

Abstract
Transcription factors involved in the specification and differentiation of neurons often continue to be expressed in the adult brain, but remarkably little is known about their late functions. Nurr1, one such transcription factor, is essential for early differentiation of midbrain dopamine (mDA) neurons but continues to be expressed into adulthood. In Parkinson's disease, Nurr1 expression is diminished and mutations in the Nurr1 gene have been identified in rare cases of disease; however, the significance of these observations remains unclear. Here, a mouse strain for conditional targeting of the Nurr1 gene was generated, and Nurr1 was ablated either at late stages of mDA neuron development by crossing with mice carrying Cre under control of the dopamine transporter locus or in the adult brain by transduction of adeno-associated virus Cre-encoding vectors. Nurr1 deficiency in maturing mDA neurons resulted in rapid loss of striatal DA, loss of mDA neuron markers, and neuron degeneration. In contrast, a more slowly progressing loss of striatal DA and mDA neuron markers was observed after ablation in the adult brain. As in Parkinson's disease, neurons of the substantia nigra compacta were more vulnerable than cells in the ventral tegmental area when Nurr1 was ablated at late embryogenesis. The results show that developmental pathways play key roles for the maintenance of terminally differentiated neurons and suggest that disrupted function of Nurr1 and other developmental transcription factors may contribute to neurodegenerative disease.
Introduction
Adaptation to a changing environment requires plasticity in the adult CNS. However, to ensure that neurons are properly maintained, such plasticity must be balanced against mechanisms that counteract phenotypic instability. Studies of how neurons develop may help to unravel functions important for the stability of nerve cells as factors promoting their differentiation may also contribute to their maintenance. Indeed, many transcription factors identified for their critical roles during neuronal development continue to be expressed in the postnatal nervous system, raising the possibility that they contribute to the integrity of already differentiated neurons (Hendricks et al., 1999; Vult von Steyern et al., 1999; Kang et al., 2007; Alavian et al., 2008). However, the consequences of adult gene ablation of any of these factors have not yet been reported, and very little is known of their functions in differentiated neurons.
From a clinical perspective, it is of particular interest to identify factors that maintain stability of neurons that are affected in neurodegenerative disorders as loss of phenotype would likely cause or contribute to disease. Parkinson's disease (PD) is characterized by progressive pathology of midbrain dopamine (mDA) neurons of substantia nigra pars compacta (SNc) and the ventral tegmental area (VTA), typically involving deposition of α-synuclein-rich cytoplasmic protein aggregates termed Lewy bodies. During development, early signaling events induce transcription factors that control the specification and differentiation of mDA neurons (Smidt and Burbach, 2007). Several of these factors, including Nurr1, Lmx1a, Lmx1b, Pitx3, FoxA2, and En1/2, continue to be expressed in the postnatal and adult brain (Zetterström et al., 1996; Smidt et al., 1997, 2000; Albéri et al., 2004; Simon et al., 2004; Kittappa et al., 2007). Nurr1, belonging to a family of ligand-independent nuclear receptors (Wang et al., 2003; Perlmann and Wallén-Mackenzie, 2004), becomes expressed in developing mDA neurons that have just exited the cell cycle and is essential for mDA neuron development because mDA neurons of both the SNc and VTA fail to express dopaminergic markers and newborn Nurr1-null mice lack mDA neuron cell bodies and their striatal projections (Zetterström et al., 1997; Castillo et al., 1998; Saucedo-Cardenas et al., 1998). How Nurr1 regulates target genes in mDA neuron development remains essentially unknown but may involve a functional interaction with the homeobox transcription factor Pitx3 (Jacobs et al., 2009).
Determining the role of Nurr1 also in the adult brain is of particular importance because previous studies suggested an association of this protein with PD pathology. Nurr1 expression is diminished in neurons with α-synuclein inclusions in postmortem PD brain tissue, and Nurr1 mutations and polymorphisms have been identified in rare cases of PD (Xu et al., 2002; Le et al., 2003; Zheng et al., 2003; Grimes et al., 2006). However, the significance of genetic lesions remain unclear (Wellenbrock et al., 2003; Hering et al., 2004; Tan et al., 2004). These observations emphasize the importance of elucidating the role of Nurr1 in more mature mDA neurons by analyzing the consequences of conditional Nurr1 gene ablation in mice.
Materials and Methods
Conditional Nurr1 gene-targeted mice.
Mouse 129SV genomic library constructed in bacterial artificial chromosome (BAC) was screened by PCR. A BAC clone containing the entire Nurr1 gene was selected, and a BamHI–MunI fragment containing exon 1 to exon 5 was recloned into a pBluescript II vector. A floxed neomycin cassette was inserted into an internal EcoRI site located in intron 3, and a synthetic loxP sequence was inserted at SalI site located in intron 2. Mouse embryonic stem (ES) cells were electroporated with the targeting vector, and the homologously recombinated clones were screened by PCR and Southern blot analysis. ES clones with three loxP sites were selected, and a plasmid expressing Cre DNA recombinase was transiently transfected into the cells. ES cells with two loxP sites without a neomycin cassette were selected by PCR and used for production of chimeric mice.
Animals.
Mice were kept in rooms with controlled 12 h light/dark cycles, temperature, and humidity, with food and water provided ad libitum. All animal experiments were performed with permission from the local animal ethics committee. The generation of dopamine transporter (DAT)–Cre mutant mice has been described previously (Ekstrand et al., 2007). Mice were mated during the night, and the females were checked for vaginal plugs in the morning [day of vaginal plug considered as embryonic day 0.5 (E0.5)].
l-3,4-Dihydroxyphenylalanine treatment.
Methyl l-3,4-dihydroxyphenylalanine (l-DOPA) hydrochloride and the peripheral DOPA decarboxylase inhibitor benserazide-HCl (Sigma-Aldrich) were dissolved in Ringer's solution immediately before use. l-DOPA was intraperitoneally given every second day at the dose of 2.5 mg/kg combined with 0.625 mg/kg benserazide. Chronic treatment with l-DOPA/benserazide was administered for 50 d, starting at postnatal day 15 (P15). During this period, the mice were carefully observed and weight was measured regularly. Reported hyperactivity was observed in cNurr1DATCre mice when given a single higher dose of l-DOPA (25 mg/kg l-DOPA, 6.25 mg/kg benserazide).
Histological analyses.
At embryo stages, embryos were fixed for 2–24 h in 4% phosphate-buffered paraformaldehyde (PFA), cryopreserved in 30% sucrose before being embedded in OCI (Sakura Finetek), and cryosectioned at a thickness of 10–20 μm onto slides (SuperFrostPlus; Menzel Gläser). For the isolation of brains for immunolabeling at P15 and onward, animals were anesthetized with Avertin (tribromoethanol; 0.5 mg/g) and perfused through the left ventricle with body-temperature PBS, followed by ice-cold 4% PFA. The brains were dissected and postfixed overnight in 4% paraformaldehyde and subsequently cryoprotected for 24–48 h in 30% sucrose at 4°C. The brains were serially sectioned on a cryostat or sliding microtome at 10–30 μm. Littermates were used in an all comparative experiments.
For immunohistochemistry, sections were preincubated for 1 h in blocking solution containing either 10% normal goat sera or 5–10% bovine serum albumin, 0.25% Triton X-100, and 0.01% Na-azide in PBS. Primary antibodies diluted in blocking solution were applied overnight at 4°C. After rinses with PBS, biotinylated- or fluorophore-conjugated secondary antibodies diluted in PBS were applied for 1 h at room temperature. Biotinylated secondary antibodies were followed by incubation with streptavidin–horseradish peroxidase complex (ABC elite kit, Vectastain) for 1 h and subsequent exposure to diaminobenzidine (DAB kit; Vector Laboratories). Primary antibodies and dilution factors were as follows: rabbit anti-Nurr1 (1:100; M196; Santa Cruz Biotechnology), anti-Nurr1 (1:250; E20; Santa Cruz Biotechnology), rabbit anti-tyrosine hydroxylase (TH) (1:500; Pel-Freeze), rat anti-DAT (1:2000; Millipore Bioscience Research Reagents), mouse anti-TH (1:200; Millipore Bioscience Research Reagents), rabbit anti-vesicular monoamine transporter (VMAT) (1:500; Millipore Bioscience Research Reagents), rabbit anti-l-DOPA decarboxylase (AADC) (1:500; Millipore Bioscience Research Reagents), rabbit anti-Cre (1:10,000; Covance Research Products), guinea pig anti-Lmx1b (1:1000) (Andersson et al., 2006), and rabbit anti-Pitx3 (Smidt et al., 2004). In some cases (anti-AADC, anti-VMAT, and anti-Nurr1), the blocking steps were performed after antigen retrieval (Dako). Finally, expression was detected by secondary antibodies from Jackson ImmunoResearch. Section images were collected by confocal microscopy (Leica DMIRE2) and bright-field microscopy (Eclipse E1000K; Nikon). Cell counting was performed by counting all SNc DA neurons detected by immunohistochemistry (DAB) in a total of three sections per animal (every 12th tissue section) within the ventral midbrain of animals taken at 4 months after vector injection in both wild-type wtAAVCre and cNurr1AAVCre animals. The mean of counted cells per animal was established from both the injected and non-injected sides in each animal, and the relative decrease was calculated as a percentage as described in Results.
AAV–Cre injections.
Two and a half- to 5-month-old animals received one unilateral stereotaxic injection in the right striatum using a 10 μl Hamilton microsyringe fitted with a glass pipette tip. The animals were anesthetized with isoflurane, 1 μl was injected during 5 min, and the cannula was left in place for an additional 2 min before being slowly retracted. The anteroposterior and mediolateral coordinates from bregma were −2.8 and −1.1 mm, respectively, and the dorsoventral coordinates from the dura were −4.3 mm. Animals were killed 0.5, 1.5, and 4 months after injection, and the brains were isolated.
Measurement of tissue content for dopamine, serotonin, and their metabolites.
In supplemental Tables 1–3 (available at www.jneurosci.org as supplemental material), tissues were collected from P1, P7, P14, and adult (P48) wtDATCre and cNurr1DATCre mice. One- to 14-d-old mice were killed by decapitation, and P48 mice were killed by cervical dislocation. Brains were rapidly removed, chilled in saline (4°C), dissected, frozen on dry ice, and stored at −80°C until use. To process tissues for HPLC and electrochemical detection of monoamines and metabolites, samples were homogenized by sonication in 5 vol or in 30 μl of 0.1 m perchloric acid, followed by centrifugation. Endogenous levels of noradrenaline, DA, 3,4-dihydroxyphenylacetic acid, homovanillic acid (HVA), serotonin (5-HT), and 5-hydroxyindoleacetic acid were determined in the supernatants. A reverse column (BAS, C-18, 100.0 × 3.2 mm, 3 μm particle diameter) was used for separation. The mobile phase consisted of 0.05 m sodium phosphate/0.03 m citric acid buffer containing 0.1 mm EDTA and was adjusted with various amounts of methanol and sodium-l-octane sulfonic acid. The flow rate was 0.3 ml/min. Monoamines and metabolites were detected using a glassy-carbon electrode detector, which as set at +0.7 V versus an Ag/AgCl reference electrode. Resultant peaks were measured and compared with repeated control samples containing fixed mixed amounts of compounds of interest.
In supplemental Table 4 (available at www.jneurosci.org as supplemental material), all animals were killed, and striata and cortex were rapidly dissected out, frozen on dry ice, and stored at −80°C. To determine monoamines, tissue was homogenized in 0.1 m perchloric acid and centrifuged at 10,000 rpm for 10 min before filtering though minispin filters for an additional 3 min at 10,000 rpm. The tissue extract were then analyzed by HPLC as described previously (Carta et al., 2007) with minor modifications. Briefly, 25 μl of each sample were injected by a cooled autosampler (Midas) into an ESA Coulochem III coupled with an electrochemical detector. The mobile phase (5 g/L sodium acetate, 30 mg/L Na2-EDTA, 100 mg/L octane-sulfonic acid, and 10% methanol, pH 4.2) was delivered at a flow rate of 500 μl/min to a reverse-phase C18 column (4.6 mm diameter, 150 mm).
Fluorogold retrograde tracing.
Animals received one unilateral stereotaxic injection in the right striatum using a 5 μl Hamilton microsyringe (22 gauge steel cannula) filled with the retrograde tracer Fluorogold (hydroxystilbamidine, 4%, Biotium). The animals were anesthetized with isoflurane, 0.5 μl was injected during 1 min, and the cannula was left in place for an additional 2 min before slowly being retracted. The anteroposterior and mediolateral coordinates from bregma were 0.27 and −2.10 mm, respectively, and the dorsoventral coordinates from the dura were −2.60 mm. Animals were killed 4 d after injection, and the brains were isolated. Fresh-frozen sections (14 μm) were cut with a cryostat and examined under a epifluorescence microscope (Eclipse E1000K; Nikon) coupled to an RTke spot camera.
Open-field test.
This test was used to monitor overall activity and rearing behavior. The open field consisted of a white plastic box (55 × 35 × 30 cm) with lines (squares of 7 × 35 cm) painted on its floor. The animals were put in the center of the box, habituated for 10 min, and filmed 15 min thereafter while rearing was scored. The video recordings were used to measure the number of lines crossed during the monitoring period. A line crossing was counted when the mouse moved its whole body from one square to another.
Stepping test.
Forelimb akinesia was monitored in a modified version of the stepping test, as described previously for rats (Schallert et al., 1992; Kirik et al., 1998). The test was performed three times daily over 3 consecutive days. In this test, the mouse was held firmly by the experimenter with both hindlimbs and one forelimb immobilized, and the mouse was passively moved with the free limb contacting a table surface. The number of adjusting steps, performed by the free forelimb when moved in the forehand and backhand directions, over a distance of 30 cm, was recorded. Results are presented as data collected on the third testing day.
Results
Selective Nurr1 ablation in late developing mDA neurons
A mouse strain containing a Nurr1 allele for conditional gene ablation was generated by insertion of two loxP sequences in the second and third introns so that the coding sequence, including the first coding exon 3, is excised by Cre-mediated recombination (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). To analyze the consequences of Nurr1 ablation at late stages of mDA neuron development, we crossed floxed Nurr1 mice with mice carrying Cre inserted in the locus of the DAT gene (Ekstrand et al., 2007). Crosses generated Nurr1 mice that were homozygous for the conditional targeted Nurr1 allele and heterozygous for the DAT–Cre allele (Nurr1L2/L2;DATCre/wt; hereafter referred to as cNurr1DATCre mice). Littermates of genotype Nurr1L2/L2;DATwt/wt or Nurr1w/w;DATCre/wt were used as controls. Although we cannot exclude that a small number of cells escape Nurr1 gene deletion, immunohistochemistry using an antibody against Nurr1 showed that DAT–Cre-mediated Nurr1 ablation resulted in the expected delayed loss of Nurr1 expression in mDA neurons beginning from approximately E13.5 and becomes essentially complete at E15.5 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). At this stage of normal development, cells express pan-neuronal properties as well as many mDA neuron markers, and axons are growing toward the developing striatum (Smidt and Burbach, 2007).
cNurr1DATCre mice were born at the expected Mendelian frequency of ∼25% (of a total n = 159); however, cNurr1DATCre mice were less active than controls and did not survive beyond 3 weeks after birth. If litters were allowed to remain with their mothers after weaning, perinatal death was avoided in ∼50% of cNurr1DATCre pups. These surviving mice were, however, ∼40% smaller than controls at the age of 2 months (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Although no significant change in spontaneous light-phase locomotor activity could be observed in adult cNurr1DATCre mice, rearing was dramatically decreased (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). l-DOPA treatment of mutant mice did not improve viability and did not induce any weight gain. Instead, cNurr1DATCre mice display a pronounced and severe hypersensitivity to l-DOPA treatment characterized by an acute phase of hyperactivity and repetitive behaviors (including repetitive gnawing, excessive grooming, and self-injury) in all tested mutant (n = 9) but not in any wild-type controls (n = 7) (see Materials and Methods). These behaviors resemble those that have been observed in neonatal 6-hydoxydopadmine lesioned rats treated with l-DOPA (Breese et al., 2005). In conclusion, late embryonic mDA neuron-selective Nurr1 ablation is associated with decreased weight, rearing, and viability, and mice show an altered response to l-DOPA.
Reduced levels of TH and DA in brains of cNurr1DATCre mice
The observed abnormalities are consistent with a dopaminergic deficiency. To analyze the possible cellular basis for the phenotype, brain sections from controls and cNurr1DATCre mice were analyzed by immunohistochemistry using an antibody against TH (Fig. 1). A progressive loss of TH immunostaining in SNc was observed in the cNurr1DATCre mice (Fig. 1A–J). TH levels were significantly decreased already at E15.5, soon after Nurr1 is lost, and decreased further until adulthood when only scattered TH-positive neurons could be detected. TH was diminished also within the VTA at later stages, but a significant number of cells remained even in adult animals (Fig. 1A–J). These cells were counted in four nonconsecutive sections for each analyzed brain. In adult control VTA, a mean of 74.5 ± 7.1 cells per section were counted in cNurr1DATCre mice (n = 4) and 499.3 ± 5.2 cells in controls (n = 3) (Student's t test, 4.4 × 10−7). TH immunostaining within the caudatus putamen (CPu) was completely lost (Fig. 1K–T). However, weak immunoreactivity remained in nucleus accumbens (NAc) innervated preferentially by VTA neurons (Fig. 1K–T). We also noted the appearance of ectopic TH-positive cell bodies within the striatal parenchyma in cNurr1DATCre mice (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). These cells were more frequent in regions in which striatal TH had been most severely depleted as a consequence of Nurr1 ablation and resemble TH-positive neurons appearing in rodent and primate DA-depletion models (Huot and Parent, 2007). Decreased levels of TH immunostaining were paralleled by decreased DA levels, as measured by HPLC (supplemental Tables 1–3, available at www.jneurosci.org as supplemental material). Striatal DA was dramatically reduced to 14% of controls at P1 in cNurr1DATCre mice and was almost completely lost by P60. An increased ratio of HVA to DA at P14 indicated increased turnover of DA in remaining cells at this stage (supplemental Table 2, available at www.jneurosci.org as supplemental material). DA was more severely decreased in CPu compared with NAc (supplemental Table 3, available at www.jneurosci.org as supplemental material). In contrast, 5-HT was significantly increased in both CPu and NAc, consistent with previous findings showing increased serotonergic innervation after striatal DA depletion (Snyder et al., 1986). Thus, a severe neurotransmitter deficiency of the mesostriatal DA system is apparent in cNurr1DATCre mice. Together, measurements of TH immunoreactivity and DA levels demonstrate that Nurr1 is critically required for maintaining TH expression and DA synthesis from late stages of mDA neuron differentiation.
Figure 1.
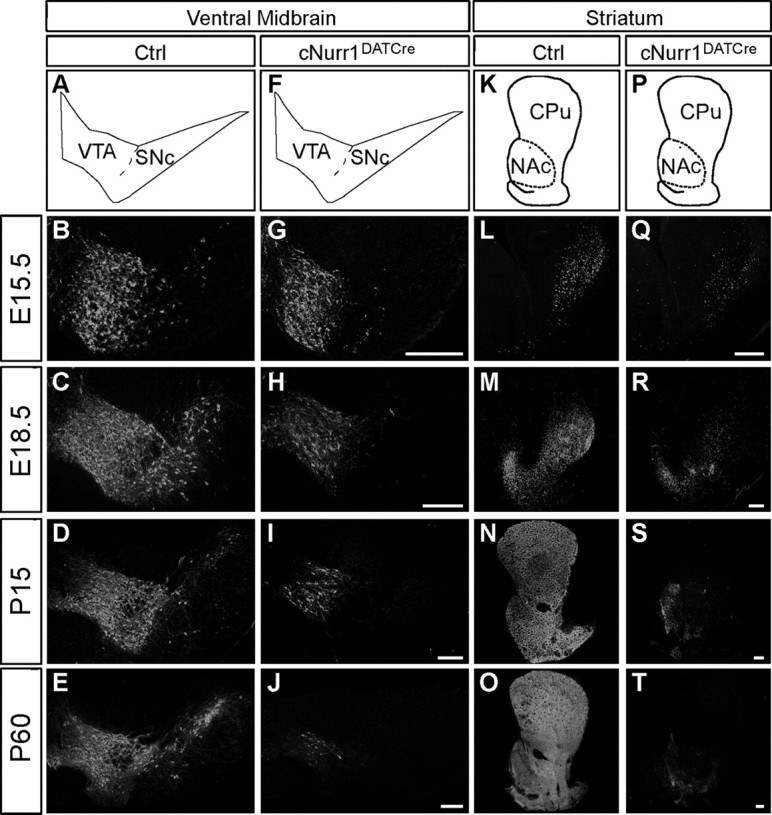
TH is progressively lost in both the ventral midbrain and striatum of cNurr1DATCre mice. A–T, Confocal microscopy showing TH immunohistochemistry in control (ctrl) and cNurr1DATCre mice as indicated. A–J, Sections were analyzed at the levels of ventral midbrain (as indicated in A and F) and in the striatum (as indicated in K and P). TH immunofluorescence was analyzed at both embryonal and postnatal stages as indicated. Results demonstrate a progressive loss of TH immunoreactivity in the ventral midbrain. Note that TH immunoreactivity was more drastically downregulated at more lateral regions compared with the prospective medial VTA. K–T, TH immunoreactivity in the striatum. In cNurr1DATCre mice, TH was lost in the CPu and diminished in the NAc. Scale bars, 250 μm.
Cellular deficiency within the ventral midbrain of cNurr1DATCre mice
To investigate whether the phenotype is a consequence of a more limited disruption of DA synthesis or a more severe cellular deficiency, a number of additional mDA neuron markers were analyzed. All analyzed mDA neuron markers were diminished or absent within SNc in cNurr1DATCre mice already at E15.5 (Fig. 2). DAT was completely lost at E15.5 and therefore, consistent with previous data (Sacchetti et al., 1999), stands out as being a likely direct target of Nurr1 (Fig. 2A–D). Additional control experiments showed that DAT and other markers, including TH and Nurr1, were not visibly decreased in mice heterozygous for the DAT–Cre allele (supplemental Fig. 5, available at www.jneurosci.org as supplemental material) (data not shown). In contrast to DAT, AADC, VMAT2, Pitx3, or Lmx1b were not reduced within the most medial ventral midbrain at this early stage and, with the exception of DAT, markers were not completely downregulated at P1 (Fig. 2E–T). The progressive loss of markers indicates a severe loss of phenotype within the SNc, whereas cells within the VTA appear more resilient. Importantly, most TH-positive cells within the VTA have lost any detectable expression of DAT, indicating that these cells have not escaped Nurr1 gene targeting (supplemental Fig. 6, available at www.jneurosci.org as supplemental material).
Figure 2.
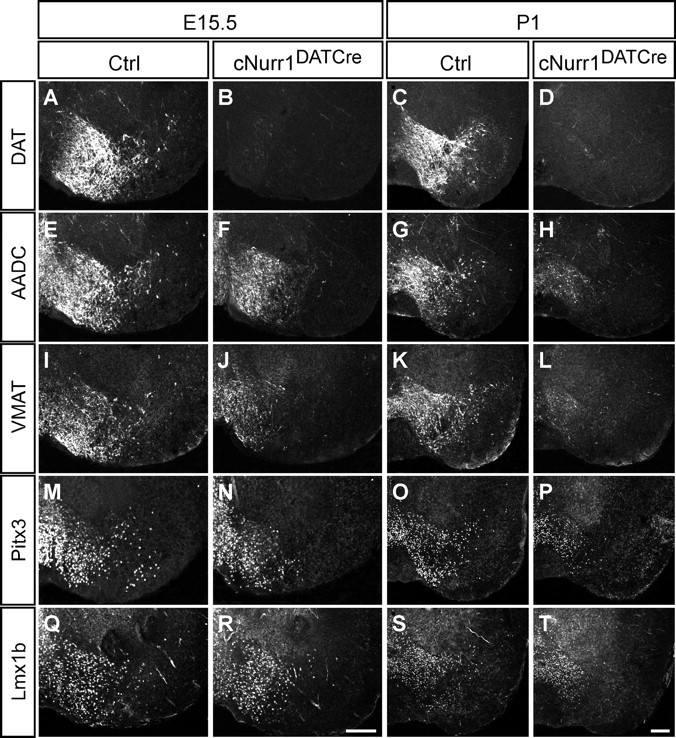
All analyzed mDA neuron markers are lost or diminished in cNurr1DATCre mice. A–T, Confocal microscopy showing immunohistochemistry of several different mDA neuron markers in control (ctrl) and in cNurr1DATCre mice at E15.5 or at P1, as indicated. The following markers were analyzed: DAT, AADC, VMAT, Pitx3, and Lmx1b. Results show a progressive loss of markers that is more substantial in the lateral SNc, whereas more medial VTA cells are lost more slowly. DAT is completely absent already at E15.5 in cNurr1DATCre mice, whereas all other markers are decreased more slowly in this area (compare A, B). Scale bars, 200 μm.
To further assess the extent of a cellular deficiency, striatal target innervation was analyzed by Fluorogold retrograde tracing after injection into the striatum of live 8- to 9-week-old controls and cNurr1DATCre mice. Fluorogold was transported into SNc cell bodies of control mice; however, fluorescence was entirely undetected within the SNc of Fluorogold-injected cNurr1DATCre mice (Fig. 3, compare D, E with G, H). In addition, characteristic large and densely packed TH-immunoreactive mDA neurons within the SNc were virtually absent in cNurr1DATCre mice (Fig. 3I). In conclusion, Nurr1 ablation in cNurr1DATCre mice results in rapid loss of SNc cell bodies; however, scattered VTA neurons remained even in adult cNurr1DATCre mice.
Figure 3.
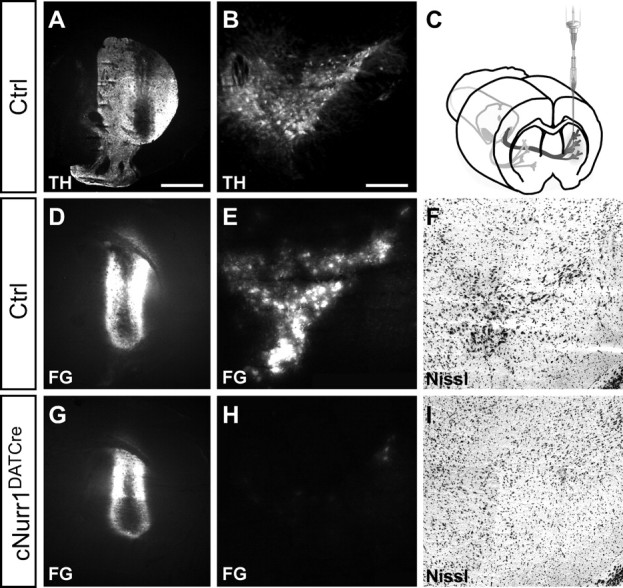
Cell bodies and striatal innervation are lost in cNurr1DATCre mice as determined by Fluorogold (FG) retrograde tracing of fibers extending from cell bodies in SNc to the striatum. A–C, After Fluorogold injection, injected into the left striatum in either adult (1.5 months old) control (Ctrl) or cNurr1DATCre mice as indicated in C, mice were killed after 4 d and analyzed for TH immunofluorescence in the striatum (A) or ventral midbrain (B). D–I, Analysis for Fluorogold (FG) or by Nissl staining. Strong Fluorogold staining in both striatum (D) and in the ventral midbrain (E) was consistently seen in all control (Ctrl) animals (n = 7). In contrast, Fluorogold fluorescence was only detected in the striatum (G) in cNurr1DATCre mice (n = 5), indicating that fibers from the SNc (H) had been lost in these animals. Moreover, large, densely packed cell bodies are only visualized by Nissl staining in the ventral midbrain of control animals (F) but are completely absent from cNurr1DATCre mice (I). Striatal site of Fluorogold injection is marked by asterisk in A, D, and G. Scale bars: A, B, D, E, G, H, 1 μm; F, I, 1200 μm.
Adeno-associated virus–Cre-mediated Nurr1 ablation in adult mice
In cNurr1DATCre mice, Nurr1 is ablated well before full mDA neuron maturity and before targets in the striatum have become innervated; thus, it remained possible that the phenotype is a consequence of a developmental dysfunction. Therefore, we proceeded to inactivate Nurr1 specifically in ventral midbrain of adult mice, using an adeno-associated virus (AAV)–Cre vector driven by the neuron-specific synapsin promoter. AAV–Cre was administered by unilateral stereotaxic microinjection above the right SNc. Cre immunohistochemistry and β-galactosidase expression was analyzed after intranigral AAV–Cre injection into reporter mice in which the ROSA26 locus is targeted with a LacZ reporter gene (Soriano, 1999). Results show widespread Cre expression around the site of injection, spreading into both SNc and VTA, and robust recombination of the LacZ reporter construct (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). Moreover, except for a small necrotic area around the site of injection, virus administration did not affect tissue morphology, expression of mDA neuron markers, or microglia activation (data not shown).
AAV–Cre was unilaterally injected above the SNc of adult mice homozygous for the floxed Nurr1 allele to generate adult conditional gene-targeted mice (cNurr1AAVCre) or into wild-type control mice (wtAAVCre). In addition, a vector encoding the green fluorescent protein (GFP) driven by the synapsin promoter (AAV–GFP) was injected in mice homozygous for the floxed Nurr1 allele (cNurr1AAV–GFP mice) to ensure that the floxed animals are not more sensitive to nonspecific toxicity induced by AAV transduction. Histological analyses were performed from animals killed at 0.5, 1.5, and 4 months after injection.
Reduction of TH and DA in adult Nurr1-ablated mice
TH immunohistochemistry at the level of the ventral midbrain was analyzed to assess the consequences of adult Nurr1 ablation. Within SNc, TH immunoreactivity was unaffected at 0.5 months but was progressively reduced at 1.5 and 4 months in the injected SNc in cNurr1AAVCre mice (Fig. 4E–H). In contrast, TH immunoreactivity was unaffected in SNc of control wtAAVCre and cNurr1AAV–GFP mice (Fig. 4A–D) (data not shown). TH was also reduced in the VTA at 1.5 and 4 months; however, at 4 months, the reduction in VTA was less dramatic compared with SNc (Fig. 4E–H).
Figure 4.

TH expression in both the ventral midbrain and striatum is progressively lost in the injected, but not non-injected, side of cNurr1AAVCre mice. A–H, Sections from 0.5, 1.5, and 4 month (mo; as indicated) old AAV–Cre-injected controls (wtAAVCre) or cNurr1AAVCre mice were used for analyses by nonfluorescent DAB TH immunostaining in the ventral midbrain. The analyzed region within the ventral midbrain is schematically illustrated in A and E. The site of injection, marked by an asterisk in B–D and F–H, was verified in all animals by high-power magnification microscopy and was identified as a small area of injection-induced necrosis. Results show that TH immunostaining is not drastically altered at 0.5 months but is progressively decreased at 1.5 and 4 months in the injected SNc and VTA. I–L, DAB TH staining at the level of striatum. Analyzed regions are indicated in I. TH staining is progressively decreased at 1.5 and 4 months in the side that is ipsilateral to the side of AAV–Cre injection in cNurr1AAVCre mice (J–L). OT, Olfactory tubercle. Scale bars: A–H, 600 μm; I–L, 1 mm.
Decreased striatal TH immunoreactivity paralleled the reduction in the ventral midbrain. Thus, although no signs of degenerating striatal TH-stained fibers (swollen axons or dystrophic neurites) were detected, striatal sections ipsilateral to the side of AAV–Cre injection showed clearly reduced TH in cNurr1AAVCre mice but not in controls (wtAAVCre or cNurr1AAV–GFP) (Fig. 4I–L) (supplemental Fig. 8, available at www.jneurosci.org as supplemental material). Diminished TH immunoreactivity was observed in regions innervated by both SNc and VTA (CPu and NAc, respectively), consistent with the reduced TH immunoreactivity in both SNc and VTA mDA neuron cell bodies. Measurement of DA and metabolites by HPLC from dissected tissue at 4 months confirmed this picture as a significant reduction in DA and DA metabolites noted both within the dorsolateral striatum and in areas mostly innervated by the VTA (cortex and ventromedial striatum) (supplemental Table 4, available at www.jneurosci.org as supplemental material). Thus, TH, DA, and DA metabolites are clearly reduced as a result of adult Nurr1 ablation.
Loss of mDA neuron characteristics in adult Nurr1-ablated mice
To further analyze the fate of Nurr1-ablated neurons, cells were counted within the SNc and VTA in cNurr1AAVCre and wtAAVCre mice. Within SNc, the number of TH-positive cells was significantly decreased at 4 months (58.1 ± 8.3 and 95.4 ± 6.3% in the injected vs non-injected sides of cNurr1AAVCre and wtAAVCre mice, respectively; p = 0.0053). In contrast, the numbers of TH-positive cells was not significantly reduced within the VTA (104.1 ± 4.7 and 101.6 ± 10.5% in the injected versus non-injected sides of cNurr1AAVCre and wtAAVCre mice, respectively). Also, the numbers of TH-positive cells were not significantly changed in cNurr1AAVCre mice at 1.5 months (data not shown).
To assess the integrity of neurons, cellular analysis was extended by analyzing Cre-immunolabeled sections that were superimposed on adjacent TH-labeled sections (Fig. 5). Notably, in cNurr1AAVCre mice, Cre expression was clearly detected within the area of SNc at both 0.5 and 1.5 months but was lost at 4 months in the region in which mDA neurons should normally be localized (Fig. 5, compare E–G with B–D). Cre expression is driven by a general neuronal promoter (synapsin), suggesting that loss of Nurr1 may eventually affect some pan-neuronal properties at 4 months after ablation.
Figure 5.
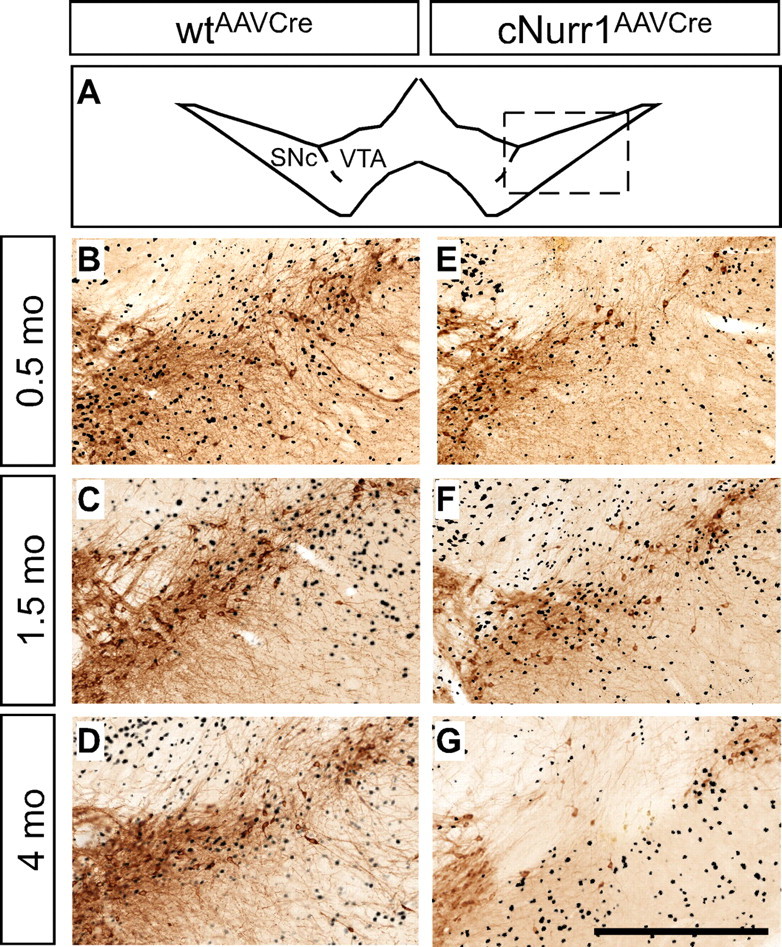
Cre-expressing cells are lost at 4 months within the SNc after Nurr1 ablation. A, Higher magnification showing TH by DAB staining (brown) at the level of the injected SNc in wtAAVCre and cNurr1AAVCre mice. The region that has been magnified is boxed in A. B–G, Adjacent sections were immunostained for Cre (black) and are superimposed on the DAB-stained TH sections in all micrographs. Cre staining is widespread in the area in which mDA neurons are normally localized at 0.5 and 1.5 months (E, F) but almost completely absent in this area at 4 months (G). Scale bar, 500 μm.
Confocal microscopy confirmed the loss of TH at 1.5 and 4 months after Nurr1 ablation and the loss of Cre at 4 months (Fig. 6A–D). At 1.5 months, DAT expression was weak but cell bodies were readily identified (Fig. 6E,F and inset in F). DAT staining remained also at 4 months, but, at this stage, high-power magnification indicated that some of the staining appeared confined to fibers and/or dystrophic cells (Fig. 6G,H and inset in H). Nonetheless, at 4 months, most cells with decreased TH stained positive for AADC, showing that not all mDA neuron characteristics were affected (Fig. 7A–F). Moreover, VMAT2 is yet another marker that was severely decreased in cNurr1AAVCre mice, but remaining weakly stained cells were positive for the general neuronal marker Hu (Fig. 7G–L). The observed changes were not correlated to increased number of apoptotic cells because increased activated Caspase 3 could not be detected (data not shown). Also, we found no evidence for nigral inflammation or α-synucleinopathy because activated microglia and α-synuclein-rich inclusions were not detected at any stage after Nurr1 ablation in cNurr1DATCre mice (data not shown). Finally, TH and DAT expression in VTA was also affected, without any apparent loss of the neuronal marker Hu or any signs of dystrophic cells (supplemental Fig. 9, available at www.jneurosci.org as supplemental material) (data not shown). Thus, Nurr1 ablation results in a progressive dysfunction characterized by a partial loss of the mDA neuron phenotype. Although we see few signs of neuronal degeneration, we cannot exclude a limited cell loss.
Figure 6.

Decreased expression of DAT and signs of dystrophic cells in cNurr1AAVCre mice. A–H, Confocal analysis of SNc in wtAAVCre and cNurr1AAVCre mice at 1.5 and 4 months, as indicated. Confocal images show double staining of Cre (red) and TH (green; A–D) and staining for DAT (green; E–H). Micrographs show that there is a loss of TH and DAT and a progressive loss of synapsin-driven Cre at 4 months. At 4 months, DAT staining appears fragmented and stains scattered fibers, whereas very few intact cell profiles (marked with arrowheads in F and H) can be identified in cNurr1AAVCre mice (compare insets in F, H). Scale bar, 200 μm.
Figure 7.
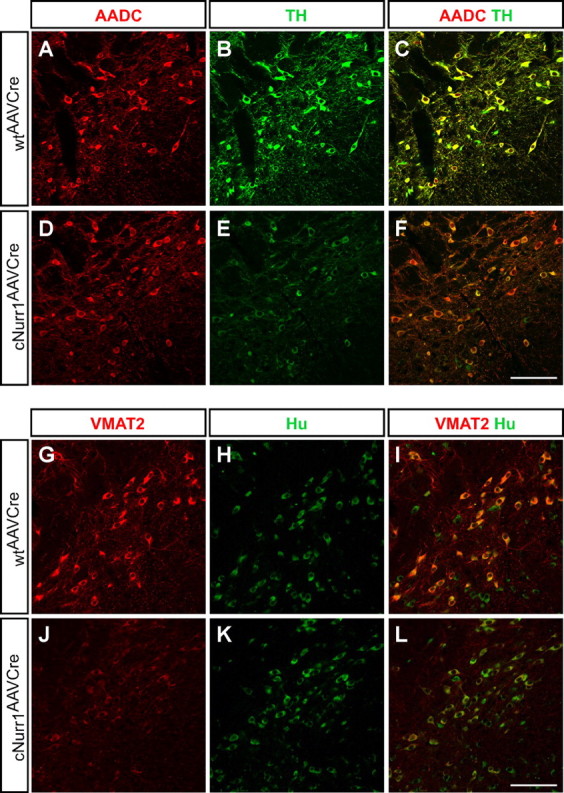
Decreased levels of VMAT2 but not AADC in cNurr1AAVCre mice. Confocal analysis of SNc in wtAAVCre and cNurr1AAVCre mice at 4 months, as indicated. A–F, Confocal images show staining of AADC (red; A, D), TH (green; B, E), and double staining of both markers (C, F). Micrographs show that AADC expression appears expressed at normal levels in most cells with decreased levels of TH. G–L, Confocal images show staining for VMAT2 (red; G, J), Hu (H, K), and double staining of both markers (I, L). Micrographs show that VMAT2 is severely decreased in cNurr1AAVCre mice, whereas Hu is maintained at normal levels in essentially all cells with decreased VMAT2. Scale bar, 200 μm.
To assess whether the observed dysfunction was paralleled by an altered motor behavior, cNurr1AAVCre mice were subjected to a stepping test at 3 and 4 months (Schallert et al., 1992; Kirik et al., 1998). Performance of the left forelimb (i.e., the limb contralateral to the vector injection) was impaired at both time points (Fig. 8). Additional behavioral testing, including amphetamine-induced rotations and a corridor test, indicated that individual mutant animals appeared affected; however, the Nurr1-ablated group did not show alterations that were statistically significant (supplemental Fig. 10, available at www.jneurosci.org as supplemental material). Our results demonstrate progressive mDA neuron dysfunction, leading to a more severe deficiency at 3–4 months after Nurr1 ablation.
Figure 8.

Forelimb akinesia in the stepping test. The performance of the left paw (contralateral to the vector injection) was significantly impaired in the fl/fl mice (n = 16) but not the wild-type mice (n = 13). Although the impairment was significant at both time points, 3 and 4 months after vector injection, their performance got significantly worse over time (p < 0.01, Student's paired t test): 15 of the 16 mice in the fl/fl group showed a decline in their stepping scores between the two tests, and at 4 months, 14 of the fl/fl mice had scores below 5 compared with 6 in the 3 month test. Scores give the means of steps recorded in the forehand and backhand direction for each paw (see Materials and Methods). *p < 0.001, Student's paired t test.
Discussion
This study provides definitive evidence that Nurr1 is not only critical for early differentiation but also for the maintenance of functional mDA neurons. Conditional gene targeting at late embryogenesis, when characteristic features of mDA neurons are already apparent, results in a rapid and close to complete mDA neuron loss. Only few TH-positive cells remain within the VTA also in the absence of Nurr1. Removal of Nurr1 leads to a severe dysfunction also in adult mDA neurons. It should be noted that reduction of striatal DA and the behavioral effects after adult ablation most likely underestimate the importance of Nurr1 in the adult brain because AAV injection only transduced a proportion of all mDA neurons in the injected side of treated animals. Thus, these data emphasize the importance of studying developmental mechanisms for elucidating neuron maintenance mechanisms. An analogous example is provided by the glial cell line-derived neurotrophic factor (GDNF). GDNF is known to promote neuronal survival under development, but only recently has conditional gene targeting enabled studies that interrogate the role of GDNF and other factors signaling via Ret for maintenance of midbrain dopamine neurons in the adult brain (Oo et al., 2003; Jain et al., 2006; Kramer et al., 2007; Pascual et al., 2008).
Data presented here have implications for our understanding of how mature differentiated cell types are maintained. Previous studies have indicated that the differentiated state is not irreversible because even mature specialized cells, including for example, olfactory neurons and mature T- and B-cells, can be reprogrammed into undifferentiated pluripotent cells by either somatic cell nuclear transfer or using the recently developed methodology for the generation of induced pluripotent stem cells (Takahashi and Yamanaka, 2006; Gurdon and Melton, 2008). Nevertheless, under normal nonmanipulated conditions in vivo, differentiated cells are remarkably stable, indicating the importance of mechanisms that maintain cells in their appropriate differentiated state. Gene targeting in non-neural cell types has revealed how transcription factors functioning in development can be important for the maintenance of terminally differentiated cell types, e.g., Pax5 in B-lymphocytes and Prox1 in lymphatic endothelial cells (Cobaleda et al., 2007; Johnson et al., 2008). In CNS, transcription factors identified for their key roles in early neuron development often continue to be expressed in the adult brain and may therefore guard against loss of phenotype or drift into alternative states (Smidt et al., 1997, 2000; Zetterström et al., 1997; Hendricks et al., 1999; Vult von Steyern et al., 1999; Albéri et al., 2004; Simon et al., 2004; Kang et al., 2007; Kittappa et al., 2007; Smidt and Burbach, 2007; Alavian et al., 2008). However, remarkably little is known of how these factors function at late stages of development or in the adult. Although examples of adult mDA neuron loss has been reported in mice haploinsufficient for transcription factor genes such as Engrailed and FoxA2, it remains possible that defects originate during embryonic development (Albéri et al., 2004; Zhao et al., 2006; Kittappa et al., 2007; Sonnier et al., 2007). Importantly, FoxA2 and Engrailed are critical for the establishment of the floor plate and for early midbrain/hindbrain development, respectively, and they are directly and indirectly affecting many cell fates along the entire neuraxis. Thus, haploinsufficiency may cause embryonal deficiencies that do not become manifest until adult stages, a possibility that emphasizes the importance of temporally controlled conditional gene targeting to rigorously test how transcription factors function in terminally differentiated neurons.
We do not yet understand why Nurr1 is required in already differentiated mDA neurons. However, data presented here provide compelling evidence for the existence of “terminal selector genes” in mammalian CNS development. Such genes, defined from studies of Caenorhabditis elegans neuronal development, are continuously expressed throughout the life of neurons and are essential for both the establishment and maintenance of distinct neuronal phenotypes (Hobert, 2008). Thus, Nurr1, which probably regulates typical mDA neuron markers such as TH, DAT, AADC, and VMAT2 (Sakurada et al., 1999; Sacchetti et al., 2001; Hermanson et al., 2003; Kim et al., 2003), is likely required for both early differentiation and maintenance by regulating genes that distinguish mDA neurons from other neuron types. Presumably, such regulation is critical throughout the life of mDA neurons and would depend on additional components, such as Pitx3, in a core transcription factor network (Jacobs et al., 2009).
How may dysregulated Nurr1 activity contribute to PD? Studies in PD patients have shown that, in early stages of the disease, SNc cell bodies are relatively spared compared with the loss of DA in the putamen (Fearnley and Lees, 1991) and that a significant fraction of the surviving, pigmented, DA somata in the SNc have much reduced expression of the TH enzyme (Hirsch et al., 1988; Chu et al., 2006). This suggests that, during early stages of disease, nigral DA neurons may survive in a dysfunctional state characterized by a downregulated neurotransmitter machinery. An interesting possibility supported by our data is that reduced expression of Nurr1 contributes to such symptoms. Indeed, Nurr1 is severely reduced in neurons with signs of pathology in PD brain tissue, and reduced Nurr1 expression in patients' peripheral blood lymphocytes indicates that diminished Nurr1 activity may be a systemic feature of disease (Chu et al., 2006; Le et al., 2008). Although such correlations do not determine whether reduced Nurr1 expression is a cause or a consequence of disease, progressive cell dysfunction in Nurr1-ablated mice provides a clear indication that diminished Nurr1 expression in PD should have deleterious consequences for patients. This view is supported by the identification of Nurr1 gene variants that have been associated with rare cases of familial and sporadic PD (Xu et al., 2002; Le et al., 2003; Zheng et al., 2003; Jankovic et al., 2005; Grimes et al., 2006; Jacobsen et al., 2008). Although other studies have failed to identify such mutations and indicated that Nurr1 gene variants as a cause of PD must be very rare, the combined data from genetics, postmortem PD brain tissue analyses, and the ablation experiments reported here strongly imply that, if and when Nurr1 function is reduced, it will exaggerate PD progression and severity (Wellenbrock et al., 2003; Hering et al., 2004; Tan et al., 2004).
The results indicate that therapies that can restore Nurr1 activity in diseased but not yet degenerated mDA neurons could be of clinical relevance. We envision several strategies whereby Nurr1 activity could be increased. (1) Nurr1 belongs to the nuclear receptor family whose members are commonly regulated by small lipophilic ligands. The putative ligand binding domain of Nurr1 is unconventional and lacks a ligand-binding pocket, but Nurr1 forms heterodimers with retinoid X receptors (RXRs), and ligands activating these receptors can protect mDA neurons in culture (Wallen-Mackenzie et al., 2003; Wang et al., 2003). Thus, RXR may be a relevant target for ligand modulation of Nurr1-regulated processes. It will be important to investigate to what extent Nurr1–RXR heterodimers versus Nurr1 alone are important in pathways associated with the phenotype described in this paper. (2) Nurr1 activity is possible to modulate, for example, by the leukemia drug 6-mercaptopurine (Ordentlich et al., 2003). Although such drugs are pleiotropic and serious side effects are likely, other compounds with higher specificity may be possible to identify. (3) Therapies directed at increasing Nurr1 activity would be effective only as long as sufficient levels of Nurr1 are expressed in diseased neurons. Thus, treatments aimed at restoration of Nurr1 expression by gene delivery may prove advantageous. Using similar AAV vectors as administered in this study may be of particular interest because they have properties that are clinically favorable and are already used in clinical trials in PD patients (Check, 2007).
In conclusion, loss of Nurr1 at stages when characteristic features of mDA neurons are already apparent in the developing embryo or in fully differentiated adult neurons results in loss of mDA neuron-specific gene expression and neuron degeneration. These findings highlight the importance of developmental mechanisms also in the adult brain and clearly indicate that they may be critical for the understanding of cell maintenance and neurodegeneration. How Nurr1 and other transcription factors operate in adult neurons to control and prevent loss or drift in phenotype remains a challenge for future studies.
Footnotes
This work was supported by grants from the Michael J. Fox Foundation (T.P., A.B., L.O.), Vetenskapsrådet via Linné Center DBRM (T.P.), Grants-in-Aid for Human Frontier Science Program (P.C., D.M., H.I.), Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, the Ministry of Health, Labor, and Welfare of Japan, and the Japan Science and Technology Agency, Core Research for Evolutional Science and Technology (S.-I.M., H.I.), Vetenskapsrådet (L.O.), Swedish Brain Power (L.O.), Swedish Brain Foundation (L.O.), and the Swedish Parkinson Foundatin (L.O.). We are grateful to Johan Ericson and members of the Perlmann and Ericson laboratories for valuable discussions. We thank Andrée Dierich and Jean-Marc Bornert for their help in mouse mutagenesis, Noriko Ihira for help in construction of the targeting vector and screening of ES clones, Björn Anzelius and Naomi Takino for their help with AAV vectors, Ulla Jarl for help with histology, and Marie-Louise Alun for advice on mice handling.
References
- Alavian KN, Scholz C, Simon HH. Transcriptional regulation of mesencephalic dopaminergic neurons: the full circle of life and death. Mov Disord. 2008;23:319–328. doi: 10.1002/mds.21640. [DOI] [PubMed] [Google Scholar]
- Albéri L, Sgadò P, Simon HH. Engrailed genes are cell-autonomously required to prevent apoptosis in mesencephalic dopaminergic neurons. Development. 2004;131:3229–3236. doi: 10.1242/dev.01128. [DOI] [PubMed] [Google Scholar]
- Andersson E, Tryggvason U, Deng Q, Friling S, Alekseenko Z, Robert B, Perlmann T, Ericson J. Identification of intrinsic determinants of midbrain dopamine neurons. Cell. 2006;124:393–405. doi: 10.1016/j.cell.2005.10.037. [DOI] [PubMed] [Google Scholar]
- Breese GR, Knapp DJ, Criswell HE, Moy SS, Papadeas ST, Blake BL. The neonate-6-hydroxydopamine-lesioned rat: a model for clinical neuroscience and neurobiological principles. Brain Res Brain Res Rev. 2005;48:57–73. doi: 10.1016/j.brainresrev.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Castillo SO, Baffi JS, Palkovits M, Goldstein DS, Kopin IJ, Witta J, Magnuson MA, Nikodem VM. Dopamine biosynthesis is selectively abolished in substantia nigra ventral tegmental area but not in hypothalamic neurons in mice with targeted disruption of the Nurr1 gene. Mol Cell Neurosci. 1998;11:36–46. doi: 10.1006/mcne.1998.0673. [DOI] [PubMed] [Google Scholar]
- Check E. Second chance. Nat Med. 2007;13:770–771. doi: 10.1038/nm0707-770. [DOI] [PubMed] [Google Scholar]
- Chu Y, Le W, Kompoliti K, Jankovic J, Mufson EJ, Kordower JH. Nurr1 in Parkinson's disease and related disorders. J Comp Neurol. 2006;494:495–514. doi: 10.1002/cne.20828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobaleda C, Jochum W, Busslinger M. Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature. 2007;449:473–477. doi: 10.1038/nature06159. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, Hoffer B, Cullheim S, Mohammed AH, Olson L, Larsson NG. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnley JM, Lees AJ. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain. 1991;114:2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- Grimes DA, Han F, Panisset M, Racacho L, Xiao F, Zou R, Westaff K, Bulman DE. Translated mutation in the Nurr1 gene as a cause for Parkinson's disease. Mov Disord. 2006;21:906–909. doi: 10.1002/mds.20820. [DOI] [PubMed] [Google Scholar]
- Gurdon JB, Melton DA. Nuclear reprogramming in cells. Science. 2008;322:1811–1815. doi: 10.1126/science.1160810. [DOI] [PubMed] [Google Scholar]
- Hendricks T, Francis N, Fyodorov D, Deneris ES. The ETS domain factor Pet-1 is an early and precise marker of central serotonin neurons and interacts with a conserved element in serotonergic genes. J Neurosci. 1999;19:10348–10356. doi: 10.1523/JNEUROSCI.19-23-10348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering R, Petrovic S, Mietz EM, Holzmann C, Berg D, Bauer P, Woitalla D, Müller T, Berger K, Krüger R, Riess O. Extended mutation analysis and association studies of Nurr1 (NR4A2) in Parkinson disease. Neurology. 2004;62:1231–1232. doi: 10.1212/01.wnl.0000118285.18383.90. [DOI] [PubMed] [Google Scholar]
- Hermanson E, Joseph B, Castro D, Lindqvist E, Aarnisalo P, Wallén A, Benoit G, Hengerer B, Olson L, Perlmann T. Nurr1 regulates dopamine synthesis and storage in MN9D dopamine cells. Exp Cell Res. 2003;288:324–334. doi: 10.1016/s0014-4827(03)00216-7. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Graybiel AM, Agid YA. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature. 1988;334:345–348. doi: 10.1038/334345a0. [DOI] [PubMed] [Google Scholar]
- Hobert O. Regulatory logic of neuronal diversity: terminal selector genes and selector motifs. Proc Natl Acad Sci U S A. 2008;105:20067–20071. doi: 10.1073/pnas.0806070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huot P, Parent A. Dopaminergic neurons intrinsic to the striatum. J Neurochem. 2007;101:1441–1447. doi: 10.1111/j.1471-4159.2006.04430.x. [DOI] [PubMed] [Google Scholar]
- Jacobs FM, van Erp S, van der Linden AJ, von Oerthel L, Burbach JP, Smidt MP. Pitx3 potentiates Nurr1 in dopamine neuron terminal differentiation through release of SMRT-mediated repression. Development. 2009;136:531–540. doi: 10.1242/dev.029769. [DOI] [PubMed] [Google Scholar]
- Jacobsen KX, MacDonald H, Lemonde S, Daigle M, Grimes DA, Bulman DE, Albert PR. A Nurr1 point mutant, implicated in Parkinson's disease, uncouples ERK1/2-dependent regulation of tyrosine hydroxylase transcription. Neurobiol Dis. 2008;29:117–122. doi: 10.1016/j.nbd.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Jain S, Golden JP, Wozniak D, Pehek E, Johnson EM, Jr, Milbrandt J. RET is dispensable for maintenance of midbrain dopaminergic neurons in adult mice. J Neurosci. 2006;26:11230–11238. doi: 10.1523/JNEUROSCI.1876-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic J, Chen S, Le WD. The role of Nurr1 in the development of dopaminergic neurons and Parkinson's disease. Prog Neurobiol. 2005;77:128–138. doi: 10.1016/j.pneurobio.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Johnson NC, Dillard ME, Baluk P, McDonald DM, Harvey NL, Frase SL, Oliver G. Lymphatic endothelial cell identity is reversible and its maintenance requires Prox1 activity. Genes Dev. 2008;22:3282–3291. doi: 10.1101/gad.1727208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang BJ, Chang DA, Mackay DD, West GH, Moreira TS, Takakura AC, Gwilt JM, Guyenet PG, Stornetta RL. Central nervous system distribution of the transcription factor Phox2b in the adult rat. J Comp Neurol. 2007;503:627–641. doi: 10.1002/cne.21409. [DOI] [PubMed] [Google Scholar]
- Kim KS, Kim CH, Hwang DY, Seo H, Chung S, Hong SJ, Lim JK, Anderson T, Isacson O. Orphan nuclear receptor Nurr1 directly transactivates the promoter activity of the tyrosine hydroxylase gene in a cell-specific manner. J Neurochem. 2003;85:622–634. doi: 10.1046/j.1471-4159.2003.01671.x. [DOI] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Björklund A. Characterization of behavioral and neurodegenerative changes following partial lesions of the nigrostriatal dopamine system induced by intrastriatal 6-hydroxydopamine in the rat. Exp Neurol. 1998;152:259–277. doi: 10.1006/exnr.1998.6848. [DOI] [PubMed] [Google Scholar]
- Kittappa R, Chang WW, Awatramani RB, McKay RD. The foxa2 gene controls the birth and spontaneous degeneration of dopamine neurons in old age. PLoS Biol. 2007;5:e325. doi: 10.1371/journal.pbio.0050325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer ER, Aron L, Ramakers GM, Seitz S, Zhuang X, Beyer K, Smidt MP, Klein R. Absence of Ret signaling in mice causes progressive and late degeneration of the nigrostriatal system. PLoS Biol. 2007;5:e39. doi: 10.1371/journal.pbio.0050039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le W, Pan T, Huang M, Xu P, Xie W, Zhu W, Zhang X, Deng H, Jankovic J. Decreased NURR1 gene expression in patients with Parkinson's disease. J Neurol Sci. 2008;273:29–33. doi: 10.1016/j.jns.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le WD, Xu P, Jankovic J, Jiang H, Appel SH, Smith RG, Vassilatis DK. Mutations in NR4A2 associated with familial Parkinson disease. Nat Genet. 2003;33:85–89. doi: 10.1038/ng1066. [DOI] [PubMed] [Google Scholar]
- Oo TF, Kholodilov N, Burke RE. Regulation of natural cell death in dopaminergic neurons of the substantia nigra by striatal glial cell line-derived neurotrophic factor in vivo. J Neurosci. 2003;23:5141–5148. doi: 10.1523/JNEUROSCI.23-12-05141.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordentlich P, Yan Y, Zhou S, Heyman RA. Identification of the antineoplastic agent 6-mercaptopurine as an activator of the orphan nuclear hormone receptor Nurr1. J Biol Chem. 2003;278:24791–24799. doi: 10.1074/jbc.M302167200. [DOI] [PubMed] [Google Scholar]
- Pascual A, Hidalgo-Figueroa M, Piruat JI, Pintado CO, Gómez-Díaz R, López-Barneo J. Absolute requirement of GDNF for adult catecholaminergic neuron survival. Nat Neurosci. 2008;11:755–761. doi: 10.1038/nn.2136. [DOI] [PubMed] [Google Scholar]
- Perlmann T, Wallén-Mackenzie A. Nurr1, an orphan nuclear receptor with essential functions in developing dopamine cells. Cell Tissue Res. 2004;318:45–52. doi: 10.1007/s00441-004-0974-7. [DOI] [PubMed] [Google Scholar]
- Sacchetti P, Brownschidle LA, Granneman JG, Bannon MJ. Characterization of the 5′-flanking region of the human dopamine transporter gene. Brain Res Mol Brain Res. 1999;74:167–174. doi: 10.1016/s0169-328x(99)00275-2. [DOI] [PubMed] [Google Scholar]
- Sacchetti P, Mitchell TR, Granneman JG, Bannon MJ. Nurr1 enhances transcription of the human dopamine transporter gene through a novel mechanism. J Neurochem. 2001;76:1565–1572. doi: 10.1046/j.1471-4159.2001.00181.x. [DOI] [PubMed] [Google Scholar]
- Sakurada K, Ohshima-Sakurada M, Palmer TD, Gage FH. Nurr1, an orphan nuclear receptor, is a transcriptional activator of endogenous tyrosine hydroxylase in neural progenitor cells derived from the adult brain. Development. 1999;126:4017–4026. doi: 10.1242/dev.126.18.4017. [DOI] [PubMed] [Google Scholar]
- Saucedo-Cardenas O, Quintana-Hau JD, Le WD, Smidt MP, Cox JJ, De Mayo F, Burbach JP, Conneely OM. Nurr1 is essential for the induction of the dopaminergic phenotype and the survival of ventral mesencephalic late dopaminergic precursor neurons. Proc Natl Acad Sci U S A. 1998;95:4013–4018. doi: 10.1073/pnas.95.7.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schallert T, Norton D, Jones TA. A clinically relevant unilateral rat model of Parkinsonian akinesia. J Neural Transpl Plast. 1992;3:332–333. [Google Scholar]
- Simon HH, Thuret S, Alberi L. Midbrain dopaminergic neurons: control of their cell fate by the engrailed transcription factors. Cell Tissue Res. 2004;318:53–61. doi: 10.1007/s00441-004-0973-8. [DOI] [PubMed] [Google Scholar]
- Smidt MP, Burbach JP. How to make a mesodiencephalic dopaminergic neuron. Nat Rev Neurosci. 2007;8:21–32. doi: 10.1038/nrn2039. [DOI] [PubMed] [Google Scholar]
- Smidt MP, van Schaick HS, Lanctôt C, Tremblay JJ, Cox JJ, van der Kleij AA, Wolterink G, Drouin J, Burbach JP. A homeodomain gene Ptx3 has highly restricted brain expression in mesencephalic dopaminergic neurons. Proc Natl Acad Sci U S A. 1997;94:13305–13310. doi: 10.1073/pnas.94.24.13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smidt MP, Asbreuk CH, Cox JJ, Chen H, Johnson RL, Burbach JP. A second independent pathway for development of mesencephalic dopaminergic neurons requires Lmx1b. Nat Neurosci. 2000;3:337–341. doi: 10.1038/73902. [DOI] [PubMed] [Google Scholar]
- Smidt MP, Smits SM, Burbach JP. Homeobox gene Pitx3 and its role in the development of dopamine neurons of the substantia nigra. Cell Tissue Res. 2004;318:35–43. doi: 10.1007/s00441-004-0943-1. [DOI] [PubMed] [Google Scholar]
- Snyder AM, Zigmond MJ, Lund RD. Sprouting of serotoninergic afferents into striatum after dopamine-depleting lesions in infant rats: a retrograde transport and immunocytochemical study. J Comp Neurol. 1986;245:274–281. doi: 10.1002/cne.902450209. [DOI] [PubMed] [Google Scholar]
- Sonnier L, Le Pen G, Hartmann A, Bizot JC, Trovero F, Krebs MO, Prochiantz A. Progressive loss of dopaminergic neurons in the ventral midbrain of adult mice heterozygote for Engrailed 1. J Neurosci. 2007;27:1063–1071. doi: 10.1523/JNEUROSCI.4583-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tan EK, Chung H, Chandran VR, Tan C, Shen H, Yew K, Pavanni R, Puvan KA, Wong MC, Teoh ML, Yih Y, Zhao Y. Nurr1 mutational screen in Parkinson's disease. Mov Disord. 2004;19:1503–1505. doi: 10.1002/mds.20246. [DOI] [PubMed] [Google Scholar]
- Vult von Steyern F, Martinov V, Rabben I, Njå A, de Lapeyrière O, Lømo T. The homeodomain transcription factors Islet 1 and HB9 are expressed in adult alpha and gamma motoneurons identified by selective retrograde tracing. Eur J Neurosci. 1999;11:2093–2102. doi: 10.1046/j.1460-9568.1999.00631.x. [DOI] [PubMed] [Google Scholar]
- Wallen-Mackenzie A, Mata de Urquiza A, Petersson S, Rodriguez FJ, Friling S, Wagner J, Ordentlich P, Lengqvist J, Heyman RA, Arenas E, Perlmann T. Nurr1-RXR heterodimers mediate RXR ligand-induced signaling in neuronal cells. Genes Dev. 2003;17:3036–3047. doi: 10.1101/gad.276003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Benoit G, Liu J, Prasad S, Aarnisalo P, Liu X, Xu H, Walker NP, Perlmann T. Structure and function of Nurr1 identifies a class of ligand-independent nuclear receptors. Nature. 2003;423:555–560. doi: 10.1038/nature01645. [DOI] [PubMed] [Google Scholar]
- Wellenbrock C, Hedrich K, Schafer N, Kasten M, Jacobs H, Schwinger E, Hagenah J, Pramstaller PP, Vieregge P, Klein C. NR4A2 mutations are rare among European patients with familial Parkinson's disease. Ann Neurol. 2003;54:415. doi: 10.1002/ana.10736. [DOI] [PubMed] [Google Scholar]
- Xu PY, Liang R, Jankovic J, Hunter C, Zeng YX, Ashizawa T, Lai D, Le WD. Association of homozygous 7048G7049 variant in the intron six of Nurr1 gene with Parkinson's disease. Neurology. 2002;58:881–884. doi: 10.1212/wnl.58.6.881. [DOI] [PubMed] [Google Scholar]
- Zetterström RH, Williams R, Perlmann T, Olson L. Cellular expression of the immediate early transcription factors Nurr1 and NGFI-B suggests a gene regulatory role in several brain regions including the nigrostriatal dopamine system. Brain Res Mol Brain Res. 1996;41:111–120. doi: 10.1016/0169-328x(96)00074-5. [DOI] [PubMed] [Google Scholar]
- Zetterström RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science. 1997;276:248–250. doi: 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- Zhao ZQ, Scott M, Chiechio S, Wang JS, Renner KJ, Gereau RW, 4th, Johnson RL, Deneris ES, Chen ZF. Lmx1b is required for maintenance of central serotonergic neurons and mice lacking central serotonergic system exhibit normal locomotor activity. J Neurosci. 2006;26:12781–12788. doi: 10.1523/JNEUROSCI.4143-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K, Heydari B, Simon DK. A common NURR1 polymorphism associated with Parkinson disease and diffuse Lewy body disease. Arch Neurol. 2003;60:722–725. doi: 10.1001/archneur.60.5.722. [DOI] [PubMed] [Google Scholar]