

Abstract
Successful regeneration of damaged neurons depends on the coordinated expression of neuron-intrinsic genes. At present however, there is no comprehensive view of the transcriptional regulatory mechanisms underlying neuronal regeneration. We used high-content cellular screening to investigate the functional contribution of 62 transcription factors to regenerative neuron outgrowth. Ten transcription factors are identified that either increase or decrease neurite outgrowth. One of these, NFIL3, is specifically upregulated during successful regeneration in vivo. Paradoxically however, knockdown of NFIL3 and overexpression of dominant-negative NFIL3 both increase neurite outgrowth. Our data show that NFIL3, together with CREB, forms an incoherent feedforward transcriptional regulatory loop in which NFIL3 acts as a negative regulator of CREB-induced regeneration-associated genes.
Introduction
Neurons in the CNS do normally not regenerate after injury, whereas in the peripheral nervous system (PNS) damaged axons spontaneously regrow and reinnervate their targets. Current strategies to promote regeneration of injured neurons are often aimed at manipulating the molecular and cellular environment of damaged neurons (Yiu and He, 2006; Cafferty et al., 2008). Importantly, successful axonal regeneration also depends on the intrinsic ability of neurons to translate growth-promoting signals into an appropriate gene expression response (Raivich and Makwana, 2007). Dorsal root ganglion (DRG) neurons are an attractive model to study neuron-intrinsic mechanisms of regeneration. DRG neurons extend one peripheral axon into the spinal nerve which regenerates spontaneously after damage, and one central axon into the dorsal root which shows little regenerative capacity (for review, see Teng and Tang, 2006). Successful regeneration following peripheral nerve crush of DRG neurons is transcription-dependent (Smith and Skene, 1997) and requires retrograde transport of injury-induced signals from the lesion site to the nuclei of the injured neurons (Chong et al., 1999; Neumann and Woolf, 1999; Hanz et al., 2003). Injured DRG neurons show increased expression of many regeneration-associated genes, including Gap43, Cap23, Arg1 and Sprr1a, and overexpression of these genes stimulates axonal outgrowth in injured neurons (Frey et al., 2000; Bomze et al., 2001; Bonilla et al., 2002; Cai et al., 2002). Injury-induced expression of regeneration-associated genes requires the coordinated activity of regeneration-associated transcription factors (TFs). To date, several injury-responsive TFs have been identified that promote neuron outgrowth, including CREB (Gao et al., 2004), STAT3 (Qiu et al., 2005), ATF3 (Seijffers et al., 2006; Seijffers et al., 2007), c-JUN (Broude et al., 1997; Raivich et al., 2004), SOX11 (Jankowski et al., 2006), Smad1 (Zou et al., 2009), and several KLF family members (Moore et al., 2009). At present however there is no comprehensive view of the transcriptional regulatory mechanisms underlying neuronal regeneration.
Recent advances in gene regulation analysis have made it possible to map the gene regulatory networks underlying complex physiological processes. Large scale application of gene expression profiling, RNA interference, TF binding site prediction and TF-promoter binding analysis, allow accurate reconstruction of gene regulatory networks and prediction of key components within these networks (Lee et al., 2002; Blais and Dynlacht, 2005; Tegnér and Björkegren, 2007). Here, we applied these methods to uncover novel aspects of the gene regulatory network underlying successful neuronal regeneration. Specifically, we performed an RNAi-based screen on a large set of TFs that were previously shown to be early and differentially regulated in DRG neurons following either peripheral or central nerve crush (Stam et al., 2007). Ten transcription factors were identified that robustly and significantly affect neurite outgrowth. One of these, NFIL3, is specifically upregulated during successful regeneration in vivo. Paradoxically however, knockdown of NFIL3 and overexpression of dominant-negative NFIL3 both increase neurite outgrowth. Our data show that NFIL3, together with CREB, forms an incoherent feedforward transcriptional regulatory loop in which NFIL3 acts as a negative regulator of the CREB-induced expression of regeneration-associated genes.
Materials and Methods
Cell culture and transfections.
F11 and HEK293 cells were maintained as described previously (Stam et al., 2007). F11 cells were transfected with Dharmacon siGENOME siRNA SMARTpools using the DharmaFECT 3 transfection reagent according to the manufacturer's instructions (Dharmacon). For transfection with DNA plasmids Lipofectamine 2000 Reagent (Invitrogen) was used. Dissection and dissociation of primary adult DRG neurons was performed as described previously (Stam et al., 2007). Dissociated neurons were transfected using the Nucleofector 96-well system (Amaxa Biosystems) according to the manufacturer's protocol. After nucleofection, neurons were immediately plated on poly-l-lysine- and laminin-coated 8-well chambers (Lab-Tek). After 40 h in culture neurons were fixed and immunostained. The longest neurite of each of 100–200 neurons was measured. For survival assays, DRG cultures were incubated 30 min before fixation in medium containing 2 μm ethidium homodimer-1 (EthD-1; Invitrogen). GFP-expressing neurons were inspected and the number of EthDI-negative neurons was assessed as a measure for the percentage of viable neurons.
High-content screening.
F11 cells were cultured in 96-well plates and transfected with Dharmacon siGENOME siRNA SMARTpools, including three negative controls (siCONTROL nontargeting pool; siGLO RISC-free siRNA; transfection without siRNA) as well as one positive control (siATF3). Outgrowth was induced 4 h after transfection by replacing the medium with DMEM containing 0.5% FCS and 10 μm forskolin. After 2 d cells were fixed and stained. Neurite outgrowth was quantified using a Cellomics KineticScan HCS Reader and the Cellomics Neuronal Profiling Bioapplication. Per well 500–1000 cells were analyzed and neurite total length per cell (cell-based analysis) and the percentage of cells per well having a neurite average length of >25 μm (population-based analysis) were calculated.
Statistics and target selection.
Statistical significance was determined per plate by One-Way ANOVA and Kruskal–Wallis test for cell-based features and by One-Way ANOVA only for well based features. A Dunnett's post hoc test was used for ANOVA analyses. Post hoc multiple-comparisons tests for Kruskal Wallis analyses were performed as described by Siegel and Castellan (1988). siRNA effects were compared with one of the controls (usually siGLO) and deemed significant when p < 0.01. In addition to the statistical significance criterion, hits were only selected when the size effect of the siRNA was larger than 1 SD of all combined negative controls throughout the screen. All positive hits were replicated 2–3 times using the siRNA pools, and a selection of 10 positive hits were also replicated using the four individual siRNAs that comprise each siRNA pool.
Expression constructs.
Full-length rat NFIL3 cDNA was PCR amplified from rat whole-brain cDNA and inserted into the pcDNA3.1 expression vector (Invitrogen). The pCMV-Myc-CREB plasmid was kindly provided by Dr. A. Riccio (Johns Hopkins University School of Medicine, Baltimore, MD). For generation of the NFIL3 dominant-negative inhibitor, the C-terminal portion of NFIL3 including the leucine zipper was inserted into pcDNA3.1, and was modified to contain an N-terminal Flag epitope followed by a Φ10 sequence and an acidic extension as described previously (Ahn et al., 1998).
Animals and surgical procedures.
Adult male Wistar rats were subjected to either sciatic nerve or dorsal root crush for indicated time-points. L4-6 DRGs were isolated and stored at −80°C until use for quantitative PCR (qPCR), Western blot or in situ hybridization. All procedures were performed as previously described (Stam et al., 2007).
RNA isolation and quantitative real-time PCR.
Total RNA was isolated using Trizol (Invitrogen) and reverse transcribed with Moloney murine leukemia virus reverse transcriptase (MMLV-RT; Invitrogen). Quantitative RT-PCR was performed on the ABI 7900HT detection system (Applied Biosystems) with the 2× SYBR green ready reaction mix (Applied Biosystems). GAPDH and NSE transcripts were measured for normalization.
Western blot analysis.
Cells were directly lysed in 1× Laemmli sample buffer and run on a 10% SDS-PAGE gel. Proteins were blotted onto a PVDF membrane (Bio-Rad Laboratories), blocked with 5% low-fat milk, 1% Tween 20 in PBS. Membranes were incubated with phospho-CREB (Ser133), anti-CREB (both from Cell Signaling Technology) or anti-NFIL3 antibody (V19; Santa Cruz Biotechnology), washed three times with PBS-T (PBS with 1% Tween 20) and incubated with alkaline phosphatase-conjugated secondary antibodies (DAKO). Immunoreactivity was analyzed using the ECF detection system (GE Healthcare).
Luciferase assays.
The pTK-EBPRE vector was a kind gift from Dr. S. Tetradis (UCLA School of Dentistry, Los Angeles, CA). The SST-Luciferase vector was a kind gift from Dr. S. Herzig (University of Kiel, Germany). The Nfil3-, Gap43- and Arg1-luciferase constructs were created by inserting an ∼1 kb fragment encompassing the predicted EBPREs into the pGL2-BASIC-luciferase plasmid (Invitrogen). F11 or HEK293 cells were transfected with indicated constructs/siRNAs and medium was replaced with DMEM containing 0.5% FCS and antibiotics with or without 10 μm forskolin the next day. Primary DRG neurons were transfected with indicated constructs and siRNAs using nucleofection as described. After 2 d, cells were lysed with Steady-Glo luciferase lysis buffer (Promega) and luciferase activity was analyzed with a luminometer (Wallac Victor 1420 Luminometer; PerkinElmer). The luminescent signal was corrected for transfection efficiency using lacZ measurement. Experiments were performed in triplicate and each experiment was repeated 2–3 times.
Chromatin immunoprecipitation.
Chromatin immunoprecipitation (ChIP) experiments were performed on F11 cells transiently expressing NFIL3 and on rat L4–6 DRGs that had received a sciatic nerve crush. Cells/tissues were isolated and chromatin was cross-linked with 1% formaldehyde for 10 min and subsequently quenched with 125 mm glycine for 5 min. DRGs were then homogenized in homogenization buffer (0.35 m sucrose, 5 mm HEPES/NaOH, pH7.4) using a Dounce homogenizer. Cells were pelleted and washed with cold PBS, nuclei were lysed with SDS lysis buffer (1% SDS, 10 mm EDTA in 20 mm Tris-HCl). F11 cells were washed and lysed without homogenization. Cross-linked chromatin was sheared with 4 pulses of 30 s each, yielding products of 200–1000 bp long. Immunoprecipitation was performed with NFIL3 (C18 and V19, Santa Cruz Biotechnology) or phospho-CREB (Ser133) (Cell Signaling Technology) antibodies overnight with rotation at 4°C. Immunocomplexes were captured with protein A/G beads (Santa Cruz Biotechnology) preincubated with sonicated salmon sperm DNA, washed, and eluted with elution buffer (1% SDS, 100 mm NaHCO3). The eluates were proteinase K treated (215 μg/ml) and incubated overnight at 65°C. DNA was purified by phenol/chloroform extraction and subsequent ethanol precipitation. IP and input fractions were analyzed by PCR with primers flanking the predicted EBPRE/CRE sites of Arg1 (5′-CAAAGCTGTTCGGTTCTTGA-3 and 5′-GCTTTGGTCTCCTGAATCGT-3′), Nfil3 (5′-GTTTGATGGTGAGGCCAGAG-3′ and 5′-CTACAACGGCGACCAAAAC-3′), Gap43 (5′-CAGGGTCCTTCCTTCTTCAT-3′ and 5′-GTCACCTTCCAGGGTTTCTT-3′) and Actb as a negative control region (5′-AGAGCAAGAGAGGCATCCTG-3′ and 5′-GGGTCATCTTTTCACGGTTGG-3′). DNA was amplified using HotStart Taq polymerase (Eppendorf), using the following cycling conditions: 10 min at 95°C, then 35–40 cycles of 30 s, 95°C; 30 s, 58°C; and 30 s, 65°C. Resulting PCR products were run on 3% agarose gels. Real-time qPCR analysis was performed as described above. Enrichment was calculated as (Cttarget,mock − Cttarget,IP) − (CtActb,mock − CtActb,IP).
Results
High-content screening identifies novel transcriptional regulators of DRG neuron outgrowth
F11 cells were used to test 62 TFs for their ability to regulate neurite outgrowth. These 62 TFs are differentially regulated in DRG neurons following sciatic nerve injury compared with dorsal root injury (Stam et al., 2007), and include bona fide TFs as well as many putative transcriptional regulators based on gene ontology or subcellular localization (see supplemental Table 1, available at www.jneurosci.org as supplemental material, for selection criteria). F11 cells are neuroblastoma cells derived from rat embryonic DRG neurons (Platika et al., 1985). They express many DRG neuron markers (Francel et al., 1987; Boland and Dingledine, 1990) and display cAMP-induced neurite outgrowth (Ghil et al., 2000). We used F11 cells for screening because high transfection efficiencies (>90%) can reproducibly be obtained and neurite outgrowth can be quantified in an automated and accurate manner (Fig. 1A–C). Automated neurite tracing accuracy was validated by manual tracing (data not shown). Systematic knockdown of all 62 TFs followed by high-throughput automated analysis of neurite lengths identified TFs that affect neurite outgrowth. Examples of reduced neurite outgrowth in ATF3 knockdown cells and enhanced neurite outgrowth in NFIL3 knockdown cells are shown in Figure 1D–F. Knockdown of 19 TFs significantly affected the neurite total length per neuron (Fig. 1G), whereas 10 of these 19 TFs also significantly affected the proportion of outgrowth-positive cells per well after knockdown (Fig. 1H). All significant effects were observed in at least two independent experiments; representative data for all TFs are summarized in supplemental Figure 1 (available at www.jneurosci.org as supplemental material). For the 10 candidate TFs that scored positive in both analyses, the four individual siRNAs constituting the siRNA pool were tested separately to further validate the screening results (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In eight cases, the pool-induced effect was observed for at least two individual siRNAs. For two TFs (BHLHB3 and RTEL1) the pool-induced effect was observed for only one of the individual siRNAs.
Figure 1.
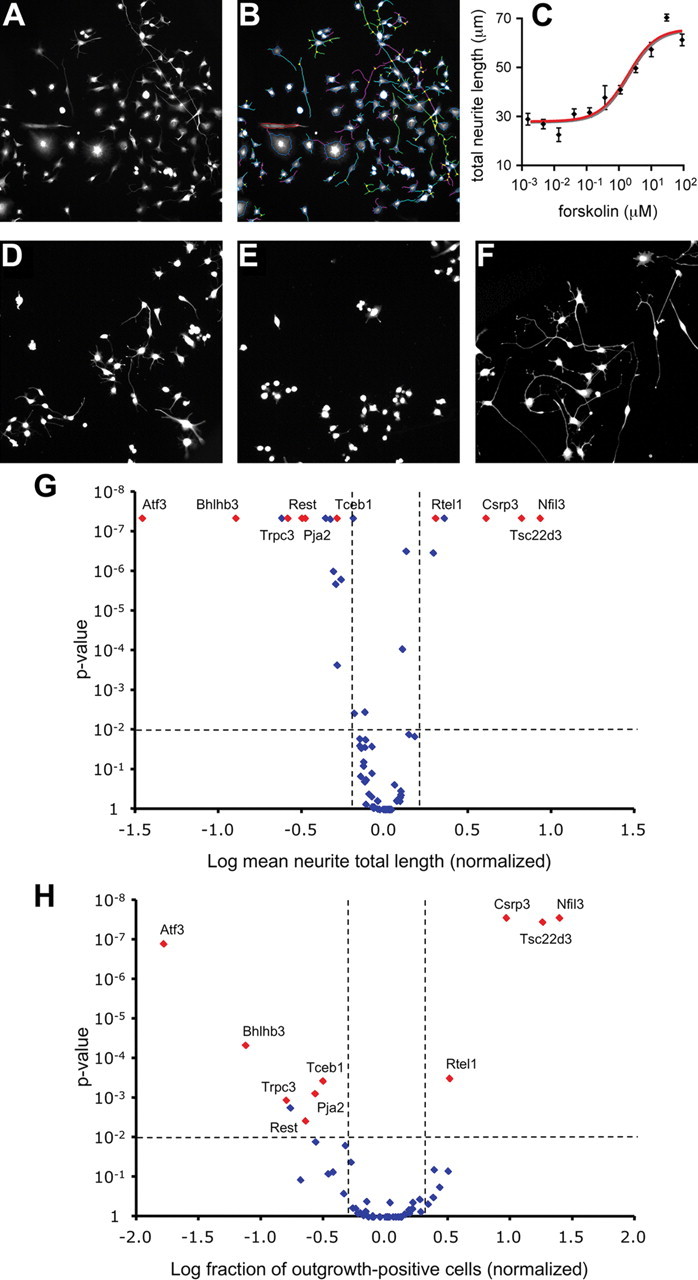
High-content screening identifies TFs involved in regenerative neurite outgrowth. A, Cellomics KineticScan HCS Reader-obtained images of F11 cells stained with anti-neurofilament showing forskolin-induced neurite outgrowth. B, The same image as in A showing how the Cellomics Neuronal Profiling algorithm accurately traces neurites and calculates neurite lengths based on anti-neurofilament staining. C, Cellomics quantification of forskolin-induced neurite outgrowth from F11 cells showing a dose-dependent increase in neurite total length. Data points represent means ± SEM; n = 6 wells for each concentration of forskolin. D–F, Examples of forskolin-stimulated F11 cells transfected with control siRNA (D), siATF3 (E), and siNFIL3 (F), showing reduced neurite outgrowth after knockdown of ATF3 and enhanced neurite outgrowth after knockdown of NFIL3. G, H, Volcano plots summarizing the screening results for all 62 TFs. Data points represent neurite total length per cell (G) and fraction of outgrowth-positive cells per well (H) after TF knockdown. Ten TFs showing effects that are statistically significant (p < 0.01; horizontal dotted lines) and biologically relevant (effect size >1 SD of the combined negative controls; vertical dotted lines) in both assays are indicated in red.
NFIL3 expression correlates with DRG neurite outgrowth in vitro and in vivo
Functional screening identified the bZIP TF NFIL3 as the strongest repressor of neurite outgrowth (Fig. 1). This appeared paradoxical since NFIL3 expression is specifically induced in DRG neurons following sciatic nerve injury and not after dorsal root injury (Stam et al., 2007). Therefore we decided to further characterize NFIL3 expression in injured DRG neurons in vivo, and in forskolin-stimulated F11 cells and primary adult DRG neurons in vitro. Quantitative PCR analysis revealed a robust and early induction of Nfil3 mRNA in DRGs in vivo following sciatic nerve crush, but not after dorsal root crush (Fig. 2A), corroborating our earlier microarray data (Stam et al., 2007). Western blot analysis confirmed the upregulation of NFIL3 protein after sciatic nerve crush (Fig. 2B). To determine the cellular source of Nfil3 mRNA we performed in situ hybridization. Nfil3 mRNA is almost absent in DRG neurons of control animals, but is abundantly expressed in most neurons at 24 h after sciatic nerve crush (Fig. 2C). Elevation of intracellular cAMP levels in F11 cells and in primary adult DRG neurons resulted in a similar rapid induction of Nfil3 mRNA as observed in injured DRGs in vivo (Fig. 2D,G). In DRG neurons, the forskolin-induced mRNA upregulation was almost completely abolished when neurons were pretreated with the protein kinase A (PKA) inhibitor H89 (Fig. 2G). Because cAMP, PKA and CREB all promote successful regeneration of injured DRG neurons in vivo (Qiu et al., 2002; Gao et al., 2004), we also compared the temporal patterns of CREB activation and NFIL3 protein expression in DRGs after sciatic nerve crush in vivo, in forskolin-stimulated F11 cells, and in primary adult DRG neurons in culture. In each case we observed a rapid activation of CREB, while NFIL3 protein levels only started to increase several hours later (Fig. 2B,E,H). Finally, we find that NFIL3 protein is primarily expressed in the nucleus of F11 cells and primary adult DRG neurons in culture (Fig. 2F,I), confirming the neuronal expression of NFIL3. Together, these results show that NFIL3 is downstream of the cAMP-PKA-CREB pathway, and that Nfil3 mRNA expression, protein synthesis and localization are consistent with a transcriptional regulatory role in injured DRG neurons.
Figure 2.

NFIL3 expression correlates with neurite outgrowth in vivo and in vitro. A, qPCR analysis demonstrates a robust and specific upregulation of Nfil3 mRNA after sciatic nerve (SN) crush, and not after dorsal root (DR) crush, corroborating previously reported microarray data. B, Western blot analysis shows upregulation of NFIL3 protein in DRGs in vivo 12–24 h after SN crush. NFIL3 induction is preceded by CREB activation as indicated by phospho-CREB (P-CREB) levels. Total CREB levels are measured for comparison. C, In situ hybridization confirms that expression of Nfil3 is induced in DRG neurons by peripheral axonal lesion. D, Nfil3 mRNA expression is induced in forskolin-stimulated F11 cells as measured by qPCR. Data points represent means ± SEM; n = 5 for each time-point. E, Western blot analysis shows upregulation of NFIL3 protein after 4 h in forskolin stimulated F11 cells. NFIL3 induction is preceded by CREB activation as indicated by phospho-CREB levels. F, Confocal images showing nuclear localization of NFIL3 in F11 cells. G, Nfil3 mRNA expression is induced in cultured primary adult DRG neurons as measured by qPCR. Addition of the PKA inhibitor H89 blocks Nfil3 mRNA induction. Data points represent means ± SEM; n = 3 for each time-point. H, Western blot analysis shows upregulation of NFIL3 protein in primary adult DRG neurons after 4 h in culture. NFIL3 induction is preceded by CREB activation as indicated by phospho-CREB levels. I, Confocal images showing nuclear localization of NFIL3 in cultured DRG neurons.
NFIL3 is a suppressor of neurite outgrowth in DRG neurons
In F11 cells, siRNA-mediated knockdown of NFIL3 resulted in a significant increase in total neurite length (Fig. 1). To demonstrate a similar role for NFIL3 in DRG neurons we used RNA interference and dominant-negative expression. First, we confirmed that NFIL3 siRNAs are able to knockdown NFIL3 protein levels. Both the pool as well as two of the four individual siRNAs significantly reduced NFIL3 protein levels in HEK293 cells overexpressing NFIL3 (Fig. 3A). Next, adult primary DRG neurons were transfected using nucleofection, which resulted in >50% siRNA transfection efficiency. qPCR analysis confirmed efficient knockdown of Nfil3 mRNA levels in DRG neurons (Fig. 3B). After transfection, neurons were cultured for 40 h and neurite lengths were measured. Consistent with our results obtained in F11 cells, we found a significant increase in DRG neurite length when NFIL3 expression was knocked down using the pool of siRNAs (Fig. 3C,D). This effect could be replicated with three of the four individual siRNAs (Fig. 3E). Notably, the increase in neurite length induced by each individual siRNA in DRG neurons correlated well with their efficacy to knockdown NFIL3 protein levels in HEK293 cells (Fig. 3A). No effects on neurite number were observed since all DRG neurons extended neurites, independent of the experimental condition.
Figure 3.

NFIL3 knockdown increases neurite outgrowth from primary adult DRG neurons. A, siRNA-mediated knockdown of NFIL3 protein levels was analyzed in HEK293 cells. Western blotting confirmed that siNFIL3 causes knockdown of NFIL3 protein in HEK293 cells overexpressing rat NFIL3. The siNFIL3 pool and two individual siRNAs (#2 and #3) significantly reduced NFIL3 protein levels; control siRNAs (siGLO and siCONTROL) did not affect NFIL3 protein levels. B, qPCR measurements confirm a 50–60% knockdown of Nfil3 mRNA levels in siNFIL3-transfected DRG neurons. C–E, Primary adult DRG neurons transfected with the siNFIL3 pool show a 51% increase in neurite length. Three of the four individual NFIL3 siRNAs (#2, #3 and #4) also induce a significant increase in neurite outgrowth. The length of the longest neurite was measured for 100–150 cells per condition. Note that the individual siRNAs resulting in the strongest increase in neurite length (E) also give the highest reduction in protein expression in HEK293 cells (A). Bars represent means ± SEM; *p < 0.01. Scale bar, 100 μm.
To exclude the possibility that the siRNA-induced increase in neurite outgrowth is due to off-target effects, we also overexpressed a dominant-negative NFIL3 (DN-NFIL3) protein in primary adult DRG neurons. DN-NFIL3 lacks the basic DNA-binding domain and the nuclear localization sequence, which are replaced by an acidic region and an N-terminal Flag tag (Fig. 4A). A similar dominant-negative CREB protein has previously been used to specifically inhibit CREB function and reduce DRG neuron outgrowth (Ahn et al., 1998; Gao et al., 2004). Coimmunoprecipitation experiments show that DN-NFIL3 specifically interacts with NFIL3 and not with CREB (Fig. 4B). As expected, DN-NFIL3 resides in the cytoplasm and fails to translocate to the nucleus after overexpression in primary adult DRG neurons (Fig. 4C). Overexpression of DN-NFIL3 in primary adult DRG neurons resulted in a similar increase in neurite length as observed in siRNA-transfected neurons (Fig. 4D). This finding shows that in addition to knockdown of NFIL3 expression, functional inactivation of NFIL3 also induces neurite length.
Figure 4.
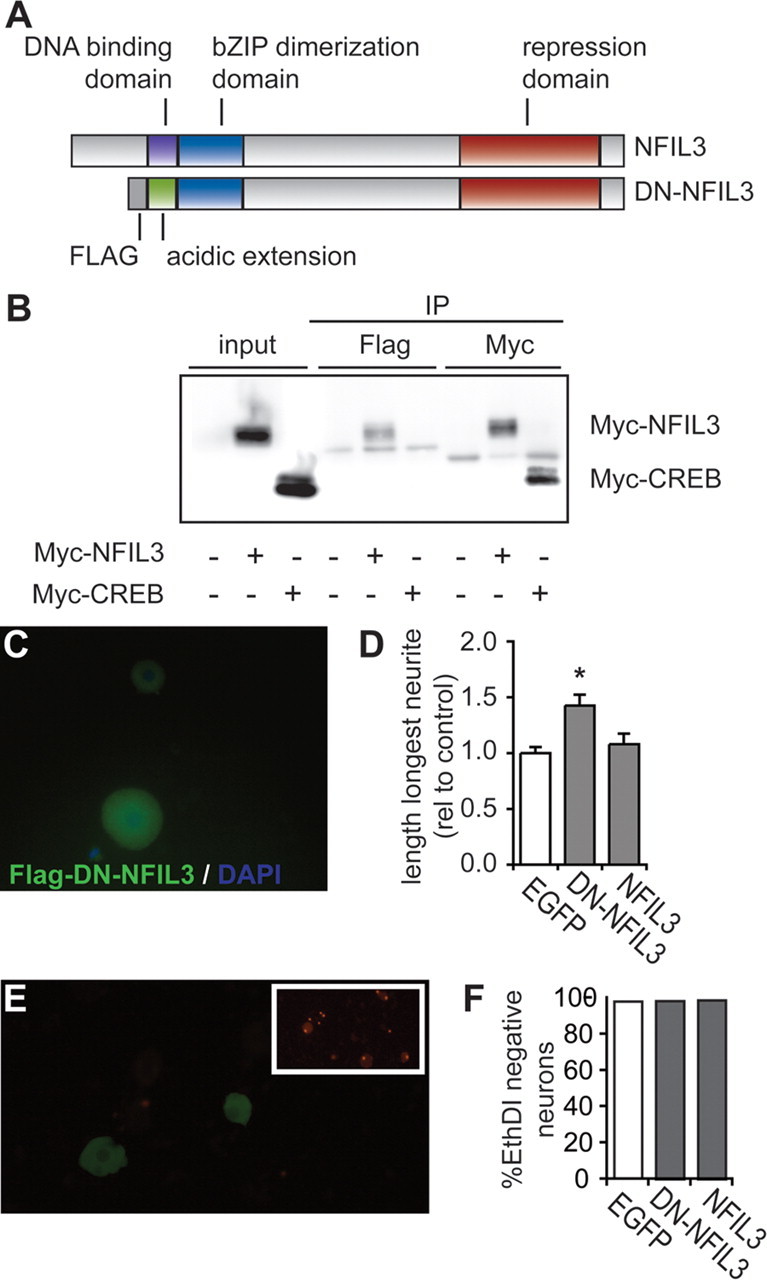
Dominant-negative NFIL3 expression increases neurite outgrowth from primary adult DRG neurons. A, Schematic representation of full-length and dominant-negative NFIL3 (DN-NFIL3) protein. DN-NFIL3 lacks the DNA binding domain, which is replaced by an acidic amphipathic amino acid sequence resulting in a higher affinity for the endogenous full-length protein (see also Ahn et al., 1998). B, Western blot analysis shows specific coimmunoprecipitation of Flag-tagged DN-NFIL3 with Myc-tagged NFIL3 when coexpressed in HEK293 cells. All cells were transfected with Flag-DN-NFIL3 and cotransfected with Myc-NFIL3 or Myc-CREB as indicated. Note that CREB does not coimmunoprecipitate with DN-NFIL3. C, Immunofluorescence staining shows cytoplasmic localization of Flag-DN-NFIL3 expressed in primary adult DRG neurons. D, Overexpression of DN-NFIL3 causes a 42% increase in neurite length from adult DRG neurons in culture. Overexpression of full-length NFIL3 has no effect on neurite outgrowth. Bars represent means ± SEM; *p < 0.01. E, EthD-1 uptake assays of transfected primary adult DRG neurons indicate that most cells are viable after transfection. As a control ethanol treatment induces EthD-1 uptake in most cells (inset). F, Quantification of EthD-1-negative neurons shows no difference in cell viability between GFP-, DN-NFIL3- and NFIL3-expressing neurons.
Overexpression of NFIL3 had no effect on DRG neurite outgrowth compared with GFP-transfected controls (Fig. 4D). Also, in F11 cells we observed no effect of NFIL3 overexpression on forskolin-induced neurite outgrowth (data not shown). These findings seem to suggest that forskolin stimulation (F11 cells) or dissociation (DRG neurons) result in maximally effective levels of NFIL3, and that further increasing NFIL3 levels has no additive effect. Previous studies showed that overexpression of NFIL3 promotes the survival of embryonic chicken spinal cord motor neurons (Junghans et al., 2004). Overexpression of either wild type NFIL3 or dominant-negative NFIL3 had no effect on the percentage of viable DRG neurons (Fig. 4E,F). Together, these observations demonstrate that NFIL3 represses cAMP- and lesion-induced neurite outgrowth in DRG neurons without affecting neuronal survival.
NFIL3 and CREB compete for the same binding sites and control gene transcription together
NFIL3 binds to the E4BP4 response element (EBPRE; TTA[CT]GTAA). To study NFIL3-mediated transcription we used a luciferase reporter construct containing three repeats of the consensus E4BP4 response element (Ozkurt and Tetradis, 2003). Because the EBPRE consensus sequence is very similar to the cAMP response element (CRE; TGACGT[AC]A) to which CREB binds, we also used a luciferase reporter construct containing the CREB-responsive part of the rat somatostatin gene promoter (Montminy et al., 1986) to monitor CRE-mediated transcription.
Forskolin treatment of F11 cells transfected with either the EBPRE-luciferase construct or the CRE-luciferase construct resulted in an induction of luciferase activity showing peak levels at 24 h after stimulation (Fig. 5A). To discriminate between the effects of NFIL3 and CREB in this experiment, we combined expression of the luciferase constructs with overexpression of NFIL3 or CREB in HEK293 and in F11 cells. Interestingly, NFIL3 overexpression repressed the activity of both the EBPRE- and the CRE-reporter, whereas both reporters were strongly induced by CREB (Fig. 5B). Importantly, when we coexpressed CREB with increasing amounts of NFIL3, we observed a decrease in CREB-induced EBPRE-reporter activation (Fig. 5C), indicating that NFIL3 represses CREB-mediated transcription in a dose-dependent manner. Consistent with these data we found that in F11 cells the forskolin-stimulated EBPRE-reporter activity is further enhanced by NFIL3 knockdown, and repressed by CREB knockdown (Fig. 5D). These results show that cAMP-induced DRG neuron outgrowth correlates with dynamic EBPRE/CRE-mediated gene transcription, which is controlled by NFIL3 and CREB. Specifically, NFIL3 and CREB bind to the same promoter elements, but have opposite effects on gene transcription.
Figure 5.

NFIL3 and CREB have opposite effects on CRE/EBPRE-mediated gene transcription. A, Forskolin induces EBPRE- and CRE-mediated transcriptional activity in F11 cells. F11 cells were transfected with either the EBPRE or the CRE reporter construct and stimulated with forskolin for indicated times. Normalized luciferase activities are plotted (means ± SEM; n = 3 for each condition). B, Luciferase assays in HEK293 cells showing the effects of overexpression of CREB and NFIL3 on forskolin-stimulated transcriptional activity. CREB activates both CRE and EBPRE sites, whereas NFIL3 represses both sites. Bars represent means ± SEM; n = 3 for each condition; *p ≤ 0.01. C, Coexpression of CREB with increasing amounts of NFIL3 shows a dose-dependent repression by NFIL3 of CREB-mediated EBPRE-luciferase activity. D, Luciferase assays in forskolin-stimulated F11 cells showing that EBPRE activity is repressed by overexpression of NFIL3 and enhanced by overexpression of CREB. Moreover, knockdown of endogenous NFIL3 and CREB expression by siRNAs produced opposite effects on luciferase activity, confirming that CREB activates EBPRE sites, whereas NFIL3 represses EBPRE sites. E, In primary adult DRG neurons, both knockdown of NFIL3 and overexpression of DN-NFIL3 enhance EBPRE-luciferase activity.
To test whether NFIL3 can also inhibit EBPRE-mediated gene transcription in neurons, we transfected primary adult DRG neurons with the EBPRE-reporter plasmid and simultaneously knocked down NFIL3 using either siRNAs or DN-NFIL3 overexpression. Both knockdown of NFIL3 and overexpression of DN-NFIL3 significantly increased EBPRE-reporter activity (Fig. 5E). Together with the above observations in cell lines, these findings unequivocally confirm that NFIL3 acts as a repressor of EBPRE/CRE-mediated gene transcription in DRG neurons.
NFIL3 binds to regeneration-associated genes in vivo and represses their transcription
We next tested whether NFIL3 is also able to bind to and repress expression of known regeneration-associated target genes. Of the genes that were previously shown to be regulated during regeneration (Stam et al., 2007), 67 contain predicted CRE or EBPRE sites, including Arg1, Gap43 and Nfil3 itself. To establish direct binding of NFIL3 to these sites, we first overexpressed NFIL3 in F11 cells and performed chromatin immunoprecipitation using two antibodies against NFIL3. Coprecipitated DNA was amplified using primers specific for the sequences surrounding each predicted binding site. Both antibodies coprecipitated the promoters of Arg1, Gap43, and Nfil3, but not the promoter of the β-actin gene (Actb), which does not contain EBPRE or CRE sites (Fig. 6A). We next tested whether both CREB and NFIL3 could play a role in the regulation of Arg1, Gap43 and Nfil3 gene expression in regenerating DRG neurons in vivo. Rats were subjected to a sciatic nerve crush and lesioned DRGs were collected 48 h later. Chromatin immunoprecipitation was performed using either anti-phospho-CREB or anti-NFIL3. Both phospho-CREB and NFIL3 were found to be present at the promoters of Arg1, Gap43, and Nfil3, but not at the Actb promoter (Fig. 6A). Quantification of phospho-CREB and NFIL3 binding at earlier time points after sciatic nerve crush showed that binding at the Arg1 and Gap43 promoters was not detected in unlesioned control DRGs, but became detectable at 6–24 h after sciatic nerve crush (Fig. 6B). Thus, Arg1, Gap43, and Nfil3 are CREB and NFIL3 target genes in regenerating DRG neurons in vivo.
Figure 6.

NFIL3 binds to and represses transcription of regeneration-associated genes. A, ChIP assay showing NFIL3 binding to promoter regions of Nfil3, Arg1 and Gap43 in F11 cells overexpressing NFIL3 (left panel) and in DRG neurons 2 d after sciatic nerve crush in vivo (right panel). Two different NFIL3 antibodies were used that both precipitated all three promoters. No immunoprecipitation was observed in the absence of antibodies (control) or for a negative control gene (Actb). B, Quantitative PCR analysis of ChIP enrichment showing phospho-CREB and NFIL3 occupancy at the Arg1 and Gap43 promoters at 6–24 h after sciatic nerve crush. No binding was observed in unlesioned control DRGs. C, Schematic diagrams of the Nfil3, Gap43 and Arg1 genes indicating predicted EBPRE sites (black boxes). Indicated are coding exon regions (dark gray boxes), untranslated exon regions (light gray boxes), introns (black lines) and transcription start sites (arrows). Genomic fragments of the Nfil3, Gap43 and Arg1 genes containing predicted EBPRE sites were cloned into the pGL2-B-luciferase plasmid. Luciferase assays show that these constructs are transcriptionally active in HEK293 cells (white bars), and that transcriptional activity is repressed when NFIL3 is cotransfected (gray bars). Activity of the P0 promotor is not repressed by NFIL3. Bars represent means ± SEM; n = 3 for each condition; *p < 0.01.
To determine the effect of NFIL3 binding to the promoters of Arg1, Gap43, and Nfil3, we cloned the predicted binding site regions of all three genes in luciferase reporter plasmids (Fig. 6C). These reporter plasmids were introduced in HEK293 cells, which lack endogenous NFIL3 expression. Expression of the reporters alone resulted in an increase in luciferase activity compared with empty luciferase constructs, but coexpression of NFIL3 almost completely reduced luciferase activity to basal levels (Fig. 6C). Importantly, a peripheral myelin P0 promoter-luciferase construct which lacks EBPRE sites (Brown and Lemke, 1997) was not repressed by NFIL3. These data show that in addition to the direct binding of NFIL3 to EBPRE sites in these genes, NFIL3 represses gene transcription mediated by these sites, indicating that NFIL3 is a repressor of regeneration-associated gene expression. The observed transcriptional repression of the Nfil3 gene by NFIL3 furthermore suggests the presence of a direct negative feedback loop.
Discussion
Axonal damage to neurons in the PNS activates a gene response which enables the injured neuron to successfully regenerate and reinnervate targets (Skene, 1989; Smith and Skene, 1997; Gao et al., 2004; Raivich and Makwana, 2007). The coordination of this gene expression program probably requires tight transcriptional regulation, but the underlying transcriptional regulatory mechanisms remain largely unknown. Here, we identify NFIL3 as a novel transcriptional repressor of CREB-mediated gene transcription, and we demonstrate that NFIL3 and CREB form a transcriptional network motif, which is involved in the regulation of neuronal regeneration-associated genes.
Based on previously published in vivo gene expression data (Stam et al., 2007) we investigated the role of 62 TFs in neurite outgrowth from DRG-like F11 cells. This resulted in the identification of 10 TFs that significantly affect neurite outgrowth following siRNA-mediated knockdown. Nine of these have not previously been implicated in neuronal regeneration. The number of positive hits (10 of 62) may seem relatively low given that all 62 TFs were selected based on in vivo gene expression data. This might be due to (1) the fact that regulation of some TFs is the result rather than the cause of regeneration, (2) limitations of F11 cells as a model for in vivo neuronal regeneration, (3) redundancy or synergism in TF functions that prevented their detection because we did not knockdown TFs in combination, and (4) the stringent selection criteria that were applied to eliminate false-positives. Nevertheless, the finding of nine novel putative transcriptional regulators of neuronal outgrowth is exciting and may shed new light on transcriptional regulatory mechanisms underlying neuronal regeneration.
Paradoxically, we did not always observe a positive correlation between TF gene regulation in vivo and the siRNA-induced effect on neurite outgrowth in vitro. For instance, knockdown of both NFIL3 and CSRP3 enhanced neurite outgrowth, even though these factors are upregulated during successful regeneration. Little if anything is known about the possible roles of these TFs in neuronal outgrowth, but it is intriguing that regenerating neurons upregulate TFs that have inhibitory roles in the process of neurite outgrowth. To further address this issue in detail, we studied the transcriptional regulatory functions of NFIL3 in cell lines, in adult primary DRG neurons, and in vivo after sciatic nerve crush. Interestingly, NFIL3 belongs to the same class of TFs as CREB, which is known to stimulate DRG neuron regeneration (Qiu et al., 2002; Gao et al., 2004). Our data show that NFIL3 expression follows phospho-CREB induction in DRG neurons. The Nfil3 gene contains two functional EBPRE sites, which according to our data can also bind CREB. Although recent genome-wide promoter binding studies failed to identify the Nfil3 gene as a direct CREB target in PC12 and HEK293 cells (Impey et al., 2004; Zhang et al., 2005), we suggest that in DRG neurons Nfil3 gene expression is regulated by the cAMP–CREB pathway. In line with this suggestion, we found that PKA inhibition suppressed Nfil3 mRNA levels in primary adult DRG neurons and that CREB binds the Nfil3 promoter in vivo after a sciatic nerve crush. Another important finding in our study is that NFIL3 competes with CREB for EBPRE and CRE motifs in the promoters of regeneration-associated genes. These sites appear to be functionally equivalent: CREB enhances gene expression via both sites, whereas NFIL3 represses gene expression via both sites. These results are consistent with a model in which, as part of the cAMP-regulated gene program, NFIL3 is upregulated to exert control over the CREB-mediated transcriptional response (Fig. 7).
Figure 7.

Model for the role of NFIL3 in regulating regeneration-associated gene expression. Our data indicate that CREB and NFIL3 form a type 1 incoherent transcriptional feedforward loop. Elevation of cAMP levels triggered by peripheral neuronal injury activates PKA and CREB. CREB then activates regeneration-associated genes (RAGs) containing CRE/EBPRE sites, including Nfil3. NFIL3 acts as a negative regulator on CRE/EBPRE-mediated transcription. At this moment we cannot exclude the possibility that NFIL3 in parallel regulates the expression of other regeneration-associated genes, independent of CREB.
A gene regulatory network motif in which one TF in parallel activates a set of target genes, and a negative regulator that subsequently represses the same target genes, is referred to as a type 1 incoherent feedforward loop and is frequently detected in genetic model organisms (Mangan and Alon, 2003; Alon, 2007). Computational modeling and experimental studies in bacteria both showed that type 1 incoherent feedforward loops efficiently control the dynamics of target gene expression by accelerating target gene response time and generating nonmonotonic pulses of target gene expression (Mangan and Alon, 2003; Mangan et al., 2006; Kaplan et al., 2008). Thus, the paradoxical upregulation of NFIL3 during successful neuronal regeneration, even though it suppresses neurite outgrowth, can be explained by the fact that NFIL3 and CREB coevolved to form a functionally important gene regulatory network motif. Apparently, appropriate temporal regulation of growth-promoting genes by the cAMP–CREB pathway requires the induction of NFIL3 as a feedforward repressor.
Individual components of gene regulatory network motifs often acquire different functions in different species and cellular systems (Madan Babu et al., 2007; Tuch et al., 2008). Indeed, NFIL3 shows functional diversity in different organisms and cell types. This was first described in C. elegans, where the NFIL3 orthologue CES-2 (cell death selector-2) has differential effects on target genes and cell morphogenesis or apoptosis in different cell types (Metzstein et al., 1996; Wang and Chamberlin, 2002; Wang et al., 2006). In the mammalian circadian clock, NFIL3 acts as a transcriptional repressor, and regulates the complex diurnal oscillating expression patterns of clock genes (Doi et al., 2001; Mitsui et al., 2001). In lymphoid tissues NFIL3 has a strong anti-apoptotic effect (Ikushima et al., 1997; Kuribara et al., 1999), and in developing chick spinal cord motor neurons NFIL3 overexpression promotes survival and axonal growth (Junghans et al., 2004). These latter findings seem contradictory to ours; however, we clearly show with two independent methods (RNA interference and dominant-negative expression) that loss of NFIL3 function stimulates outgrowth of adult DRG neurons, and that neither overexpression nor inhibition of NFIL3 affects DRG neuron survival. Together, we conclude that the precise role of NFIL3 in gene regulatory networks appears to depend on the cell type and the developmental stage. Also, we cannot exclude that the function of NFIL3 differs in different species.
In conclusion, we show that NFIL3 is a suppressor of regenerative DRG neuron outgrowth. Together with CREB, NFIL3 forms an incoherent transcriptional feedforward loop controlling the expression of regeneration-associated genes. These findings provide for the first time insight into the architecture of transcriptional regulatory networks that underlie neuronal regeneration, and highlight the importance of transcriptional repressors in determining a neuron's regenerative response. Other regeneration-associated TFs may be subject to similar negative regulatory control mechanisms. For instance, suppressor of cytokine signaling-3 (SOCS3) is upregulated by injured DRG neurons, and negatively regulates STAT3 function and neurite outgrowth (Miao et al., 2006). Clearly, further insight into transcription regulatory network interactions is indispensable for our understanding of how neuronal regeneration is regulated at the level of gene expression.
Footnotes
This work received financial support from the Dutch Ministry of Economic Affairs (SenterNovem Grant ISO52022) and from the Center for Medical Systems Biology (CMSB) in the framework of the Netherlands Genomics Initiative (NGI). We thank Ruben Eggers for experimental assistance.
References
- Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, Vinson C. A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol Cell Biol. 1998;18:967–977. doi: 10.1128/mcb.18.2.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet. 2007;8:450–461. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- Blais A, Dynlacht BD. Constructing transcriptional regulatory networks. Genes Dev. 2005;19:1499–1511. doi: 10.1101/gad.1325605. [DOI] [PubMed] [Google Scholar]
- Boland LM, Dingledine R. Expression of sensory neuron antigens by a dorsal root ganglion cell line, F-11. Brain Res Dev Brain Res. 1990;51:259–266. doi: 10.1016/0165-3806(90)90284-6. [DOI] [PubMed] [Google Scholar]
- Bomze HM, Bulsara KR, Iskandar BJ, Caroni P, Skene JH. Spinal axon regeneration evoked by replacing two growth cone proteins in adult neurons. Nat Neurosci. 2001;4:38–43. doi: 10.1038/82881. [DOI] [PubMed] [Google Scholar]
- Bonilla IE, Tanabe K, Strittmatter SM. Small proline-rich repeat protein 1A is expressed by axotomized neurons and promotes axonal outgrowth. J Neurosci. 2002;22:1303–1315. doi: 10.1523/JNEUROSCI.22-04-01303.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broude E, McAtee M, Kelley MS, Bregman BS. c-Jun expression in adult rat dorsal root ganglion neurons: differential response after central or peripheral axotomy. Exp Neurol. 1997;148:367–377. doi: 10.1006/exnr.1997.6665. [DOI] [PubMed] [Google Scholar]
- Brown AM, Lemke G. Multiple regulatory elements control transcription of the peripheral myelin protein zero gene. J Biol Chem. 1997;272:28939–28947. doi: 10.1074/jbc.272.46.28939. [DOI] [PubMed] [Google Scholar]
- Cafferty WB, McGee AW, Strittmatter SM. Axonal growth therapeutics: regeneration or sprouting or plasticity? Trends Neurosci. 2008;31:215–220. doi: 10.1016/j.tins.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Deng K, Mellado W, Lee J, Ratan RR, Filbin MT. Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron. 2002;35:711–719. doi: 10.1016/s0896-6273(02)00826-7. [DOI] [PubMed] [Google Scholar]
- Chong MS, Woolf CJ, Haque NS, Anderson PN. Axonal regeneration from injured dorsal roots into the spinal cord of adult rats. J Comp Neurol. 1999;410:42–54. [PubMed] [Google Scholar]
- Doi M, Nakajima Y, Okano T, Fukada Y. Light-induced phase-delay of the chicken pineal circadian clock is associated with the induction of cE4bp4, a potential transcriptional repressor of cPer2 gene. Proc Natl Acad Sci U S A. 2001;98:8089–8094. doi: 10.1073/pnas.141090998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francel PC, Harris K, Smith M, Fishman MC, Dawson G, Miller RJ. Neurochemical characteristics of a novel dorsal root ganglion × neuroblastoma hybrid cell line, F-11. J Neurochem. 1987;48:1624–1631. doi: 10.1111/j.1471-4159.1987.tb05711.x. [DOI] [PubMed] [Google Scholar]
- Frey D, Laux T, Xu L, Schneider C, Caroni P. Shared and unique roles of CAP23 and GAP43 in actin regulation, neurite outgrowth, and anatomical plasticity. J Cell Biol. 2000;149:1443–1454. doi: 10.1083/jcb.149.7.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Deng K, Hou J, Bryson JB, Barco A, Nikulina E, Spencer T, Mellado W, Kandel ER, Filbin MT. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron. 2004;44:609–621. doi: 10.1016/j.neuron.2004.10.030. [DOI] [PubMed] [Google Scholar]
- Ghil SH, Kim BJ, Lee YD, Suh-Kim H. Neurite outgrowth induced by cyclic AMP can be modulated by the alpha subunit of Go. J Neurochem. 2000;74:151–158. doi: 10.1046/j.1471-4159.2000.0740151.x. [DOI] [PubMed] [Google Scholar]
- Hanz S, Perlson E, Willis D, Zheng JQ, Massarwa R, Huerta JJ, Koltzenburg M, Kohler M, van-Minnen J, Twiss JL, Fainzilber M. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron. 2003;40:1095–1104. doi: 10.1016/s0896-6273(03)00770-0. [DOI] [PubMed] [Google Scholar]
- Ikushima S, Inukai T, Inaba T, Nimer SD, Cleveland JL, Look AT. Pivotal role for the NFIL3/E4BP4 transcription factor in interleukin 3-mediated survival of pro-B lymphocytes. Proc Natl Acad Sci U S A. 1997;94:2609–2614. doi: 10.1073/pnas.94.6.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G, Goodman RH. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119:1041–1054. doi: 10.1016/j.cell.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Jankowski MP, Cornuet PK, McIlwrath S, Koerber HR, Albers KM. SRY-box containing gene 11 (Sox11) transcription factor is required for neuron survival and neurite growth. Neuroscience. 2006;143:501–514. doi: 10.1016/j.neuroscience.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junghans D, Chauvet S, Buhler E, Dudley K, Sykes T, Henderson CE. The CES-2-related transcription factor E4BP4 is an intrinsic regulator of motoneuron growth and survival. Development. 2004;131:4425–4434. doi: 10.1242/dev.01313. [DOI] [PubMed] [Google Scholar]
- Kaplan S, Bren A, Dekel E, Alon U. The incoherent feed-forward loop can generate non-monotonic input functions for genes. Mol Syst Biol. 2008;4:203. doi: 10.1038/msb.2008.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuribara R, Kinoshita T, Miyajima A, Shinjyo T, Yoshihara T, Inukai T, Ozawa K, Look AT, Inaba T. Two distinct interleukin-3-mediated signal pathways, Ras-NFIL3 (E4BP4) and Bcl-xL, regulate the survival of murine pro-B lymphocytes. Mol Cell Biol. 1999;19:2754–2762. doi: 10.1128/mcb.19.4.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, Zeitlinger J, Jennings EG, Murray HL, Gordon DB, Ren B, Wyrick JJ, Tagne JB, Volkert TL, Fraenkel E, Gifford DK. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- Madan Babu M, Balaji S, Aravind L. General trends in the evolution of prokaryotic transcriptional regulatory networks. Genome Dyn. 2007;3:66–80. doi: 10.1159/000107604. [DOI] [PubMed] [Google Scholar]
- Mangan S, Alon U. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci U S A. 2003;100:11980–11985. doi: 10.1073/pnas.2133841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan S, Itzkovitz S, Zaslaver A, Alon U. The incoherent feed-forward loop accelerates the response-time of the gal system of Escherichia coli. J Mol Biol. 2006;356:1073–1081. doi: 10.1016/j.jmb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Metzstein MM, Hengartner MO, Tsung N, Ellis RE, Horvitz HR. Transcriptional regulator of programmed cell death encoded by Caenorhabditis elegans gene ces-2. Nature. 1996;382:545–547. doi: 10.1038/382545a0. [DOI] [PubMed] [Google Scholar]
- Miao T, Wu D, Zhang Y, Bo X, Subang MC, Wang P, Richardson PM. Suppressor of cytokine signaling-3 suppresses the ability of activated signal transducer and activator of transcription-3 to stimulate neurite growth in rat primary sensory neurons. J Neurosci. 2006;26:9512–9519. doi: 10.1523/JNEUROSCI.2160-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui S, Yamaguchi S, Matsuo T, Ishida Y, Okamura H. Antagonistic role of E4BP4 and PAR proteins in the circadian oscillatory mechanism. Genes Dev. 2001;15:995–1006. doi: 10.1101/gad.873501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montminy MR, Sevarino KA, Wagner JA, Mandel G, Goodman RH. Identification of a cyclic-AMP-responsive element within the rat somatostatin gene. Proc Natl Acad Sci U S A. 1986;83:6682–6686. doi: 10.1073/pnas.83.18.6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, Goldberg JL. KLF family members regulate intrinsic axon regeneration ability. Science. 2009;326:298–301. doi: 10.1126/science.1175737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S, Woolf CJ. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron. 1999;23:83–91. doi: 10.1016/s0896-6273(00)80755-2. [DOI] [PubMed] [Google Scholar]
- Ozkurt IC, Tetradis S. Parathyroid hormone-induced E4BP4/NFIL3 down-regulates transcription in osteoblasts. J Biol Chem. 2003;278:26803–26809. doi: 10.1074/jbc.M212652200. [DOI] [PubMed] [Google Scholar]
- Platika D, Boulos MH, Baizer L, Fishman MC. Neuronal traits of clonal cell lines derived by fusion of dorsal root ganglia neurons with neuroblastoma cells. Proc Natl Acad Sci U S A. 1985;82:3499–3503. doi: 10.1073/pnas.82.10.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Cai D, Dai H, McAtee M, Hoffman PN, Bregman BS, Filbin MT. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron. 2002;34:895–903. doi: 10.1016/s0896-6273(02)00730-4. [DOI] [PubMed] [Google Scholar]
- Qiu J, Cafferty WB, McMahon SB, Thompson SW. Conditioning injury-induced spinal axon regeneration requires signal transducer and activator of transcription 3 activation. J Neurosci. 2005;25:1645–1653. doi: 10.1523/JNEUROSCI.3269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich G, Makwana M. The making of successful axonal regeneration: genes, molecules and signal transduction pathways. Brain Res Rev. 2007;53:287–311. doi: 10.1016/j.brainresrev.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Da Costa C, Iwata O, Galiano M, Hristova M, Nateri AS, Makwana M, Riera-Sans L, Wolfer DP, Lipp HP, Aguzzi A, Wagner EF, Behrens A. The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron. 2004;43:57–67. doi: 10.1016/j.neuron.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Seijffers R, Allchorne AJ, Woolf CJ. The transcription factor ATF-3 promotes neurite outgrowth. Mol Cell Neurosci. 2006;32:143–154. doi: 10.1016/j.mcn.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Seijffers R, Mills CD, Woolf CJ. ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J Neurosci. 2007;27:7911–7920. doi: 10.1523/JNEUROSCI.5313-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel S, Castellan NJ. Nonparametric statistics for the behavioral sciences. Ed 2. New York: McGraw-Hill; 1988. [Google Scholar]
- Skene JH. Axonal growth-associated proteins. Annu Rev Neurosci. 1989;12:127–156. doi: 10.1146/annurev.ne.12.030189.001015. [DOI] [PubMed] [Google Scholar]
- Smith DS, Skene JH. A transcription-dependent switch controls competence of adult neurons for distinct modes of axon growth. J Neurosci. 1997;17:646–658. doi: 10.1523/JNEUROSCI.17-02-00646.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam FJ, MacGillavry HD, Armstrong NJ, de Gunst MC, Zhang Y, van Kesteren RE, Smit AB, Verhaagen J. Identification of candidate transcriptional modulators involved in successful regeneration after nerve injury. Eur J Neurosci. 2007;25:3629–3637. doi: 10.1111/j.1460-9568.2007.05597.x. [DOI] [PubMed] [Google Scholar]
- Tegnér J, Björkegren J. Perturbations to uncover gene networks. Trends Genet. 2007;23:34–41. doi: 10.1016/j.tig.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Teng FY, Tang BL. Axonal regeneration in adult CNS neurons–signaling molecules and pathways. J Neurochem. 2006;96:1501–1508. doi: 10.1111/j.1471-4159.2006.03663.x. [DOI] [PubMed] [Google Scholar]
- Tuch BB, Li H, Johnson AD. Evolution of eukaryotic transcription circuits. Science. 2008;319:1797–1799. doi: 10.1126/science.1152398. [DOI] [PubMed] [Google Scholar]
- Wang X, Chamberlin HM. Multiple regulatory changes contribute to the evolution of the Caenorhabditis lin-48 ovo gene. Genes Dev. 2002;16:2345–2349. doi: 10.1101/gad.996302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Jia H, Chamberlin HM. The bZip proteins CES-2 and ATF-2 alter the timing of transcription for a cell-specific target gene in C. elegans. Dev Biol. 2006;289:456–465. doi: 10.1016/j.ydbio.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, Kadam S, Ecker JR, Emerson B, Hogenesch JB, Unterman T, Young RA, Montminy M. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 2005;102:4459–4464. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Ho C, Wong K, Tessier-Lavigne M. Axotomy-induced Smad1 activation promotes axonal growth in adult sensory neurons. J Neurosci. 2009;29:7116–7123. doi: 10.1523/JNEUROSCI.5397-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]