

Abstract
Aversive memories of drug withdrawal can generate a motivational state leading to compulsive drug taking. Changes in synaptic plasticity may be involved in the formation of aversive memories. Dynamic rearrangement of the cytoskeletal actin, a major structural component of the dendritic spine, regulates synaptic plasticity. Here, the potential involvement of actin rearrangements in the induction of aversive memories of morphine withdrawal was examined. We found that lesions of the amygdala or dorsal hippocampus (DH) but not nucleus accumbens (NAc) impaired conditioned place aversion (CPA) of acute morphine-dependent rats. Accordingly, conditioned morphine withdrawal induced actin rearrangements in the amygdala and the DH but not in the NAc. In addition, we found that conditioned morphine withdrawal also increased activity-regulated cytoskeletal-associated protein (Arc) expression in the amygdala but not in the DH, although actin rearrangements were observed in both areas. We further found that inhibition of actin rearrangements by intra-amygdala or intra-DH injections of latrunculin A, an inhibitor of actin polymerization, significantly attenuated CPA. Furthermore, we found that manipulation of amygdala β-adrenoceptor activity by its antagonist propranolol and agonist clenbuterol differentially altered actin rearrangements in the DH. Therefore, our findings reveal that actin rearrangements in the amygdala and the DH are required for the acquisition and consolidation of the aversive memories of drug withdrawal and that the β-noradrenergic system within the amygdala modulates aversive memory consolidation by regulating actin rearrangements but not Arc protein expression in the DH, which is distinct from its role in modulation of inhibitory avoidance memory.
Introduction
Opiate addiction is the chronic relapsing disorder characterized by compulsive drug seeking and taking (De Vries and Shippenberg, 2002). Recently, the negative reinforcement mechanisms underlying drug seeking and taking have received considerable attention (Koob, 2000; Hutcheson et al., 2001). It has been suggested that the maintenance of compulsive use of the drugs is substantially associated with the aversive properties of withdrawal in dependent subjects (Rodríguez de Fonseca and Navarro, 1998; Koob, 2000). The aversive memories of drug withdrawal play a key role in the induction of a motivational state leading to drug seeking and taking. However, the mechanisms underlying generation of drug withdrawal memories remain unclear.
In rodents, the negative affective component of opiate dependence could be reflected by several behavioral alterations (Schaefer and Michael, 1986; Stinus et al., 1990; Higgins and Sellers, 1994), among which conditioned place aversion (CPA) is the most sensitive measurement for the negative motivational state produced by opioid withdrawal in both chronically dependent animals (Stinus et al., 1990) and acutely dependent animals (Azar et al., 2003). Essentially, CPA is a pavlovian conditioning paradigm in which drug withdrawal is paired with a particular environment, which triggers the onset of association between the negative affective consequences of withdrawal with context (aversive memory formation), and then the animals are reexposed to the paired environment in drug-free state, leading to avoidance of the paired environment (aversive memory retrieval). Thus, CPA can be used to explore the neurobiological mechanisms underlying the formation and retrieval of aversive memories of opioid withdrawal in opiate-dependent animals.
Synaptic plasticity contributes to a wide variety of brain functions, including learning and memory. Changes in dendritic spine morphology are thought to be involved in synaptic plasticity (Segal, 2005). Cytoskeletal actin is the major structural component of the dendritic spine (Matus et al., 1982; Fifková and Morales, 1992) and has been shown to play a role in synaptic plasticity by maintaining characteristically and highly dynamic transformation between G-actin (monomeric globular actin) and F-actin (polymeric filamentous actin) through polymerization and depolymerization (actin rearrangement) (Fukazawa et al., 2003; Zhou et al., 2004; Zito et al., 2004). Recent studies support a role for rearrangement of actin in long-term potentiation (LTP) maintenance and behavioral adaptations (Fukazawa et al., 2003; Lisman, 2003; Fischer et al., 2004) and demonstrate that changes in the actin cytoskeleton in certain neuronal cell types in flies, mice, and rats alter behavioral responses to alcohol and cocaine (Offenhäuser et al., 2006; Rothenfluh et al., 2006; Toda et al., 2006). Therefore, we hypothesized that rearrangement of the actin cytoskeleton in brain regions, particularly within limbic structures [e.g., amygdala, nucleus accumbens (NAc), and hippocampus] which could specifically mediate the negative motivational component of opiate withdrawal (Frenois et al., 2002, 2005), might play a crucial role in the aversive memories of opioid withdrawal. The present study was undertaken to test this hypothesis by examining actin rearrangements in the amygdala, NAc, as well as the dorsal hippocampus (DH) and determining the role of actin rearrangements in the induction of CPA.
Materials and Methods
Subjects
Sprague Dawley male rats weighing 220–250 g were obtained from the Laboratory Animal Center, Chinese Academy of Sciences (Shanghai, China). Rats were housed two to three per cage and maintained on a 12 h light/dark cycle with access to food and water ad libitum. All experimental procedures were in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Drugs and antibodies
Morphine hydrochloride was purchased from Qinghai Pharmaceutical General Factory. Naloxone hydrochloride, ibotenic acid, clenbuterol hydrochloride, and dl-propranolol hydrochloride were supplied by Sigma-Aldrich. Latrunculin A was obtained by Calbiochem. The antibodies of anti-actin were purchased from Sigma-Aldrich and diluted 1:3000, and the antibodies of anti-Arc were obtained from Santa Cruz Biotechnology and diluted 1:1000 for Western blot analysis or 1:500 for immunohistochemistry analysis.
Conditioned place aversion
Apparatus.
The CPA apparatus [56 cm (length) × 22 cm (width) × 37 cm (height)] made of Plexiglas was divided into two equal-sized compartments by a removable partition with an opening (10 × 10 cm) at one end, which allowed rats free access to each compartment. Two compartments distinguished by visual and tactile cues: the one was grays walls with a textured floor, whereas the other was black-and-white vertical striped walls with a smooth floor. Another partition without an opening was used to confine rats to a given compartment in the conditioning phase of CPA procedure.
Procedures.
The method to establish conditioned place aversion was similar to that used by White et al. (2005). The CPA procedure consisted of three phases: preconditioning, conditioning, and testing. In the preconditioning phase, rats were allowed to freely explore the entire apparatus for 15 min. Time spent in each compartment was recorded, and those showing a strong unconditioned aversion (<180 s) for either compartment were eliminated from the study. There was no significant difference between the time spent in the smooth floor (442.3 ± 6.8 s, n = 306) and the textured floor (457.7 ± 6.8 s, n = 306) compartment during the preconditioning phase. Conditioning took place over the next 2 d. On the first day, the rats were injected with saline (1 ml/kg, s.c.) and then returned to home cages. Four hours later, they were again given saline and then confined to either compartment in a counterbalanced manner for 30 min. On the second day, the rats were injected with either morphine (10 mg/kg, s.c.) or saline (1 ml/kg, s.c.) and then returned to their home cages. Four hours later, they were injected with either naloxone (0.3 mg/kg, s.c.) or saline and then confined to the compartment opposite to that on the first day for 30 min. This compartment will be referred to as the “drug treatment-paired compartment.” According to drug treatments, the rats were divided into four groups: morphine + naloxone, morphine + saline, saline + naloxone, and saline + saline. In the testing phase (24 h after the conditioning trial), all rats were allowed to freely explore the entire apparatus for 15 min, and the amount of time spent in each compartment was recorded. CPA score represents the time in the drug treatment-paired compartment during the testing phase minus that during the preconditioning phase.
Excitotoxic lesions.
Rats were anesthetized using sodium pentobarbital (50 mg/kg, i.p.), treated with atropine sulfate (0.2 mg/kg, i.p.), and then placed in a stereotaxic apparatus (Narishige) with the incisor bar set at 3.3 mm. The scalp was incised and retracted to expose the skull. Small burr holes were drilled through the skull to place a 26 gauge guide cannula using the following coordinates: for amygdala lesions, anteroposterior (AP), −2.8 mm from bregma, mediolateral (ML), ±4.2 mm from midline, dorsoventral (DV), −7.0 mm from skull; for dorsal hippocampus lesions (1), AP, −2.8 mm, ML, ±1.6 mm, DV, −2.6 mm; (2) AP, −4.2 mm, ML, ±2.6 mm, DV, −2.4 mm; for nucleus accumbens lesions, AP, +1.6 mm, ML, ±1.4 mm, DV, −7.0 and −6.0 mm. Ibotenic acid (10 μg/μl) was infused in the amygdala, nucleus accumbens (0.2 μl/site), and dorsal hippocampus (0.15 μl/site) over 2 min with a 31 gauge injection cannula (1.0 mm beyond the tip of guide cannula) that was connected to a 10 μl microsyringe mounted in the microinfusion pump (Harvard Apparatus). After each infusion, the injection cannula was left in place for 2 min and then removed slowly. Rats in the sham group were given the same surgical procedures but did not receive microinfusion. All rats were given 7–10 d for postsurgical recovery before behavioral training.
Intracerebral microinjection.
Rats were implanted bilaterally with guide cannulae in the amygdala (AP, −2.8 mm; ML, ±4.2 mm; DV, −7.0 mm) or the dorsal hippocampus (AP, −3.8 mm; ML, ±2.5 mm; DV, −2.5 mm). Bilateral microinfusions were made through 31 gauge injection cannulae (1.0 mm beyond the tip of guide cannulae) over 3 min and given an additional 2 min for drug diffusion. Latrunculin A was dissolved in DMSO (25 μg/μl stock solution) and diluted in PBS to a final concentration of 0.5 μg/μl. Latrunculin A was bilaterally microinjected into the amygdala 10 min before pairing or into the DH immediately after pairing. Propranolol and clenbuterol were dissolved in PBS before use and were bilaterally microinjected into the amygdala 10 min before pairing. The doses of latrunculin A, propranolol, and clenbuterol were chosen based on pilot experiments and previous studies (Watanabe et al., 2003; McIntyre et al., 2005; Toda et al., 2006).
Histology.
After behavior testing, rats were deeply anesthetized with sodium pentobarbital and perfused transcardially with PBS, followed by 10% Formalin. The brains were removed and stored in a 30% sucrose/10% Formalin solution for 2–3 d. Coronal sections (50 μm thick) were cut on a cryostat (Leica), stained with cresyl violet, and then examined by light microscopy to determine lesion locations or injection sites.
Immunohistochemistry of Arc.
Immunohistochemical assay was performed as we described previously (Li et al., 2009). Briefly, brain sections from different groups were processed in parallel to minimize the variations in immunohistochemical labeling. The sections of brain were cut at a thickness of 30 μm and washed with 0.1 mol/L phosphate buffer. Brian sections were blocked with 10% normal goat serum for 2 h at room temperature and then incubated overnight with primary antibody (mouse anti-Arc antibody, 1:500 dilution in 10% normal goat serum) at 4°C. Next, brain sections were incubated with secondary antibody (biotinylated goat anti-mouse IgG, 1:200 dilution in 10% normal goat serum) for 2 h at room temperature. Arc-positive sites were visualized using a streptavidin–ABC kit and a DAB kit using 0.1% 3,3′-diaminobenzidine as the chromogen. In control sections in which the primary antibodies were omitted or replaced by non-immune rabbit or goat serum, no stained cells were seen. The brain slices were subsequently dehydrated in alcohol and xylene, coverslipped, and imaged on an Olympus IX51 microscope.
Subcellular fractionation and immunoblotting.
After rats were decapitated, brains were rapidly removed, frozen in liquid nitrogen, and stored in a −80°C freezer before dissection. Coronal brain sections (0.5 mm thick) were obtained using a rat brain slicer (Braintree Scientific). Both sides of the amygdala, dorsal hippocampus, and nucleus accumbens were punched from brain slices using a blunt-end, 17 gauge syringe needle (1 mm inner diameter). The tissue of amygdala punched included most of the central (CeA), lateral, and basolateral (BLA) nuclei of the amygdala. The tissue of dorsal hippocampus punched included most of CA1 and dentate gyrus areas. Subcellular fractionation and actin analysis of the dissected brain tissue was performed as described previously (Toda et al., 2006). Briefly, homogenate of the tissue in the 0.32 m sucrose buffer was centrifuged at 1000 × g for 10 min, and the supernatant was further centrifuged at 10,000 × g for 15 min to obtain a crude synaptosome fraction (P2) that was subsequently dissolved in hypoosmotic buffer and centrifuged at 25,000 × g for 25 min to precipitate a synaptosomal membrane fraction (LP1). Before centrifugation, protein concentration of P2 solution was determined by Bradford analysis, and then samples of equal protein amounts was used to precipitate LP1. To separate F-actin and G-actin, LP1 was lysed in 100 μl of buffer A (1% Triton X-100, 20 mm HEPES, 100 mm NaCl, 2 mm EDTA, 5 mm NaF, 1 mm Na3VO4, 1 mm aprotinin, 1 mm leupeptin, and 1 mm PMSF, pH 7.2) for 1 h, then 50 μl of solution was centrifuged at 10,000 × g for 20 min, and the rest (50 μl) was collected to determine total actin fraction. Pellets was dissolved in 50 μl of buffer B (15 mm HEPES, 0.15 mm NaCl, 1% SDS, 10 mm EDTA, 1 mm DTT, 5 mm NaF, 1 mm Na3VO4, 1 mm aprotinin, 1 mm leupeptin, and 1 mm PMSF, pH 7.5) for 1 h and centrifuged at 10,000 × g for 20 min. The G-actin fraction (the first supernatant) and the F-actin fraction (the second supernatant) were collected. Equal volume samples were used to detect immunoreactivity of total actin (20 μl), F-actin (25 μl), and G-actin (30 μl) by Western blot analysis. Arc expression analysis was performed as described previously (McIntyre et al., 2005). Equal protein amount (20 μg) of sample was used to detected immunoreactivity of Arc and actin. The immunopositive signals were quantified by Quantity One software (Bio-Rad).
Data analysis.
The data were analyzed with one- or two-way ANOVA or Student's t tests, followed by Bonferroni's post hoc tests when appropriate. Differences with p < 0.05 were considered statistically significant. The results are presented as mean ± SEM.
Results
Conditioned place aversion is elicited by one pairing with low dose of naloxone in rats exposed to a single dose of morphine
Previous studies showed that a significant place aversion could be conditioned by naloxone-precipitated withdrawal after either acute or chronic morphine treatments. A recent study demonstrated that conditioned place aversion was able to be elicited by low doses of naloxone after a single dose of morphine after even only one pairing (Azar et al., 2003). In the present study, we used a similar training paradigm in which CPA was induced by administration of naloxone (0.3 mg/kg, s.c.) 4 h after a single exposure to morphine (10 mg/kg, s.c.). Consistent with previous studies, one pairing with naloxone after a single dose of morphine exposure produced significant aversion for the withdrawal-associated environment in morphine-treated rats. As presented in Figure 1, rats pretreated with a single dose of morphine, followed by one pairing with naloxone, spent significantly less time in the compartment paired with morphine withdrawal (CPA score: −407.3 ± 26.8 s; p < 0.01 vs other groups). Such enhanced aversion is thought to reflect the negative motivational and affective state associated with morphine withdrawal (Mucha and Walker, 1987). However, rats pretreated with a single dose of saline, followed by one pairing with naloxone, did not produce a significant place aversion compared with rats pretreated with saline, followed by pairing with saline (CPA score: naloxone, −117.2 ± 55.0 s; saline, −69.7 ± 52.5 s; p > 0.05), indicating that the dose of naloxone used was not aversive by itself in our experimental conditions. In addition, rats pretreated with a single dose of morphine, followed by one pairing with saline, also failed to produce a significant CPA (CPA score: −60.5 ± 34.3 s; p > 0.05).
Figure 1.

Place aversion was induced by conditioned naloxone-precipitated drug withdrawal in the rats exposed to a single dose of morphine. After the preconditioning, rats were conditioned over 2 d using saline or morphine (10 mg/kg, s.c) pretreatment, followed by saline and naloxone (0.3 mg/kg, s.c.) challenges. Testing (15 min) occurred 24 h after conditioning. CPA score is defined as time in the drug treatment-paired compartment during the testing phase minus that during the preconditioning phase. Values are expressed as mean ± SEM. **p < 0.01, compared with the saline-treated control group, one-way ANOVA with Bonferroni's post hoc test. Sal, Saline; Mor, morphine; Nal, naloxone.
Conditioned place aversion induced by naloxone precipitation in single-dose morphine-pretreated rats was attenuated by excitotoxic lesions of the amygdala or the dorsal hippocampus but not nucleus accumbens
Studies on brain regions involved in emotional responses by lesions have found that the limbic structures, such as amygdala and bed nucleus of the stria terminalis (BNST), play a crucial role in the negative affective component of morphine withdrawal (Kelsey and Arnold, 1994; Watanabe et al., 2003; Nakagawa et al., 2005). However, the role of NAc in the negative affective component of morphine withdrawal is controversial (Stinus et al., 1990; Kelsey and Arnold, 1994; Caillé et al., 2003), although it has been well established to play a crucial role in regulation of rewarding responses. In addition, studies on opiate withdrawal-induced c-Fos immunoreactivity in the limbic circuit have also suggested that the hippocampus is involved in mediating morphine withdrawal-induced CPA (Frenois et al., 2002, 2005), although direct in vivo behavioral evidence supporting this role of the hippocampus is lacking. To determine the brain regions in limbic circuit that mediate CPA elicited by one pairing with naloxone precipitation after a single dose of morphine exposure, we examined the effects of bilateral excitotoxic lesions of the amygdala, NAc, and the DH with ibotenic acid on CPA induced by naloxone-precipitated morphine withdrawal in rats.
Schematic illustrations of lesion areas in 33 of the rats that received bilateral infusion of ibotenic acid into the amygdala, the DH, and the NAc are shown in Figure 2, A, C, and E, respectively. Photomicrographs of representative lesions of the amygdala, the DH, and the NAc were shown in supplemental Figure S1, B, D, and F, respectively (available at www.jneurosci.org as supplemental material). Amygdala lesions encompassed the central, lateral, and basolateral nuclei of the amygdala. A few lesions extended dorsally into caudate–putamen (CPu) in some cases. Bilateral dorsal hippocampus was mostly damaged with minimal effects on the overlying cortex (most probably by mechanical effects of the injection cannula; not shown in the figure). NAc lesions were primarily confined to the NAc core and shell with minor damage to the overlying CPu.
Figure 2.

Conditioned place aversion was attenuated by lesions of the amygdala or the DH but not the NAc. Left column, Schematic representation of bilateral excitotoxic lesions of the amygdala (A), the DH (C), and the NAc (E). The black areas represent the smallest extent of neuronal damage, and the gray areas represent largest extent of neuronal damage. Right column, Effects of lesions of the amygdala (B), the DH (D), or the NAc (F) on place aversion induced by conditioned naloxone-precipitated morphine withdrawal in acute morphine-dependent rats. The open bar represents saline treatment, and the filled bar represents morphine treatment. Values are expressed as mean ± SEM (n = 5–6). **p < 0.01, compared with corresponding saline-treated group; ##p < 0.01, compared with corresponding sham-operated group, two-way ANOVA with Bonferroni's post hoc test. Amy, Amygdala.
The effects of bilateral excitotoxic lesions of the amygdala or the DH on naloxone-precipitated morphine withdrawal-induced CPA are shown in Figure 2, B and D, respectively. Two-way ANOVA revealed the significant main effect of operation (sham vs lesion: amygdala, F(1,17) = 22.80, p < 0.001; DH, F(1,17) = 13.59, p < 0.01) and treatment (saline vs morphine: amygdala, F(1,17) = 13.42, p < 0.001; DH, F(1,17) = 5.83, p < 0.01) on CPA scores, as well as a significant interaction between these two factors (amygdala, F(1,17) = 8.35, p < 0.05; DH, F(1,17) = 12.80, p < 0.01). The following post hoc test showed a significant difference in CPA scores between the saline and morphine treatment in amygdala and dorsal hippocampus sham-operated groups (p < 0.01) but not in amygdala- and dorsal hippocampus-lesioned groups. Furthermore, there was a significant difference between the sham and lesion operation in morphine treatment groups (p < 0.01) but not in saline treatment groups. However, bilateral excitotoxic lesions of the NAc had no significant effect on the naloxone-precipitated morphine withdrawal-induced CPA (Fig. 2F). A two-way ANOVA indicated a nonsignificant effect of NAc lesion (F(1,19) = 0.21, p > 0.05) and a significant effect of treatment (F(1,19) = 43.10, p < 0.001) on CPA score, consistent with previous studies (Kelsey and Arnold, 1994; Caillé et al., 2003). These results demonstrated that bilateral excitotoxic lesions of the amygdala or the DH but not the NAc significantly attenuated the naloxone-precipitated morphine withdrawal-induced CPA behaviors.
Conditioned place aversion was accompanied by actin rearrangements in the amygdala and the dorsal hippocampus
Place aversion is the reflection of the aversive memories formed by the negative affective consequences of morphine withdrawal with a particular environment. Given a growing body of evidence that actin rearrangements play a crucial role in synaptic plasticity associated with learning and memory, we determined whether actin rearrangements were involved in the formation of aversive memories of conditioned morphine withdrawal. To do this, we measured the levels of the monomeric actin (G-actin) and the polymerized actin (F-actin) in the amygdala and the dorsal hippocampus after conditioned naloxone-precipitated morphine withdrawal and compared them with those in the same regions after unconditioned naloxone-precipitated morphine withdrawal. A significant elevation of the ratio of F-actin to G-actin was observed in the amygdala at 0.5 h (p < 0.05 vs control group) (Fig. 3A) and in the DH at 1 h (p < 0.05 vs control group) (Fig. 3B), respectively, after conditioned naloxone-precipitated morphine withdrawal, indicative of occurrence of actin rearrangements in two tested regions. However, the levels of total actin were not altered in both regions at the tested time. In contrast to conditioned naloxone-precipitated morphine withdrawal, unconditioned naloxone-precipitated morphine withdrawal failed to induce actin rearrangement in the amygdala and the DH (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). In contrast to the amygdala and the DH, conditioned naloxone-precipitated morphine withdrawal did not alter the ratio of F-actin to G-actin in the NAc at either 0.5 or 1 h (Fig. 3C), suggesting that NAc may not be involved in the formation of aversive memories of conditioned morphine withdrawal, which parallels with the finding that lesions of the NAc had no significant effect on conditioned morphine withdrawal-induced CPA (Fig. 2F). These results suggest that actin rearrangements in the amygdala and the dorsal hippocampus may be involved in the acquisition and consolidation of CPA.
Figure 3.
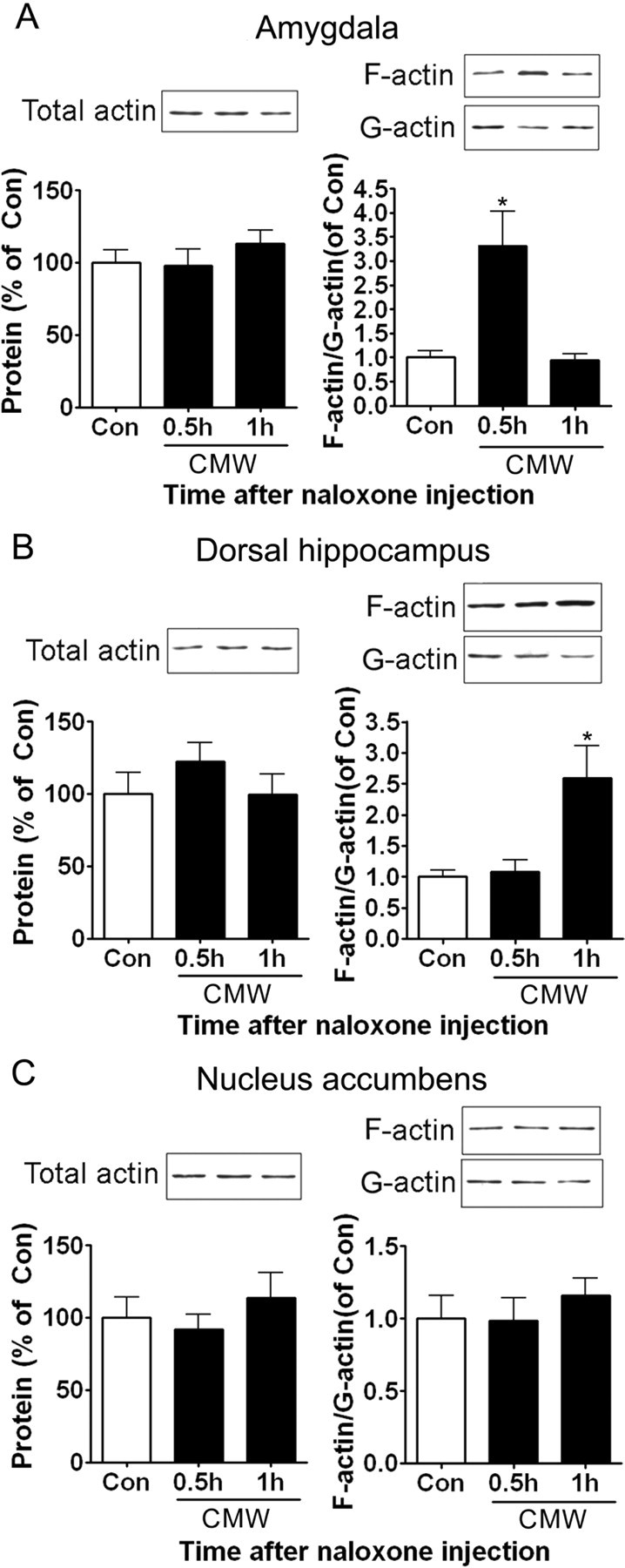
Effects of conditioned naloxone-precipitated morphine withdrawal on actin rearrangements in the amygdala, the DH, and the NAc. A, Conditioned morphine withdrawal induced an increase in the ratio of F-actin to G-actin in the amygdala at 0.5 h after naloxone injection. B, Conditioned morphine withdrawal induced an increase in the ratio of F-actin to G-actin in the DH at 1 h after naloxone injection. C, Conditioned morphine withdrawal failed to induce an increase in the ratio of F-actin to G-actin in the NAc. Values are expressed as mean ± SEM (n = 4). *p < 0.05, compared with the corresponding saline-treated control group, one-way ANOVA with Bonferroni's post hoc test. Con, Control; CMW, conditioned morphine withdrawal.
Elevation of Arc protein expression in the amygdala but not in the dorsal hippocampus in response to conditioned morphine withdrawal
The immediate-early gene Arc mRNA and protein are found at recently activated synapses and interacts with the cytoskeletal protein actin (Lyford et al., 1995; Steward et al., 1998). Increasing evidence also shows that Arc is critically involved in processes of synaptic plasticity underlying certain forms of behavioral memories (e.g., inhibitory avoidance and contextual fear memories) (McIntyre et al., 2005; Huff et al., 2006). To determine whether Arc protein is also involved in aversive memory of morphine withdrawal, we examined the effects of conditioned naloxone-precipitated morphine withdrawal on Arc protein expression in the amygdala and the DH by immunohistochemistry (Li et al., 2009) and immunoblot. Representative immunohistochemical staining of Arc protein in the amygdala and the DH is shown in Figure 4, A and C. Conditioned naloxone-precipitated morphine withdrawal induced a robust increase in the staining of Arc protein in the amygdala (BLA and CeA) (Fig. 4A). Positive staining of Arc protein was clearly visualized at the dendrites and soma of neurons (Li et al., 2009). Surprisingly, in contrast to the amygdala, the DH showed undetectable changes in the staining of Arc protein in response to conditioned naloxone-precipitated morphine withdrawal (Fig. 4C). Immunoblot assay also revealed a remarkable increase of Arc protein expression in the amygdala but not in the DH in response to conditioned naloxone-precipitated morphine withdrawal (Fig. 4B,D). This is inconsistent with the contextual fear conditioning reported by Huff et al. (2006) and inhibitory avoidance conditioning report by McIntyre et al. (2005) in that Arc protein is elevated in the DH after contextual fear conditioning and inhibitory avoidance conditioning.
Figure 4.

Conditioned morphine withdrawal induced Arc protein expression in the amygdala but not in the dorsal hippocampus. Left column, Photomicrographs illustrating Arc protein expression in the amygdala (A) and the DH (C) after either conditioned morphine withdrawal treatment or saline control treatment. Right column, Effects of conditioned morphine withdrawal on the Arc expression of the amygdala (B) and the dorsal hippocampus (D) were determined by immunoblot. The level of Arc expression was normalized to actin level. Values are expressed as mean ± SEM (n = 4–5). *p < 0.05, **p < 0.01, compared with the saline-treated control group, one-way ANOVA with Bonferroni's post hoc test. CMW, Conditioned morphine withdrawal; Con, control; Nal, naloxone; DG, dentate gyrus.
Actin rearrangements in the amygdala and the dorsal hippocampus were required for morphine withdrawal-induced conditioned place aversion
To determine whether the changes in actin rearrangements are responsible for the induction of CPA, effects of intra-amygdala and intra-DH injections of latrunculin A, an inhibitor of actin polymerization (Morton et al., 2000; Yarmola et al., 2000), on CPA induced by reexposure of rats to a withdrawal-paired environment were determined. The microinjection tips located in the amygdala and the DH were illustrated in Figure 5, A and C, respectively. Histological analysis by cresyl violet staining showed that microinjection of latrunculin A into the amygdala and the DH did not produce greater damage to the targeted tissue compared with vehicle microinjection (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Bilateral microinjection of latrunculin A (250 ng/0.5 μl per side) into the amygdala 10 min before pairing with morphine withdrawal markedly attenuated CPA compared with the vehicle microinjection (p < 0.01) (Fig. 5B). In contrast, bilateral microinjection of latrunculin A into the amygdala immediately after pairing had no significant impact on CPA (p > 0.05 vs vehicle-microinjected control group), suggesting that actin rearrangement was involved with the acquisition of CPA. Because the DH plays a unique role in memory consolidation (Bernabeu et al., 1997; Ahi et al., 2004) and alteration of actin rearrangement was found in the DH as shown above, we next determined whether inhibition of actin rearrangement in the DH with latrunculin A could impair CPA behaviors. As shown in Figure 5D, bilateral microinjection of latrunculin A (500 ng/1 μl per side) into the DH immediately after pairing significantly impaired CPA (t(1,13) = 5.476; p < 0.01), suggesting that actin rearrangement in the dorsal hippocampus may be involved in the consolidation of CPA. In addition, we further determined the effect of microinjection of latrunculin A into the amygdala or DH on actin rearrangements induced by conditioned morphine withdrawal and found that such manipulation diminished the elevation of the ratio of F-actin to G-actin (supplemental Fig. S4, available at www.jneurosci.org as supplemental material), indicating that actin rearrangements in the amygdala or DH were involved in conditioned morphine withdrawal-induced CPA behavior.
Figure 5.

Effects of intra-amygdala or intra-DH injections of latrunculin A, an inhibitor of actin polymerization, on place aversion induced by conditioned drug withdrawal after exposure to a single dose of morphine. Left column, Schematic representation of injection sites in the amygdala (A) and the DH (C) for rats used in the experiments (○, control; •, latrunculin A injected before pairing; ▴, latrunculin A injected after pairing). Right column, B, Prepairing but not postpairing intra-amygdala injection of latrunculin A attenuated place aversion induced by conditioned naloxone-precipitated morphine withdrawal in the acute morphine-dependent rats. Latrunculin A (250 ng/0.5 μl per side) or vehicle (0.5 μl/side) was bilaterally microinjected into the amygdala 10 min before pairing or immediately after pairing with morphine withdrawal. D, Postpairing intra-DH injection of latrunculin A attenuated place aversion induced by conditioned naloxone-precipitated morphine withdrawal in the acute morphine-dependent rats. Latrunculin A (500 ng/1 μl per side) or vehicle (1 μl/side) was bilaterally microinjected into the DH immediately after pairing with morphine withdrawal. Values are represented as the mean ± SEM. **p < 0.01, compared with the corresponding vehicle-microinjected control group, Student's t test or one-way ANOVA with Bonferroni's post hoc test.
Lesions of the amygdala reduced actin rearrangements in the dorsal hippocampus induced by conditioned naloxone-precipitated morphine withdrawal
Many studies show that the amygdala (e.g., BLA) has a role in modulating memory consolidation by interacting with other brain regions (McGaugh et al., 2002). Moreover, considerable evidence has shown that the amygdala is functionally and anatomically connected to the hippocampus (Akirav and Richter-Levin, 2002; McIntyre et al., 2005; Huff et al., 2006). On the basis of the findings that actin rearrangement occurred in the amygdala is earlier than that occurred in the DH, we hypothesized that the amygdala might exert a regulatory effect on actin rearrangement in the DH. Thus, we examined the effects of bilateral excitotoxic lesions of the amygdala or the DH on conditioned naloxone-precipitated morphine withdrawal-induced actin rearrangements in the DH or the amygdala. As presented in Figure 6A, lesions of the amygdala eliminated actin rearrangements in the DH at 1 h after naloxone injection (p > 0.05 vs control), and this effect was not attributable to mechanical damage to amygdala in surgery because actin rearrangement in the DH was still elicited in sham-operated rats (p < 0.05 vs control). Conversely, lesions of the DH did not affect actin rearrangements in the amygdala at 0.5 h after naloxone injection. A significant enhancement of actin rearrangements in the amygdala was observed in both the DH sham- and lesion-operated rats (p < 0.05 vs control) (Fig. 6B). These results suggest that neuronal input from the amygdala is critical in enabling actin rearrangement in the DH. Recent studies also show that the amygdala can modulate the expression of Arc mRNA or protein in the hippocampus in response to contextual fear conditioning and inhibitory avoidance learning (McIntyre et al., 2005; Huff et al., 2006).
Figure 6.

Amygdala regulates actin rearrangements in the DH associated with conditioned drug withdrawal from acute morphine dependence. A, Lesions of the amygdala prevented the enhancement of the ratio of F-actin to G-actin in the DH at 1 h after naloxone injection. B, Lesions of the DH had no effect on the enhancement of the ratio of F-actin to G-actin in the amygdala at 0.5 h after naloxone injection. Values are expressed as the mean ± SEM (n = 4). *p < 0.05, compared with the corresponding saline-treated sham-operated rats, one-way ANOVA with Bonferroni's post hoc test. Con, Control; Nal, naloxone; Amy, amygdala; CMW, conditioned morphine withdrawal.
Intra-amygdala injections of the β-adrenoceptor antagonist propranolol blocked CPA and abolished actin rearrangements in the dorsal hippocampus induced by conditioned naloxone-precipitated morphine withdrawal
Extensive evidence has demonstrated that the amygdala modulates memory-related processes in the hippocampus by the noradrenergic system (Ferry et al., 1999; Roozendaal et al., 1999). Activation of β-adrenoceptor in the amygdala (e.g., BLA) modulates memory storage and plasticity in the hippocampus (Ikegaya et al., 1997; Frey et al., 2001) and enhances inhibitory avoidance and contextual fear conditioning retention (Ferry et al., 1999; LaLumiere et al., 2003). To determine whether noradrenergic systems in the amygdala are involved in CPA formation through changes in actin rearrangements in the DH, we examined the effects of microinjection of β-adrenoceptor antagonist propranolol into the amygdala on the morphine withdrawal-induced CPA, as well as actin rearrangements in DH. Bilateral injection of propranolol (15 nmol/0.5 μl per side) into the amygdala at 10 min before pairing with morphine withdrawal significantly impaired CPA (CPA score: vehicle, −297.5 ± 38.1 s; propranolol, −121.6 ± 34.3 s; t(1,13) = 3.389; p < 0.01), consistent with previous reports that intra-amygdala infusion of propranolol diminished naloxone-precipitated CPA in chronic morphine-dependant rats (Harris and Aston-Jones, 1993; Watanabe et al., 2003). Correspondingly, identical injection of propranolol into the amygdala also decreased actin rearrangements in the DH induced by conditioned morphine withdrawal (p > 0.05 vs control group) (Fig. 7B), whereas a significant actin rearrangement was observed in the vehicle-injected rats (p < 0.05 vs control group) (Fig. 7B). These results demonstrate that injections of propranolol into the amygdala induced parallel effects on CPA formation and actin rearrangements, supporting that actin rearrangements in the DH is involved in CPA formation, consistent with the findings that attenuation of actin rearrangements in the DH with latrunculin A inhibited CPA formation (Fig. 5D).
Figure 7.

Effects of intra-amygdala injection of β-adrenoceptor antagonists propranolol on actin rearrangements in the DH and CPA behaviors induced by conditioned naloxone-precipitated morphine withdrawal. A, Schematic representation of injection sites in the amygdala of rats used to determine the effect of intra-amygdala injection of propranolol on morphine withdrawal-induced CPA behaviors (○, control; •, propranolol). B, Intra-amygdala injection of propranolol prevents actin rearrangements in the DH induced by conditioned morphine withdrawal in acute morphine-dependent rats. Propranolol (15 nmol/0.5 μl per side) or vehicle (0.5 μl/side) was bilaterally microinjected into the amygdala 10 min before naloxone-precipitated morphine withdrawal. Values are expressed as the mean ± SEM. **p < 0.01, compared with the corresponding vehicle-microinjected control group, one-way ANOVA with Bonferroni's post hoc test. Nal, Naloxone; CMW, conditioned morphine withdrawal.
Pretraining intra-amygdala injections of the β-adrenoceptor agonist clenbuterol induced dorsal hippocampal actin rearrangements and CPA behavior without morphine treatment and naloxone precipitation
A previous study showed that naloxone-precipitated morphine withdrawal resulted in the release of norepinephrine in the amygdala, which contributes to naloxone-precipitated morphine withdrawal-induced CPA (Watanabe et al., 2003). To further confirm the role of the noradrenergic system within the amygdala in dorsal hippocampal actin rearrangement and CPA induction after conditioned morphine withdrawal, we examined the effect of pretraining intra-amygdala injection of β-adrenoceptor agonist clenbuterol on actin rearrangement in the DH and induction of CPA under the condition of without morphine treatment and naloxone precipitation. Clenbuterol was microinjected into the DH 10 min before training. The effects of bilateral intra-amygdala injection of clenbuterol on actin rearrangement in the DH were shown in Figure 8A. Two-way ANOVA revealed the significant main effect of training (F(1,15) = 6.337; p < 0.05) and clenbuterol (F(1,15) = 11.63; p < 0.01) on the ratio of F-actin to G-actin in the DH but did not reveal a significant interaction between these two factors (F(1,15) = 3.475; p > 0.05). The following post hoc test showed that pretraining intra-amygdala injection of clenbuterol significantly increased the ratio of F-actin to G-actin in the DH (p < 0.01), whereas intra-amygdala injection of clenbuterol without conditioned training failed to increase the ratio of F-actin to G-actin. This is consistent with the findings that conditioned morphine withdrawal but not unconditioned morphine withdrawal elicited actin rearrangement in the dorsal hippocampus. Accordingly, pretraining intra-amygdala injection of clenbuterol also produced a significant CPA under the condition of without morphine treatment and naloxone precipitation (CPA score: vehicle, −70.0 ± 44.1 s; clenbuterol, −209.9 ± 42.8 s; t(1.12) = 2.277; p < 0.05). These findings further support that the β-noradrenergic system within the amygdala can modulate dorsal hippocampal actin rearrangement and CPA induction.
Figure 8.

Effects of pretraining intra-amygdala injection of β-adrenoceptor agonist clenbuterol on actin rearrangement in the DH and CPA behavior without morphine treatment and naloxone precipitation. A, Intra-amygdala injection of β-adrenoceptor agonist clenbuterol in combination with conditioned training induced actin rearrangement in the DH. Clenbuterol (10 ng/0.5 μl per side) was bilaterally microinjected into the amygdala 10 min before paring with or without conditioned training. The tissue of dorsal hippocampus was collected for actin analysis 70 min after the intra-amygdala injection. The open bar represents vehicle injection, and the filled bar represents clenbuterol injection. B, Schematic representation of injection sites in the amygdala of rats used to determine whether intra-amygdala injection of clenbuterol in combination with conditioned training induced CPA behaviors (○, control; •, clenbuterol). Clenbuterol (10 ng/0.5 μl per side) or vehicle (0.5 μl/side) was bilaterally microinjected into the amygdala 10 min before paring. Values are expressed as the mean ± SEM. **p < 0.01, compared with the corresponding vehicle-microinjected group, two-way ANOVA with Bonferroni's post hoc test.
Discussion
Amygdala and dorsal hippocampus, but not nucleus accumbens, are critically involved in mediating the negative affective component of morphine withdrawal in acute morphine-dependent rats
Studies using brain lesions, intracerebral injections of opiate antagonists, or immediate-early genes as markers of neuronal reactivity have shown that the negative motivational component of opioid withdrawal appears to be centrally mediated by limbic structures, such as the amygdala, NAc (Stinus et al., 1990; Heinrichs et al., 1995), BNST (Delfs et al., 2000), and CA1 of the hippocampus (Frenois et al., 2002, 2005) in chronic opioid-dependent animals. In this study, we found that lesions of the amygdala impaired the induction of CPA, indicating that the amygdala also played a key role in mediating the negative motivational component of morphine withdrawal in acute opioid-dependent rats. In addition, we showed that lesions of the DH also significantly attenuated the CPA in acute morphine-dependent rats, thus providing a first direct evidence to support that the hippocampus is crucially involved in CPA induction. Although the hippocampus has been suggested to participate in CPA induction by studies using immediate-early genes as markers of neuronal reactivity, the direct evidence of involvement of the hippocampus in CPA induction in opioid-dependent animals is lacking. Distinct from previous observations on chronic morphine-dependent rats in which NAc is implicated in CPA induction (Stinus et al., 1990), this study suggests that the NAc may not play an important role in mediating the negative motivational component of morphine withdrawal in acute opioid-dependent rats, because lesions of the NAc has no significant effect on CPA induction in response to acute morphine treatment. This is consistent with previous studies using chronic dependence models in which the involvement of the NAc in conditioned place aversion was not seen (Kelsey and Arnold 1994; Caillé et al., 2003).
Actin rearrangements in the amygdala and the dorsal hippocampus underlies the aversive memories of conditioned morphine withdrawal in acute morphine-dependent rats
Synaptic plasticity is thought to contribute to a wide variety of brain functions, including learning and memory. The dendritic spine, a specialized structure in the neurons on which the majority of excitatory synapses is formed, appears to be a critical site for synaptic plasticity (Matus et al., 1982; Matus, 2000). Actin is the major structural component of the dendritic spine (Matus et al., 1982; Fifková and Morales, 1992). Actin rearrangements have been shown to modulate synaptic plasticity (Fukazawa et al., 2003; Zhou et al., 2004; Zito et al., 2004) and contribute to behavioral adaptations (Fischer et al., 2004; Offenhäuser et al., 2006; Rothenfluh et al., 2006; Toda et al., 2006). In support of these findings, the present study demonstrated that conditioned withdrawal from acute morphine dependence resulted in changes in actin rearrangements in the amygdala and the DH characterized by a net increase in F-actin content in the synapse. We also found that the actin rearrangements in the amygdala and the DH in response to conditioned drug withdrawal are involved in aversive memories of morphine withdrawal. This involvement was supported by the following observations. First, actin rearrangements specifically occurred in the regions involved in CPA formation (e.g., amygdala and DH) but not in the regions not involved in CPA formation (e.g., NAc). Second, inhibition of actin rearrangements in these two regions by intra-amygdala or intra-hippocampus injections of latrunculin A significantly inhibited CPA formation. Finally, manipulation of actin rearrangements in the DH by intra-amygdala injections of the β-adrenoceptor agonist clenbuterol or antagonist propranolol differentially modulated CPA induced by conditioned morphine withdrawal. Thus, these results strongly suggest that actin rearrangements in the amygdala and the DH are required for induction of the aversive memories of morphine withdrawal. Furthermore, our findings suggest that actin rearrangements in two regions may be implicated in different stages of CPA. Actin rearrangement in the amygdala may be involved in the acquisition of aversive withdrawal memory, because intra-amygdala injection of latrunculin A after pairing fails to attenuate CPA induction. However, actin rearrangement in the DH may be implicated in the consolidation of aversive withdrawal memories, because intra-hippocampus injections of latrunculin A after pairing significantly attenuates CPA induction. This is consistent with previous studies showing that disruption of actin rearrangements in the hippocampus impaired consolidation of memory and eliminated long-term but not short-term memory (Dash et al., 2004; Kramár et al., 2006). Although our findings indicate that actin rearrangements in the amygdala and the DH are involved in synaptic plasticity associated with aversive morphine withdrawal memories, how the transient actin rearrangements induced by conditioned withdrawal from acute morphine dependence mediate synaptic plasticity remains to be established.
Amygdala modulates actin rearrangements in the dorsal hippocampus induced by conditioned drug withdrawal from acute morphine dependence through activation of β-adrenoceptors
The amygdala is generally thought to be a key neural substrate underlying emotional responses (Gallagher and Chiba, 1996). It directly mediates aspects of emotional learning and facilitates memory storage in other brain regions (McGaugh et al., 2002). An important target that the amygdala modulates is the DH, a critical region for memory consolidation (Bernabeu et al., 1997; Ahi et al., 2004). There is considerable evidence that the amygdala is functionally and anatomically connected to the hippocampus (Pikkarainen et al., 1999; Pitkanen et al., 2000; Akirav and Richter-Levin, 2002). Recent studies demonstrate that the amygdala modulates hippocampus-dependent memories and gene products associated with the behavioral experience that produces the memory (McIntyre et al., 2005; Huff et al., 2006). The noradrenergic system appears to play a critical role in amygdala modulating hippocampal memories consolidation (Roozendaal et al., 1999; McIntyre et al., 2002, 2005; LaLumiere et al., 2003). Consistently, we found that amygdala modulates actin rearrangements in the DH in response to conditioned morphine withdrawal, because lesions of the amygdala abolished actin rearrangements in the DH. Furthermore, we found that intra-amygdala injections of the β-adrenoceptor antagonist propranolol attenuated and agonist clenbuterol induced actin rearrangements in the DH. An additional indirect evidence to suggest that the amygdala modulates actin rearrangement in the DH is that actin rearrangements occurred in the amygdala is earlier than those occurred in the DH. Altogether, our findings indicate that the amygdala modulates actin rearrangements in the DH through activation of β-adrenoceptor. Thus, our data add to the growing body of evidence that the amygdala is involved in the acquisition or consolidation of fear, drug, and incentive memories (Nader et al., 2000; Lee et al., 2005, 2006; Wang et al., 2005) and extend these findings by reporting a potential mechanism underlying aversive memories of opiate withdrawal.
Currently, the mechanisms by which the amygdala modulates actin rearrangements in the DH remain unclear. One way in which the amygdala might exert its regulatory effect on actin rearrangements in the DH is perhaps by modulating expression of Arc, as reported by a recent study showing that Arc controls LTP consolidation through regulation of local actin polymerization in the dentate gyrus in vivo (Messaoudi et al., 2007). In support of this speculation, it has been reported that the amygdala modulates inhibitory avoidance and contextual fear memories by regulating expression of Arc gene and/or protein in the hippocampus (McIntyre et al., 2005; Huff et al., 2006), and activation of the β-adrenoceptor in the amygdala is involved in this regulation (McIntyre et al., 2005). However, the results of the present study suggest that alteration of Arc protein expression in the DH may not be involved in the modulation of aversive memory of morphine withdrawal, because elevation of Arc protein expression in the DH was not detected by both immunohistochemistry and Western blot. Our findings suggest that the amygdala modulates aversive memory of morphine withdrawal by regulating actin rearrangements but not Arc protein expression in the DH, which is distinct from modulation of inhibitory avoidance and contextual fear memories by the amygdala. Our findings also suggest that the β-noradrenergic system within the amygdala can modulate hippocampal memory function by different mechanisms, depending on distinct conditioning. It should be noticed that, although a robust elevation of Arc protein expression was detected in the amygdala in response to conditioned morphine withdrawal, its roles in actin rearrangements and induction of CPA remain to be established. Additional work is required to address these issues.
Conclusions
Changes in dendritic spine morphology are thought to play a critical role in psychostimulant-induced behavioral plasticity and addiction (Robinson and Kolb, 1997, 1999). Actin rearrangements have been suggested to contribute to altering dendritic morphology and function. Although morphological studies have shown that addictive drugs alter dendritic spine morphology, the evidence showing alterations of actin rearrangements by addictive drugs are sparse in general (Toda et al., 2006). The present study is the first to demonstrate that the aversive morphine withdrawal memories involve actin rearrangements in the amygdala and the hippocampus. Given the growing body of evidence that the negative affective state of opiate withdrawal could enhance the incentive value of the drug and contribute to the maintenance of drug-seeking behavior, our work will then contribute to understand a potential molecular mechanism associated with the maintenance of drug-seeking behavior.
Footnotes
This study was supported by National Basic Research Program Grants G2003CB515400 and 2009CB522000 from the Ministry of Science and Technology of China, National Science Fund for Distinguished Young Scholar from the National Natural Science Foundation of China Grant 30425002, and Chinese Academy of Sciences Grant KSCXI/YW/R/68.
References
- Ahi J, Radulovic J, Spiess J. The role of hippocampal signaling cascades in consolidation of fear memory. Behav Brain Res. 2004;149:17–31. doi: 10.1016/s0166-4328(03)00207-9. [DOI] [PubMed] [Google Scholar]
- Akirav I, Richter-Levin G. Mechanisms of amygdala modulation of hippocampal plasticity. J Neurosci. 2002;22:9912–9921. doi: 10.1523/JNEUROSCI.22-22-09912.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azar MR, Jones BC, Schulteis G. Conditioned place aversion is a highly sensitive index of acute opioid dependence and withdrawal. Psychopharmacology (Berl) 2003;170:42–50. doi: 10.1007/s00213-003-1514-y. [DOI] [PubMed] [Google Scholar]
- Bernabeu R, Bevilaqua L, Ardenghi P, Bromberg E, Schmitz P, Bianchin M, Izquierdo I, Medina JH. Involvement of hippocampal cAMP/cAMP-dependent protein kinase signaling pathways in a late memory consolidation phase of aversively motivated learning in rats. Proc Natl Acad Sci U S A. 1997;94:7041–7046. doi: 10.1073/pnas.94.13.7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillé S, Rodriguez-Arias M, Minarro J, Espejo EF, Cador M, Stinus L. Changes in dopaminergic neurotransmission do not alter somatic or motivational opiate withdrawal-induced symptoms in rats. Behav Neurosci. 2003;117:995–1005. doi: 10.1037/0735-7044.117.5.995. [DOI] [PubMed] [Google Scholar]
- Dash PK, Orsi SA, Moody M, Moore AN. A role for hippocampal Rho-ROCK pathway in long-term spatial memory. Biochem Biophys Res Commun. 2004;322:893–898. doi: 10.1016/j.bbrc.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Delfs JM, Zhu Y, Druhan JP, Aston-Jones G. Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature. 2000;403:430–434. doi: 10.1038/35000212. [DOI] [PubMed] [Google Scholar]
- De Vries TJ, Shippenberg TS. Neural systems underlying opiate addiction. J Neurosci. 2002;22:3321–3325. doi: 10.1523/JNEUROSCI.22-09-03321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferry B, Roozendaal B, McGaugh JL. Basolateral amygdala noradrenergic influences on memory storage are mediated by an interaction between beta- and alpha1-adrenoceptors. J Neurosci. 1999;19:5119–5123. doi: 10.1523/JNEUROSCI.19-12-05119.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fifková E, Morales M. Actin matrix of dendritic spines, synaptic plasticity, and long-term potentiation. Int Rev Cytol. 1992;139:267–307. doi: 10.1016/s0074-7696(08)61414-x. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Schrick C, Spiess J, Radulovic J. Distinct roles of hippocampal de novo protein synthesis and actin rearrangement in extinction of contextual fear. J Neurosci. 2004;24:1962–1966. doi: 10.1523/JNEUROSCI.5112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenois F, Cador M, Caillé S, Stinus L, Le Moine C. Neural correlates of the motivational and somatic components of naloxone-precipitated morphine withdrawal. Eur J Neurosci. 2002;16:1377–1389. doi: 10.1046/j.1460-9568.2002.02187.x. [DOI] [PubMed] [Google Scholar]
- Frenois F, Stinus L, Di Blasi F, Cador M, Le Moine C. A specific limbic circuit underlies opiate withdrawal memories. J Neurosci. 2005;25:1366–1374. doi: 10.1523/JNEUROSCI.3090-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey S, Bergado-Rosado J, Seidenbecher T, Pape HC, Frey JU. Reinforcement of early long-term potentiation (early-LTP) in dentate gyrus by stimulation of the basolateral amygdala: heterosynaptic induction mechanisms of late-LTP. J Neurosci. 2001;21:3697–3703. doi: 10.1523/JNEUROSCI.21-10-03697.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron. 2003;38:447–460. doi: 10.1016/s0896-6273(03)00206-x. [DOI] [PubMed] [Google Scholar]
- Gallagher M, Chiba AA. The amygdala and emotion. Curr Opin Neurobiol. 1996;6:221–227. doi: 10.1016/s0959-4388(96)80076-6. [DOI] [PubMed] [Google Scholar]
- Harris GC, Aston-Jones G. Beta-adrenergic antagonists attenuate somatic and aversive signs of opiate withdrawal. Neuropsychopharmacology. 1993;9:303–311. doi: 10.1038/npp.1993.66. [DOI] [PubMed] [Google Scholar]
- Heinrichs SC, Menzaghi F, Schulteis G, Koob GF, Stinus L. Suppression of corticotropin-releasing factor in the amygdala attenuates aversive consequences of morphine withdrawal. Behav Pharmacol. 1995;6:74–80. [PubMed] [Google Scholar]
- Higgins GA, Sellers EM. Antagonist-precipitated opioid withdrawal in rats: evidence for dissociations between physical and motivational signs. Pharmacol Biochem Behav. 1994;48:1–8. doi: 10.1016/0091-3057(94)90489-8. [DOI] [PubMed] [Google Scholar]
- Huff NC, Frank M, Wright-Hardesty K, Sprunger D, Matus-Amat P, Higgins E, Rudy JW. Amygdala regulation of immediate-early gene expression in the hippocampus induced by contextual fear conditioning. J Neurosci. 2006;26:1616–1623. doi: 10.1523/JNEUROSCI.4964-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheson DM, Everitt BJ, Robbins TW, Dickinson A. The role of withdrawal in heroin addiction: enhances reward or promotes avoidance? Nat Neurosci. 2001;4:943–947. doi: 10.1038/nn0901-943. [DOI] [PubMed] [Google Scholar]
- Ikegaya Y, Nakanishi K, Saito H, Abe K. Amygdala beta-noradrenergic influence on hippocampal long-term potentiation in vivo. Neuroreport. 1997;8:3143–3146. doi: 10.1097/00001756-199709290-00027. [DOI] [PubMed] [Google Scholar]
- Kelsey JE, Arnold SR. Lesions of the dorsomedial amygdala, but not the nucleus accumbens, reduce the aversiveness of morphine withdrawal in rats. Behav Neurosci. 1994;108:1119–1127. doi: 10.1037//0735-7044.108.6.1119. [DOI] [PubMed] [Google Scholar]
- Koob GF. Neurobiology of addiction. Toward the development of new therapies. Ann N Y Acad Sci. 2000;909:170–185. doi: 10.1111/j.1749-6632.2000.tb06682.x. [DOI] [PubMed] [Google Scholar]
- Kramár EA, Lin B, Rex CS, Gall CM, Lynch G. Integrin-driven actin polymerization consolidates long-term potentiation. Proc Natl Acad Sci U S A. 2006;103:5579–5584. doi: 10.1073/pnas.0601354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLumiere RT, Buen TV, McGaugh JL. Post-training intra-basolateral amygdala infusions of norepinephrine enhance consolidation of memory for contextual fear conditioning. J Neurosci. 2003;23:6754–6758. doi: 10.1523/JNEUROSCI.23-17-06754.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JL, Di Ciano P, Thomas KL, Everitt BJ. Disrupting reconsolidation of drug memories reduces cocaine-seeking behavior. Neuron. 2005;47:795–801. doi: 10.1016/j.neuron.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Lee JL, Milton AL, Everitt BJ. Cue-induced cocaine seeking and relapse are reduced by disruption of drug memory reconsolidation. J Neurosci. 2006;26:5881–5887. doi: 10.1523/JNEUROSCI.0323-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Hou YY, Lu B, Chen J, Chi ZQ, Liu JG. Expression pattern of neural synaptic plasticity marker-Arc in different brain regions induced by conditioned drug withdrawal from acute morphine-dependent rats. Acta Pharmacol Sin. 2009;30:282–290. doi: 10.1038/aps.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J. Actin's actions in LTP-induced synapse growth. Neuron. 2003;38:361–362. doi: 10.1016/s0896-6273(03)00257-5. [DOI] [PubMed] [Google Scholar]
- Lyford GL, Yamagata K, Kaufmann WE, Barnes CA, Sanders LK, Copeland NG, Gilbert DJ, Jenkins NA, Lanahan AA, Worley PF. Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron. 1995;14:433–445. doi: 10.1016/0896-6273(95)90299-6. [DOI] [PubMed] [Google Scholar]
- Matus A. Actin-based plasticity in dendritic spines. Science. 2000;290:754–758. doi: 10.1126/science.290.5492.754. [DOI] [PubMed] [Google Scholar]
- Matus A, Ackermann M, Pehling G, Byers HR, Fujiwara K. High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sci U S A. 1982;79:7590–7594. doi: 10.1073/pnas.79.23.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaugh JL, McIntyre CK, Power AE. Amygdala modulation of memory consolidation: interaction with other brain systems. Neurobiol Learn Mem. 2002;78:539–552. doi: 10.1006/nlme.2002.4082. [DOI] [PubMed] [Google Scholar]
- McIntyre CK, Hatfield T, McGaugh JL. Amygdala norepinephrine levels after training predict inhibitory avoidance retention performance in rats. Eur J Neurosci. 2002;16:1223–1226. doi: 10.1046/j.1460-9568.2002.02188.x. [DOI] [PubMed] [Google Scholar]
- McIntyre CK, Miyashita T, Setlow B, Marjon KD, Steward O, Guzowski JF, McGaugh JL. Memory-influencing intra-basolateral amygdala drug infusions modulate expression of Arc protein in the hippocampus. Proc Natl Acad Sci U S A. 2005;102:10718–10723. doi: 10.1073/pnas.0504436102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messaoudi E, Kanhema T, Soulé J, Tiron A, Dagyte G, da Silva B, Bramham CR. Sustained Arc/Arg3.1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J Neurosci. 2007;27:10445–10455. doi: 10.1523/JNEUROSCI.2883-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton WM, Ayscough KR, McLaughlin PJ. Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nat Cell Biol. 2000;2:376–378. doi: 10.1038/35014075. [DOI] [PubMed] [Google Scholar]
- Mucha RF, Walker MJ. Aversive property of opioid receptor blockade in drug-naive mice. Psychopharmacology (Berl) 1987;93:483–488. doi: 10.1007/BF00207239. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Yamamoto R, Fujio M, Suzuki Y, Minami M, Satoh M, Kaneko S. Involvement of the bed nucleus of the stria terminalis activated by the central nucleus of the amygdala in the negative affective component of morphine withdrawal in rats. Neuroscience. 2005;134:9–19. doi: 10.1016/j.neuroscience.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Offenhäuser N, Castelletti D, Mapelli L, Soppo BE, Regondi MC, Rossi P, D'Angelo E, Frassoni C, Amadeo A, Tocchetti A, Pozzi B, Disanza A, Guarnieri D, Betsholtz C, Scita G, Heberlein U, Di Fiore PP. Increased ethanol resistance and consumption in Eps8 knockout mice correlates with altered actin dynamics. Cell. 2006;127:213–226. doi: 10.1016/j.cell.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Pikkarainen M, Rönkkö S, Savander V, Insausti R, Pitkänen A. Projections from the lateral, basal, and accessory basal nuclei of the amygdala to the hippocampal formation in rat. J Comp Neurol. 1999;403:229–260. [PubMed] [Google Scholar]
- Pitkänen A, Pikkarainen M, Nurminen N, Ylinen A. Reciprocal connections between the amygdala and the hippocampal formation, perirhinal cortex, and postrhinal cortex in rat. A review. Ann N Y Acad Sci. 2000;911:369–391. doi: 10.1111/j.1749-6632.2000.tb06738.x. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Kolb B. Persistent structural modifications in nucleus accumbens and prefrontal cortex neurons produced by previous experience with amphetamine. J Neurosci. 1997;17:8491–8497. doi: 10.1523/JNEUROSCI.17-21-08491.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Kolb B. Morphine alters the structure of neurons in the nucleus accumbens and neocortex of rats. Synapse. 1999;33:160–162. doi: 10.1002/(SICI)1098-2396(199908)33:2<160::AID-SYN6>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Rodríguez de Fonseca F, Navarro M. Role of the limbic system in dependence on drugs. Ann Med. 1998;30:397–405. doi: 10.3109/07853899809029940. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Nguyen BT, Power AE, McGaugh JL. Basolateral amygdala noradrenergic influence enables enhancement of memory consolidation induced by hippocampal glucocorticoid receptor activation. Proc Natl Acad Sci U S A. 1999;96:11642–11647. doi: 10.1073/pnas.96.20.11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenfluh A, Threlkeld RJ, Bainton RJ, Tsai LT, Lasek AW, Heberlein U. Distinct behavioral responses to ethanol are regulated by alternate RhoGAP18B isoforms. Cell. 2006;127:199–211. doi: 10.1016/j.cell.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Schaefer GJ, Michael RP. Changes in response rates and reinforcement thresholds for intracranial self-stimulation during morphine withdrawal. Pharmacol Biochem Behav. 1986;25:1263–1269. doi: 10.1016/0091-3057(86)90121-8. [DOI] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Steward O, Wallace CS, Lyford GL, Worley PF. Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron. 1998;21:741–751. doi: 10.1016/s0896-6273(00)80591-7. [DOI] [PubMed] [Google Scholar]
- Stinus L, Le Moal M, Koob GF. Nucleus accumbens and amygdala are possible substrates for the aversive stimulus effects of opiate withdrawal. Neuroscience. 1990;37:767–773. doi: 10.1016/0306-4522(90)90106-e. [DOI] [PubMed] [Google Scholar]
- Toda S, Shen HW, Peters J, Cagle S, Kalivas PW. Cocaine increases actin cycling: effects in the reinstatement model of drug seeking. J Neurosci. 2006;26:1579–1587. doi: 10.1523/JNEUROSCI.4132-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SH, Ostlund SB, Nader K, Balleine BW. Consolidation and reconsolidation of incentive learning in the amygdala. J Neurosci. 2005;25:830–835. doi: 10.1523/JNEUROSCI.4716-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Nakagawa T, Yamamoto R, Maeda A, Minami M, Satoh M. Involvement of noradrenergic system within the central nucleus of the amygdala in naloxone-precipitated morphine withdrawal-induced conditioned place aversion in rats. Psychopharmacology (Berl) 2003;170:80–88. doi: 10.1007/s00213-003-1504-0. [DOI] [PubMed] [Google Scholar]
- White DA, Hwang ML, Holtzman SG. Naltrexone-induced conditioned place aversion following a single dose of morphine in the rat. Pharmacol Biochem Behav. 2005;81:451–458. doi: 10.1016/j.pbb.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Yarmola EG, Somasundaram T, Boring TA, Spector I, Bubb MR. Actin-latrunculin A structure and function. Differential modulation of actin-binding protein function by latrunculin A. J Biol Chem. 2000;275:28120–28127. doi: 10.1074/jbc.M004253200. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Zito K, Knott G, Shepherd GM, Shenolikar S, Svoboda K. Induction of spine growth and synapse formation by regulation of the spine actin cytoskeleton. Neuron. 2004;44:321–334. doi: 10.1016/j.neuron.2004.09.022. [DOI] [PubMed] [Google Scholar]