

Abstract
Expression of the prion protein (PrPC) is a requirement for host susceptibility to the transmissible spongiform encephalopathies (TSEs) and thought to be necessary for the replication and transport of the infectious agent. The mechanism of TSE neuroinvasion is not fully understood, although the routing of infection has been mapped through the peripheral nervous system (PNS) and Schwann cells have been implicated as a potential conduit for transport of the TSE infectious agent. To address whether Schwann cells are a requirement for spread of the TSE agent from the site of infection to the CNS, PrPC expression was selectively removed from Schwann cells in vivo. This dramatically reduced total PrPC within peripheral nerves by 90%, resulting in the selective loss of glycosylated PrPC species. Despite this, 139A and ME7 mouse-passaged scrapie agent strains were efficiently replicated and transported to the CNS following oral and intraperitoneal exposure. Thus, the myelinating glial cells within the PNS do not appear to play a significant role in TSE neuroinvasion.
Introduction
Transmissible spongiform encephalopathies (TSEs) are neurodegenerative diseases characterized by neuronal loss, gliosis, amyloid deposition, and vacuolar pathological hallmarks. TSE diseases target and affect the CNS and usually present with characteristic deposition of a protease-resistant isoform of the host-encoded protein PrPC. TSEs are naturally acquired peripherally (Taylor et al., 1996; Bartz et al., 2003) and are associated with prolonged incubation periods, during which the infectious agent travels to the CNS (Kimberlin and Walker, 1980; Glatzel et al., 2002). Precisely which cells are involved in TSE neuroinvasion is currently unknown, though the infectious agent has been traced through the peripheral nervous system (PNS) (Schulz-Schaeffer et al., 2000; McBride et al., 2001) and Schwann cells have been implicated in this process (Follet et al., 2002).
In experimental models of mouse-passaged scrapie, a lag phase occurs after infection, during which the agent replicates in the periphery, accumulating upon the follicular dendritic cells in secondary lymphoid organs such as the spleen (Brown et al., 1999). The invasion of the CNS either from the spleen or directly from the site of infection has been proposed to be dependent on a continuous chain of PrPC-expressing cells (Blättler et al., 1997; Glatzel and Aguzzi, 2000; Race et al., 2000). Time course studies of several experimental TSE strains have shown that PrPSc is first detectable in the CNS within the spinal cord and medulla oblongata, concurrent with transport via vagal and splanchnic nerves, and TSE infectious agent has been detected by bioassay in the PNS throughout the time course of infection (Cole and Kimberlin, 1985; Beekes et al., 1998; McBride et al., 2001). Further studies traced PrPSc deposition at various time points along these nerves from the periphery to the CNS (Groschup et al., 1999) and the PNS has been shown to meet the requirements for the neuroinvasive route (Kimberlin et al., 1983). Transgenic knock-out (KO) of PrPC revealed that expression is required for a host to be susceptible to TSE disease (Büeler et al., 1993; Manson et al., 1994). Subsequent studies revealed that the TSE agent is not transported within a PrPC-deficient CNS (Aguzzi et al., 1997) and expression within the PNS appears critical for neuroinvasion (Glatzel and Aguzzi, 2000).
The presence of PrPC in Schwann cells has been demonstrated (Lemaire-Vieille et al., 2000; Follet et al., 2002) and it has been suggested that this may be dependent upon PrP expression in nerve axons (Ford et al., 2002). Moreover, Schwann cells have been implicated as a possible route of transport following in vitro studies which showed cultured Schwann cells capable of amplification and serial propagation of PrPSc (Follet et al., 2002; Archer et al., 2004). Here we examined whether the expression of PrP in Schwann cells is a requirement for TSE neuroinvasion. To address this, we have developed a transgenic mouse model that specifically lacks PrP in Schwann cells. These transgenic mice were challenged both peripherally and intracerebrally with two well characterized mouse-passaged scrapie agent strains, ME7 and 139A. We demonstrate that Schwann cells express the majority of PrP in the peripheral nerve but this expression is not an absolute requirement for TSE neuroinvasion.
Materials and Methods
Production of Schwann cell PrP knock-out mice.
To produce Schwann cell PrP knock-out mice the Cre/LoxP recombination system was used. Cre recombinase (Cre) is a 38 kDa protein isolated from the bacteriophage P1. Cre mediates site-specific recombination of DNA sequences between LoxP (locus of X-over of P1) sites. LoxP sites are 34 bp sequences consisting of two 13 bp inverted repeats with an 8 bp asymmetric spacer region which bestows directionality to the sequence [for review, see Sauer (1993)]. These elements can be combined to excise a functional endogenous gene in vivo (Gu et al., 1994). Expression of Cre was controlled by the myelin Protein Zero (P0) promoter and intragenic regulatory elements. P0 is an adhesion molecule of the Ig superfamily and constitutes 50% of the protein content of peripheral myelin (Greenfield et al., 1973). The murine promoter for the P0 gene (Mpz) has been successfully used in transgenic mice to drive Schwann-cell-specific expression of reporter genes (Feltri et al., 1996, 1999b) and Cre (Feltri et al., 1999a, 2002; Saito et al., 2003). The P0-Cre transgene was combined via selective breeding with gene-targeted transgenic mice in which exon 3 of the Prnp gene has been flanked by unidirectional LoxP sequences (floxed), and the resultant transgenic allele was named PrPfl (Tuzi et al., 2004). Compound transgenic mice possessing both PrPfl and P0-Cre transgenes therefore undergo Cre-mediated excision of the Prnp exon 3, including the complete PrPC coding sequence, in Schwann cells and as such are referred to as Schwann cell PrP knock-out mice. The inclusion of the LoxP sequences into the Prnp gene has been extensively investigated previously and shown to have no effect on PrPC expression or TSE disease in the absence of Cre (Tuzi et al., 2004).
Generation and genotyping of compound transgenic mousemodel.
PrPfl mice were interbred with P0-Cre mice for successive generations to produce compound transgenic mice homozygous for PrPfl at the Prnp gene locus and also possessing the P0-Cre transgene. Mice devoid of PrPfl [i.e., homozygous Prnp wild type (wt)], P0-Cre, or both transgenic elements were also produced during the breeding process and were assigned to various control groups based on their genotype (Table 1). All animal studies and breeding were performed via standard methods and conducted under the provisions of the UK Animals Scientific Procedures Act 1986 and approved by the Neuropathogenesis Division Ethical Review Committee. For genotyping of transgenic mice, DNA was extracted from tail tip tissue for PCR analysis. Tail biopsy samples (0.5 cm) were taken under anesthesia from all animals at 28 (±7) d of age. DNA was extracted from tail biopsy samples via DNeasy blood and tissue kit (Qiagen). Postmortem tail tissue from inoculated animals was also collected and digested overnight at 37°C in lysis buffer [300 mm sodium acetate, 1% SDS, 10 mm Tris, pH 8, 1 mm EDTA, 200 μg/ml proteinase K (PK)] and subsequent extraction was performed with an equal volume of 1:1 phenol/chloroform. DNA was precipitated with isopropanol, washed with 70% ethanol, and resuspended in 100 μl of TE buffer [containing the following (in mm): 10 Tris, 1 EDTA, pH 7.4]. PCR analysis was performed for both PrPfl (Tuzi et al., 2004) and P0-Cre (Feltri et al., 1999a) transgenes as described previously. All animal genotypes were confirmed before and after TSE challenge.
Table 1.
Experimental animal groups, showing genotype and resulting effect upon PrP expression pattern
| Group | Genotype |
Phenotype (PrP expression pattern) | |
|---|---|---|---|
| Prnp | P0-Cre | ||
| Schwann cell PrP KO | PrPfl homozygous | Positive | Removed only from Schwann cells |
| Floxed PrP control | PrPfl homozygous | Negative | Normal |
| Cre control | PrP wild type | Positive | Normal |
| Nontransgenic control | PrP wild type | Negative | Normal |
Transmissible spongiform encephalopathy exposure.
Inocula were produced as 10% brain homogenates from pools of tissue collected from either terminal ME7- or 139A-infected mice. Groups of animals of confirmed genotype (as displayed in Table 1) were inoculated at 20 (±10) weeks of age with 20 μl of homogenate either intracerebrally under anesthesia or intraperitoneally. Oral TSE exposure of mice was achieved with 100 μl of homogenate presoaked onto a single standard food pellet. Mice were isolated into individual cages during oral exposure period and free food was withdrawn until the inoculation pellet was fully consumed.
Clinical scoring, observation, and neuropathology.
Mice were observed for signs of clinical illness as described previously (Bruce et al., 1991). Clinical scoring of experiments was performed blind and animals were regenotyped postchallenge for confirmation and assignation to groups. Disease incubation times were calculated as the interval between inoculation and positive clinical assessment of terminal TSE disease. Tissue for neuropathological assessment was fixed in 10% formal saline for a minimum of 48 h, processed, and embedded in paraffin. Six-micrometer sections were cut and stained with hematoxylin and eosin and scored for spongiform vacuolar degeneration on a scale of 0–5 for gray matter or 0–3 for white matter in 12 standard areas as previously described (Fraser and Dickinson, 1967).
Data analysis.
Group mean scores were plotted against area to give representative lesion profiles using Excel software (Microsoft). Group mean incubation periods were calculated and compared for both 139A and ME7 experiments. Incubation period data were analyzed by one-way ANOVA between genotype groups for each inoculum and route separately. Statistical analysis was performed using Minitab software (Minitab). Clinically and pathologically confirmed scrapie cases were used to derive the incubation times and plotted as Kaplan–Meier survival curves.
Western blot analysis.
Ten-percent (w/v) brain and sciatic nerve homogenates were prepared in Nonidet P-40 (NP40) buffer (0.5% NP40, 0.5% sodium deoxycholate, 150 mm NaCl, 50 mm Tris-HCl, pH 7.5, and 1 mm PMSF). Chemicals were sourced from VWR International, unless stated otherwise. Tissues were homogenized in Dounce glass/glass homogenizers and centrifuged at 16,000 × g for 15 min at 4°C, and supernatant was isolated. Total protein assessed via Coomassie total protein assay (Perbio Science). N-linked glycan chains were removed with peptide-N-glycosidase F (New England Biolabs) for 1 h at 37°C. PK-resistant PrP was isolated with 20 μg/ml PK (Roche) for 1 h at 37°C. Twenty-microgram total protein samples were denatured at 90°C for 15 min in SDS sample buffer (Invitrogen), separated on 12% Novex Tris-glycine polyacrylamide gels (Invitrogen), and transferred onto polyvinylidene fluoride membrane (Millipore) by semidry electroblotting in transfer buffer (20% methanol, 39 mm glycine, 48 mm Tris, and 0.0375% SDS) performed at 2 mA/cm2. Membranes blocked for 1 h in Western blocking reagent (Roche). PrP was detected with antibodies 7A12 (Li et al., 2000) and 8H4 (Zanusso et al., 1998) at 50 ng/ml. Primary antibody detected via horseradish peroxidase-conjugated rabbit anti-mouse IgG (Stratech Scientific) at 200 ng/ml and BM chemiluminescent blotting substrate (Roche) exposed to Lumi-Film (Roche).
PrP densitometry and quantification.
Western blots were scanned using a Kodak Image Station IS440 (Kodak) and analyzed with Kodak 1D software for PrP quantification. Statistical analyses were performed using Excel (Microsoft). PrP expression data were calculated as a percentage change via comparison against standard control samples and are quoted as approximate values due to calculation from densitometric analysis of semiquantitative Western blot analysis.
PrPSc immunocytochemistry.
Tissues were fixed in formal saline, paraffin embedded, and microtome sectioned at 6 μm. Sections were autoclaved at 121°C for 15 min and immersed in 98% formic acid for 5 min for antigen retrieval. Sections were then incubated for 20 min with normal goat serum (Stratech Scientific) diluted 1:20 in PBS and PrP was detected with anti-PrP antibody 6H4 (Prionics) overnight diluted to a final concentration of 1 μg/ml in PBS/0.1% BSA (Sigma). Antibody binding was detected with biotinylated goat anti-mouse IgG (Stratech Scientific) at 2 μg/ml in PBS/BSA and the Vectastain Elite ABC Kit (Vector Laboratories), visualized with diaminobenzidine. Non-neural tissues were processed with alkaline phosphatase-conjugated goat anti-mouse IgG (Stratech Scientific) and visualized with Vector Red (Vector Laboratories). Sections were counterstained with hematoxylin. Pictures were taken using an Eclipse E800 microscope (Nikon) and Image Pro plus software (Media Cybernetics).
PrPC immunofluorochemistry.
Samples were immersion fixed in 4% paraformaldehyde/0.25% glutaraldehyde in PBS, embedded in optimal cryotomy temperature (O.C.T.) compound, and cryosectioned on a Leica CM1850 (Leica) at 30 μm. Sections were blocked for 1 h with PBS/0.5%BSA/mouse on mouse Ig blocking reagent (Vector Laboratories). Mouse on mouse Ig blocking reagent was removed by washing with PBS/1× protein concentrate (Vector Laboratories) incubated for 2 h with anti-PrP antibodies 7A12 and 8H4 at 1 μg/ml in PBS/0.5× protein concentrate. Primary antibody binding was detected with biotinylated goat anti-mouse IgG (Stratech Scientific) secondary antibody at 2.5 μg/ml in PBS/0.5% BSA/0.5× protein concentrate. Secondary antibody binding was detected with streptavidin, Alexa Fluor 594 conjugate (Invitrogen) at 2 μg/ml. Sections were analyzed and imaged via laser scanning 5 Pa confocal microscope (Carl Zeiss).
X-Gal histochemistry.
To confirm Cre expression pattern, P0-Cre mice were crossed with ROSA26 reporter mice (Mao et al., 1999). Tissues from P0-Cre and ROSA26 double-positive or ROSA26-positive-only mice were dissected into ice-cold PBS and fixed in 2% paraformaldehyde/0.2% glutaraldehyde in 5 mm ethylene glycol bis(2-aminoethyl ether)-N,N,N′N′-tetraacetic acid (EGTA), 2 mm MgCl2, 0.02% NP40, and 0.01% sodium deoxycholate. Postfixation, tissues were washed in 2 mm MgCl2, 0.02% NP40, and 0.01% sodium deoxycholate in PBS and incubated at 4°C overnight in 15% and then 30% sucrose in PBS. Tissues were embedded in O.C.T. compound and cryosectioned on a Leica CM1850 (Leica) at 30 μm. Slides were stained by incubation at 37°C in 1 mg/ml 5-bromo-4-chloro-3-indolyl- β-d-galactopyranoside (X-Gal) dissolved in DMSO (Sigma), 5 mm potassium ferricyanide, 5 mm potassium ferrocyanide, 2 mm MgCl2, 0.02% P40, and 0.01% sodium deoxycholate in PBS from 2 to 48 h. The staining reaction was stopped in 20 mm EDTA in PBS for 5 min and slides were counterstained with nuclear fast red (Vector Laboratories).
Myelin morphological analysis.
Tissues were perfusion fixed in 2% paraformalydehyde/2% glutaraldehyde in 0.1 m sodium cacodylate, pH 7.4, and postfixed for 1 h with 1% osmium tetroxide in 0.1 m sodium cacodylate buffer, pH 7.4. Samples were embedded in Araldite 502 (all chemicals from Electron Microscopy Sciences) for sectioning and use in electron microscopy. Figures were prepared using Photoshop CS3.
Results
PrPC expression in Schwann cell PrP knock-out mice
Cre-mediated recombination of the PrP transgene with exon 3 flanked by LoxP sites (PrPfl) in PNS tissue from Schwann cell PrP knock-out mice was detected via standard PCR methods, as described previously (Tuzi et al., 2004), revealing excision of the PrPC coding region had occurred as expected in DNA extracted from peripheral nerve (Fig. 1a). Routine PCR screening of tail tip tissue from the same subjects revealed no detectable recombination. Expression level of PrPC was examined via Western blot analysis and densitometry. The brain of Schwann cell PrP knock-out mice was shown to be identical to control groups of floxed PrP, Cre, and nontransgenic littermate control mice, unlike the sciatic nerve in which PrP was dramatically reduced (Fig. 1b). Further investigation of the sciatic nerve revealed that in wild-type mice, total PrPC approximates to 25% the amount found in brain. PrPC in the sciatic nerve favors the unglycosylated form with 16% diglycosylated, 20% monoglycosylated, and 64% unglycosylated PrP species as opposed to brain with 66% diglycosylated, 14% monoglycosylated, and 20% unglycosylated when detected with anti-PrP antibody 7A12. Densitometric analysis of PrP expression revealed ∼90% reduction in total PrPC in the PNS of Schwann cell PrP knock-out mice when compared with wild-type PrP-expressing mice (Fig. 1d). Reduction of PrPC expression occurs in all glycoforms, to the extent that monoglycosylated and diglycosylated PrPC were rarely visualized following Western analysis of Schwann cell PrP knock-out nerves. Unglycosylated PrPC species within these nerves were detectable after prolonged film exposure times (Fig. 1b). This result was confirmed by PNGase-F treatment and densitometry analysis (Fig. 1c).
Figure 1.

a, PCR analysis of DNA extracted from sciatic nerve tissue revealing Cre-mediated recombination in Schwann cell PrP knock-out mice only. Lanes: 1, 1 kb DNA ladder (Invitrogen); 2, wt sciatic nerve; 3, Cre control sciatic nerve; 4 and 5, Schwann cell PrP knock-out sciatic nerve; 6, wt tail tip PCR control; 7, PrPfl/fl tail tip PCR control; 8, PrPfl/wt Cre-Deleter tail tip PCR control; 9, H2O PCR control. Band sizes indicated in base pairs: 167 bp 5′ region upstream of PrP exon 3 (wt allele); 210 bp 5′ region upstream of Prnp exon 3 including loxP site (PrPfl allele); 315 bp 5′ and 3′ PrP after Cre-mediated excision of exon 3 (Cre-mediated recombined PrPfl allele). b, Western blot analysis of PrP expression in brain (top) and sciatic nerve (bottom) tissue using anti-PrP antibody 7A12. Samples: 1, 129/Ola brain; 2, floxed PrP control; 3, Schwann cell PrP knock-out, revealing reduction of total PrP and loss of PrP glycoforms in PNS tissue; 4, Cre control; and 5, nontransgenic control. Approximate protein molecular weight markers given in kilodaltons. c, Western blot analysis of wild-type brain (1) and sciatic nerve (3) and Schwann cell PrP KO sciatic nerve (2) following N-glycosidase F (F) treatment (+), when not treated (nt), or without enzyme treatment (−). d, Relative PrP expression and glycoform profile in brain and sciatic nerve of wild-type mice and Schwann cell PrP knock-out mice, normalized to wild-type brain expression level; diglycosylated (light gray), monoglycosylated (hatched), and unglycosylated (dark gray) species are indicated. Error bars represent total expression level ± SEM.
The use of ROSA26 reporter mice confirmed the localization of Cre activity in P0-Cre mice solely to Schwann cells in peripheral nerve (Fig. 2b) as reported previously (Feltri et al., 2002). Localization of PrPC within cross-sectioned PNS nerve axons revealed that PrPC was normally present in compact myelin and outer Schwann cell cytoplasmic regions (Fig. 2c). PrPC was absent from these structures in nerves from Schwann cell PrP knock-out mice (Fig. 2d). Morphological analysis revealed normal myelination of peripheral nerve axons in Schwann cell PrP knock-out mice (Fig. 2f), thus no overt phenotype was detected due to Schwann cell PrP knock-out other than the effects on PrP expression and glycoform levels.
Figure 2.

Phenotype investigation of Schwann cell PrP knock-out in transverse sciatic nerve sections. a, b, Detection of Cre-mediated β-galactosidase expression via X-Gal histochemistry (blue) counterstained with nuclear fast red in Cre control ROSA26-positive mice (b) but not ROSA26 mice lacking Cre (a). Images taken at ×20 magnification. c, d, Detection of PrPC expression (red) via immunofluorescence in myelin structures of Cre control mice (c) but not Schwann cell PrP knock-out mice (d). Scale bars, 20 μm. e, f, Normal myelin morphology observed in both floxed PrP control mice (e) and Schwann cell PrP knock-out transgenic mice (f). Scale bars, 5 μm.
Effect of Schwann cell PrP knock-out on TSE disease incubation period
Following oral inoculation with both 139A and ME7 strains of agent, incubation period data in the Schwann cell PrP knock-out group revealed no statistically significant difference from control groups (Tables S1 and S2). A statistically significant difference was detected in group mean incubation period between the Schwann cell PrP knock-out group and the control genotype groups following intracerebral and intraperitoneal 139A and ME7 infection with the Schwann cell PrP knock-out group having the longest mean incubation period, with intracerebral incubation times 4–8% longer and intraperitoneal incubation times 13–16% longer. Following intraperitoneal 139A infection and intracerebral ME7 inoculation a statistically significant variation, around the 5% level, was observed between the control groups. In these experiments the floxed PrP group has the shortest mean incubation period among the control genotype groups, with intraperitoneal incubation times 10–15% shorter than other control groups despite expressing PrP normally (Table 2). A wide range of incubation periods were observed within genotype groups in these experiments. Distribution analysis of these data revealed statistically outlying individuals, these occurred rarely in only 5 mice of a total of 191, in a non-genotype-specific manner and only after peripheral challenge (Fig. 3). Such outliers are not unusual in peripheral TSE challenges using mice of mixed genetic background and the age range within the experimental design.
Table 2.
Statistical analysis of TSE incubation period
| Inoculum | Route | Variance ratio (within controls) | % Difference in incubation period (floxed PrP vs controls) | Variance ratio (Schwann cell PrPknock-out vs controls) | % Difference in incubation period (Schwann cell PrP knock-out vs controls) |
|---|---|---|---|---|---|
| 139A | Intracerebral | 0.66ns | +1.52 | 5.50* | +4.09 |
| 139A | Intraperitoneal | 3.22* | −14.89 | 9.31** | +15.84 |
| 139A | Oral | 0.46ns | +5.94 | 0.78ns | −5.65 |
| ME7 | Intracerebral | 3.24* | −9.35 | 16.15*** | +8.36 |
| ME7 | Intraperitoneal | 2.51ns | −10.00 | 10.65** | +13.32 |
| ME7 | Oral | 7.23** | −11.44 | 3.43ns | +4.65 |
Following ANOVA of incubation period data, the statistical significance of the variance ratios produced were calculated using F tests. The levels of statistical significance are indicated: ns Not significant (p > 0.05);
*p < 0.05;
**p < 0.01;
***p < 0.001.
The variance ratios and percentage differences in mean incubation periods are displayed between Schwann cell PrP knock-out mice and combined control groups. For comparison, the percentage difference in mean incubation periods within the control groups was calculated as the difference between the largest outlying control group (floxed PrP) and the remaining control groups. The difference in incubation periods observed between Schwann cell PrP knock-out mice and controls, following peripheral routes of inoculation (intraperitoneal and oral), is comparable to that observed among groups of animals expressing PrP normally, with similar statistical significance.
Figure 3.

a–f, Kaplan–Meier survival curves following transmission of mouse-adapted scrapie strains 139A (a, c, e) and ME7 (b, d, f) via intracerebral (a, b), intraperitoneal (c, d), and oral (e, f) routes. Legend: ♦, Schwann cell PrP knock-out; ■, floxed PrP control; ▲, Cre control; ●, nontransgenic control.
Effect of Schwann cell PrP knock-out on PrPSc deposition
Total amounts of PrPSc deposition were quantified via Western blot analysis with prior PK digestion of whole brain homogenates. The amount of PrPSc in the brains from Schwann cell PrP knock-out mice is equivalent to the amount in brains from control group mice, occurring within the same incubation period (Fig. 4). The detection of PrPSc, as a marker of the disease process, revealed that Schwann cell PrP knock-out mice are equally capable of supporting the disease process as mice expressing PrPC normally. The pattern of PrPSc deposition in the CNS and peripheral lymphoid tissues were also examined via standard immunocytochemical methods. No differences in localization, pattern or intensity of PrPSc deposition were observed between Schwann cell PrP knock-out and control genotype mice. Following intracerebral inoculation PrPSc deposition was observed throughout the brain and spinal cord cervical, thoracic and lumbar sections as well as in the germinal centers of the spleen, suggesting that either anterograde transport of agent or hematogenous spread were unaffected (Fig. 5). PrPSc deposition was also observed in germinal centers of the spleen, gray matter of the spinal cord and throughout the CNS via oral and intraperitoneal inoculation route (Fig. 6). Staining within the CNS was differentiated solely by agent, with 139A resulting in a uniform deposition throughout the neuropil and ME7 revealing a stellate perineuronal pattern, for review of prion strains see (Bruce, 2003; Morales et al., 2007). No host-specific effect was observed therefore discounting any difference due to genotype.
Figure 4.

Western blot analysis of PrPSc deposition load in terminal brain samples. Samples: 1, Uninfected 129/Ola; 2, Schwann cell PrP knock-out; 3, floxed PrP control; 4, Cre control; and 5, nontransgenic control. Densitometry revealed no significant differences in the amount of PK-resistant PrP detectable between genotypes; similar results were obtained following oral and intraperitoneal challenges. Approximate protein molecular weight markers are given in kilodaltons.
Figure 5.
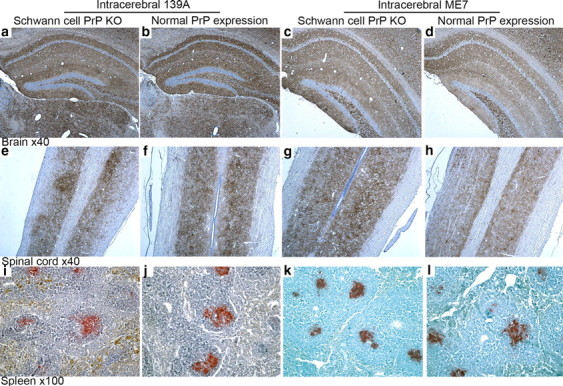
a–l, Immunocytochemical detection of PrPSc deposition in tissues from terminally infected Schwann cell PrP KO mice (a, c, e, g, i, k) and normal PrP-expressing control mice (b, d, f, h, j, l) following intracerebral 139A or ME7 challenge. Tissue sections: brain (hippocampus), longitudinal spinal cord showing deposition in gray matter tracts, and spleen showing deposition in germinal centers. Anti-PrP antibody was visualized with DAB (brown) in CNS tissues and Vector Red (red) in spleen; tissue sections were counterstained with hematoxylin (blue). PrPSc deposition pattern in Schwann cell PrP knock-out animals was indistinguishable from control genotype mice in all tissues investigated.
Figure 6.

a–p, Immunocytochemical detection of PrPSc deposition in terminal tissue sections from Schwann cell PrP KO mice (a, c, e, g, i, k, m, o) and normal PrP-expressing control mice (b, d, f, h, j, l, n, p) following oral (a–l) or intraperitoneal (m–p) 139A or ME7 challenge. Tissue sections: brain (hippocampus), longitudinal spinal cord showing deposition in gray matter tracts, and spleen showing deposition in germinal centers. Following peripheral infection, the PrPSc deposition pattern in Schwann cell PrP knock-out animals was again indistinguishable from control genotype mice in all tissues investigated.
Pathological examination of PrPSc deposition patterns revealed that terminal Schwann cell PrP knock-out mice were indistinguishable from control group mice and also 129/Ola or C57BL “parental” inbred mouse strains following both ME7 and 139A challenges. PrPSc deposition was also investigated during the course of both intraperitoneal and oral challenges. Time-points were selected representing the earliest time that PrPSc deposition is observed in the CNS of both 129/Ola and C57BL mouse strains. Following oral challenge 2 mice from each genotype group were killed at 126 d after 139A infection and 216 d after ME7 infection. These time-points were selected based on previous observations of early deposition of PrPSc occurring within the CNS following oral challenge by these agents. PrPSc deposition in the brains of 139A orally challenged mice was first observed in the red nucleus as expected (Fig. 7a,c), with no difference observed between Schwann cell PrP knock-out mice and controls. This data suggests that transport of the infectious agent to the CNS from the site of infection was occurring at the same rate in Schwann cell PrP knock-out mice as control mice expressing PrPC normally. Little or no PrPSc deposition was observed in ME7 challenged mice at 216 d postinfection, preventing further analysis of these data. These data were taken as an indication that progression of disease in the mixed genetic background mice used in this study was occurring slower than expected compared with known data on disease progression in the parental inbred strains namely 129/Ola and C57BL, and that the deposition of PrPSc in the CNS in this model is a very late stage event. PrPSc deposition was observed in peripheral lymphoid tissue such as ileal Peyer's patches and lymph nodes associated with the celiac–superior mesenteric ganglion complex at these time points with both agent strains (Fig. 7i,k). This demonstrates that peripheral concentration or amplification of infection is effective in lymphoid tissues of both wild-type and Schwann cell PrP knock-out mice.
Figure 7.
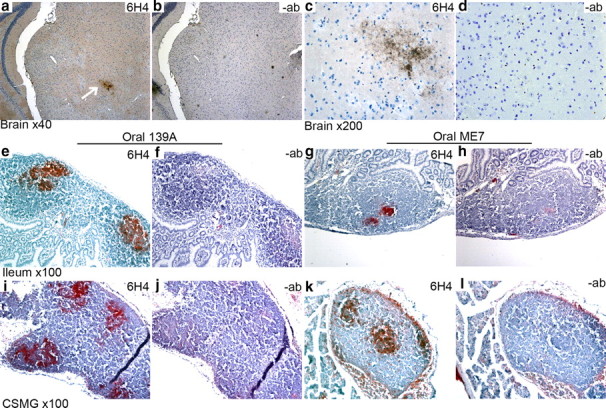
a–l, Immunocytochemical detection of PrPSc deposition in preclinical tissues following oral challenge (a, c, e, g, i, k); serial sections screened without anti-PrP antibody (−ab) are shown for comparison (b, d, f, h, j, l). At 126 d after 139A oral challenge, PrPSc deposition was detected in the brain in the red nucleus (arrow in a), surrounding but not inside magnocellular neurons (c). PrPSc deposition was detected only within Peyer's patches of ileal tissue (e, g). Investigation of the celiac–superior mesenteric ganglion (CSMG) complex revealed PrPSc deposition in associated lymph nodes (i, k).
Effect of Schwann cell PrP knock-out on vacuolar pathology
Vacuolation in standard brain areas was assessed and lesion profile analyses constructed to determine whether the removal of PrP expression from Schwann cells would affect strain targeting abilities. Although there is slight variation in the results obtained between genotype groups, no overt differences were observed specifically between Schwann cell PrP knock-out mice and control group mice (Fig. 8). These data reveal that disease-dependent vacuolar spongiform change progresses with equivalent severity in the same brain areas of Schwann cell PrP knock-out mice as control group mice. This result reveals that, similar to PrPSc deposition pattern, strain targeting processes were unaffected.
Figure 8.

a–f, Pathological vacuolar change was assessed following transmission of mouse-adapted scrapie strains 139A (a, c, e) and ME7 (b, d, f) via intracerebral (a, b), intraperitoneal (c, d), and oral (e, f) routes. Brains were scored on a scale of 0–5 in nine gray matter areas and 0–3 in three white matter areas, animals were grouped following postmortem confirmation of genotype, and were mean scores calculated (error bars representing ±SEM). Brain scoring areas: 1, dorsal medulla; 2, cerebellar cortex; 3, superior colliculus; 4, hypothalamus; 5, medial thalamus; 6, hippocampus; 7, septum; 8, cerebral cortex; 9, forebrain cerebral cortex; 10, cerebellar white matter; 11, midbrain white matter; 12, cerebral peduncle. Legend: ♦, Schwann cell PrP knock-out; ■, floxed PrP control; ▲, Cre control; ●, nontransgenic control.
Discussion
The majority of PrPC in peripheral nerves has been postulated to be derived solely from neuronal expression (Bendheim et al., 1992), transported via fast axonal transport (Borchelt et al., 1994), and localized specifically to the synapse and possibly nodes of Ranvier (Kretzschmar et al., 2000). Studies have suggested that glial cells do not express PrPC, as it could not be detected following axon degeneration with Schwann cells still present (Moya et al., 2005). In contrast PrPC expression in glial cells (Moser et al., 1995) and specifically myelin of the CNS has previously been reported (Mironov et al., 2003). Data from this study clearly attributes the majority of PrPC expression in peripheral nerves to myelin sheaths and myelinating cells. The removal of the PrP coding sequence from Schwann cells has a very profound effect on the expression of PrP in the PNS. A reduction of 90% total PrPC was observed in the sciatic nerve compared with the wild type. PrPC was recently postulated to have a pivotal role in the maintenance of myelin integrity (Baumann et al., 2007). The removal of PrPC expression from myelin forming cells in the study presented here, however, revealed no overt deficit in myelin integrity or morphology. Transmission frequencies of the genetic elements suggest no prenatal mortality. Aging of Schwann cell PrP knock-out mice to >700 d old revealed no overt phenotype. The function of PrP within Schwann cells therefore remains to be defined.
Analysis of PrP expression in peripheral nerves from Schwann cell PrP knock-out mice also revealed a striking loss of glycosylated PrP species. The PrPC glycoform profile in sciatic nerve is very different from that of the CNS, with a predominance of unglycosylated PrP species. Removal of PrP expression solely from Schwann cells reduced both monoglycosylated and diglycosylated PrP species to below definable and measurable levels in the nerve as a whole. These data suggests that in the PNS the processing of PrPC to complex glycoforms is predominantly performed by Schwann cells and that nerve axons and other endoneurial cells appear to glycosylate little of the PrPC they may express. This finding was consistently observed in all adult Schwann cell PrP knock-out mice investigated with ages ranging from 21 to 700 d old. The knock-out of PrP in this model likely occurs during embryonic development and the regulation of axonal protein expression and processing by Schwann cell glycoproteins has been shown previously (Dashiell et al., 2002); therefore we cannot rule out the possibility that removal of PrP expression from Schwann cells may affect PrP expression in associated nerve axons.
Removal of PrP expression from Schwann cells did not prevent TSE neuroinvasion. Following oral infection with ME7 or 139A agent strains, no statistically significant differences in incubation period were observed between Schwann cell PrP knock-out mice and controls. A lengthening of incubation period was observed in Schwann cell PrP knock-out mice following intraperitoneal challenge. The statistical significance of these results however, is rendered obsolete due to the variation observed between control groups of mice via this route. Following direct intracerebral challenge Schwann cell PrP knock-out groups revealed a slight increase in group mean incubation period when compared with control groups. These results suggest that obligatory events occurring within the PNS may regulate disease pathogenesis to a small degree. PrPSc deposition is observed in the spleen at the terminal stage of disease; confirming that infection is not limited to the CNS following intracerebral inoculation as described previously (Beekes and McBride, 2007). Via all inoculation routes, no differences in other disease criteria, such as vacuolation or PrPSc deposition, were observed between Schwann cell PrP knock-out mice and controls. The absence of any overt effect on TSE disease neuroinvasion and pathogenesis following removal of PrPC from Schwann cells and peripheral challenge leads to a number of possible conclusions.
The first of these is that the PNS is not a conduit for the transport of infectivity from the periphery to the CNS following peripheral inoculation. However, much evidence exists to refute this conclusion. The PNS has been implicated as an important route of transport for the infectious agent to the CNS following peripheral challenge (Kimberlin and Walker, 1980). PrPSc deposition has been observed in the PNS from a variety of species infected with TSE disease both naturally and experimentally particularly in experiments designed to trace the infectious routing (Beekes et al., 1998; McBride et al., 2001) with early deposition of PrPSc within the CNS finely targeted to specific areas. The localization of PrPSc deposition within the PNS appears to be between the nerve axon and myelin sheath (Groschup et al., 1999) thereby implicating both neuronal and glial cells in the transport of infection. Schwann-like cells in culture are capable of serial propagation of the agent (Follet et al., 2002), suggesting that these cells may be capable of agent propagation. The minimal effect following intraperitoneal inoculation and absence of any increase in incubation time following oral challenge however, question the role of Schwann cells as an important routing to the CNS.
Another possibility is that PrPC expression is not a requirement for a cell to facilitate transport of infectivity. A number of experimental approaches have implicated PrPC expression as a requirement for neuroinvasion and transport of the infectious TSE agent. Invasion of the CNS by the agent either from the spleen, or directly from the site of infection is thought to be dependent upon a continuous chain of PrPC-expressing cells (Blättler et al., 1997; Glatzel and Aguzzi, 2000; Race et al., 2000). The observation of neuritic transport of PrPSc in PrP-null cultured neuronal cells (Magalhães et al., 2005), suggests that in vivo there may be little requirement for PrPC expression in the PNS for the transport of TSE infection. Moreover, axonal degeneration and incomplete nerve regeneration does not affect TSE agent transport along peripheral nerve conduits (Kratzel et al., 2007), questioning whether nerve axons or PrPC are required for transport of the infectious agent through the peripheral nervous system. Again the alterations observed via the intraperitoneal route would at best argue for the peripheral nerves being only a minor component of the route to the CNS. We have established in parallel studies that while mice expressing only unglycosylated PrP can become infected with a TSE agent following intracerebral challenge (Tuzi et al., 2008), they are not capable of TSE neuroinvasion following peripheral routes of inoculation (EC, BMB and JCM unpublished data) thus implicating glycosylated PrP as important in the peripheral phases of infection (replication and/or transport). It may be the case that fully glycosylated PrP is a requirement for agent replication in the periphery but is not a requirement for agent transport, or vice versa.
The experiments described here have raised further significant questions concerning the role of the various cell types within the PNS in uptake, transport, or replication of the infectious TSE agent. It may be the case that previous experiments have shown only part of the overall picture of replication and transport of the infectivity in the periphery and that other routes such as blood may be significantly more important than has been previously recognized. We are addressing this possibility in ongoing studies. If the PrPC expressed and glycosylated by Schwann cells is not required for TSE neuroinvasion, then nerve axons alone may constitute the neuroinvasive route. The highly reproducible targeting of TSE agent strains to specific areas of the CNS following peripheral infection suggests that little lateral spread of infection occurs between nerve axons during neuroinvasion. The ability of a particular cell to become infected, or contribute to the disease process, may be unrelated to the relative amount or glycoform of PrPC it expresses. The factors determining the susceptibility of a particular cell type to TSE infection are therefore yet to be determined.
Footnotes
This work was supported by Medical Research Council Grants G9721848/D42740 and BBSRC RI/ISPG3. We thank Dr. Lawrence Wrabetz (San Raffaele Scientific Institute, Milan, Italy) for the kind gift of the mP0TOT mice; Dr. Man-Sun Sy (Case Western Reserve University, Cleveland, OH) for the kind gift of anti-PrP antibodies 7A12 and 8H4; and Drs. Peter Brophy and Diane Sherman and Mr. Steven Mitchell (University of Edinburgh, Edinburgh, UK) for assistance with myelin morphology studies. We also thank the staff of The Roslin Institute Neuropathogenesis Division, in particular Aileen Boyle and Sandra Mack, for brain vacuolation scoring, Val Thomson and Kris Hogan for care and clinical assessment of transgenic animals, Dr. Patricia McBride for assistance and collaboration with oral 139A challenge work in inbred mouse strains, and Dr. Robert Somerville and Dr. Neil Mabbott for reading and reviewing this manuscript.
References
- Aguzzi A, Blättler T, Klein MA, Räber AJ, Hegyi I, Frigg R, Brandner S, Weissmann C. Tracking prions: the neurografting approach. Cell Mol Life Sci. 1997;53:485–495. doi: 10.1007/s000180050060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer F, Bachelin C, Andreoletti O, Besnard N, Perrot G, Langevin C, Le Dur A, Vilette D, Baron-Van Evercooren A, Vilotte JL, Laude H. Cultured peripheral neuroglial cells are highly permissive to sheep prion infection. J Virol. 2004;78:482–490. doi: 10.1128/JVI.78.1.482-490.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartz JC, Kincaid AE, Bessen RA. Rapid prion neuroinvasion following tongue infection. J Virol. 2003;77:583–591. doi: 10.1128/JVI.77.1.583-591.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, Heikenwalder M, Rülicke T, Bürkle A, Aguzzi A. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007;26:538–547. doi: 10.1038/sj.emboj.7601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beekes M, McBride PA. The spread of prions through the body in naturally acquired transmissible spongiform encephalopathies. FEBS J. 2007;274:588–605. doi: 10.1111/j.1742-4658.2007.05631.x. [DOI] [PubMed] [Google Scholar]
- Beekes M, McBride PA, Baldauf E. Cerebral targeting indicates vagal spread of infection in hamsters fed with scrapie. J Gen Virol. 1998;79:601–607. doi: 10.1099/0022-1317-79-3-601. [DOI] [PubMed] [Google Scholar]
- Bendheim PE, Brown HR, Rudelli RD, Scala LJ, Goller NL, Wen GY, Kascsak RJ, Cashman NR, Bolton DC. Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology. 1992;42:149–156. doi: 10.1212/wnl.42.1.149. [DOI] [PubMed] [Google Scholar]
- Blättler T, Brandner S, Raeber AJ, Klein MA, Voigtländer T, Weissmann C, Aguzzi A. PrP-expressing tissue required for transfer of scrapie infectivity from spleen to brain. Nature. 1997;389:69–73. doi: 10.1038/37981. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Koliatsos VE, Guarnieri M, Pardo CA, Sisodia SS, Price DL. Rapid anterograde axonal transport of the cellular prion glycoprotein in the peripheral and central nervous systems. J Biol Chem. 1994;269:14711–14714. [PubMed] [Google Scholar]
- Brown KL, Stewart K, Ritchie DL, Mabbott NA, Williams A, Fraser H, Morrison WI, Bruce ME. Scrapie replication in lymphoid tissues depends on prion protein-expressing follicular dendritic cells. Nat Med. 1999;5:1308–1312. doi: 10.1038/15264. [DOI] [PubMed] [Google Scholar]
- Bruce ME. TSE strain variation: an investigation into prion disease diversity. Br Med Bull. 2003;66:99–108. doi: 10.1093/bmb/66.1.99. [DOI] [PubMed] [Google Scholar]
- Bruce ME, McConnell I, Fraser H, Dickinson AG. The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: implications for the nature of the agent and host control of pathogenesis. J Gen Virol. 1991;72:595–603. doi: 10.1099/0022-1317-72-3-595. [DOI] [PubMed] [Google Scholar]
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- Cole S, Kimberlin RH. Pathogenesis of mouse scrapie—dynamics of vacuolation in brain and spinal-cord after intraperitoneal infection. Neuropathol Appl Neurobiol. 1985;11:213–227. doi: 10.1111/j.1365-2990.1985.tb00019.x. [DOI] [PubMed] [Google Scholar]
- Dashiell SM, Tanner SL, Pant HC, Quarles RH. Myelin-associated glycoprotein modulates expression and phosphorylation of neuronal cytoskeletal elements and their associated kinases. J Neurochem. 2002;81:1263–1272. doi: 10.1046/j.1471-4159.2002.00927.x. [DOI] [PubMed] [Google Scholar]
- Feltri ML, Arona M, Messing A, Wrabetz L. The mouse P0 gene as a vector for high-level expression of cDNAs specifically in Schwann cells. J Neurochem. 1996;66:S49. [Google Scholar]
- Feltri ML, D'Antonio M, Previtali S, Fasolini M, Messing A, Wrabetz L. P0-Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann N Y Acad Sci. 1999a;883:116–123. [PubMed] [Google Scholar]
- Feltri ML, D'antonio M, Quattrini A, Numerato R, Arona M, Previtali S, Chiu SY, Messing A, Wrabetz L. A novel P-0 glycoprotein transgene activates expression of lacZ in myelin-forming Schwann cells. Eur J Neurosci. 1999b;11:1577–1586. doi: 10.1046/j.1460-9568.1999.00568.x. [DOI] [PubMed] [Google Scholar]
- Feltri ML, Graus Porta D, Previtali SC, Nodari A, Migliavacca B, Cassetti A, Littlewood-Evans A, Reichardt LF, Messing A, Quattrini A, Mueller U, Wrabetz L. Conditional disruption of beta 1 integrin in Schwann cells impedes interactions with axons. J Cell Biol. 2002;156:199–209. doi: 10.1083/jcb.200109021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follet J, Lemaire-Vieille C, Blanquet-Grossard F, Podevin-Dimster V, Lehmann S, Chauvin JP, Decavel JP, Varea R, Grassi J, Fontès M, Cesbron JY. PrP expression and replication by Schwann cells: implications in prion spreading. J Virol. 2002;76:2434–2439. doi: 10.1128/jvi.76.5.2434-2439.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford MJ, Burton LJ, Morris RJ, Hall SM. Selective expression of prion protein in peripheral tissues of the adult mouse. Neuroscience. 2002;113:177–192. doi: 10.1016/s0306-4522(02)00155-0. [DOI] [PubMed] [Google Scholar]
- Fraser H, Dickinson AG. Distribution of experimentally induced scrapie lesions in the brain. Nature. 1967;216:1310–1311. doi: 10.1038/2161310a0. [DOI] [PubMed] [Google Scholar]
- Glatzel M, Aguzzi A. PrPC expression in the peripheral nervous system is a determinant of prion neuroinvasion. J Gen Virol. 2000;81:2813–2821. doi: 10.1099/0022-1317-81-11-2813. [DOI] [PubMed] [Google Scholar]
- Glatzel M, Gottwein J, Aguzzi A. The role of prions in transmissible spongiform encephalopathies. Schweiz Arch Tierheilkd. 2002;144:633–638. doi: 10.1024/0036-7281.144.12.633. [DOI] [PubMed] [Google Scholar]
- Greenfield S, Brostoff S, Eylar EH, Morell P. Protein composition of myelin of the peripheral nervous system. J Neurochem. 1973;20:1207–1216. doi: 10.1111/j.1471-4159.1973.tb00089.x. [DOI] [PubMed] [Google Scholar]
- Groschup MH, Beekes M, McBride PA, Hardt M, Hainfellner JA, Budka H. Deposition of disease-associated prion protein involves the peripheral nervous system in experimental scrapie. Acta Neuropathol. 1999;98:453–457. doi: 10.1007/s004010051108. [DOI] [PubMed] [Google Scholar]
- Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–106. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- Kimberlin RH, Walker CA. Pathogenesis of mouse scrapie: evidence for neural spread of infection to the CNS. J Gen Virol. 1980;51:183–187. doi: 10.1099/0022-1317-51-1-183. [DOI] [PubMed] [Google Scholar]
- Kimberlin RH, Hall SM, Walker CA. Pathogenesis of mouse scrapie—evidence for direct neural spread of infection to the Cns after injection of sciatic-nerve. J Neurol Sci. 1983;61:315–325. doi: 10.1016/0022-510x(83)90165-x. [DOI] [PubMed] [Google Scholar]
- Kratzel C, Krüger D, Beekes M. Prion propagation in a nerve conduit model containing segments devoid of axons. J Gen Virol. 2007;88:3479–3485. doi: 10.1099/vir.0.83187-0. [DOI] [PubMed] [Google Scholar]
- Kretzschmar HA, Tings T, Madlung A, Giese A, Herms J. Function of PrPC as a copper-binding protein at the synapse. Arch Virol Suppl. 2000;16:239–249. doi: 10.1007/978-3-7091-6308-5_23. [DOI] [PubMed] [Google Scholar]
- Lemaire-Vieille C, Schulze T, Podevin-Dimster V, Follet J, Bailly Y, Blanquet-Grossard F, Decavel JP, Heinen E, Cesbron JY. Epithelial and endothelial expression of the green fluorescent protein reporter gene under the control of bovine prion protein (PrP) gene regulatory sequences in transgenic mice. Proc Natl Acad Sci U S A. 2000;97:5422–5427. doi: 10.1073/pnas.080081197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Liu T, Wong BS, Pan T, Morillas M, Swietnicki W, O'Rourke K, Gambetti P, Surewicz WK, Sy MS. Identification of an epitope in the C terminus of normal prion protein whose expression is modulated by binding events in the N terminus. J Mol Biol. 2000;301:567–573. doi: 10.1006/jmbi.2000.3986. [DOI] [PubMed] [Google Scholar]
- Magalhães AC, Baron GS, Lee KS, Steele-Mortimer O, Dorward D, Prado MA, Caughey B. Uptake and neuritic transport of scrapie prion protein coincident with infection of neuronal cells. J Neurosci. 2005;25:5207–5216. doi: 10.1523/JNEUROSCI.0653-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J. 129/Ola mice carrying a null mutation in Prp that abolishes messenger-Rna production are developmentally normal. Mol Neurobiol. 1994;8:121–127. doi: 10.1007/BF02780662. [DOI] [PubMed] [Google Scholar]
- Mao X, Fujiwara Y, Orkin SH. Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proc Natl Acad Sci U S A. 1999;96:5037–5042. doi: 10.1073/pnas.96.9.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride PA, Schulz-Schaeffer WJ, Donaldson M, Bruce M, Diringer H, Kretzschmar HA, Beekes M. Early spread of scrapie from the gastrointestinal tract to the central nervous system involves autonomic fibers of the splanchnic and vagus nerves. J Virol. 2001;75:9320–9327. doi: 10.1128/JVI.75.19.9320-9327.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov A, Jr, Latawiec D, Wille H, Bouzamondo-Bernstein E, Legname G, Williamson RA, Burton D, DeArmond SJ, Prusiner SB, Peters PJ. Cytosolic prion protein in neurons. J Neurosci. 2003;23:7183–7193. doi: 10.1523/JNEUROSCI.23-18-07183.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales R, Abid K, Soto C. The prion strain phenomenon: molecular basis and unprecedented features. Biochim Biophys Acta. 2007;1772:681–691. doi: 10.1016/j.bbadis.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser M, Colello RJ, Pott U, Oesch B. Developmental expression of the prion protein gene in glial-cells. Neuron. 1995;14:509–517. doi: 10.1016/0896-6273(95)90307-0. [DOI] [PubMed] [Google Scholar]
- Moya KL, Hässig R, Breen KC, Volland H, Di Giamberardino L. Axonal transport of the cellular prion protein is increased during axon regeneration. J Neurochem. 2005;92:1044–1053. doi: 10.1111/j.1471-4159.2004.02940.x. [DOI] [PubMed] [Google Scholar]
- Race R, Oldstone M, Chesebro B. Entry versus blockade of brain infection following oral or intraperitoneal scrapie administration: role of prion protein expression in peripheral nerves and spleen. J Virol. 2000;74:828–833. doi: 10.1128/jvi.74.2.828-833.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito F, Moore SA, Barresi R, Henry MD, Messing A, Ross-Barta SE, Cohn RD, Williamson RA, Sluka KA, Sherman DL, Brophy PJ, Schmelzer JD, Low PA, Wrabetz L, Feltri ML, Campbell KP. Unique role of dystroglycan in peripheral nerve myelination, nodal structure, and sodium channel stabilization. Neuron. 2003;38:747–758. doi: 10.1016/s0896-6273(03)00301-5. [DOI] [PubMed] [Google Scholar]
- Sauer B. Manipulation of transgenes by site-specific recombination—use of Cre recombinase. Guide to techniques in mouse development, Methods Enzymol. 1993;225:890–900. doi: 10.1016/0076-6879(93)25056-8. [DOI] [PubMed] [Google Scholar]
- Schulz-Schaeffer WJ, McBride PA, Beekes M, Kretzschmar HA. Spread of PrPSc in orally infected animals during the incubation time of prion disease. Brain Pathol. 2000;10:663. [Google Scholar]
- Taylor DM, McConnell I, Fraser H. Scrapie infection can be established readily through skin scarification in immunocompetent but not immunodeficient mice. J Gen Virol. 1996;77:1595–1599. doi: 10.1099/0022-1317-77-7-1595. [DOI] [PubMed] [Google Scholar]
- Tuzi NL, Clarke AR, Bradford B, Aitchison L, Thomson V, Manson JC. Cre-loxP mediated control of PrP to study transmissible spongiform encephalopathy diseases. Genesis. 2004;40:1–6. doi: 10.1002/gene.20046. [DOI] [PubMed] [Google Scholar]
- Tuzi NL, Cancellotti E, Baybutt H, Blackford L, Bradford B, Plinston C, Coghill A, Hart P, Piccardo P, Barron RM, Manson JC. Host PrP glycosylation: a major factor determining the outcome of prion infection. PLoS Biol. 2008;6:872–882. doi: 10.1371/journal.pbio.0060100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanusso G, Liu D, Ferrari S, Hegyi I, Yin X, Aguzzi A, Hornemann S, Liemann S, Glockshuber R, Manson JC, Brown P, Petersen RB, Gambetti P, Sy MS. Prion protein expression in different species: analysis with a panel of new mAbs. Proc Natl Acad Sci U S A. 1998;95:8812–8816. doi: 10.1073/pnas.95.15.8812. [DOI] [PMC free article] [PubMed] [Google Scholar]