of the OptiPrep gradient was taken as lysosomes.
Cell surface biotinylation
A confluent monolayer of HEK293-D2R cells transiently transfected with NCAM was washed twice with ice-cold PBS containing 2 mm MgCl2 and 0.5 mm CaCl2 (PBS2+), and incubated with 0.5 mg/ml Sulfo-NHS-LC-biotin (Pierce) for 10 min at 4°C. Excess biotin was quenched by incubating cells with 20 mm glycine for 5 min at 4°C. Cells were rinsed twice with PBS2+ and lysed with RIPA buffer for 30 min at 4°C. After centrifugation at 700 × g for 10 min at 4°C, the supernatant was taken as cell lysates and incubated with streptavidin beads at 4°C overnight under constant agitation. After extensive washes with RIPA buffer, precipitated biotinylated proteins were eluted from beads with 2× SDS sample buffer and subjected to Western blot analysis.
Antibody-feeding immunocytochemistry
Antibody-feeding immunocytochemistry was performed according to the protocol from Bartlett et al. (2005). Live HEK293-D2R cells transfected with NCAM were incubated with monoclonal antibody against myc for 5 min at 37°C in a CO2 incubator, washed, and stimulated with dopamine (10 μm; 60 min; 37°C). Cells were fixed with 2% formaldehyde for 5 min and blocked with 5% nonimmune goat serum for 30 min at room temperature before application of Cy3-conjugated secondary antibody to label cell surface D2Rs. Internalized D2Rs were detected by Cy5-conjugated secondary antibody after permeabilizing cells with 0.2% Triton X-100. Polyclonal antibody against NCAM was applied and incubated overnight at 4°C, followed by incubation with Cy2-conjugated secondary antibody after washes.
Confocal images were acquired using a Leica confocal microscope (Leica SP2; Leica Microsystems). For quantification of internalization, the confocal settings for image acquisition were maintained for all cells. Image stacks were flattened into a single image by a maximum projection and analyzed with ImageJ. Surface and internal D2R fluorescence intensities were measured as the integrated pixel intensities in the red and blue channels, respectively. Total D2R fluorescence was determined as the sum of the surface (red) and the internal (blue) fluorescence intensities. For each cell, the internalization index was defined as the ratio of the internalized fluorescence intensity to the total fluorescence intensity.
Immunocytochemistry
Cells were fixed in 4% ice-cold formaldehyde and 4% sucrose for 15 min, and blocked in 5% nonimmune goat serum containing 0.2% Triton X-100 for 30 min at room temperature. Cells were then incubated with primary antibodies overnight at 4°C in a humidified chamber. After several washes with PBS, appropriate secondary antibodies coupled with Cy2 or Cy3 were applied and incubated for 1 h at room temperature in the dark. After extensive washes, nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich), and coverslips were mounted on glass slides with Fluoromount G (Southern Biotechnology).
Drug administration
The D1R-specific antagonist 8-bromo-2,3,4,5-tetrahydro-3-methyl-5-phenyl-1H-3-benzazepin-7-ol hydrobromide (SKF83566) (Tocris Bioscience) or D2R-specific antagonist raclopride (Sigma-Aldrich) were dissolved in saline and injected intraperitoneally before behavioral testing. To investigate the effect of dopamine receptor agonists in dopamine-depleted mice, a combination of reserpine (5 mg/kg, i.p.; 20 h before the test; Sigma-Aldrich) and methyl-dl-tyrosine (250 mg/kg, i.p.; 1 h before the test; Sigma-Aldrich) were applied to deplete dopamine in mice (Gainetdinov et al., 2003), followed by administration of the D1R/D2R agonist apomorphine (1 mg/kg, s.c.; Sigma-Aldrich). Reserpine, methyl-dl-tyrosine and apomorphine were dissolved in distilled water with drops of glacial acetic acid, hydrochloric acid, or 0.1% ascorbic acid, respectively. All injections were given in a volume of 0.1 ml/30 g of body weight.
Measurement of locomotor activity
Locomotion was evaluated in the open field as described previously (Law et al., 2003). The open field consisted of a wooden box (50 × 50 × 40 cm) laminated with rough, matted, light-gray resin and illuminated by a white bulb (100 lux). After drug administration, mice were gently introduced into a cylinder placed at the corner of the box for 5 s. As the cylinder was lifted, mice could move freely in the arena for a duration of 30 min. Locomotor activity was measured at 5 min intervals and cumulative counts were taken for data analysis with the software EthoVision (Noldus).
Statistical analysis
Values in graphs are presented as mean + SEM. Data were analyzed by unpaired t test. The threshold value for acceptance of differences between group mean values was 5%.
Results
NCAM interacts with the D2R at amino acids 296-320 via the N-terminal segment of NCAM-ICD
In search of interaction partners of NCAM-ICD, we performed a phage display screening of a random 12-mer peptide library using NCAM180-ICD as a bait, and obtained a phage expressing an NCAM180-ICD binding peptide that showed similarity to a sequence stretch within the third intracellular domain (IC3) of D2R (Fig. 1A). This short stretch comprising amino acids 296-310 is present in the IC3-D2R. To ascertain the result from phage display screening, pull-down experiments were performed by using recombinant NCAM180-ICD and GST-IC3-D2R. NCAM180-ICD was pulled down by GST-IC3-D2R in a concentration-dependent manner (Fig. 1B). No such pull down of NCAM180-ICD was observed when NCAM180-ICD was incubated with the GST control (Fig. 1B).
Figure 1.

Identification of an interaction of NCAM with D2R. A, The sequence of a peptide selected by screening a phage library using the recombinant intracellular domain of NCAM180 (NCAM180-ICD) as a bait shows significant similarity to a sequence in the third intracellular domain of the dopamine D2 receptor (IC3-D2R). Identical amino acids (|) are shown. Numbers designate amino acid positions. B, Increasing amounts of NCAM180-ICD (1–10 μg/ml) were incubated with GST-IC3-D2R, followed by pull down with glutathione beads. Incubation of 10 μg/ml NCAM180-ICD with GST alone was served as control. Pulled down proteins were detected by NCAM antibody 5B8 and GST antibody. C, Schematic representation of NCAM180-ICD structure. The gray-marked sequence highlights N-terminal truncated fragment of NCAM140-ICD (140-ICDΔN, lacking amino acids 730-772). The dashed line indicates exon 18 encoded sequence of NCAM. Numbers designate amino acid positions. D, GST pull-down assay was performed with recombinant His-tagged NCAM180-ICD, exon 18 encoded protein, and NCAM140-ICDΔN with GST-IC3-D2R or GST as control. Precipitated proteins were detected by anti-Penta His antibody. E, GST pull-down assay with NCAM180-ICD and GST-IC3-D2R in the presence of NCAM peptide 1 (P1) and peptide 2 (P2). The minus sign (−) designates no application of NCAM peptides. F, Schematic representation of D2R structure. The line highlights the D2R peptide that shows similarity to the peptide identified by phage library screening, and numbers designate amino acid positions. Pull down was performed with NCAM180-ICD and GST-IC3-D2R in the presence of increasing concentrations of D2R peptide (2 and 8 μg/ml). The minus sign (−) designates no application of D2R peptide. G, GST pull-down assay was performed by incubation NCAM180-ICD with GST-IC3-D2R or mutated GST-IC3-D2R (S311C).
Additional pull-down assays were performed to clarify the binding sites on NCAM and D2R that could mediate their interaction. NCAM180-ICD differs from NCAM140-ICD by the presence of additional 261 aa, which are encoded by exon 18 (Fig. 1C). Since both NCAM180-ICD (Fig. 1B) and NCAM140-ICD (data not shown) interacted with D2R in the GST pull-down assays, it was unlikely that NCAM180-specific exon 18 sequences are the binding domain for D2R. Pull-down assays using the recombinant protein encoding exon 18 of NCAM180 revealed no pull down of exon 18 with GST-IC3-D2R (Fig. 1D), verifying that the exon 18 indeed did not mediate the interaction between NCAM and D2R. Similarly, recombinant N-terminally truncated NCAM140-ICD lacking amino acids 730-772 (NCAM140-ICDΔN) (Fig. 1C) did not show pull down with GST-IC3-D2R either (Fig. 1D), indicating that the binding region is present in the membrane-proximal terminus of NCAM-ICD. To further narrow down the binding site, two peptides matching the N-terminal sequences of NCAM-ICD (peptide 1, 730-750; peptide 2, 748-765) (Fig. 1C) were applied for competition in the pull-down assay. NCAM peptide 2 showed strong competition with the binding of NCAM-ICD to GST-IC3-D2R, whereas peptide 1 exhibited no competition (Fig. 1E). This result indicates that the membrane-proximal part of NCAM-ICD carries the binding site for D2R.
To confirm the phage display result showing that amino acids 296-310 of D2R mediate the binding to NCAM, the D2R peptide (296-320) (Fig. 1F) was used as competitor in the GST pull-down assay. The D2R peptide reduced the binding of NCAM180-ICD to IC3-D2R in a concentration-dependent manner (Fig. 1F), indicating that the binding site of D2R to NCAM localizes to the D2R peptide region. These results demonstrate that NCAM interacts with IC3-D2R at amino acids 296-320 via an intracellular segment of NCAM close to the transmembrane domain.
The polymorphism of serine to cysteine at the 311 residue of D2R, which locates within the binding region for NCAM, has been implicated as a risk factor in schizophrenia (Itokawa et al., 1993). To investigate the effect of this mutation on the NCAM/D2R interaction, mutated GST-IC3-D2R (S311C) was generated by site-directed mutagenesis and used in the pull-down assay. Compared with wild-type IC3-D2R, mutated IC3-D2R (S311C) showed a ∼35% decrease of the binding to NCAM180-ICD (65.4 ± 7.3% in mutated D2R, when compared with wild-type D2R, which was set to 100%; n = 4; p < 0.01 by unpaired t test) (Fig. 1G), indicating that the mutation at 311-serine of D2R disrupts the NCAM/D2R interaction, and confirming that this D2R sequence stretch mediates the interaction with NCAM.
Cell-biological indications for the NCAM/D2R interaction
To obtain indications for the NCAM/D2R interaction in neural cells, we first examined the subcellular localization of NCAM and D2R in cultured hippocampal neurons using immunofluorescence colabeling. NCAM partially colocalized with D2R at the cell body and along neurites (Fig. 2A), suggesting these two proteins associate with each other in neurons.
Figure 2.
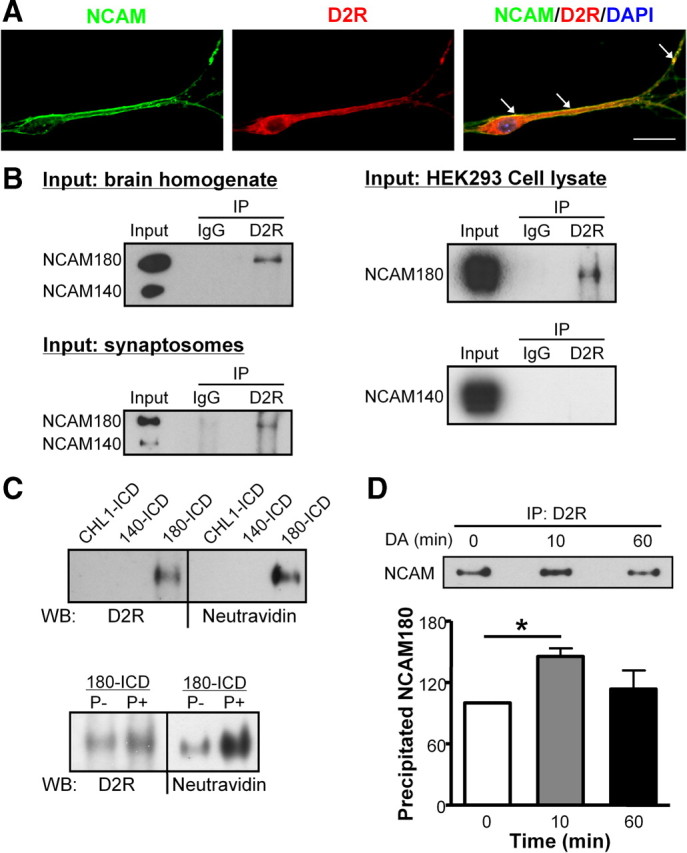
Interaction between NCAM and D2R in cells and tissue. A, Cultured hippocampal neurons were subjected to immunostaining with NCAM antibody and D2R antibody; nucleus was stained with DAPI. Colocalization of NCAM and D2R in cell soma and neurites is indicated by the white arrows. Scale bar, 20 μm. B, Brain homogenate, synaptosomal fraction, or HEK293 cell lysate transfected with D2R and NCAM were subjected to immunoprecipitation (IP) using mouse anti-D2R antibody or nonspecific mouse IgG. Precipitated proteins were analyzed by Western blot with NCAM antibody 5B8. Coimmunoprecipitation of NCAM180 and D2R was observed. C, NCAM180-ICD, NCAM140-ICD, or CHL1-ICD were coupled to the biotin containing cross-linker Sulfo-SBED and incubated as baits with brain homogenate under dephosphorylation (P−) or phosphorylation (P+) conditions. After UV cross-linking and denaturation under reducing conditions, the biotin moiety was transferred from the cross-linker to the molecules bound to the baits. Bound proteins were analyzed by Western blot (WB) using Neutravidin and anti-D2R antibody. D, Myc-D2R-expressing HEK293 cells were transfected with NCAM180 and stimulated with 10 μm DA for 0, 10, or 60 min. Cell lysates were prepared and subjected to immunoprecipitation (IP) using mouse anti-D2R antibody. Precipitated proteins were analyzed by Western blot with NCAM antibody 5B8. Coimmunoprecipitation of NCAM180 and D2R was observed. The amount of coimmunoprecipitated NCAM was quantified and the amount obtained after 0 min stimulation was set to 100%. Means + SEM are shown. *p < 0.05 by unpaired t test (n = 3).
Next, coimmunoprecipitation experiments were performed using brain homogenate, synaptosomal fractions, or cell lysate from transfected cells. When D2R antibody was used for immunoprecipitation, Western blot analysis of immunoprecipitates using an NCAM antibody that recognizes NCAM140 and NCAM180 showed that NCAM180, but not NCAM140, was coimmunoprecipitated from brain homogenate, synaptosomes, and D2R-transfected cells, whereas no such coimmunoprecipitation was observed with a control antibody (Fig. 2B). This specific coimmunoprecipitation suggests that, in brain tissue and cultured cells, only the NCAM180 isoform interacts with D2R.
To further characterize the interaction between NCAM and D2R, a chemical cross-linking experiment was performed. The trifunctional cross-linker Sulfo-SBED containing a biotin moiety was coupled to the ICDs of NCAM180 and NCAM140, or CHL1 as a control, followed by incubation with brain homogenate. After UV cross-linking, the samples were separated by SDS-PAGE under reducing conditions, which leads to the transfer of the biotin moiety from the cross-linker to the molecules that bound to the ICD–cross-linker conjugates. Biotinylated D2Rs were detected when NCAM180-ICD was used for cross-linking, whereas D2Rs were not isolated when using NCAM140-ICD or CHL1-ICD (Fig. 2C). In line with results from coimmunoprecipitation experiments, cross-linking revealed that NCAM180, but not NCAM140 interacted with D2R.
Furthermore, since phosphorylation plays an important role in protein– protein interactions, we performed the cross-linking experiment under condition that favors either phosphorylation or dephosphorylation. We found that the NCAM/D2R interaction was enhanced under phosphorylation conditions compared with dephosphorylation conditions (Fig. 2C).
Together, these observations indicate that NCAM180, but not NCAM140, interacts with D2R in cultured neurons and transfected HEK293 cells, brain tissue, and synaptosomes.
NCAM/D2R interaction is regulated by dopamine stimulation
Agonist-stimulated receptor internalization is a common feature of G-protein-coupled receptors in the regulation of receptor responsiveness to its ligand (Bohm et al., 1997). To investigate whether the formation of the NCAM/D2R complex depends on dopamine stimulation, HEK293 cells transfected with NCAM and D2R were stimulated with dopamine followed by immunoprecipitation with D2R antibody. Quantitative Western blot analysis of immunoprecipitates showed a significant increase in the levels of NCAM that were coimmunoprecipitated with D2R after 10 min of dopamine stimulation. Sixty minutes after dopamine stimulation, the level of NCAM that was coimmunoprecipitated with D2R was comparable with those obtained under nonstimulated conditions (Fig. 2D). This result indicates that the NCAM/D2R interaction is enhanced on dopamine stimulation and suggests that NCAM modulates the D2R function after dopamine stimulation.
NCAM reduces the cell surface localization of D2R and promotes its internalization and degradation
In response to the stimulation of dopamine, D2Rs undergo internalization in both cultured cells and brain tissue (Itokawa et al., 1996; Sun et al., 2003). To analyze whether NCAM plays a role in D2R internalization, we first determined the surface expression of D2Rs in dopamine-stimulated HEK293-D2R cells by a cell surface biotinylation approach. The levels of surface-exposed and total D2Rs were determined by Western blot using D2R antibody. The levels of D2Rs at the cell surface in mock-transfected and NCAM180-transfected cells were comparable in the absence of dopamine (Fig. 3A). However, after dopamine stimulation, the amount of D2Rs at the cell surface in NCAM180-transfected cells was significantly decreased compared with the mock-transfected cells (Fig. 3A). Quantification of the amount of cell surface D2Rs normalized to the total amount of D2Rs revealed a reduction by ∼50%. Another transmembrane protein, amyloid precursor protein (APP), remained at a constant level on the cell surface independently on NCAM expression or dopamine stimulation (Fig. 3A). However, no such effect was observed in NCAM140-transfected cells, which showed the same levels of D2Rs at the cell surface as obtained in mock-transfected group with dopamine stimulation (data not shown). These results indicate that NCAM180 reduces the cell surface D2R localization on dopamine stimulation. At the same time, NCAM levels at the cell surface were not altered by dopamine stimulation (84.8 ± 9.9% with dopamine stimulation, when the NCAM levels obtained in the absence of dopamine were set to 100%; n = 3; p > 0.05 by unpaired t test) (Fig. 3A), suggesting that NCAM does not internalize together with D2Rs on dopamine treatment.
Figure 3.
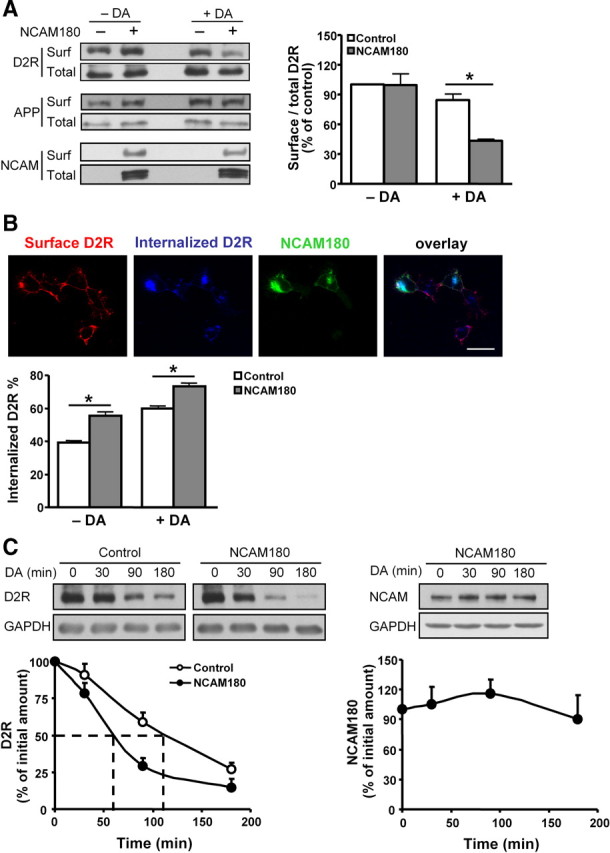
Regulation of D2R trafficking by NCAM. A, Myc-D2R-expressing HEK293 cells were mock-transfected or transfected with NCAM180. Two days later, cells were stimulated with 10 μm DA for 30 min, followed by cell surface biotinylation with Sulfo-NHS-LC-biotin. Biotin-labeled surface proteins (surf) and total proteins (total) were detected by Western blot analysis using antibodies against D2R, APP, or NCAM, respectively. The levels of selected proteins at the cell surface relative to their total levels were determined, with the ratio obtained from the mock-transfected cells in the absence of dopamine being set to 100%. *p < 0.05 by unpaired t test (n = 3). B, Myc-D2R-expressing HEK293 cells transfected with NCAM180 were subjected to live staining with myc antibody to label cell surface D2Rs. After treatment with 10 μm dopamine, cells were stained with antibodies against D2R before permeabilization (surface D2Rs) and after permeabilization (internalized D2Rs). Percentage of internalized D2Rs, defined as the ratio of internalized to total fluorescence intensities, was presented as mean + SEM for 50 cells. Results are representative of two independent experiments. *p < 0.05 by unpaired t test. Scale bar, 25 μm. C, Myc-D2R-expressing HEK293 cells transiently transfected with NCAM180 were stimulated with 10 μm DA for 0, 30, 90, or 180 min in the presence of 10 μg/ml cycloheximide to block de novo protein synthesis. The levels of D2R and NCAM were evaluated by Western blot analysis with D2R and NCAM antibodies, respectively. Determination of GAPDH levels served as control for loading. The proteins levels obtained by dopamine stimulation for 0 min were set to 100%. The dashed lines indicate 50% degradation of D2Rs. Means + SEM are shown here (n = 3).
We then investigated whether the decreased cell surface D2R localization in NCAM180-expressing cells was attributable to enhanced D2R internalization. An antibody-feeding immunocytochemistry approach was taken to label cell surface D2Rs with antibodies on live cells, followed by exposure to dopamine to trigger internalization of receptor–antibody complexes in live cells. Afterward, remaining surface receptors and internalized receptors were immunostained in the same cell under nonpermeabilization or permeabilization conditions, respectively. Quantification of fluorescence intensity revealed that NCAM180-positive cells, rather than NCAM140-positive cells, showed significantly higher levels of internalized D2Rs relative to NCAM-negative cells both without and with dopamine stimulation (Fig. 3B) (data for NCAM140 are not shown), indicating that NCAM180, but not NCAM140, promotes the internalization of D2R in a dopamine-independent manner.
It has been shown that most internalized D2Rs are transported to lysosomes for degradation and do not recycle back to the cell surface (Bartlett et al., 2005). We therefore examined whether NCAM regulates the degradation of D2R by affecting the internalization of D2R on dopamine stimulation. HEK293-D2R cells were treated with dopamine to stimulate the internalization and degradation of D2Rs in the presence of cycloheximide, which blocks protein synthesis (Siegel and Sisler, 1963). Levels of D2Rs were then determined by quantitative Western blot analysis. Compared with mock-transfected cells, degradation of D2Rs was accelerated in NCAM180-transfected, but not in NCAM140-transfected, cells: 50% of total D2Rs disappeared after ∼60 min of dopamine stimulation in NCAM180-transfected cells and after ∼110 min in mock-transfected and NCAM140-transfected cells (Fig. 3C) (data for NCAM140 are not shown), respectively. However, NCAM180 was not degraded during the time phase and remained at a constant level on dopamine stimulation (Fig. 3C), indicating that NCAM serves as a “chaperone” molecule for D2R internalization and degradation. Together, NCAM180 reduces the level of cell surface D2R and promotes its internalization and subsequent degradation.
NCAM deficiency in mice leads to altered subcellular distribution of D2R
Since NCAM regulates D2R internalization, we asked whether the ablation of NCAM in NCAM−/− mice alters the internalization of D2R in vivo by investigating the subcellular distribution of D2R in brains of NCAM−/− versus NCAM+/+ mice. Western blot analysis revealed no difference in total D2R expression when comparing the D2R levels in brain homogenates or synaptosomes of NCAM−/− and NCAM+/+ mice (108.7 ± 7.5% in total brain homogenates and 97.2 ± 6.9% in synaptosomes of NCAM−/− mice, respectively, when compared with NCAM+/+ mice, which were set to 100%; n = 5; p > 0.05 by unpaired t test) (Fig. 4A). To analyze the cell surface expression of D2R, plasma membrane fractions were subjected to Western blot analysis. The plasma membrane fraction from NCAM−/− brains showed a twofold (2.05 ± 0.13) higher amount of D2Rs relative to that found from NCAM+/+ brains (n = 5; p < 0.001 by unpaired t test) (Fig. 4B), whereas no difference was observed for the D1R (100.2 ± 10.2% in NCAM−/− mice with D1R levels in NCAM+/+ mice being set to 100%; n = 5; p > 0.05 by unpaired t test) (Fig. 4B), indicating that the level of D2Rs at the plasma membrane is dysregulated in NCAM−/− mice. Additional analyses of fractions enriched in endosomes, lysosomes, endoplasmic reticulum (ER), and Golgi apparatus were performed. Compared with fractions from NCAM+/+ brains, a significant reduction in the levels of D2Rs was found in endosomal and lysosomal fractions from NCAM−/− brains (60.1 ± 7.5% in endosomes and 60.4 ± 5.4% in lysosomes of NCAM−/− mice, respectively, when compared with D2R levels in NCAM+/+ mice, which were set to 100%; n = 5; p < 0.01 by unpaired t test) (Fig. 4C). No difference was observed with ER or Golgi fractions between the two genotypes (data not shown).
Figure 4.

Subcellular distribution of D2R is altered in NCAM-deficient mouse brains. The amounts of D2Rs (A–C) and D1Rs (B) were determined in total brain homogenates and synaptosomal fractions (A), in plasma membrane fractions (B), and in fractions enriched in endosomes and lysosomes (C) prepared from adult wild-type (NCAM+/+) and NCAM-deficient (NCAM−/−) mouse brains.
The decrease of D2R levels in endosomal and lysosomal fractions and the elevated levels of D2R in plasma membrane-derived fractions indicate that NCAM deficiency leads to an abnormal subcellular D2R distribution and confirms that NCAM regulates internalization and degradation of D2R.
NCAM−/− mice show augmented D2R-mediated signaling
Since the levels of neurotransmitter receptors at the plasma membrane dictate the strength of receptor responses (Gainetdinov et al., 2003), elevated amounts of D2Rs at the plasma membrane of NCAM−/− brains could indicate an alteration in D2R-mediated signaling pathways. Activation of the long D2R isoform (D2L), which is predominantly postsynaptic, leads to inhibition of the phosphorylation of DARPP32 at the Thr34 residue, whereas activation of the short D2R isoform (D2S), which is located in presynaptic boutons and functions as an autoreceptor, inhibits the phosphorylation of TH at the Ser40 residue (Giros et al., 1989; Lindgren et al., 2003) (Fig. 5A). Both pathways were subjected to Western blot analysis with antibodies recognizing the phosphorylated Thr34 of DARPP32 or Ser40 of TH in brain homogenates of NCAM+/+ and NCAM−/− mice. Compared with NCAM+/+ brains, the amount of phospho-Thr34-DARPP32 was significantly reduced in NCAM−/− mice (Fig. 5B). Quantification of the levels revealed a ∼40% decrease of phospho-Thr34-DARPP32 in NCAM−/− versus NCAM+/+ mouse brains (61.1 ± 11.6% in NCAM−/− mice, when compared with phospho-Thr34-DARPP32 levels in NCAM+/+ mice, which were set to 100%; n = 6; p < 0.05 by unpaired t test) (Fig. 5B). However, the levels of phospho-Ser40-TH were similar in the two genotypes (94.7 ± 11.2% in NCAM−/− mice, when compared with phospho-Ser40-TH levels in NCAM+/+ mice, which were set to 100%; n = 6; p > 0.05 by unpaired t test) (Fig. 5C). These results indicate that D2R-mediated signaling transduction is enhanced in the absence of NCAM. Because of the differential localization of D2R isoforms and specialized downstream effects mediated by D2R isoforms, we would suggest that the ablation of NCAM leads to excessive D2R signaling, particularly in postsynaptic spines.
Figure 5.

D2R-mediated signaling is altered in the absence of NCAM. A, Schematic representation of signaling pathways mediated by presynaptic short isoform of D2R (D2S) and postsynaptic long isoform of D2R (D2L). The activation of D2S and D2L leads to the inhibition of phosphorylation in TH and DARPP32, respectively. B, C, The levels of phosphorylated DARPP32 (pDARPP32) (B) and phosphorylated TH (pTH) (C) were analyzed by Western blot and normalized to total DARPP32 and TH levels in brain homogenates of adult NCAM+/+ and NCAM−/− mice.
The behavioral response to dopamine receptor activation is enhanced in dopamine-depleted NCAM−/− mice
Since D2R-mediated signaling is dysregulated in the absence of NCAM, we investigated whether dopamine-related locomotor activity, which was controlled by nigrostriatal dopaminergic transmission (Hu et al., 1990), was influenced in NCAM−/− mice. NCAM, known as a signaling receptor of GDNF, plays important roles in the development and survival of midbrain dopaminergic neurons (Lin et al., 1993; Chao et al., 2003; Paratcha et al., 2003). Stereological assessment of TH-positive (TH+) dopaminergic neurons revealed a 23% reduction of TH+ cells in the substantia nigra of NCAM−/− mice compared with NCAM+/+ mice (6637 ± 366 vs 8674 ± 320 TH+ cells in the substantial nigra for NCAM−/− and wild-type mice, respectively; n = 5; p < 0.05 by unpaired t test). However, no difference of dopamine levels in total brain and striatum was observed between the two genotypes (209.0 ± 5.5 vs 222.9 ± 6.4 in total brain and 1484.0 ± 217.5 vs 1500.0 ± 221.4 nmol of dopamine per gram of proteins in the striatum for NCAM+/+ and NCAM−/− mice, respectively; n = 5; p > 0.05 by unpaired t test), suggesting that the loss of TH+ neurons in the substantia nigra did not lead to the abnormality of dopamine levels in the striatum of NCAM−/− mice.
Although NCAM deficiency did not cause alterations of dopamine levels in brain tissue, it is possible that the release of dopamine is altered in NCAM−/− mice. To assess direct sensitivities of dopamine receptors in NCAM−/− mice, endogenous dopamine was ablated by treatment with reserpine, which depletes intracellular storage of monoamines and thus dopamine, and with methyl-dl-tyrosine to inhibit dopamine synthesis. Dopamine-depleted mice were then challenged with the nonselective dopamine receptor agonist apomorphine to assess locomotor responses to dopamine receptor activation. After dopamine depletion, locomotor activity was dramatically reduced both in NCAM+/+ and NCAM−/− mice. Administration of apomorphine partially restored the locomotor activity in dopamine-depleted mice. Furthermore, compared with dopamine-depleted NCAM+/+ mice, dopamine-depleted NCAM−/− mice showed a markedly enhanced response to apomorphine (924.5 ± 138.9 vs 572.3 ± 175.2 cm/30 min for NCAM−/− and NCAM+/+ mice, respectively; n = 5; p < 0.05 by paired t test) (Fig. 6), demonstrating that dopamine receptor responsiveness is enhanced in NCAM−/− mice.
Figure 6.

Behavioral responses to dopaminergic agonists are altered in dopamine-depleted NCAM−/− mice. A combination of reserpine (5 mg/kg, i.p.) and methyl-dl-tyrosine (250 mg/kg, i.p.) was applied to deplete dopamine in NCAM+/+ and NCAM−/− mice. After injection of the dopamine receptor agonist apomorphine (1 mg/kg, s.c.), dopamine-depleted NCAM+/+ and NCAM−/− mice were placed into the open field, and locomotor activity was immediately monitored for 30 min at 5 min intervals. Time course of the effect of apomorphine on the locomotor activity of dopamine-depleted NCAM+/+ and NCAM−/− mice is shown. Distance moved was counted at 5 min intervals. Mean + SEM values are shown (n = 5).
NCAM−/− mice show hyperactivity of locomotion resulting from abnormal D2R activity
Since the dopaminergic response was enhanced in dopamine-depleted NCAM−/− mice, we further investigated which class of dopamine receptor was responsible for this hyperactivity of locomotion in NCAM−/− mice by using dopamine receptors antagonists. To test the locomotor responses to dopamine receptor antagonists, the D1R-specific antagonist SKF83566 or the D2R-specific antagonist raclopride were injected into NCAM−/− mice and NCAM+/+ littermates. Immediately after drug administration, mice were placed in open-field boxes and then monitored for locomotor activity. In line with the observation from the dopamine depletion experiment, higher locomotor activity was found in vehicle-treated NCAM−/− mice when compared with NCAM+/+ mice. After treatment with dopamine receptor antagonists, both D1R antagonist SKF83566 and D2R antagonist raclopride reduced locomotor activity in NCAM−/− mice as well as NCAM+/+ mice. No significant difference of locomotor activity was observed between genotypes after SKF83566 treatment, and SKF83566 led to comparable and low activity levels of locomotion in both genotypes (Fig. 7). However, after administration of raclopride, locomotor activity was significantly higher in NCAM−/− mice when compared with NCAM+/+ mice (Fig. 7), indicating enhanced activity of D2R-triggered signaling in NCAM−/− mice. These observations on locomotor activity strongly suggest that dysregulated D2R levels contribute to the hyperactivity of locomotion observed in NCAM−/− mice.
Figure 7.

Behavioral responses to dopaminergic antagonists are changed in NCAM−/− mice. NCAM+/+ and NCAM−/− mice were placed into the open field after injection of D1R-specific antagonist SKF83566, D2R-specific antagonist raclopride, or vehicle control (0.1 mg/kg, i.p.), and locomotor activity was monitored immediately after injection for 30 min at 5 min intervals. A, Time course of the effect of dopamine receptor antagonists on the locomotor activity of NCAM+/+ and NCAM−/− mice. Distance moved was counted at 5 min intervals. Mean + SEM values are shown (n = 7–9). B, Total distance moved was measured in NCAM+/+ or NCAM−/− mice for a period of 30 min after injection of dopamine receptor antagonists. Means + SEM are shown. *p < 0.05 by unpaired t test (n = 7–9).
Discussion
NCAM180, but not NCAM140, interacts with D2R
DRIPs (dopamine receptor interacting proteins), including scaffold and trafficking proteins, signaling molecules, synaptic proteins, ion channels, and other receptors, appear to be essential in regulating key aspects of receptor signaling and functions (Kabbani and Levenson, 2007). Emerging evidence reveals extensive associations between cell adhesion molecules and neurotransmitter receptors (Stork et al., 1999; Fux et al., 2003; Chih et al., 2005; Kohsaka et al., 2007; Li et al., 2009). Here, we identified the transmembrane cell adhesion molecule NCAM as a novel binding partner of D2R and a new modulator of D2R signaling. Pull-down assays provide in vitro evidence that the interaction between NCAM and D2R is mediated by a membrane-proximal part of NCAM-ICD and the IC3-D2R. The binding regions on NCAM and D2R were narrowed down to amino acids 748-765 and 296-320, respectively.
Although the recombinant proteins NCAM140-ICD and NCAM180-ICD both show binding to the IC3-D2R in pull-down assays, immunoprecipitation and chemical cross-linking experiments using brain homogenates clearly demonstrate that only the NCAM180 isoform, which mainly localizes postsynaptically (Persohn et al., 1989), but not NCAM140, interacts with D2R. Immunoprecipitation using D2R-expressing cells transfected with either NCAM isoform confirms that NCAM180, but not NCAM140, is associated with D2R in a cellular context. Moreover, the interaction of NCAM180 with D2R is regulated by dopamine stimulation and/or phosphorylation of NCAM180 and/or D2R, because increased association of NCAM180 with D2R is observed by coimmunoprecipitation shortly after dopamine stimulation, or by biochemical cross-linking under phosphorylation conditions. According to these findings, we propose that NCAM180 interacts with D2R at postsynaptic sites on dopamine stimulation and that this interaction triggers D2R- and NCAM-dependent cellular responses (Fig. 8).
Figure 8.

Working model of the modulation of D2R signaling by NCAM. The transmembrane proteins NCAM and D2R are expressed at the cell surface of the postsynaptic terminus (1). On DA stimulation, cell surface NCAM forms a complex with D2R by interaction between intracellular domains of the two molecules (2), promotes D2R internalization (3) to endocytic compartments, and leads to the attenuation of D2R-mediated signal transduction (4).
NCAM180 regulates D2R internalization and trafficking to endosomal and lysosomal compartments
G-protein-coupled receptors like D2R undergo constitutive and agonist-stimulated internalization, which modulate receptor responsiveness by regulating receptor availability at the cell surface (Rankin et al., 2006). Recent studies suggest a modulatory role of cell adhesion molecules in the trafficking and assembly of presynaptic vesicles and postsynaptic neurotransmitter receptors (Polo-Parada et al., 2001; Nuriya and Huganir, 2006; Sytnyk et al., 2006; Saglietti et al., 2007). Here, we show by in vitro and in vivo investigations that NCAM regulates D2R internalization and thus affects D2R-mediated signaling. Data from cell surface biotinylation using HEK293 cells expressing D2R and NCAM180 indicate that NCAM is involved in agonist-induced D2R internalization, since enhanced internalization of D2R in NCAM-expressing cells was observed only on dopamine stimulation. However, antibody-feeding internalization assays show that NCAM exerts its function on D2R internalization both without and with dopamine stimulation. An explanation for this discrepancy of the results obtained from the two different approaches may be that the application of antibody to live cells in the antibody-feeding assay leads to the clustering of receptors (Diestel et al., 2007), which are likely triggering NCAM and D2R internalization even in the absence of dopamine and thus mimic agonist stimulation. Thus, we assume that NCAM mainly functions in agonist-induced D2R internalization. However, we do not exclude the possibility that NCAM might be also involved in constitutive internalization of D2R. The finding that D2R levels, but not D1R levels in a plasma membrane-enriched fraction are significantly increased in brains of NCAM−/− mice confirms that NCAM specifically modulates the internalization of D2R.
Internalized D2Rs are targeted to late endosomes and lysosomes for degradation, and, as a consequence, D2Rs fail to resensitize after agonist stimulation (Bartlett et al., 2005). Here, we show that the levels of D2R in endosomal and lysosomal fractions are dramatically reduced in brains from NCAM−/− mice. Moreover, D2R degradation is accelerated in NCAM-transfected cells on dopamine stimulation, indicating that NCAM regulates both D2R internalization and subsequent degradation in lysosomes. Additional studies will be needed to analyze the mechanisms by which NCAM regulates the D2R endocytosis and degradation.
NCAM modulates dopamine-related signaling and locomotor activity in mice
Dopamine signaling is extensively modulated by membrane internalization processes, which reduce receptor responsiveness to agonists and protect against receptor overstimulation. Disruption in dopamine receptor internalization leads to dopaminergic supersensitivity, which has been related to several psychiatric disorders (Carlsson, 2001; Nestler, 2001). In the present study, we show that NCAM deficiency leads to a reduction in D2R internalization, a corresponding increased cell surface localization of D2R, and concomitantly enhanced D2R signal transduction, which is demonstrated by the reduction in phosphorylation level of the downstream target protein DARPP32, but not of TH. Because of the differential pathways mediated by D2R isoforms at presynaptic and postsynaptic terminus (Lindgren et al., 2003), the changes in the phosphorylation level of DARPP32 indicate that the intracellular signal response at postsynaptic terminals mediated by the postsynaptic long isoform of D2R is regulated by NCAM. This is consistent with our results, which show that only the postsynaptically localized NCAM180 (Persohn et al., 1989) binds to D2R, whereas presynaptic NCAM140 does not interact with D2R. We thus propose that the interaction and concomitant internalization of NCAM180 and the long isoform of D2R regulate the dopaminergic signaling pathways and cellular responses at postsynaptic terminals.
Furthermore, we show that NCAM not only functions as a modulator of D2R internalization and D2R-mediated signaling but also exerts a regulatory effect on dopamine-related locomotor behavior of mice. Dopamine-depleted NCAM−/− mice show a more pronounced behavioral response to the dopamine receptor agonist apomorphine than dopamine-depleted NCAM+/+ mice, proving that the level of postsynaptic dopamine receptors is dysregulated in the absence of NCAM and that NCAM modulates the dopamine-related locomotion of mice by regulating dopamine receptor sensitivity. Dopamine receptors are classified into D1-like (D1, D5) and D2-like (D2, D3, D4) based on their physiological and pharmacological properties (Missale et al., 1998). Our investigations on locomotor responses to selective antagonists of dopamine receptors show attenuated inhibitory response to a D2-like receptor antagonist in NCAM−/− mice in comparison with NCAM+/+ mice, whereas no significant difference is seen with a D1-like receptor antagonist. These results demonstrate that both signaling and locomotor behavior related to D2-like, but not D1-like, receptors are modulated by NCAM. However, since there is no known specific D2R antagonist and the antagonist we applied is against D2-like receptors, the present data do not rule out the possibility that D3 and D4 receptors might be also responsible for the higher locomotor activity in NCAM−/− mice.
In vivo, the increase of D2Rs at the cell surface in the absence of NCAM could be attributable to dysregulation of two pathways. On the one hand, since NCAM directly promotes D2R internalization and reduces the amount of D2Rs at the cell surface as observed in several in vitro experiments, the absence of NCAM results in an increase of D2Rs at the cell surface. On the other hand, the NCAM-mediated GDNF signaling is crucial for the survival of dopaminergic neurons (Lin et al., 1993; Paratcha et al., 2003), and, accordingly, the number of dopaminergic neurons is reduced in the substantia nigra of NCAM−/− mice. Nevertheless, the reduced number of dopaminergic neurons does not alter total dopamine levels in the striatum of NCAM−/− mice. However, we cannot exclude the possibility that extracellular dopamine levels may be reduced, which in turn could lead to a compensatory upregulation of D2Rs at the cell surface of NCAM−/− mice. Thus, it is conceivable that the increased number of D2Rs at the cell surface and the higher locomotor activity in NCAM−/− mice is attributable to the direct regulatory effect of NCAM on D2R internalization, or indirect effect of NCAM on extracellular dopamine levels, or a combination of both effects.
Hyperactivity of dopamine receptors has been implicated in the pathogenesis of schizophrenia, and dysregulation of the agonist-induced internalization of D2Rs was recently suggested to be responsible for this disorder (Iizuka et al., 2007). In the present study, we obtained indications that the interaction between NCAM and D2R is disturbed when D2R carries the Ser311Cys polymorphism found in schizophrenia patients (Itokawa et al., 1993). Thus, we speculate that, in schizophrenia, the internalization of D2R (S311C) is impaired because of its disturbed interaction with NCAM, which results in enhanced D2R signaling. Since a number of different studies suggest that alteration of the NCAM expression is linked to schizophrenia-like phenotypes (Barbeau et al., 1995; van Kammen et al., 1998; Vawter et al., 1998), it is likely that dysregulation of NCAM expression leads to alterations in D2R internalization and D2R-dependent signaling, and thus affects the D2R-related neurological and emotional abnormalities in humans.
In conclusion, we identified a novel molecular mechanism by which D2R internalization and signaling and dopamine-related behavior is modulated by NCAM via direct cis-interaction in the postsynaptic plasma membrane (Fig. 8). NCAM deficiency increases the level of cell surface D2Rs and enhances the D2R signaling, which in turn leads to hyperactivity of dopamine-related behavior. These observations provide a new alternative pathway by which the dopaminergic system is modulated. Investigations on the functions and mechanisms of this regulatory pathway in dopaminergic transmission should provide new insights into the understanding of neuropsychiatric disorders, thereby leading to novel views on therapeutic approaches.
Footnotes
We are grateful to the Deutsche Forschungsgemeinschaft for support (Scha185/51-1). We are particularly grateful to Dr. Kim A. Neve for providing the myc-D2R-expressing cell line and Dr. David R. Sibley for the vector encoding D2R. We thank Eva Kronberg for animal care; Dr. Fabio Morellini, Dr. Iryna Leshchyns'ka, and Achim Dahlmann for mice breeding and genotyping; Jens Lüttjohann for phage display screening; Dr. Harold Cremer for NCAM-deficient mice; Dr. Burkhard Schlosshauer for the D3 antibody; Dr. Patricia Maness for the expression vectors pcDNA3-NCAM140 and pcDNA3-NCAM180; and Dr. Gabriele Loers and Dr. Edgar Kramer for helpful discussions and comments on this manuscript.
References
- Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, Weiss R, Cooper TB, Mann JJ, Van Heertum RL, Gorman JM, Laruelle M. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci U S A. 2000;97:8104–8109. doi: 10.1073/pnas.97.14.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeau D, Liang JJ, Robitalille Y, Quirion R, Srivastava LK. Decreased expression of the embryonic form of the neural cell adhesion molecule in schizophrenic brains. Proc Natl Acad Sci U S A. 1995;92:2785–2789. doi: 10.1073/pnas.92.7.2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett SE, Enquist J, Hopf FW, Lee JH, Gladher F, Kharazia V, Waldhoer M, Mailliard WS, Armstrong R, Bonci A, Whistler JL. Dopamine responsiveness is regulated by targeted sorting of D2 receptors. Proc Natl Acad Sci U S A. 2005;102:11521–11526. doi: 10.1073/pnas.0502418102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Bohm SK, Grady EF, Bunnett NW. Regulatory mechanisms that modulate signalling by G-protein-coupled receptors. Biochem J. 1997;322:1–18. doi: 10.1042/bj3220001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennaman LH, Maness PF. NCAM in neuropsychiatric and neurodegenerative disorders. Neurochem Res. Advance online publication. 2008 doi: 10.1007/s11064-008-9630-z. [DOI] [PubMed] [Google Scholar]
- Carlsson A. A paradigm shift in brain research. Science. 2001;294:1021–1024. doi: 10.1126/science.1066969. [DOI] [PubMed] [Google Scholar]
- Chao CC, Ma YL, Chu KY, Lee EH. Integrin alphav and NCAM mediate the effects of GDNF on DA neuron survival, outgrowth, DA turnover and motor activity in rats. Neurobiol Aging. 2003;24:105–116. doi: 10.1016/s0197-4580(02)00047-7. [DOI] [PubMed] [Google Scholar]
- Chih B, Engelman H, Scheiffele P. Control of excitatory and inhibitory synapse formation by neuroligins. Science. 2005;307:1324–1328. doi: 10.1126/science.1107470. [DOI] [PubMed] [Google Scholar]
- Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–483. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- Cremer H, Lange R, Christoph A, Plomann M, Vopper G, Roes J, Brown R, Baldwin S, Kraemer P, Scheff S, Barthels D, Rajewsky K, Wille W. Inactivation of the N-CAM gene in mice results in size reduction of the olfactory bulb and deficits in spatial learning. Nature. 1994;367:455–459. doi: 10.1038/367455a0. [DOI] [PubMed] [Google Scholar]
- Diestel S, Schaefer D, Cremer H, Schmitz B. NCAM is ubiquitylated, endocytosed and recycled in neurons. J Cell Sci. 2007;120:4035–4049. doi: 10.1242/jcs.019729. [DOI] [PubMed] [Google Scholar]
- Dodd J, Morton SB, Karagogeos D, Yamamoto M, Jessell TM. Spatial regulation of axonal glycoprotein expression on subsets of embryonic spinal neurons. Neuron. 1988;1:105–116. doi: 10.1016/0896-6273(88)90194-8. [DOI] [PubMed] [Google Scholar]
- Edelman GM. Cell adhesion molecule expression and the regulation of morphogenesis. Cold Spring Harb Symp Quant Biol. 1985;50:877–889. doi: 10.1101/sqb.1985.050.01.105. [DOI] [PubMed] [Google Scholar]
- Fux CM, Krug M, Dityatev A, Schuster T, Schachner M. NCAM180 and glutamate receptor subtypes in potentiated spine synapses: an immunogold electron microscopic study. Mol Cell Neurosci. 2003;24:939–950. doi: 10.1016/j.mcn.2003.07.001. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Bohn LM, Sotnikova TD, Cyr M, Laakso A, Macrae AD, Torres GE, Kim KM, Lefkowitz RJ, Caron MG, Premont RT. Dopaminergic supersensitivity in G protein-coupled receptor kinase 6-deficient mice. Neuron. 2003;38:291–303. doi: 10.1016/s0896-6273(03)00192-2. [DOI] [PubMed] [Google Scholar]
- Giros B, Sokoloff P, Martres MP, Riou JF, Emorine LJ, Schwartz JC. Alternative splicing directs the expression of two D2 dopamine receptor isoforms. Nature. 1989;342:923–926. doi: 10.1038/342923a0. [DOI] [PubMed] [Google Scholar]
- Gray JA, Roth BL. The pipeline and future of drug development in schizophrenia. Mol Psychiatry. 2007;12:904–922. doi: 10.1038/sj.mp.4002062. [DOI] [PubMed] [Google Scholar]
- Greengard P. The neurobiology of slow synaptic transmission. Science. 2001;294:1024–1030. doi: 10.1126/science.294.5544.1024. [DOI] [PubMed] [Google Scholar]
- Hu XT, Wachtel SR, Galloway MP, White FJ. Lesions of the nigrostriatal dopamine projection increase the inhibitory effects of D1 and D2 dopamine agonists on caudate-putamen neurons and relieve D2 receptors from the necessity of D1 receptor stimulation. J Neurosci. 1990;10:2318–2329. doi: 10.1523/JNEUROSCI.10-07-02318.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman C, Hofer M, Barde YA, Juhasz M, Yancopoulos GD, Squinto SP, Lindsay RM. BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature. 1991;350:230–232. doi: 10.1038/350230a0. [DOI] [PubMed] [Google Scholar]
- Iizuka Y, Sei Y, Weinberger DR, Straub RE. Evidence that the BLOC-1 protein dysbindin modulates dopamine D2 receptor internalization and signaling but not D1 internalization. J Neurosci. 2007;27:12390–12395. doi: 10.1523/JNEUROSCI.1689-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Haga T, Lameh J, Sadée W. Sequestration of dopamine D2 receptors depends on coexpression of G-protein-coupled receptor kinases 2 or 5. Eur J Biochem. 1999;260:112–119. doi: 10.1046/j.1432-1327.1999.00125.x. [DOI] [PubMed] [Google Scholar]
- Itokawa M, Arinami T, Futamura N, Hamaguchi H, Toru M. A structural polymorphism of human dopamine D2 receptor, D2(Ser311→Cys) Biochem Biophys Res Commun. 1993;196:1369–1375. doi: 10.1006/bbrc.1993.2404. [DOI] [PubMed] [Google Scholar]
- Itokawa M, Toru M, Ito K, Tsuga H, Kameyama K, Haga T, Arinami T, Hamaguchi H. Sequestration of the short and long isoforms of dopamine D2 receptors expressed in Chinese hamster ovary cells. Mol Pharmacol. 1996;49:560–566. [PubMed] [Google Scholar]
- Kabbani N, Levenson R. A proteomic approach to receptor signaling: molecular mechanisms and therapeutic implications derived from discovery of the dopamine D2 receptor signalplex. Eur J Pharmacol. 2007;572:83–93. doi: 10.1016/j.ejphar.2007.06.059. [DOI] [PubMed] [Google Scholar]
- Kabbani N, Negyessy L, Lin R, Goldman-Rakic P, Levenson R. Interaction with neuronal calcium sensor NCS-1 mediates desensitization of the D2 dopamine receptor. J Neurosci. 2002;22:8476–8486. doi: 10.1523/JNEUROSCI.22-19-08476.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohsaka H, Takasu E, Nose A. In vivo induction of postsynaptic molecular assembly by the cell adhesion molecule Fasciclin2. J Cell Biol. 2007;179:1289–1300. doi: 10.1083/jcb.200705154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law JW, Lee AY, Sun M, Nikonenko AG, Chung SK, Dityatev A, Schachner M, Morellini F. Decreased anxiety, altered place learning, and increased CA1 basal excitatory synaptic transmission in mice with conditional ablation of the neural cell adhesion molecule L1. J Neurosci. 2003;23:10419–10432. doi: 10.1523/JNEUROSCI.23-32-10419.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levivier M, Przedborski S, Bencsics C, Kang UJ. Intrastriatal implantation of fibroblasts genetically engineered to produce brain-derived neurotrophic factor prevents degeneration of dopaminergic neurons in a rat model of Parkinson's disease. J Neurosci. 1995;15:7810–7820. doi: 10.1523/JNEUROSCI.15-12-07810.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, Huang BS, Vishwasrao H, Sutedja N, Chen W, Jin I, Hawkins RD, Bailey CH, Kandel ER. Dscam mediates remodeling of glutamate receptors in Aplysia during de novo and learning-related synapse formation. Neuron. 2009;61:527–540. doi: 10.1016/j.neuron.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- Lindgren N, Usiello A, Goiny M, Haycock J, Erbs E, Greengard P, Hokfelt T, Borrelli E, Fisone G. Distinct roles of dopamine D2L and D2S receptor isoforms in the regulation of protein phosphorylation at presynaptic and postsynaptic sites. Proc Natl Acad Sci U S A. 2003;100:4305–4309. doi: 10.1073/pnas.0730708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Buck DC, Macey TA, Lan H, Neve KA. Evidence that calmodulin binding to the dopamine D2 receptor enhances receptor signaling. J Recept Signal Transduct Res. 2007;27:47–65. doi: 10.1080/10799890601094152. [DOI] [PubMed] [Google Scholar]
- Lyons F, Martin ML, Maguire C, Jackson A, Regan CM, Shelley RK. The expression of an N-CAM serum fragment is positively correlated with severity of negative features in type II schizophrenia. Biol Psychiatry. 1988;23:769–775. doi: 10.1016/0006-3223(88)90065-0. [DOI] [PubMed] [Google Scholar]
- Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10:19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- Mousavi SA, Malerød L, Berg T, Kjeken R. Clathrin-dependent endocytosis. Biochem J. 2004;377:1–16. doi: 10.1042/BJ20031000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D, Djebbara-Hannas Z, Jourdain P, Vutskits L, Durbec P, Rougon G, Kiss JZ. Brain-derived neurotrophic factor restores long-term potentiation in polysialic acid-neural cell adhesion molecule-deficient hippocampus. Proc Natl Acad Sci U S A. 2000;97:4315–4320. doi: 10.1073/pnas.070022697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung Y, Sibley DR. Protein kinase C mediates phosphorylation, desensitization, and trafficking of the D2 dopamine receptor. J Biol Chem. 2004;279:49533–49541. doi: 10.1074/jbc.M408319200. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- Niethammer P, Delling M, Sytnyk V, Dityatev A, Fukami K, Schachner M. Cosignaling of NCAM via lipid rafts and the FGF receptor is required for neuritogenesis. J Cell Biol. 2002;157:521–532. doi: 10.1083/jcb.200109059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuriya M, Huganir RL. Regulation of AMPA receptor trafficking by N-cadherin. J Neurochem. 2006;97:652–661. doi: 10.1111/j.1471-4159.2006.03740.x. [DOI] [PubMed] [Google Scholar]
- Paratcha G, Ledda F, Ibáñez CF. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell. 2003;113:867–879. doi: 10.1016/s0092-8674(03)00435-5. [DOI] [PubMed] [Google Scholar]
- Paspalas CD, Rakic P, Goldman-Rakic PS. Internalization of D2 dopamine receptors is clathrin-dependent and select to dendro-axonic appositions in primate prefrontal cortex. Eur J Neurosci. 2006;24:1395–1403. doi: 10.1111/j.1460-9568.2006.05023.x. [DOI] [PubMed] [Google Scholar]
- Persohn E, Pollerberg GE, Schachner M. Immunoelectron-microscopic localization of the 180 kD component of the neural cell adhesion molecule N-CAM in postsynaptic membranes. J Comp Neurol. 1989;288:92–100. doi: 10.1002/cne.902880108. [DOI] [PubMed] [Google Scholar]
- Pillai-Nair N, Panicker AK, Rodriguiz RM, Gilmore KL, Demyanenko GP, Huang JZ, Wetsel WC, Maness PF. Neural cell adhesion molecule-secreting transgenic mice display abnormalities in GABAergic interneurons and alterations in behavior. J Neurosci. 2005;25:4659–4671. doi: 10.1523/JNEUROSCI.0565-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo-Parada L, Bose CM, Landmesser LT. Alterations in transmission, vesicle dynamics, and transmitter release machinery at NCAM-deficient neuromuscular junctions. Neuron. 2001;32:815–828. doi: 10.1016/s0896-6273(01)00521-9. [DOI] [PubMed] [Google Scholar]
- Rankin ML, Marinec PS, Cabrera DM, Wang Z, Jose PA, Sibley DR. The D1 dopamine receptor is constitutively phosphorylated by G protein-coupled receptor kinase 4. Mol Pharmacol. 2006;69:759–769. doi: 10.1124/mol.105.019901. [DOI] [PubMed] [Google Scholar]
- Saglietti L, Dequidt C, Kamieniarz K, Rousset MC, Valnegri P, Thoumine O, Beretta F, Fagni L, Choquet D, Sala C, Sheng M, Passafaro M. Extracellular interactions between GluR2 and N-cadherin in spine regulation. Neuron. 2007;54:461–477. doi: 10.1016/j.neuron.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Schlosshauer B. Purification of neuronal cell surface proteins and generation of epitope-specific monoclonal antibodies against cell adhesion molecules. J Neurochem. 1989;52:82–92. doi: 10.1111/j.1471-4159.1989.tb10901.x. [DOI] [PubMed] [Google Scholar]
- Seeman P, Van Tol HH. Dopamine receptor pharmacology. Trends Pharmacol Sci. 1994;15:264–270. doi: 10.1016/0165-6147(94)90323-9. [DOI] [PubMed] [Google Scholar]
- Siegel MR, Sisler HD. Inhibition of protein synthesis in vitro by cycloheximide. Nature. 1963;200:675–676. doi: 10.1038/200675a0. [DOI] [PubMed] [Google Scholar]
- Stork O, Welzl H, Wotjak CT, Hoyer D, Delling M, Cremer H, Schachner M. Anxiety and increased 5-HT1A receptor response in NCAM null mutant mice. J Neurobiol. 1999;40:343–355. doi: 10.1002/(sici)1097-4695(19990905)40:3<343::aid-neu6>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Sun W, Ginovart N, Ko F, Seeman P, Kapur S. In vivo evidence for dopamine-mediated internalization of D2-receptors after amphetamine: differential findings with [3H]raclopride versus [3H]spiperone. Mol Pharmacol. 2003;63:456–462. doi: 10.1124/mol.63.2.456. [DOI] [PubMed] [Google Scholar]
- Sytnyk V, Leshchyns'ka I, Nikonenko AG, Schachner M. NCAM promotes assembly and activity-dependent remodeling of the postsynaptic signaling complex. J Cell Biol. 2006;174:1071–1085. doi: 10.1083/jcb.200604145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kammen DP, Poltorak M, Kelley ME, Yao JK, Gurklis JA, Peters JL, Hemperly JJ, Wright RD, Freed WJ. Further studies of elevated cerebrospinal fluid neuronal cell adhesion molecule in schizophrenia. Biol Psychiatry. 1998;43:680–686. doi: 10.1016/s0006-3223(97)00324-7. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Cannon-Spoor HE, Hemperly JJ, Hyde TM, VanderPutten DM, Kleinman JE, Freed WJ. Abnormal expression of cell recognition molecules in schizophrenia. Exp Neurol. 1998;149:424–432. doi: 10.1006/exnr.1997.6721. [DOI] [PubMed] [Google Scholar]
- Wong DF, Wagner HN, Jr, Tune LE, Dannals RF, Pearlson GD, Links JM, Tamminga CA, Broussolle EP, Ravert HT, Wilson AA, Toung JK, Malat J, Williams JA, O'Tuama LA, Snyder SH, Kuhar MJ, Gjedde A. Positron emission tomography reveals elevated D2 dopamine receptors in drug-naive schizophrenics. Science. 1986;234:1558–1563. doi: 10.1126/science.2878495. [DOI] [PubMed] [Google Scholar]
- Wood GK, Tomasiewicz H, Rutishauser U, Magnuson T, Quirion R, Rochford J, Srivastava LK. NCAM-180 knockout mice display increased lateral ventricle size and reduced prepulse inhibition of startle. Neuroreport. 1998;9:461–466. doi: 10.1097/00001756-199802160-00019. [DOI] [PubMed] [Google Scholar]
- Zhou QY, Palmiter RD. Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell. 1995;83:1197–1209. doi: 10.1016/0092-8674(95)90145-0. [DOI] [PubMed] [Google Scholar]