

Abstract
The network oscillation and synaptic plasticity are known to be regulated by GABAergic inhibition, but how they are affected by changes in the GABA transporter activity remains unclear. Here we show that in the CA1 region of mouse hippocampus, pharmacological blockade or genetic deletion of GABA transporter-1 (GAT1) specifically impaired long-term potentiation (LTP) induced by theta burst stimulation, but had no effect on LTP induced by high-frequency stimulation or long-term depression induced by low-frequency stimulation. The extent of LTP impairment depended on the precise burst frequency, with significant impairment at 3–7 Hz that correlated with the time course of elevated GABAergic inhibition caused by GAT1 disruption. Furthermore, in vivo electrophysiological recordings showed that GAT1 gene deletion reduced the frequency of hippocampal theta oscillation. Moreover, behavioral studies showed that GAT1 knock-out mice also exhibited impaired hippocampus-dependent learning and memory. Together, these results have highlighted the important link between GABAergic inhibition and hippocampal theta oscillation, both of which are critical for synaptic plasticity and learning behaviors.
Introduction
The functional output of principal neurons depends critically on synaptic inhibition by interneurons that release GABA. Drugs that perturb GABAergic synaptic transmission affect cognitive functions of human subjects (Barbee, 1993; Kälviäinen, 1999) and experimental animals (Sankar and Holmes, 2004). Some neurological diseases and mental disorders are also associated with changes in the GABAergic system (Wong et al., 2003; Lewis et al., 2005). At the physiological level, activity of GABAergic interneurons is known to regulate hippocampal rhythmic activities (Klausberger et al., 2003; Klausberger and Somogyi, 2008), which may be important for memory formation (Axmacher et al., 2006). Blockade of GABAA receptors (GABAARs) during picrotoxin-induced epilepsy (Mackenzie et al., 2002) or potentiation of GABAAR function during pentobarbital anesthesia (Leung, 1985; Brazhnik and Vinogradova, 1986) markedly alters the pattern of rhythmic activities. Furthermore, GABAergic inhibition exerts a powerful influence on synaptic plasticity by regulating the degree of local depolarization (Wigstrom and Gustafsson, 1983), and changes in GABAergic inhibition during development (Meredith et al., 2003) or under pathological states result in altered synaptic plasticity (Kleschevnikov et al., 2004; Liu et al., 2005).
Synaptically released GABA is removed by specific, high-affinity, Na+- and Cl−-dependent GABA transporters (GATs), among which GAT1 is predominantly expressed in GABAergic neurons (Guastella et al., 1990; Borden, 1996). Therefore, GAT1 plays a crucial role in controlling GABA spillover and modulating both phasic and tonic GABAergic inhibition (Dalby, 2000; Nusser and Mody, 2002; Semyanov et al., 2003; Keros and Hablitz, 2005). Blocking GABA uptake with the GAT1 inhibitor tiagabine impaired spatial learning of rats in Morris water maze (Schmitt and Hiemke, 2002), whereas elevating GABA uptake by overexpressing GAT1 also resulted in cognitive impairment in mice (Hu et al., 2004). Thus, how the changes in GAT1 activity affect hippocampal plasticity and network activity remains to be clarified.
In this study, we examined the effect of disrupting GABA uptake, using the GAT1 gene knock-out (KO) mice or specific GAT1 inhibitor, on activity-dependent synaptic plasticity, hippocampal oscillation, and hippocampus-dependent learning and memory. We provide evidence that GAT1 disruption selectively impairs a specific form of hippocampal long-term potentiation (LTP) induced by theta burst stimulation (TBS), i.e., multiple bursts of high-frequency (100 Hz) stimuli delivered at the theta frequency (3–7 Hz). In addition, we found that GAT1 gene deletion specifically altered hippocampal theta oscillation by reducing its frequency. Deletion of GAT1 also impaired hippocampus-dependent learning and memory. Thus, GABA uptake may serve an important function in maintaining the normal hippocampal theta activity and in so doing sets the optimal condition for LTP induction by TBS at 5 Hz.
Materials and Methods
Animals
The mGAT1 KO strain was used in this study. The details of the targeting construct, homologous recombination, and genotyping were described previously (Cai et al., 2006). Briefly, a 1.57 kb DNA fragment that contains the exon 2 and exon 3 of the mouse GAT1 gene was replaced by a 1.37 kb neomycin-resistant gene cassette (neo) to eliminate the GAT1 gene activity. Mouse embryonic stem (ES) cell (CJ7) was electroporated with the NotI-linearized targeting vector DNA. Chimeric mice were generated by injecting the recombinant ES cells into C57BL/6J blastocysts and implanted into ICR females. GAT1 KO mice were backcrossed for nine generations to C57BL/6J mice. The heterozygotes were intercrossed to generate homozygous, heterozygous, and wild-type (WT) littermate mice. They were weaned at the fourth postnatal week and their genotypes were analyzed by preparing tail DNAs and PCR assay (Cai et al., 2006). Mice were kept at a 12 h light/dark cycle, and the behavioral experiments were always done during the light phase of the cycle. Mice had access to food and water ad libitum except during tests. The care and use of animals in these experiments followed the guidelines of, and the protocols were approved by, the Institutional Animals Care and Use Committee of the Institute of Neuroscience, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. In all experiments, the investigators were blind to the genotype of mice. The experiments were performed on the mice in a randomized order.
In vitro electrophysiology
Transverse hippocampal slices (350 μm thick) were prepared from 6- to 10-week-old male WT or GAT1 KO littermate mice. After decapitation, the brain was removed and placed in oxygenated (95% O2/5% CO2) artificial CSF (ACSF) at 4°C. Slices were cut with a Leica VT1000S vibratome (Leica Instruments) and maintained at room temperature (23–25°C) in a holding chamber filled with oxygenated ACSF for at least 2 h, whereas slices for whole-cell recordings were initially incubated in warmed (32°C) ACSF for 30 min and then maintained at room temperature. Then a single slice was transferred to the recording chamber, where it was held between two nylon nets and continuously perfused with oxygenated ACSF (23–25°C) at a flow rate of 2–3 ml/min. The same ACSF was used in cutting, incubation and recording, and contained the following (in mm): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1 NaH2PO4, 1.3 MgSO4, 26.2 NaHCO3, and 11 d-glucose, saturated with 95% O2/5% CO2, pH 7.4. The osmolarity of the ACSF was 310–320 mOsm/L.
All electrophysiological recordings were performed at room temperature (23–25°C) with an Axopatch-200B amplifier (Molecular Devices) at the sampling rate of 10 kHz and filtered at 5 kHz. Data were acquired and analyzed using a Digidata 1322A interface and Clampfit 9.0 software (Molecular Devices). For extracellular recordings in the CA1 region of the hippocampus, a bipolar platinum-iridium stimulating electrode was placed in the Schaffer collateral axons to elicit field population responses. The field EPSPs (fEPSPs) were recorded via a glass micropipette filled with ACSF (1–3 MΩ) placed in the stratum radiatum. GABAAR antagonists were not present during the LTP/long-term depression (LTD) experiments. Stimuli (0.1 ms duration) were delivered every 30 s. Test pulses were recorded for 10–20 min before data collection to ensure stability of the response. To induce LTP, the stimulation intensity under control conditions was adjusted to evoke ∼30–50% of the maximum response. TBS and high-frequency stimulation (HFS) (100 Hz for 1 s) were used to induce LTP. Unless otherwise stated, TBS consisted of 5 bursts (4 pulses, 100 Hz) delivered at an interburst interval of 200 ms, and repeated once at 20 s. In some experiments, TBS was modified to multiple burst stimulations (MBS) with distinctive interburst intervals (100–1000 ms). To induce LTD, low-frequency stimulation (LFS) (1 Hz for 900 s) was used and the stimulation intensity was adjusted to evoke ∼40–60% of the maximum response. The slope of fEPSPs was determined by Clampfit 9.0 software.
Whole-cell recordings were also made from the CA1 region of hippocampal slices. The neurons were visually identified using an upright microscope (BX51WI, Olympus) equipped with differential interference contrast optics and an infrared camera. Patch pipettes were made from borosilicate glass (1.5 mm OD) with a micropipette puller (PC-830, Narishige). The internal pipette solution for voltage-clamp recording contained the following (in mm): 140 KCl, 5 NaCl, 2 MgATP, 0.3 NaGTP, 0.1 EGTA, 10 HEPES. The pH was adjusted to 7.2, and the osmolarity was 300–310 mOsm/L. To block action potentials, 2 mm QX-314 was added into the pipette solution. The resistance of the patch electrode filled with above internal solution was 3–5 MΩ. Under voltage-clamp conditions, all the cells were held at −70 mV. Series resistances were usually 10–20 MΩ. To record IPSCs, 10 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 20 μm d-2-amino-5-phosphonopentanoic acid (D-APV) were added to the ACSF to block glutamatergic responses. A bipolar platinum–iridium stimulating electrode was placed at the Schaffer collateral axons to evoke IPSCs. To mimic the burst stimulation in TBS, a burst containing four pulses at 100 Hz was applied to evoke a burst-IPSC, which had a markedly longer decay time than the IPSC induced by a single pulse (see Fig. 2B). The stability of recording was monitored by applying a 5 mV hyperpolarizing pulse shortly before the stimulus. Tonic GABAAR-mediated currents were examined by applying the selective GABAAR antagonist picrotoxin into the slice chamber in a final concentration of 100 μm. The tonic GABA current was measured as the outward shift in the holding current.
Figure 2.

GAT1 disruption robustly prolonged GABAergic inhibition during interburst intervals. To record IPSCs, 10 μm CNQX and 20 μm d-APV were added to the ACSF to block glutamatergic responses. A bipolar platinum-iridium stimulating electrode was placed at the Schaffer collateral axons to evoke IPSCs. A, Typical evoked IPSC recordings in WT, WT with NO711 (20 μm) treatment or GAT1 KO slices. To compare the decay kinetics, IPSCs were normalized to the same amplitude and superimposed (right panel). B, To mimic the burst stimulation in TBS, one burst containing four pulses at 100 Hz was applied to evoke a compound IPSC, which had a markedly longer decay time than the IPSC induced by a single pulse (A). Typical recordings were shown for the compound IPSCs recorded in slices from GAT1 KO mice, or slices from WT mice in the absence or presence of NO711 (20 μm). The GABAAR antagonist picrotoxin (PTX, 100 μm) diminished the prolonged compound IPSCs. Dashed line represents the 200 ms time point where the next burst stimulation is delivered during TBS. C, Statistical results showing the increased decay time constants of single and compound IPSCs after GAT1 disruption (WT, n = 7; NO711, n = 7; KO, n = 9). D, Statistical results showing that NO711 (20 μm) had little effect on IPSC amplitudes (n = 7). **p < 0.01; N.S., no significant difference, compared with WT group; ##p < 0.01, compared between single and compound IPSCs; Student's t test.
In vivo electrophysiology
In vivo electrophysiology was performed as described previously (Lin et al., 2006). In brief, the 96-channel recording array (in stereotrode format) was constructed and implanted onto the head of adult male WT and GAT1 KO mice. The electrodes were advanced slowly until reaching the CA1 area. The position of the electrode in the CA1 pyramidal layer was determined by the presence of fast oscillations (“ripples”) in association with synchronous discharge of neurons (Buzsáki et al., 2003). Data were obtained from the electrode with the maximum ripple amplitude. Once in the layer, maximum ripple amplitudes were used as an online reference for consistent electrode placement between animals. Electrical activity was recorded from each animal during rapid eye movement (REM) sleep and novelty exploration, two parameters closely associated with mouse's cognitive ability. The data were obtained and analyzed offline by Plexon system. Histological staining, with (1% cresyl violet) was used to confirm the electrode positions.
Behavioral tests
Adult WT and GAT1 KO male mice (3- to 5-month-old littermates) were used throughout all behavioral tests.
Morris water maze.
The Morris water maze consisted of a circular pool (100 cm diameter, 50 cm deep) filled with water at 24–26°C to a depth of 20 cm. The water surface was covered with floating black resin beads. Yellow curtains were drawn around the pool (50 cm from the pool periphery) and contained distinctive visual marks that served as distal cues. Before training, a 60 s free swim trial without the platform was run. For training, a submerged (1.5 cm below the surface of the water, invisible to the animal) platform was fixed in the center of a quadrant so that the animal had to learn the location of the platform which was the only getaway from the water. The training session consisted of 7 d (4 trials per day). A trial was terminated when the mouse had climbed onto the escape platform or when 60 s had elapsed. Each mouse was allowed to stay on the platform for 30 s. The probe test was performed on the eighth day. The platform was removed and the mouse behavior was recorded for 60 s. Swimming paths for training session and probe test was monitored using an automatic tracking system. This system was used to record the swimming trace and calculate the latency to the platform and the time spent in each quadrant.
Passive avoidance.
Mice were individually habituated to the lighted compartment before test. During training session, each mouse was placed into lighted compartment and the latency to enter the dark compartment was recorded. When the mouse entered the dark compartment with all four paws, a foot shock was delivered. During retention session 24 h later, each mouse was placed into lighted compartment again and the latency to enter the dark compartment was recorded.
Contextual fear conditioning.
The contextual fear conditioning test was performed by a Near Infrared (NIR) Video Fear Conditioning System (Med Associates). During training session, each mouse was placed into the shock chamber for 3 min and the freezing response was recorded as baseline freezing. After contextual learning, two 2 s foot shocks (0.75 mA) were delivered with a 25 s interstimulus interval. Afterward, mice remained in the chamber for 25 s before being returned to the home cage. During the retention test 24 h later, each mouse was placed back into the shock chamber and the contextual freezing response was recorded for 5 min.
All drugs and chemicals in these experiments were purchased from Sigma. In the experiments with hippocampal slices, drugs were applied to the bathing medium. All the data were shown as the mean ± SEM, with statistical significance assessed by Kolmogorov–Smirnov test or Student's t test. All statistical analysis was performed using Origin 7.0 (OriginLab).
Results
Disruption of GAT1 activity selectively impaired TBS-induced LTP
To study the function of GAT1-mediated GABA uptake in hippocampal synaptic plasticity, we used the GAT1 KO mice, which do not express GAT1 and exhibit markedly impaired GABA uptake activity in hippocampal synaptosomes (Cai et al., 2006). Using Nissl staining in coronal sections, we found no detectable differences in the gross morphology of the hippocampus between GAT1 KO and WT mice (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Furthermore, immunostaining of several markers of GABAergic and glutamatergic synapses (NMDA receptor subunits: NR1, NR2A, NR2B; AMPA receptor subunits: GluR1 and GluR2/3; GABAAR subunit β2/3; glutamic acid decarboxylases GAD65 and GAD67) did not reveal any significant difference in the expression levels of these proteins between GAT1 KO and WT mice (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Consistently, basal excitatory synaptic functions were similar between the two groups of mice, as shown by the same input–output relation of stimulus intensity versus the slope of field EPSPs (fEPSPs), and the same paired-pulse facilitation (PPF) of fEPSPs at three different interpulse intervals (50, 100, 150 ms) (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). On the other hand, consistent with impaired GABA uptake, the GAT1 KO mice showed a significantly larger tonic GABA-induced current (14.8 ± 2.5 pA, n = 6) than the WT mice (5.7 ± 1.1 pA, n = 6, p < 0.01) (supplemental Fig. S4A,C,D, available at www.jneurosci.org as supplemental material) [see also Jensen et al. (2003) and Chiu et al. (2005)]. Together, these results suggest that genetic disruption of GAT1 in mice impaired GABA uptake without significant effect on excitatory synaptic structure and function in the hippocampus.
Further experiments were performed to examine the effect of GAT1 gene deletion on hippocampal synaptic plasticity in mice. Field EPSPs were recorded from the CA1 area of hippocampal slices obtained from young adult mice in the absence of GABAAR antagonist, and changes in the slope of fEPSPs were used to monitor the induction of LTP and LTD by various patterns of Schaffer collateral stimulation, including theta burst stimulation [TBS, five bursts (four pulses at 100 Hz) delivered at 5 Hz and repeated once at 20 s], HFS (100 Hz for 1 s), and LFS (1 Hz for 900 s). We found that in hippocampal slices from WT mice, TBS induced a persistent elevation in the slope of fEPSPs for up to 60 min (149 ± 4% of the baseline level, n = 12, p < 0.01) (Fig. 1A), indicating the induction of LTP. However, the same TBS applied to the slices of GAT1 KO mice resulted in a persistent potentiation (126 ± 4% of the baseline level, n = 12, p < 0.01) (Fig. 1A), which was significantly lower than that found in the WT mice (p < 0.01, Kolmogorov–Smirnov test) (Fig. 1B). Interestingly, for LTP induced by HFS (Fig. 1C,D) and LTD induced by LFS (Fig. 1E,F), we found no significant difference in the extent of potentiation or depression between the WT and GAT1 KO mice, indicating that synaptic modification induced by TBS is particularly sensitive to disruption of GABA uptake.
Figure 1.

GAT1 disruption selectively impaired TBS-induced LTP. Field EPSPs were recorded in the CA1 area of hippocampal slices derived from WT and GAT1 KO mice, in the absence of the GABAAR antagonist. The fEPSP slope was normalized to the baseline value before LTP or LTD induction. The magnitude of LTP or LTD was measured as the averaged value from the last 10 recordings. A, Either GAT1 deletion or GAT1 inhibitor NO711 significantly impaired TBS-induced LTP in hippocampal slices (n = 12 for each group). Typical fEPSP recordings were shown before TBS (1) or 40 min after LTP induction (2). B, Cumulative probability of TBS-induced LTP magnitudes for each group. C, D, HFS-induced LTP in slices from GAT1 KO mice, or slices from WT mice in the absence or presence of NO711 (WT, n = 9; KO, n = 9; NO711, n = 7). E, F, LFS-induced LTD in slices from GAT1 KO mice, or slices from WT mice in the absence or presence of NO711 (WT, n = 9; KO, n = 9; NO711, n = 7). For all NO711 groups, 20 μm NO711 was applied throughout the entire experiments. **p < 0.01; ##p < 0.01; N.S., no significant difference; Kolmogorov–Smirnov tests. Calibration: 5 ms, 0.5 mV.
To determine whether the impairment of TBS-induced LTP is due to acute absence of GABA uptake through GAT1 or indirect chronic effect of GAT1 gene deletion, we examined the effect of acute blockade of GAT1 function with the specific GAT1 inhibitor NO711 (Suzdak et al., 1992) in hippocampal slices from WT mice. The presence of NO711 (20 μm) had no effect on the basal excitatory synaptic transmission, as shown by the input–output function and PPF of fEPSPs (supplemental Fig. S5, available at www.jneurosci.org as supplemental material), but significantly increased the tonic GABA current (from 5.7 ± 1.1 to 14.2 ± 2.0 pA, n = 6, p < 0.01) (supplemental Fig. S4A,B,D, available at www.jneurosci.org as supplemental material). As expected, we found that NO711 treatment selectively impaired TBS-induced LTP (Fig. 1A,B) to a similar extent (125 ± 2% of the baseline level, n = 12, p < 0.01) as that found in GAT1 KO mice (p > 0.1, Kolmogorov–Smirnov test) (Fig. 1B), but had no effect on either HFS-induced LTP (Fig. 1C,D) or LFS-induced LTD (Fig. 1E,F) in the WT mice. Thus, acute disruption of GAT1 activity was sufficient to mimic the effect of GAT1 gene knock-out. Furthermore, NO711 treatment had no effect on the TBS-induced LTP in GAT1 KO mice (supplemental Fig. S6, available at www.jneurosci.org as supplemental material), indicating that GAT1 KO had occluded the NO711 effect, consistent with common underlying mechanism for the impairment of TBS-induced LTP in these two conditions.
GAT1 disruption resulted in prolonged inhibition following burst stimulation
To understand how GABA uptake affects the induction of LTP by TBS, we examined the effect of GAT1 disruption on the properties of GABAergic inhibition. Whole-cell recording was made from CA1 pyramidal cells to monitor IPSCs evoked by Schaffer collateral stimulation, in the presence of glutamatergic antagonists (see Materials and Methods). Consistent with previous reports (Dingledine and Korn, 1985; Thompson and Gahwiler, 1992), inhibition of GAT1 activity by NO711 (20 μm) increased the decay time of IPSCs (WT, 49 ± 6 ms; NO711, 132 ± 25 ms; n = 7, p < 0.01) (Fig. 2A,C) without affecting the average IPSC amplitude (Fig. 2A,D). Similarly, for slices obtained from GAT1 KO mice, IPSCs decay time was longer (88 ± 5 ms, n = 9; p < 0.01) than that found for WT mice (Fig. 2A,C).
To examine GABAergic inhibition during TBS, we applied a burst stimulation containing four pulses at 100 Hz. The resultant compound IPSCs exhibited a markedly longer decay time (123 ± 22 ms, n = 7) than that found for IPSC induced by a single pulse. Furthermore, in GAT1 KO and NO711-treated slices, we found a striking (∼4–5-fold) prolongation of burst-induced compound IPSCs (Fig. 2B,C). The prolonged IPSCs were mediated by GABAARs, because they were largely abolished by GABAAR antagonist picrotoxin (100 μm, PTX) (Fig. 2B). Similar to that found for IPSCs evoked by a single stimulus, NO711 treatment did not affect the amplitude of burst-induced compound IPSCs (Fig. 2B,D). Together, these results indicate that disruption of GABA uptake greatly enhances GABAergic inhibition for a few hundreds of milliseconds following the burst stimulation by prolonging the time course of GABA action.
LTP impairment by GAT1 deletion depended on frequency of burst stimulation
For TBS at 5 Hz, the subsequent burst stimulation arrives at 200 ms following the preceding one (Fig. 2B, dashed line) when the difference in the amplitude of compound IPSCs is the largest between WT and NO711-treated or GAT1 KO slices. It is expected that the excitatory action of subsequent bursts will be inhibited by prolonged GABA action. For each burst stimulation-induced fEPSP, we analyzed the total area of burst-fEPSP, which represents its excitatory action (Fig. 3A). In slices from GAT1 KO mice or NO711-treated slices from WT mice, the decline of the burst-fEPSP area during TBS was much faster than that in WT-untreated slices (Fig. 3B,C). Thus, GAT1 disruption resulted in significant suppression of burst-induced fEPSPs, indicating a possible mechanism underlying the selective impairment of TBS-induced LTP.
Figure 3.

GAT1 disruption progressively suppressed burst-induced fEPSPs during TBS. A, Burst-induced fEPSPs were measured as the total fEPSP area marked in shadow. B, Typical burst-fEPSP recordings in slices from GAT1 KO mice, or slices from WT mice in the absence or presence of NO711 (20 μm). Note the progressive decline of the burst-fEPSPs during TBS after GAT1 disruption. C, Normalized burst-fEPSP areas for data from B (n = 12 for each group). Each burst-fEPSP was normalized to the first one. **p < 0.01 for the second, third, fourth, and fifth burst, compared between WT and GAT1 KO groups; ##p < 0.01 for the second, third, fourth, and fifth burst, compared between WT and WT plus NO711; Student's t test.
Given the time course of prolonged inhibition after burst stimulation, we expect that the excitatory action of subsequent bursts during TBS depends on the precise frequency of TBS. As shown in Figure 4A, the difference of compound IPSCs between WT and GAT1 KO, highlighted in the shaded area, reaches the maximum at ∼200 ms, but it was not evident at shorter (<100 ms) or longer (>1000 ms) time points. According to this temporal window, we redesigned the induction protocol of LTP by changing the interburst interval during TBS from 200 ms to 100, 143, 333, or 1000 ms (corresponding to 10, 7, 3, or 1 Hz). In WT hippocampal slices, this multiple burst stimulation (MBS) induced LTP with different magnitudes. We found that TBS (5 Hz) was the most effective, whereas MBS at 1 and 10 Hz was least effective, leading to a bell-shaped frequency–response relationship (Fig. 4B–F). Consistent with the prediction based on the observed differences in compound IPSCs, the difference between MBS-induced LTP in WT and GAT1 KO mice was only significant when the MBS was given at 3, 5 (TBS), and 7 Hz, but not at 1 and 10 Hz (Fig. 4B–F). Interestingly, the MBS-induced LTP in the WT mice showed a bell-shaped dependence on the frequency of stimulation, with the maximum LTP at 5 Hz (Fig. 4F). However, GAT1 deletion abolished this bell-shaped dependence (Fig. 4F). Further analysis of burst-fEPSP area (Fig. 3A) during MBS at different frequencies (Fig. 4B–E, inset) showed a similar frequency dependence in the differences between WT and GAT1 KO mice (Fig. 4G), with the maximal difference at 5 Hz. Importantly, this observed optimal range from 3 to 7 Hz agrees well with the physiological theta frequency. Thus, GABAergic inhibitory dynamics attained by GAT1 activity plays a specific role in regulating TBS-induced LTP.
Figure 4.
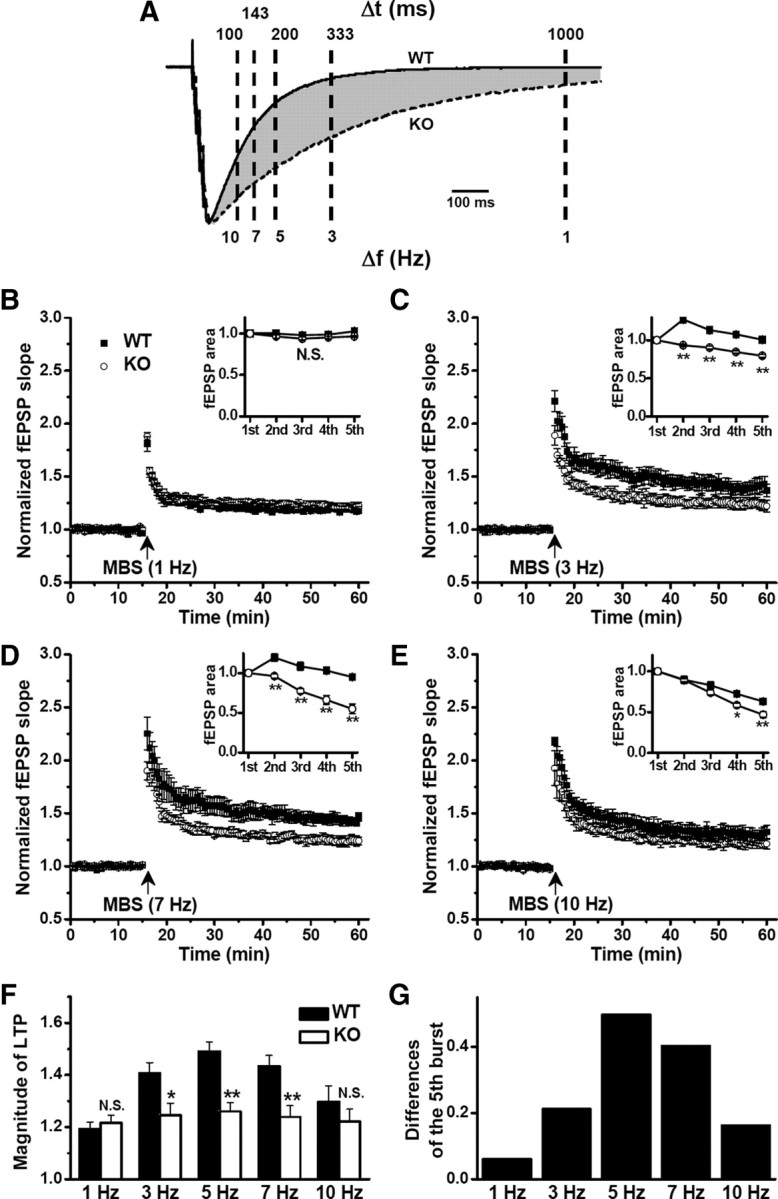
LTP impairment by GAT1 deletion depended on frequency of burst stimulation. A, Superimposed compound IPSCs recorded in slices from WT and GAT1 KO mice. Note the differences (shaded area) at the time points indicated. B–E, LTP induced by MBS given at different interburst intervals (1000 ms/1 Hz, 333 ms/3 Hz, 143 ms/7 Hz, 100 ms/10 Hz) in slices from WT and GAT1 KO mice. Note that LTP induced by 3–7 Hz burst stimulation was significantly impaired in GAT1 KO mice. Inset, comparison of the burst-fEPSP area between WT and GAT1 KO groups (n = 6 for each group). *p < 0.05; **p < 0.01; Student's t test. F, Summary data from B–E. Note the bell-shaped frequency–response relationship for WT mice with greatest LTP induced at 5 Hz (TBS). *p < 0.05; **p < 0.01; N.S., no significant difference, compared with WT group; Student's t test. G, The differences of the fifth burst–fEPSP areas between WT and GAT1 KO groups.
LTP impairment by GAT1 deletion was mediated by GABAARs
To further investigate the subtype of GABA receptors mediating the LTP impairment induced by GAT1 disruption, we first used PTX to block GABAARs in both WT and GAT1 KO mice. In the presence of 100 μm PTX, there was no difference in TBS-induced LTP between WT and GAT1 KO mice (n = 9 for each group, p > 0.1) (Fig. 5A,C). Furthermore, the facilitation of LTP by PTX was much stronger in GAT1 KO mice (from 126 ± 4 to 160 ± 8%, p < 0.01) (Fig. 5A,C) than that in the WT mice (from 149 ± 4 to 163 ± 6%, p < 0.05) (Fig. 5A,C). These results, together with the finding that PTX abolished the prolonged compound IPSCs (Fig. 2B), indicate a key role of GABAARs in regulating GABAergic inhibitory dynamics after GAT1 disruption.
Figure 5.

LTP impairment by GAT1 deletion was mediated by GABAARs. A, TBS-induced LTP in slices from WT and GAT1 KO mice in the absence (n = 12 for each group) or presence (n = 9 for each group) of 100 μm PTX. B, TBS-induced LTP in slices from WT and GAT1 KO mice in the absence (n = 12 for each group) or presence (n = 9 for each group) of 10 μm CGP 54626. C, Summary data from A and B showing that PTX rescued the impairment of LTP induction in GAT1 KO mice. *p < 0.05; **p < 0.01; N.S., no significant difference, compared with control group without PTX or CGP 54626; Student's t test. #p < 0.05; ##p < 0.01, Student's t test between WT and GAT1 KO groups. D, A proposed model illustrating the impaired GABA signaling and the functional consequence following burst stimulation when GAT1 was disrupted.
Previous studies have suggested the involvement of presynaptic GABAB receptors (GABABRs) in TBS-induced LTP (Davies et al., 1991; Mott and Lewis, 1991). However, the impairment of TBS-induced LTP in GAT1 KO mice persisted following the treatment of GABABR antagonist CGP 54626 (10 μm) (n = 9 for each group, p < 0.05) (Fig. 5B,C). We noted that CGP 54626 was more effective in reducing LTP in the WT mice (from 149 ± 4 to 136 ± 4%, p < 0.05) (Fig. 5B,C), but it had little effect on TBS-induced LTP in GAT1 KO mice (from 126 ± 4 to 124 ± 2%, p > 0.1) (Fig. 5B,C). Presynaptic blockade of inhibitory GABAB autoreceptors could facilitate the GABA release during interburst interval, resulting in the impairment of TBS-induced LTP in WT mice (Stäubli et al., 1999). Our result is consistent with the idea that GABABR blockade could not further increase the effective GABA concentration, which already reached saturation due to GAT1 disruption (Fig. 5D).
GAT1 deletion reduced the frequency of hippocampal theta oscillation in vivo
The commonly used TBS for LTP induction mimics the endogenous hippocampal theta rhythm (Otto et al., 1991), which plays an important role in learning and memory (Axmacher et al., 2006). We have recorded delta, theta, gamma, and fast ripple oscillatory field potentials from the CA1 pyramidal layer in vivo. During REM sleep and novelty exploration, spectral analysis of hippocampal local field activities revealed a significant change in theta band (4–10 Hz) activities (Fig. 6), which are involved in encoding and storing hippocampus-dependent information. We found that the peak power in theta band was not significantly altered in GAT1 KO mice during either REM sleep (WT, 7.16 ± 0.57% of total power spectral density; KO, 6.28 ± 0.48%; n = 6 recordings of 3 mice for each group, p > 0.05) (Fig. 6A) or exploration (WT, 5.75 ± 0.33%; KO, 5.75 ± 0.83%; n = 6 recordings of 3 mice for each group, p > 0.05) (Fig. 6B). However, the frequency at which theta power reached maximum was significantly shifted from 8.7 ± 0.2 Hz to 5.8 ± 0.2 Hz during REM sleep (n = 6 recordings of 3 mice for each group, p < 0.01) (Fig. 6A,C), and from 9.0 ± 0.2 Hz to 6.8 ± 0.4 Hz during exploration (n = 6 recordings of 3 mice for each group, p < 0.01) (Fig. 6B,C). Thus, GAT1 activity does not affect the physiological expression of theta oscillation activity, but modulates the precise frequency of this oscillation.
Figure 6.

GAT1 deletion reduced the frequency of hippocampal theta oscillation in vivo. Delta, theta, gamma, and fast ripple oscillatory field potentials were recorded in vivo from hippocampal CA1 pyramidal layer in free-moving mice (see Materials and Methods). A, B, Power spectra of hippocampal field potentials during REM sleep (A) and novelty exploration (B). Top, Typical recordings. Bottom, Averaged data from six recordings of three mice for each group. C, Summary data from A and B showing the decreased frequency of theta oscillation in GAT1 KO mice. **p < 0.01, compared with WT group; Student's t test.
We noted that GAT1 KO mice exhibited some motor coordination deficits, including tremor and gait abnormality, as reported by a previous study (Chiu et al., 2005). The reduction of theta oscillation frequency in the hippocampus during exploration could result from the abnormal motor activity of the animals. Further analysis of the locomotor behaviors of the mice in the open field test showed that GAT1 KO mice exhibited hyperlocomotion (distance traveled in 30 min: WT, 30.5 ± 3.8 m, n = 7; KO, 44.2 ± 3.8 m, n = 7; p < 0.01) (supplemental Fig. S7, available at www.jneurosci.org as supplemental material), consistent with a previous finding that GAT1 KO mice showed reduced anxiety and depression-like behaviors (Liu et al., 2007). Whether the reduction of theta oscillatory frequency in GAT1 KO mice during novelty exploration is causally related to the hyperlocomotor activity remains to be investigated.
GAT1 KO mice exhibited impaired hippocampus-dependent learning and memory
Synaptic plasticity is generally viewed as a cellular mechanism for learning and memory (Bliss and Collingridge, 1993; Whitlock et al., 2006), but the exact functions of different forms of synaptic plasticity remain poorly characterized. Our results on hippocampal LTP described above suggest that GAT1 KO mice may be a useful model for exploring the role of TBS-induced LTP in learning and memory. Using Morris water maze, we found that although GAT1 KO mice had motor coordination deficit (Chiu et al., 2005), their swimming speed were comparable to that of WT mice (WT, 0.16 ± 0.01 m/s, n = 12; GAT1 KO, 0.15 ± 0.01 m/s, n = 12; p > 0.1). However, after the mice were trained four trials per day for 7 d and then tested on the eighth day, GAT1 KO mice showed a significantly longer latency in finding the platform during the learning session (n = 12 for each group, p < 0.01 for each day) (Fig. 7A). During the probe test, GAT1 KO mice also showed a significantly lower preference for the target quadrant (WT, 48.2 ± 4.2% of total time, n = 12; KO, 36.2 ± 3.7%, n = 12; p < 0.05) (Fig. 7B,C) and fewer times of crossing the platform location (WT, 4.6 ± 0.9, n = 12; KO, 0.9 ± 0.2, n = 12; p < 0.01) (Fig. 7C). These results indicate that GAT1 KO mice exhibited an impaired spatial learning and memory in the Morris water maze test.
Figure 7.
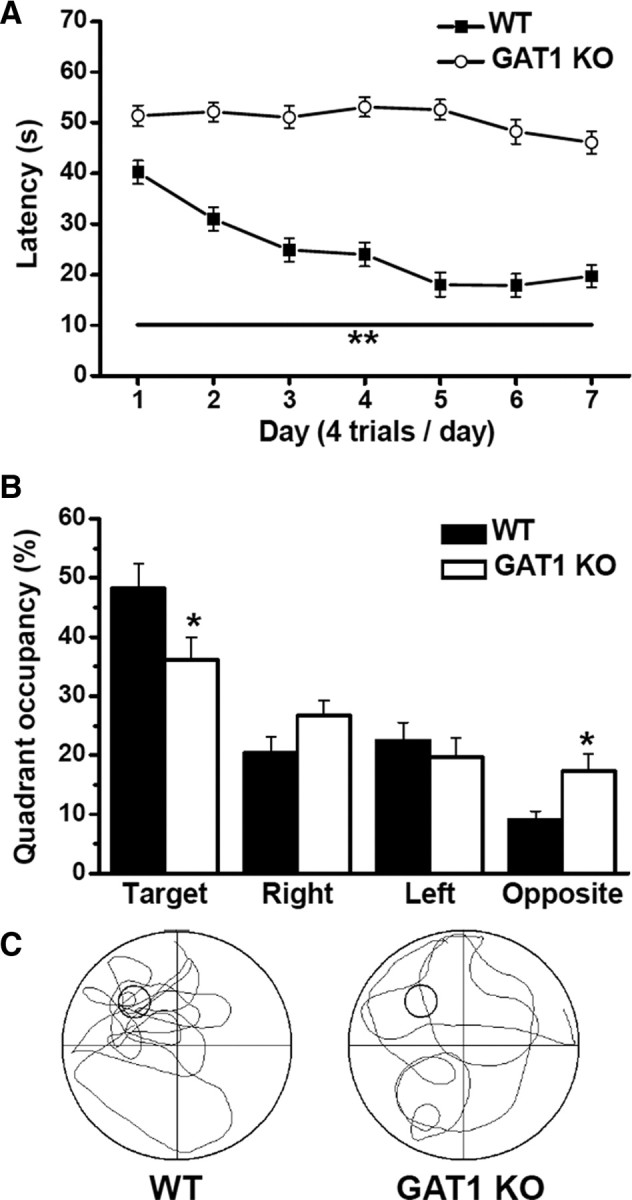
GAT1 KO mice exhibited impaired learning and memory in Morris water maze test. Mice were trained four trials per day for 7 successive days and then tested on the eighth day. A, During learning session, the latency to find the platform, plotted as function of training days, was significantly longer for GAT1 KO mice compared with WT mice. B, During probe test, GAT1 KO mice spent less time in the target quadrant but more time in the opposite quadrant than WT mice did. C, Typical swimming traces during probe test of WT and GAT1 KO mice. *p < 0.05; **p < 0.01, compared with WT group; Student's t test. In all experiments, n = 12 for each group.
Passive avoidance test was also performed to examine the hippocampus-dependent memory in GAT1 KO mice. During the training session, WT and GAT1 KO mice were placed into lighted compartment and showed a similar latency in entering the dark compartment (WT, 18.1 ± 2.5 s, n = 12; KO, 20.7 ± 3.1 s, n = 12; p > 0.1) (Fig. 8A), where they received a foot shock. In the test session 24 h after the training, mice were placed into the lighted compartment again. The GAT1 KO mice showed a much shorter latency in entering the dark compartment than the WT mice (WT, 209 ± 25 s, n = 12; KO, 44 ± 15 s, n = 12; p < 0.01) (Fig. 8A). Finally, we examined the hippocampus-dependent contextual fear conditioning. During the training session, each mouse was placed into the shock chamber for 3 min and the freezing response was recorded as baseline freezing. Consistent with the hyperlocomotion, GAT1 KO mice showed lower baseline freezing than WT mice (WT, 12.8 ± 2.0%, n = 10; KO, 6.4 ± 1.3%, n = 10; p < 0.05) (Fig. 8B). During the retention test 24 h later, each mouse was placed back into the same shock chamber and the contextual freezing response was recorded for 5 min. WT mice showed much stronger freezing responses above the baseline (32.4 ± 2.0%; n = 10; p < 0.01) (Fig. 8B), whereas the freezing responses of GAT1 KO mice were not significantly different from the baseline (6.7 ± 1.1%; n = 10; p > 0.1) (Fig. 8B), indicating significant deficit of spatial fear memory in GAT1 KO mice at 1 d after training. Together, these behavioral results indicate that GAT1 KO mice have a severe impairment of hippocampus-dependent learning and memory.
Figure 8.

GAT1 KO mice exhibited impaired fear memory in passive avoidance and contextual fear conditioning tests. A, Summary data showing the latencies for WT and GAT1 KO mice to enter the dark compartment during training and 1 d retention test in passive avoidance experiment. The GAT1 KO mice displayed a significantly shorter latency to enter the dark compartment than the WT mice did at 1 d after training. n = 12 for each group; **p < 0.01, compared with WT group; Student's t test. B, Summary data showing the percentage freezing time of WT and GAT1 KO mice during training and 1 d retention test in contextual fear conditioning. During training, GAT1 KO mice showed less baseline freezing. #p < 0.01, compared with WT group; Student's t test. During the retention test, WT but not GAT1 KO mice showed much stronger freezing responses above the baseline. **p < 0.01; N.S., no significant difference, compared with baseline freezing; Student's t test. In all experiments, n = 10 for each group.
Discussion
In this study, we demonstrated that GAT1 disruption specifically impaired TBS-induced LTP, without affecting HFS-induced LTP and LFS-induced LTD. In vivo electrophysiological recordings showed that GAT1 disruption specifically altered the temporal pattern of hippocampal theta oscillations by reducing the oscillation frequency. Furthermore, these specific changes in theta network activity and synaptic plasticity were accompanied by severe impairment of hippocampus-dependent learning and memory. Our data highlight a specific role of GABA uptake in modulating rhythmic theta activities in the hippocampus, and suggest a potential explanation of the optimal TBS frequency for inducing hippocampal LTP.
The GAT1 is primarily responsible for the removal of GABA from the synaptic cleft and the termination of GABAergic neurotransmission. Consistently, pharmacological blockade and genetic deletion of GAT1 significantly enhanced the tonic GABAAR-mediated current and prolonged the decay kinetics of GABAergic transmission. Previous studies showed that the effect of glutamate transporter blockade strongly depended on the input stimulation frequency, with larger effects found during stimulation of higher frequencies, indicating an important physiological function of glutamate transporters (Arnth-Jensen et al., 2002; Tzingounis and Wadiche, 2007). In the present study, we found that GAT1 disruption resulted in only modest prolongation of IPSCs induced by a single pulse, but much more robust prolongation of burst stimulation (four pulses at 100 Hz)-induced IPSCs. Thus, the effect of GAT1 on GABAergic transmission depends strongly on the pattern of synaptic stimulation.
Furthermore, we found that GAT1 disruption significantly impaired TBS-induced LTP, but had no effect on either HFS-induced LTP or LFS-induced LTD. Blockade of GAT1 had no significant effect on either basal fEPSPs or paired-pulse facilitation but significantly impaired the burst stimulation-induced fEPSPs during TBS and caused a faster decay of fEPSPs evoked by sequential bursts, presumably due to the robust prolongation of GABAergic inhibition that suppressed the subsequent burst-induced responses. Consistent with the temporal window determined by the GAT1 action on burst-induced IPSCs (Fig. 4A), we found that GAT1 disruption only affected LTP induced by stimulation with multiple bursts given at the theta frequency (3–7 Hz), indicating the specific role of GAT1 in modulating TBS-induced LTP. However, if GAT1 disruption exerts its effect on LTP by increasing GABAergic inhibition, why was HFS-induced LTP not affected? There are several possible explanations. First, the elevated GABA inhibition after the GAT1 disruption is too small to affect HFS-induced LTP induction. Second, HFS-induced LTP is less sensitive to GABAergic inhibition, due to, perhaps, intracellular accumulation of Cl− caused by repetitive high-frequency stimulation within a short time (Thompson and Gahwiler, 1989; Isomura et al., 2003) or activity-dependent downregulation of K+-Cl− cotransporter 2 (KCC2) (Wang et al., 2006), or due to desensitization of the inhibitory function of GABAARs. It appears that the interburst interval is critical for the GABAergic modulation of LTP.
Repetitive stimulation at high frequency (100 Hz) is a classical protocol to induce LTP, and a number of in vitro and in vivo studies have shown that LTP induction by burst stimulation is optimal when the time interval between the bursts is ∼200 ms (Larson and Lynch, 1986; Greenstein et al., 1988). Why burst stimulation at the theta frequency is particularly effective in LTP induction is largely unknown. A possible mechanism is that each burst may modulate several ion channels, e.g., inactivating A-type K+ current (Hoffman et al., 1997) or activating Ih conductance (Cobb et al., 1995), thus priming the effect of the subsequent burst. Brain-derived neurotrophic factor (BDNF) and TrkB signaling is known to selectively affect TBS-induced LTP (Kang et al., 1997), an effect that may be attributed to the specific effectiveness of TBS in stimulating BDNF secretion, although this remains to be demonstrated. Our results further showed that GABA uptake also selectively affects TBS-induced LTP. Interestingly, another component in GABAergic system, presynaptic GABAB autoreceptor, is also involved in TBS-induced LTP (Davies et al., 1991; Mott and Lewis, 1991). In TBS- but not HFS-induced LTP, a priming effect occurs among multiple bursts. During the interburst interval, GABA released from inhibitory interneurons feeds back onto presynaptic GABABRs to depress further GABA release. Thus, a common pathway for GAT1 and presynaptic GABABR to be involved in TBS-induced LTP is that they both cause the reduction of GABA level in the synaptic cleft during the interburst interval (Fig. 5D). Indeed, the effect of GABABR blockade on TBS-induced LTP appeared to be occluded by GAT1 disruption, consistent with common underlying mechanisms.
Can the blockade of GAT1 account for the modification of theta oscillation frequency? During learning, the typical firing mode of hippocampal pyramidal cells consists of several sequential high-frequency bursts (of 3–5 spikes per burst at 100–400 Hz) occurring at the theta frequency, which is mimicked by the TBS used for LTP induction (Otto et al., 1991; O'Keefe and Recce, 1993; Skaggs et al., 1996). Each high-frequency burst may cause intense activation of recurrent GABAergic inhibition and abundant release of GABA, and GAT1 disruption effectively prolongs the IPSP during each theta cycle, thus broadening the refractory period of high-frequency burst activities and decreasing the oscillation frequency of theta activities. We suggest that GABA uptake is an effective mechanism for modulating the rhythm of theta activity, which may originate from the recurrent circuit in the hippocampal CA3 region (Buzsáki, 2002). Theta oscillation is a complex phenomenon involving several brain structures and different mechanisms. Our finding does not rule out the contribution of GAT1 actions in brain areas outside hippocampus or of other mechanisms besides GABA uptake. For example, septal GABAergic neurons rhythmically hyperpolarize the hippocampal basket interneurons, and may be critically involved in the rhythm generation of theta oscillation (Buzsáki, 2002). Whether there exists endogenous regulation of GAT1 activity as a means for physiological modulation of hippocampal theta oscillation awaits further investigation.
Synaptic plasticity is known to be a cellular mechanism for learning and memory (Bliss and Collingridge, 1993; Whitlock et al., 2006), but the exact roles of different forms of synaptic plasticity remain poorly characterized. Our results on LTP induction described above suggest that GAT1 KO mice represent an excellent model for exploring the role of TBS-induced LTP in learning and memory. Indeed, we found that GAT1 KO mice showed severe impairment of learning and memory in parallel to that of TBS-induced LTP. Theta oscillations accompanying ongoing behaviors are critical for learning (Buzsáki, 2002). Given that theta oscillations were altered and similar processes to TBS-induced LTP might take place during learning, these could explain the cause of the impaired hippocampus-dependent learning and memory functions in GAT1 KO mice.
Footnotes
This study was supported by grants to T.-L. Xu from the National Natural Science Foundation of China (30621062 and 30830035), the National Basic Research Program of China (2006CB500803), the Knowledge Innovation Projects from the Chinese Academy of Sciences (KSCX2-YW-R-35 and KSCX2-YW-R-100). N.G. was a recipient of funds from the China Postdoctoral Science Foundation, the Shanghai Postdoctoral Scientific Program, and the Postdoctor Research Program of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. We thank Dr. M.-m. Poo for comments on the manuscript, Y.-Q. Cai, X. Xiao, D. Fei, and H. Cao for assistance in generating and maintaining GAT1 KO mice, M. He for assistance with Nissl staining, Y. Ding for assistance with Western blotting, and Y.-X. Yang for assistance with behavioral tests.
References
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Axmacher N, Mormann F, Fernández G, Elger CE, Fell J. Memory formation by neuronal synchronization. Brain Res Rev. 2006;52:170–182. doi: 10.1016/j.brainresrev.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Barbee JG. Memory, benzodiazepines, and anxiety: integration of theoretical and clinical perspectives. J Clin Psychiatry. 1993;54(Suppl):86–97. discussion 98–101. [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Borden LA. GABA transporter heterogeneity: pharmacology and cellular localization. Neurochem Int. 1996;29:335–356. doi: 10.1016/0197-0186(95)00158-1. [DOI] [PubMed] [Google Scholar]
- Brazhnik ES, Vinogradova OS. Control of the neuronal rhythmic bursts in the septal pacemaker of theta-rhythm: effects of anaesthetic and anticholinergic drugs. Brain Res. 1986;380:94–106. doi: 10.1016/0006-8993(86)91433-2. [DOI] [PubMed] [Google Scholar]
- Buzsáki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Buhl DL, Harris KD, Csicsvari J, Czéh B, Morozov A. Hippocampal network patterns of activity in the mouse. Neuroscience. 2003;116:201–211. doi: 10.1016/s0306-4522(02)00669-3. [DOI] [PubMed] [Google Scholar]
- Cai YQ, Cai GQ, Liu GX, Cai Q, Shi JH, Shi J, Ma SK, Sun X, Sheng ZJ, Mei ZT, Cui D, Guo L, Wang Z, Fei J. Mice with genetically altered GABA transporter subtype I (GAT1) expression show altered behavioral responses to ethanol. J Neurosci Res. 2006;84:255–267. doi: 10.1002/jnr.20884. [DOI] [PubMed] [Google Scholar]
- Chiu CS, Brickley S, Jensen K, Southwell A, Mckinney S, Cull-Candy S, Mody I, Lester HA. GABA transporter deficiency causes tremor, ataxia, nervousness, and increased GABA-induced tonic conductance in cerebellum. J Neurosci. 2005;25:3234–3245. doi: 10.1523/JNEUROSCI.3364-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378:75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- Dalby NO. GABA-level increasing and anticonvulsant effects of three different GABA uptake inhibitors. Neuropharmacology. 2000;39:2399–2407. doi: 10.1016/s0028-3908(00)00075-7. [DOI] [PubMed] [Google Scholar]
- Davies CH, Starkey SJ, Pozza MF, Collingridge GL. GABA autoreceptors regulate the induction of LTP. Nature. 1991;349:609–611. doi: 10.1038/349609a0. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Korn SJ. Gamma-aminobutyric acid uptake and the termination of inhibitory synaptic potentials in the rat hippocampal slice. J Physiol. 1985;366:387–409. doi: 10.1113/jphysiol.1985.sp015804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenstein YJ, Pavlides C, Winson J. Long-term potentiation in the dentate gyrus is preferentially induced at theta rhythm periodicity. Brain Res. 1988;438:331–334. doi: 10.1016/0006-8993(88)91358-3. [DOI] [PubMed] [Google Scholar]
- Guastella J, Nelson N, Nelson H, Czyzyk L, Keynan S, Miedel MC, Davidson N, Lester HA, Kanner BI. Cloning and expression of a rat brain GABA transporter. Science. 1990;249:1303–1306. doi: 10.1126/science.1975955. [DOI] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Hu JH, Ma YH, Jiang J, Yang N, Duan SH, Jiang ZH, Mei ZT, Fei J, Guo LH. Cognitive impairment in mice over-expressing gamma-aminobutyric acid transporter 1 (GAT1) Neuroreport. 2004;15:9–12. doi: 10.1097/00001756-200401190-00003. [DOI] [PubMed] [Google Scholar]
- Isomura Y, Sugimoto M, Fujiwara-Tsukamoto Y, Yamamoto-Muraki S, Yamada J, Fukuda A. Synaptically activated Cl− accumulation responsible for depolarizing GABAergic responses in mature hippocampal neurons. J Neurophysiol. 2003;90:2752–2756. doi: 10.1152/jn.00142.2003. [DOI] [PubMed] [Google Scholar]
- Jensen K, Chiu CS, Sokolova I, Lester HA, Mody I. GABA transporter-1 (GAT1)-deficient mice: differential tonic activation of GABAA versus GABAB receptors in the hippocampus. J Neurophysiol. 2003;90:2690–2701. doi: 10.1152/jn.00240.2003. [DOI] [PubMed] [Google Scholar]
- Kälviäinen R. Cognitive effects of GABAergic antiepileptic drugs. Electroencephalogr Clin Neurophysiol Suppl. 1999;50:458–464. [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM. Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron. 1997;19:653–664. doi: 10.1016/s0896-6273(00)80378-5. [DOI] [PubMed] [Google Scholar]
- Keros S, Hablitz JJ. Subtype-specific GABA transporter antagonists synergistically modulate phasic and tonic GABAA conductances in rat neocortex. J Neurophysiol. 2005;94:2073–2085. doi: 10.1152/jn.00520.2005. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321:53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Magill PJ, Márton LF, Roberts JD, Cobden PM, Buzsáki G, Somogyi P. Brain-state- and cell-type-specific firing of hippocampal interneurons in vivo. Nature. 2003;421:844–848. doi: 10.1038/nature01374. [DOI] [PubMed] [Google Scholar]
- Kleschevnikov AM, Belichenko PV, Villar AJ, Epstein CJ, Malenka RC, Mobley WC. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J Neurosci. 2004;24:8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Lynch G. Induction of synaptic potentiation in hippocampus by patterned stimulation involves two events. Science. 1986;232:985–988. doi: 10.1126/science.3704635. [DOI] [PubMed] [Google Scholar]
- Leung LW. Spectral analysis of hippocampal EEG in the freely moving rat: effects of centrally active drugs and relations to evoked potentials. Electroencephalogr Clin Neurophysiol. 1985;60:65–77. doi: 10.1016/0013-4694(85)90952-6. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lin L, Chen G, Xie K, Zaia KA, Zhang S, Tsien JZ. Large-scale neural ensemble recording in the brains of freely behaving mice. J Neurosci Methods. 2006;155:28–38. doi: 10.1016/j.jneumeth.2005.12.032. [DOI] [PubMed] [Google Scholar]
- Liu GX, Cai GQ, Cai YQ, Sheng ZJ, Jiang J, Mei Z, Wang ZG, Guo L, Fei J. Reduced anxiety and depression-like behaviors in mice lacking GABA transporter subtype 1. Neuropsychopharmacology. 2007;32:1531–1539. doi: 10.1038/sj.npp.1301281. [DOI] [PubMed] [Google Scholar]
- Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie L, Medvedev A, Hiscock JJ, Pope KJ, Willoughby JO. Picrotoxin-induced generalised convulsive seizure in rat: changes in regional distribution and frequency of the power of electroencephalogram rhythms. Clin Neurophysiol. 2002;113:586–596. doi: 10.1016/s1388-2457(02)00040-8. [DOI] [PubMed] [Google Scholar]
- Meredith RM, Floyer-Lea AM, Paulsen O. Maturation of long-term potentiation induction rules in rodent hippocampus: role of GABAergic inhibition. J Neurosci. 2003;23:11142–11146. doi: 10.1523/JNEUROSCI.23-35-11142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott DD, Lewis DV. Facilitation of the induction of long-term potentiation by GABAB receptors. Science. 1991;252:1718–1720. doi: 10.1126/science.1675489. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Mody I. Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. J Neurophysiol. 2002;87:2624–2628. doi: 10.1152/jn.2002.87.5.2624. [DOI] [PubMed] [Google Scholar]
- O'Keefe J, Recce ML. Phase relationship between hippocampal place units and the EEG theta rhythm. Hippocampus. 1993;3:317–330. doi: 10.1002/hipo.450030307. [DOI] [PubMed] [Google Scholar]
- Otto T, Eichenbaum H, Wiener SI, Wible CG. Learning-related patterns of CA1 spike trains parallel stimulation parameters optimal for inducing hippocampal long-term potentiation. Hippocampus. 1991;1:181–192. doi: 10.1002/hipo.450010206. [DOI] [PubMed] [Google Scholar]
- Sankar R, Holmes GL. Mechanisms of action for the commonly used antiepileptic drugs: relevance to antiepileptic drug-associated neurobehavioral adverse effects. J Child Neurol. 2004;19(Suppl 1):S6–S14. doi: 10.1177/088307380401900102. [DOI] [PubMed] [Google Scholar]
- Schmitt U, Hiemke C. Tiagabine, a gamma-amino-butyric acid transporter inhibitor impairs spatial learning of rats in the Morris water-maze. Behav Brain Res. 2002;133:391–394. doi: 10.1016/s0166-4328(02)00008-6. [DOI] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM. GABA uptake regulates cortical excitability via cell type-specific tonic inhibition. Nat Neurosci. 2003;6:484–490. doi: 10.1038/nn1043. [DOI] [PubMed] [Google Scholar]
- Skaggs WE, McNaughton BL, Wilson MA, Barnes CA. Theta phase precession in hippocampal neuronal populations and the compression of temporal sequences. Hippocampus. 1996;6:149–172. doi: 10.1002/(SICI)1098-1063(1996)6:2<149::AID-HIPO6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Stäubli U, Scafidi J, Chun D. GABAB receptor antagonism: facilitatory effects on memory parallel those on LTP induced by TBS but not HFS. J Neurosci. 1999;19:4609–4615. doi: 10.1523/JNEUROSCI.19-11-04609.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzdak PD, Frederiksen K, Andersen KE, Sørensen PO, Knutsen LJ, Nielsen EB. NNC-711, a novel potent and selective gamma-aminobutyric acid uptake inhibitor: pharmacological characterization. Eur J Pharmacol. 1992;224:189–198. doi: 10.1016/0014-2999(92)90804-d. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gähwiler BH. Activity-dependent disinhibition. I. Repetitive stimulation reduces IPSP driving force and conductance in the hippocampus in vitro. J Neurophysiol. 1989;61:501–511. doi: 10.1152/jn.1989.61.3.501. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gähwiler BH. Effects of the GABA uptake inhibitor tiagabine on inhibitory synaptic potentials in rat hippocampal slice cultures. J Neurophysiol. 1992;67:1698–1701. doi: 10.1152/jn.1992.67.6.1698. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Wadiche JI. Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci. 2007;8:935–947. doi: 10.1038/nrn2274. [DOI] [PubMed] [Google Scholar]
- Wang W, Gong N, Xu TL. Downregulation of KCC2 following LTP contributes to EPSP-spike potentiation in rat hippocampus. Biochem Biophys Res Commun. 2006;343:1209–1215. doi: 10.1016/j.bbrc.2006.03.038. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Wigström H, Gustafsson B. Facilitated induction of hippocampal long-lasting potentiation during blockade of inhibition. Nature. 1983;301:603–604. doi: 10.1038/301603a0. [DOI] [PubMed] [Google Scholar]
- Wong CG, Bottiglieri T, Snead OC., 3rd GABA, gamma-hydroxybutyric acid, and neurological disease. Ann Neurol. 2003;54(Suppl 6):S3–S12. doi: 10.1002/ana.10696. [DOI] [PubMed] [Google Scholar]