

Abstract
ALCAM [activated leukocyte cell adhesion molecule (BEN/SC-1/DM-GRASP)] is a transmembrane recognition molecule of the Ig superfamily (IgSF) containing five Ig domains (two V-type, three C2-type). Although broadly expressed in the nervous and immune systems, few of its developmental functions have been elucidated. Because ALCAM has been suggested to interact with the IgSF adhesion molecule L1, a determinant of retinocollicular mapping, we hypothesized that ALCAM might direct topographic targeting to the superior colliculus (SC) by serving as a substrate within the SC for L1 on incoming retinal ganglion cell (RGC) axons. ALCAM was expressed in the SC during RGC axon targeting and on RGC axons as they formed the optic nerve; however, it was downregulated distally on RGC axons as they entered the SC. Axon tracing with DiI revealed pronounced mistargeting of RGC axons from the temporal retina half of ALCAM null mice to abnormally lateral sites in the contralateral SC, in which these axons formed multiple ectopic termination zones. ALCAM null mutant axons were specifically compromised in medial orientation of interstitial branches, which is known to require the ankyrin binding function of L1. As a substrate, ALCAM–Fc protein promoted L1-dependent attachment of acutely dissociated retinal cells and an L1-expressing, ALCAM-negative cell line, consistent with an ALCAM–L1 heterophilic molecular interaction. Together, these results suggest a model in which ALCAM in the SC interacts with L1 on RGC axons to promote medial extension of RGC axon branches important for mediolateral axon targeting in the formation of retinocollicular maps.
Introduction
Axon guidance is orchestrated through multiple receptor/ligand interactions. In the projection of retinal ganglion cell (RGC) axons to the superior colliculus (SC), axons from graded positions within the retina are mapped to SC targets along anteroposterior and mediolateral axes, specified by A- and B-class Eph receptors and Ephrin ligands. It has been suggested that gradients of EphrinB1 in the SC (high-medial to low-lateral) and EphB2 and EphB3 on RGC axons (high-ventral to low-dorsal) help to map the dorsoventral retinal axis to the mediolateral SC (Hindges et al., 2002; McLaughlin et al., 2003). Conversely, gradients of EphrinA2 and EphrinA5 in the SC and EphA receptors (EphA5, EphA6) on RGC axons map the temporonasal retinal axis to the anteroposterior SC (Feldheim et al., 2000; Yates et al., 2001; Hansen et al., 2004). EphrinBs are chemoattractants for RGC axons with elevated EphB (Hindges et al., 2002; McLaughlin et al., 2003), whereas EphrinAs are repellents for axons with elevated EphA (Feldheim et al., 2000; Yates et al., 2001; Hansen et al., 2004). Ephrins and Eph receptors do not fully account for the map, because the phenotypes of mice with Ephrin or Eph loss-of-function mutations are not fully penetrant (Feldheim et al., 2000; Hindges et al., 2002).
Ig superfamily (IgSF) molecule L1 (Maness and Schachner, 2007) is a determinant of retinocollicular targeting, providing dynamic adhesion to the actin cytoskeleton (Demyanenko and Maness, 2003; Buhusi et al., 2008). Coupling the L1 cytoplasmic domain to filamentous actin by reversible binding to ankyrin (Garver et al., 1997) is required for mediolateral positioning of RGC axons in the SC (Buhusi et al., 2008). Tyrosine to histidine mutation of a critical residue in the L1 cytoplasmic domain in L1(Y1229H) knock-in mice disrupts ankyrin binding, causing pronounced mediolateral retinocollicular axon targeting errors (Buhusi et al., 2008). This and other L1 mutations occur in a human X-linked mental retardation syndrome with optic atrophy (Kenwrick et al., 2000).
We investigated the cell adhesion molecule ALCAM (activated leukocyte cell adhesion molecule/BEN/SC-1/DM-GRASP/Neurolin) as a potential ligand for L1 in retinotopic mapping. ALCAM is a transmembrane IgSF implicated in T-cell activation and cancer cell progression (van Kilsdonk et al., 2008) that is expressed in developing retina and brain (DeBernardo and Chang, 1996; Weiner et al., 2004). ALCAM null mice exhibit photoreceptor ectopias resembling retinal folds in human retinopathies and subtle RGC axon defasciculation within the retinal nerve fiber layer (Weiner et al., 2004). Although ALCAM orthologs have been shown to contribute to RGC axon guidance within the retina and optic tract of fish (Ott et al., 1998; Diekmann and Stuermer, 2009) and chick (DeBernardo and Chang, 1996; Avci et al., 2004), a role in topographic mapping of RGC axons, in any species, remains unexplored. We show here that ALCAM is a novel mediator of topographic targeting of retinal axons, likely by functioning as a substrate for L1 on incoming RGC axons.
Materials and Methods
Mice.
The generation and initial characterization of ALCAM null mice has been reported previously (Weiner et al., 2004). In this line, the first exon of the ALCAM gene has been replaced by a cDNA encoding green fluorescent protein (GFP), resulting in a lack of any detectable ALCAM transcripts and of the encoded protein. Heterozygotes were intercrossed to produce wild-type (WT) and homozygous null mutants in the same litters for analyses. All animals were used according to the Institutional Animal Care and Use Committee policies of The University of North Carolina at Chapel Hill and The University of Iowa and in accordance with National Institutes of Health guidelines.
Axon tracing and analysis.
Axonal tracing was performed as described previously (Simon and O'Leary, 1992; Demyanenko and Maness, 2003). ALCAM null mutant and WT littermates at postnatal day 0 (P0) to P2 or P8–P10 were anesthetized, and anterograde tracing was performed by focal injection of DiI (Invitrogen) as an 10% solution in dimethylformamide or dimethylsulfoxide into the peripheral retina using a Picospritzer II (General Valve) and glass micropipettes (internal diameter, 40 μm). After 48 h, mice were anesthetized and perfused transcardially with 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4. The eye was inspected to identify the injection site and assess its position in a given quadrant relative to extraocular muscles (lateral and inferior recti). The eyes were removed, and incisions were made to demarcate retinal quadrants. To verify the injection sites and analyze the projection areas, injected retinas, SC, and inferior colliculus (IC) were whole mounted onto glass slides and examined by epifluorescence and confocal microscopy. Analysis was restricted to mice in which retinal whole mounts displayed a single DiI injection of appropriate size and positioning relative to insertion sites of extraocular muscles. The boundaries of the SC and IC were determined by their characteristic shape and location. Termination zones (TZs) were verified by their branched appearance and density at high magnification.
To quantify branch orientation from RGC axons, DiI was injected into the ventrotemporal (VT) retina of ALCAM null and WT littermates at P2 and analyzed at P3. The SC was divided into three bins along the mediolateral axis, relative to an emerging TZ: lateral to TZ, within TZ, and medial to TZ, as described previously (Hindges et al., 2002). The total number of labeled axons and branches was counted in confocal Z-stacks, and medial or lateral branch orientation was recorded for each bin. For each bin, the number of medial branches minus the number of lateral branches was divided by the total number of branches to arrive at the branch directional coefficient (DC). A positive DC indicated more medial than lateral branches, whereas a negative DC indicated more lateral than medial branches. The mean directional coefficient for each genotype was computed and compared by a mixed ANOVA.
To evaluate the distribution of DiI-labeled axons from WT and ALCAM null littermates entering the SC at P2–P3, after retinal injections at P1, the entrance zone into the SC was divided into 10 equal bins, four medial to the principal TZ and six lateral to the TZ, as described previously (Hindges et al., 2002). The percentage of axons entering the SC in each bin was determined for each mouse and averaged over groups. The distribution of axons (expressed as percentage of total for each bin) was analyzed by a mixed ANOVA.
Dorsoventral pretarget axon sorting.
ALCAM null and WT littermates were analyzed for pretarget sorting of dorsal and ventral RGC axons in postnatal or adult mice by methods used previously (Plas et al., 2005). ALCAM mutant and WT littermates at P0–P1 were anesthetized and injected with DiI into the ventral retinal quadrant and 1,1′-dilinoleyl-3,3,3′,3′-tetra-methylindocarbocyanine, 4-chlorobenzenesulphonate (DiA) into the dorsal retinal quadrant, using a Picospritzer as described for axon tracing. After 48 h, these mice were perfused with 4% paraformaldehyde. For adult mice, DiI and DiA crystals were placed into the dorsal or ventral retinal quadrants in paraformaldehyde-fixed tissues, followed by incubation at 4°C for 75 d. Brains from each age were sectioned horizontally and analyzed for the distribution of DiI- and DiA-labeled axons in the optic tract located between the chiasm and the delta (before the LGN) and in the brachium in which axons entered the SC, by methods described previously (Plas et al., 2005). Positions of RGC axons were scored for locations in the dorsal, ventral, medial, or lateral quadrants of the optic tract from confocal images. In some cases, whole mounts of the SC were imaged to observe the distribution of axons as they passed into the SC from the brachium.
Labeling and quantifications of RGC projections to the dLGN.
WT and ALCAM−/− mice were subdued in a chamber with isoflurane. At P10, each mouse received an injection of Alexa 488-conjugated cholera toxin subunit B (green) in one eye and an injection of Alexa 594-conjugated cholera toxin subunit B (red) in the opposite eye (Alexa Fluors; Invitrogen). At P12, mice were perfused with 4% paraformaldehyde, and the brains extracted, stored in protein-free medium for 24 h, and sectioned at 200 nm. The sections were placed on microscope slides.
Fluorescent images of WT and ALCAM−/− mouse brain sections containing the dLGN were examined on a Zeiss Axioplan microscope and captured using universal gains and exposures on a Zeiss Axiocam color CCD camera. Unmodified images of the dLGN were analyzed in Adobe Photoshop using a multi-threshold protocol based on the methods described by Torborg et al. (2005). The multi-threshold protocol provides a range of signal-to-background levels for analysis of the size of the ipsilateral axon area (termed “ipsilateral patch”) relative to the total size of the dLGN, as well as any overlap between the ipsilateral patch and the contralateral axon targeting area in the dLGN. The pixel areas of the modified images were measured in NIH ImageJ for each threshold level, delivering values for both ipsilateral and overlap area. Total dLGN area was measured by selecting the unmodified contralateral image in NIH ImageJ and measuring total area. Means of ipsilateral area/dLGN area for each genotype were compared at each threshold level by the t test (two-tailed, p < 0.05).
Immunofluorescence staining and in situ hybridization.
Postnatal mice (P2 and P9) were deeply anesthetized by isoflurane and decapitated. Eyes and brain tissues were removed and fixed by immersion in 4% paraformaldehyde in PBS at 4°C overnight. For eyes, a 30 gauge needle was used to puncture the cornea to allow for better penetration of fixative; for the SC, brains were hemisected, and the front of the cerebral hemisphere was removed. Samples were cryoprotected in 30% sucrose in PBS at 4°C overnight, then embedded in Tissue-Tek OCT, snap-frozen in isopentane cooled in a dry ice/ethanol bath, and stored at −80°C. Cryostat sections (14 μm) were blocked with 2.5% bovine serum albumin (BSA) in PBS with 0.1% Triton X-100, and sections were incubated with primary antibodies (Ab) [rat anti-L1 monoclonal antibody (mAb), 1:200 (Millipore); 1:100, goat anti-ALCAM AF1172 (R & D Systems); rabbit anti-EphrinB1 (A-20), 1:100 (Santa Cruz Biotechnology); mouse anti-neurofilament (NF) mAb SMI-312, 1:1000 (Sternberger Monoclonals); rabbit anti-GFP Ab, 1:1000 (Invitrogen)] in the same solution at 4°C overnight. Sections were washed in PBS and incubated for 1 h at room temperature with secondary antibodies coupled to Alexa 488 or 568 or to cyanine 3, washed in PBS again, and coverslipped using Gel/Mount (Biomeda). Control sections were incubated with secondary antibodies only, and the specificity of the ALCAM antibody (AF1172) was confirmed by lack of staining on sections from ALCAM null mice (see Fig. 1). Images were captured on a Leica DM5000B epifluorescence microscope using nonoverlapping ET series filters (Chroma Technology). For in situ hybridization to detect ALCAM or L1 mRNA, sense and antisense probes were generated from psBluescript SK plasmid containing mouse L1 cDNA (Macias et al., 2002) or a 900 bp fragment of mouse ALCAM cDNA (Weiner et al., 2004), and hybridization was performed using digoxigenin-labeled probes as described previously (Bartsch et al., 1994; Garrett and Weiner, 2009).
Figure 1.
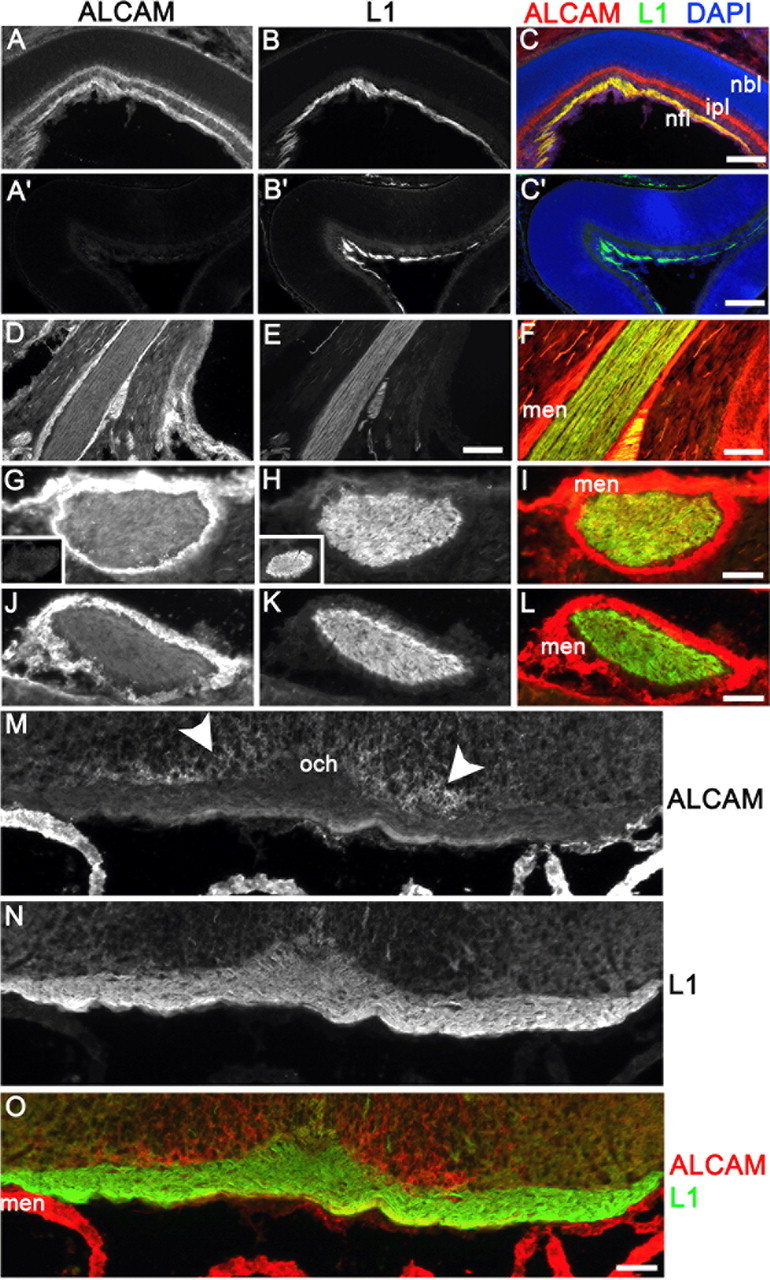
ALCAM and L1 colocalize on RGC axons proximal but not distal to the eye. Immunostaining was performed on cryostat sections from P2 WT and ALCAM null retina and optic nerve using antibodies against ALCAM (red) and L1 (green). Within the nerve fiber layer (nfl) of the retina (A–C) and in the optic nerve as it exits the eye (D–F; sagittal section), ALCAM and L1 colocalized on RGC axons. In ALCAM null retinae, no staining was observed using the anti-ALCAM antibody (A′), but staining for L1 was unaffected (B′, C′). ALCAM was also prominent in the nascent inner plexiform layer (ipl; C) and meninges surrounding the nerve (men; F, I, L). Nuclei were counterstained with DAPI (C). Coronal sections through the proximal optic nerve (G–I) and more distal optic nerve (J–L) showed progressively reduced expression of ALCAM but not of L1. Specificity of ALCAM staining was confirmed by its complete loss from the proximal optic nerve of ALCAM null mutants (inset in G), despite strong L1 expression (inset in H). ALCAM was essentially absent from RGC axons as they crossed the optic chiasm (och; M) but was expressed in some cells near the chiasm located superficially with respect to the RGC axons (arrowheads, M). Scale bars: A–C, 75 μm; D–F, 150 μm; G–O, 50 μm.
Cell adhesion assay.
Cell adhesion assays were performed using the L1-expressing human uveal melanoma cell line MUM2C (Seftor et al., 2002) or dissociated cells from the ventral or dorsal halves of retinas of WT mice at P3. MUM2B and MUM2C cell lines were subcloned from a heterogeneous uveal (iris, ciliary body, and choroid) melanoma tumor that had metastasized to the liver (Seftor et al., 2002). A complete characterization of ALCAM in these and other uveal melanoma cell lines will be reported separately (K. M. Jannie and J. A. Weiner, unpublished observations). Adhesion assays were performed essentially as described by Nelissen et al. (2000) and Buhusi et al. (2008) using chambered glass slides (Nunc). Slides were coated with poly-d-lysine without (MUM2C) or with (retinal cells) 3 μg/ml fibronectin and then treated with 4 μg/ml goat anti-human Fc-(Fab′)2 (Jackson ImmunoResearch) in TSM (in mm: 20 Tris, 150 NaCl, 1 CaCl2, and 2 MgCl2, pH 8.0) for 1 h at 37°C. After blocking with 1% BSA in TSM, slides were coated with 250 ng/ml ALCAM–Fc (R & D Systems) in TSM with 1% BSA for 1 h at 37°C. Cells (50,000 cells per chamber) in serum-free RPMI 1640 medium were plated and allowed to adhere in quadruplicate on wells with or without ALCAM–Fc for 2 h at 37°C. Cells were washed twice in TSM with 1% BSA and fixed in 4% paraformaldehyde. After washing in HBSS, cells were stained with 10 μm bis-benzimide, washed, and mounted in Vectashield. The number of attached cells in 10 random fields (0.38 cm2) was scored under epifluorescence microscopy, and the mean ± SEM number of cells per field was calculated. To distinguish L1-dependent and -independent attachment, cells were pretreated for 15 min at 4°C with function-blocking L1 monoclonal antibody (Neuro4; 20 μg/ml; gift from J. Hemperly, Becton Dickinson, Research Triangle Park, NC) or normal mouse IgG (20 μg/ml), and adhesion was measured after 2 h at 37°C. Means of adherent cells were compared by Student's t test (one-tailed, p < 0.05).
Retinal cell adhesion assay was performed in Mat-Tek glass-bottomed slides essentially as described above for MUM2C cells with few modifications. Poly-d-lysine-treated slides were first coated with 3 μg/ml human plasma fibronectin (Invitrogen) at 4°C for overnight, washed twice in 1× PBS, and then coated with 4 μg/ml goat anti-human Fc-(Fab′)2 (Jackson ImmunoResearch) in TSM for 1 h at 37°C. After two washes in TSM, the slides were blocked with 1% BSA in TSM for 30 min at 37°C. Slides were coated with 250 ng/ml ALCAM–Fc (R & D Systems) in TSM with 1% BSA for 1 h at 37°C. After two washes with TSM, Neurobasal-A medium (Invitrogen) was added to the wells until the cells were ready to be seeded. Dissociated ventral or dorsal retinal cells were prepared as described previously (Buhusi et al., 2008), and 50,000 cells per chamber were plated in Neurobasal-A medium containing 1× B27 supplement, 1× N2 supplement, and 50 ng/ml BDNF on ALCAM–Fc or Fc as substrates. For stimulating retinal cells with EphrinB1, human EphrinB1–Fc fusion protein (3 nm; R & D Systems) or normal human IgG (3 nm) as control were preclustered with 0.3 nm goat anti-human Fc antibody (Jackson ImmunoResearch) for 1 h on ice. Preclustered EphrinB1–Fc or normal IgG was added to the cells before plating. After 24 h, incubation cells were washed, fixed, stained, and analyzed as described above for MUM2C cells.
Results
ALCAM is expressed in the developing retinocollicular pathway
Topographic mapping of RGC axons to coordinates in the SC begins at approximately embryonic day 14 (E14) in the mouse, when axons arrive at the anterior SC. Axons subsequently grow posteriorly across the SC, overshooting their future TZs and covering the SC completely by P0 (McLaughlin and O'Leary, 2005). Critical to map formation, overshooting RGC axons branch interstitially at approximately P2 with a mediolateral bias for the topographically correct location of the future TZ. The map develops from P2 to P10 by a balance of branch attraction and repulsion mediated by factors, including EphrinB1 and Wnt3 gradients in the SC (Hindges et al., 2002; McLaughlin et al., 2003; Schmitt et al., 2006). We found previously that ALCAM was first detectable at E12 in mouse retina on axons of the earliest-differentiating RGCs and that expression persisted into adulthood in large subsets of RGCs and amacrine cells, as well as in the choroid (Weiner et al., 2004). Here a detailed immunofluorescence analysis in WT mice during the major stage of retinocollicular targeting (P2) confirmed the prominent expression of ALCAM on RGC axons in the nerve fiber layer and further showed that ALCAM colocalized completely with L1 (Fig. 1A–C). ALCAM was also expressed in the inner plexiform layer, in which L1 was not detectable, but neither protein was expressed in the proliferative neuroblast layer. ALCAM and L1 continued to be prominently colocalized on RGC axons exiting the eye, as shown in the proximal optic nerve (Fig. 1D–F). ALCAM labeling was also strong in the meninges (men) surrounding the optic nerve (Fig. 1D,G). The specificity of ALCAM immunoreactivity was confirmed by the absence of staining in the ALCAM null mutant retina (Fig. 1A′) and optic nerve (Fig. 1G, inset), in which L1 expression remained strong (Fig. 1B′, C′, H, inset). In contrast to the pronounced colocalization of ALCAM with L1 in RGC axons proximal to the eye at P2 (Fig. 1G–I), ALCAM, but not L1, progressively decreased as axons approached the optic chiasm (Fig. 1J–L). As shown in coronal sections at the level of the optic chiasm, little or no staining for ALCAM was seen in RGC axons, whereas L1 staining was strong (Fig. 1M–O). ALCAM remained high, however, in the meninges and in some cells superficial to the optic chiasm (Fig. 1M, arrowheads).
Analysis of mice heterozygous for the ALCAM mutant allele, in which GFP replaces the first exon of the ALCAM gene (Weiner et al., 2004), demonstrated that ALCAM was expressed by cells intrinsic to the SC. Staining of the P2 ALCAM+/− SC with antibodies against GFP revealed expression from the ALCAM locus in many cells in the SC, located mostly within deep laminae, including the stratum griseum intermediale (SGI), as well as in the stratum opticum (SO) and meninges, with less staining in the stratum griseum superficiale (SGS) (Fig. 2A). Double staining with antibodies to neurofilament (NF) (Fig. 2B) showed that cells expressing GFP from the ALCAM locus contact NF-labeled RGC axon bundles within the SO (Fig. 2C). Double immunostaining with ALCAM and L1 antibodies (Fig. 2D,E) also showed that L1 was primarily expressed on axon bundles coursing through several SC layers and that ALCAM protein could be seen to localize to neuropil that surrounded and closely apposed the L1-expressing axon bundles but was not prominent in the bundles themselves (Fig. 2F), consistent with its downregulation on RGC axons distal to the eye (Fig. 1). Double immunostaining with antibodies against L1 and NF (Fig. 2J,K) showed that most L1 protein in the SC was found on axon bundles in the SO (Fig. 2L), as well as in deeper layers; non-axonal L1 was only observed in the SGS (Fig. 2J).
Figure 2.
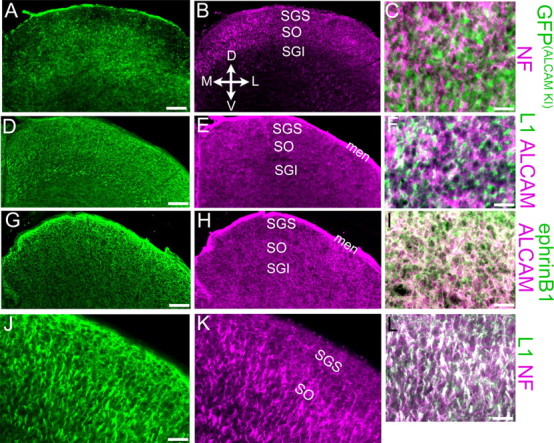
ALCAM is expressed by cells of the superior colliculus. Immunostaining of P2 WT (D–L) or ALCAM+/− (A–C) superior colliculus using antibodies against ALCAM, GFP (to detect its expression from the ALCAM locus in ALCAM+/− mice), NF, L1, or EphrinB1 is shown in representative coronal cryostat sections. Coordinates for all panels are indicated in B; midline is at or near the left edge of each panel. GFP expressed from the ALCAM locus [GFP(ALCAMKI)] in heterozygotes labels cell bodies near the SO and in deeper layers of the SC, including those in contact with NF-labeled RGC axon bundles within the SO (note apposition but little overlap in the higher magnification merge; C). D–F, In WT SC, ALCAM protein is diffusely distributed throughout the neuropil, including that innervated by L1-positive axon bundles in the SO; the merged image (F) shows ALCAM protein adjacent to, but mostly excluded from, these L1-positive axons. ALCAM and EphrinB1 exhibited extensive colocalization in the SC neuropil (seen as white staining in the merged panel; I), although EphrinB1 was more prominent in the upper SGS than was ALCAM (G, H). J–L, L1 protein was primarily found on NF-positive RGC axon bundles in the SO (white staining in the merged panel; L), although some NF-negative L1 staining, presumably from intrinsic SC cells, was observed in the SGS (J). D, Dorsal; V, ventral; M, medial; L, lateral; men, meninges. The sections in A–F are from the rostral SC; those in G–L are from approximately midway through its rostrocaudal extent. Scale bars: A, B, D, E, G, H, 100 μm; J, K, 40 μm; C, F, I, L, J, 25 μm.
Double staining of the SC at P2 for ALCAM and EphrinB1 showed that they were extensively colocalized within the neuropil of the SGC, SO, SGI, and deeper layers of the SC, as well as in the meninges (Fig. 2G–I). A high-medial/low-lateral EphrinB1 gradient has been documented in cell bodies of radial cells in ventricular zone of the mouse SC by in situ hybridization (Hindges et al., 2002), and EphrinB gradients have also been reported in the optic tectum of the chick (Braisted et al., 1997) and Xenopus (Mann et al., 2002), in which they function in retinotectal mapping. A graded pattern of EphrinB1 protein was not apparent in our immunofluorescence staining of the SC. An EphrinB1 protein gradient may be more difficult to detect in the superficial layers of the mouse SC by immunostaining, because of the shallow nature of the gradient, which declines by only ∼10% across the mediolateral SC axis (Hindges et al., 2002). At P9, ALCAM immunoreactivity in the SC had declined in most layers of the SC, although it remained high in the meninges (data not shown).
Consistent with the localization of ALCAM protein in the SC, in situ hybridization with an ALCAM antisense riboprobe demonstrated that ALCAM transcripts were abundant in cells located within the SC at P2 (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material), whereas control labeling with the sense riboprobe was negative (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). ALCAM transcripts decreased to lower levels in the SC by P9, when retinocollicular mapping neared completion (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). In situ hybridization with an L1 antisense riboprobe showed relatively low levels of L1 transcripts within the SC at P2 compared with retinal ganglion cells in the neural retina at the same stage (supplemental Fig. 1D–G, available at www.jneurosci.org as supplemental material). Together, these detailed expression analyses demonstrate that ALCAM is expressed in the SC during retinotopic map formation in a pattern primarily complementary to that of incoming L1-positive RGC axons.
Defective mediolateral targeting of retinal axons in ALCAM null mutant mice
To investigate a functional role for ALCAM in synaptic targeting of retinal axons, projections of RGC axons to the SC in ALCAM null and WT littermates were analyzed at P10–P12, when the retinocollicular map resembles its mature form. The axon tracer DiI was focally injected into the peripheral retina of live, anesthetized mice at P8, and labeled RGC TZs were analyzed after 2 d in the SC. DiI injection into the temporal retinal half of WT mice (10 of 10) labeled a single dense TZ in the anterior region of the contralateral SC as expected (Fig. 3A,B). The injection site varied in regard to the precise location and size within the temporal retinal half and thus could include some ventrotemporal RGCs. In striking contrast, DiI injection into the temporal retina of ALCAM null mutant mice revealed aberrant targeting of RGC axons along the mediolateral axis of the SC with high penetrance in 12 of 14 cases. Axons from the temporal retina of ALCAM null mice usually terminated in one or more laterally displaced ectopic TZs (eTZs), in addition to a normally positioned TZ in the mid-anterior SC (Fig. 3D–F). Often the ectopic TZs of ALCAM mutants were slightly malpositioned posteriorly in the SC as well (Fig. 3E,F). The small anteroposterior shift of ALCAM mutant axons might arise from competitive interactions with mutant axons that are displaced laterally within the SC or else indicate a minor involvement with the EphrinA/EphA system, which regulates topographic targeting along the anteroposterior SC axis (Feldheim et al., 2000). In contrast to the observed defects in SC targeting of temporal axons, the projection of RGC axons from the nasal retina was not perturbed in ALCAM mutants. Injections of DiI into the nasal retina of ALCAM null mice (4 of 4) resulted in labeling of a single TZ within the mid-posterior SC (Fig. 3I,J), which occurred at the same location as in WT SC (4 of 4) (Fig. 3G,H).
Figure 3.

Defective mediolateral retinocollicular targeting in ALCAM null mutants. A, B, DiI injections into the temporal retinal half (shown in flat mount) of WT mice at P8 labeled a single TZ in the anterior SC when analyzed at P10. C–F, DiI injections into the temporal retina of ALCAM null mice revealed eTZs displaced to lateral posterior positions in the P10 SC. Higher-magnification images of the eTZs are shown for each example. G, H, DiI injections into the nasal retina of WT mice resulted in labeling of a single TZ in the posterior SC. I, J, DiI injections into the nasal retina of ALCAM null mutant mice resulted in normal labeling of a single TZ in the posterior SC. K, L, DiI injections into the ventral retina of ALCAM null mutant mice resulted in normal labeling of a single TZ in the medial SC that was elongated in the anteroposterior axis. M, N, DiI injections into the dorsal retina of ALCAM null mutant mice resulted in normal labeling of a single TZ in the lateral SC. L, Lateral; M, medial; A, anterior; P, posterior; T, temporal; N, nasal; V, ventral; D, dorsal.
Projections of RGC axons originating in the ventral or dorsal retina of ALCAM null mutant mice terminated normally within the SC. Ventral retinal injections in ALCAM null mice (9 of 9) resulted in labeling of a single TZ within the extreme medial SC (Fig. 3K,L), similar to WT mice (4 of 4; data not shown). Elongation of the ventral retinal projection along the anteroposterior axis occurred in both WT and mutant mice and was more apparent with injections that included ventrotemporal RGCs. This elongation was shown previously for WT ventrotemporal axons and corresponded to the binocular vision field in which ipsilateral and contralateral projections terminate (Plas et al., 2005). Projections of RGC axons from the dorsal retina of ALCAM mutant mice to the lateral SC was also similar to WT. Dorsal retinal injections in ALCAM−/− mice (6 of 6) resulted in labeled TZs in the lateral SC (Fig. 3M,N), as also observed in WT (4 of 4; data not shown) (Buhusi et al., 2008).
The percentage of ALCAM nulls whose temporal axons terminated in abnormally positioned TZs was significantly greater than WT (χ2 = 18.66, p < 0.00003) (Fig. 4). Although most ALCAM mutants (11 of 14) had a correctly positioned TZ connected to one or more eTZs, one ALCAM mutant (1 of 14) had a single laterally displaced eTZ with no normally positioned TZ. Two ALCAM mutants (2 of 14) had a normal TZ in the medial SC that was similar to WT. A schematic representation of the location of TZs of ALCAM knock-out mice shows the termination positions of temporal axons from ALCAM null mice that had one correct TZ connected to at least one laterally displaced eTZ (Fig. 4).
Figure 4.

Abnormally positioned termination zones of temporal RGC axons in ALCAM null mutants. Left, Percentage of WT (n = 10) and ALCAM null mutant (n = 14) mice with abnormally positioned TZs in the SC after DiI injections into the temporal retina at P8 and analysis at P10. Right, Schematic diagram of the location of TZs (open circles) and aberrant (single displaced or multiple) eTZs (filled circles) in ALCAM null mice. The centers of TZs and eTZs from mutant mice (n = 14) are marked and connected. The range of location of TZs of WT axons in the SC is depicted by the dotted oval. L, Lateral; M, medial; A, anterior; P, posterior.
Loss of ALCAM impairs the orientation of retinal axon branches during establishment of the retinotopic map
To investigate the mechanism responsible for the formation of ectopic termination zones of RGC axons in the ALCAM mutant SC, the development of the contralateral retinocollicular projection was analyzed in mutants and WT littermates. RGC axons from different parts of the retina respond to the mediolateral SC gradient of EphrinB1 specified by relative levels of EphB receptors in RGC axons and EphrinB1 in the SC (McLaughlin et al., 2003). It has been suggested that branches arborize when attractive and repulsive forces are balanced, thus allowing formation of TZs (McLaughlin et al., 2003). RGC axons from the VT retina at P2–P3 project more medially to the anterior SC than purely temporal axons; thus, defects in mediolateral targeting would be most apparent in these axons. Therefore, we performed DiI injections into WT and ALCAM null temporal retina at P2, selected injections in which VT axons were preferentially labeled, and analyzed interstitial axon branches in the SC at P3. DiI labeling of WT VT retinal axons showed retinal axons and their branches developing within the anterior SC, including some that were located laterally (Fig. 5A). Interstitial branches extending from WT RGC axons in the lateral SC were preferentially oriented in the medial direction (Fig. 5B, arrows). Branches extending from WT VT axons in the medial SC tended to be oriented laterally (data not shown). In contrast, DiI labeling of VT axons in ALCAM null mice appeared to be more broadly distributed across the mediolateral axis of the anterior SC (Fig. 5C), and many axons in the lateral SC had interstitial branches that tended to be directed laterally, away from a correct location for the TZ (Fig. 5D, arrows).
Figure 5.
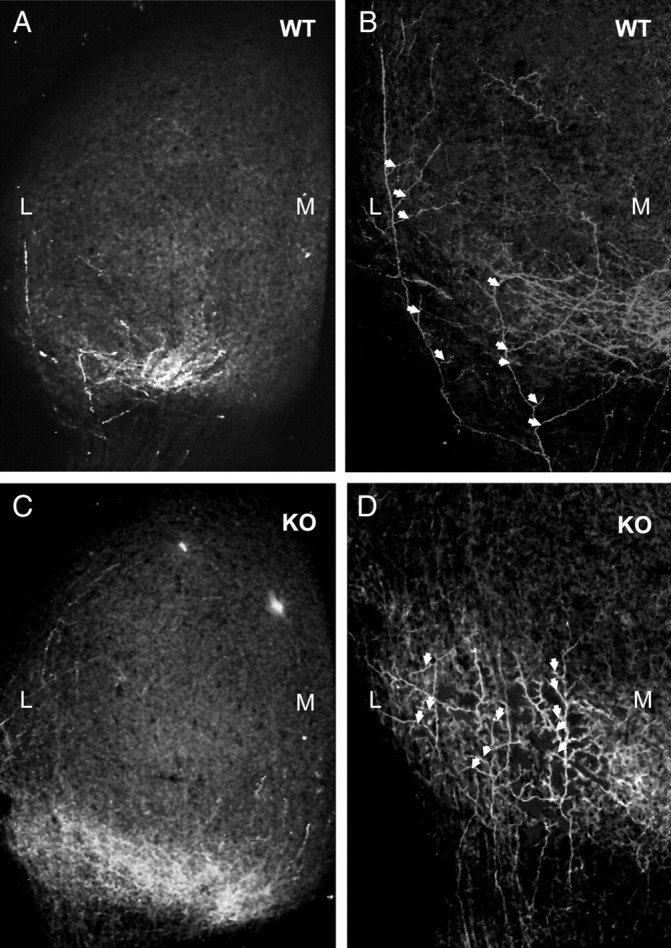
Misorientation of interstitial branches of RGC axons in ALCAM null mutants. WT and ALCAM null mutant RGC axons were labeled by DiI injection into the VT retina at P2 and analyzed at P3. A, Most branches extending from axons located lateral to the future TZ were oriented medially, as seen in a higher magnification in B (arrows) from confocal Z-stacks. C, DiI labeling of VT axons in the SC of ALCAM mutant mice showed abnormal enrichment at a lateral location, as well as at the appropriate anteromedial location. D, Many interstitial branches of mutant VT axons were abnormally oriented laterally within the SC (arrows), as shown at higher magnification of the boxed area in C. M, Medial; L, lateral; KO, knock-out.
To quantitatively evaluate axon branch orientation in WT and ALCAM mutant mice, the SC was divided into three bins along the mediolateral axis relative to the location of the emerging TZ: lateral to TZ, within TZ, and medial to TZ (Fig. 6A). The total number of labeled axons and branches was measured, and medial or lateral branch orientation was scored for each bin (Fig. 6A,B). Results were expressed as the directional coefficient (DC) as defined previously (Hindges et al., 2002), which was calculated by subtracting the number of lateral branches from the number of medial branches and dividing by the total number of branches. A positive DC indicated more medial than lateral branches, whereas a negative DC indicated more lateral than medial branches. This analysis clearly revealed that interstitial VT axon branches of WT mice (n = 10) preferentially oriented along the mediolateral axis toward the future correct TZ. The majority of branches in the lateral bin were oriented medially toward the TZ, branches in the TZ bin were primarily unbiased, and branches in the medial bin tended to be oriented laterally toward the TZ (Fig. 6A,B). In striking contrast, many VT axons of ALCAM mutants (n = 9) in the lateral bin and the TZ bin had branches oriented laterally with respect to the correctly located TZ, whereas branches of axons in the medial bins were normally oriented. Comparison of DCs by a mixed ANOVA in WT mice (n = 10 mice; 650 branches) and ALCAM mutant mice (n = 9 mice; 1076 branches) showed a significant main effect of genotype (F(1,17) = 81.9, p < 0.001), demonstrating that branch orientation in ALCAM mutant mice was different from WT. Analysis of the interaction between bin and genotype indicated a significant difference in branch orientation of WT and mutant axons in the lateral bin (F(1,17) = 125.2, p < 0.001) and TZ bin (F(1,17) = 8.6, p < 0.01) but not the medial bin (F(1,17) = 0.4, p > 0.55). These results demonstrate that loss of ALCAM alters VT axon branch directionality in the SC, such that they are not effectively oriented medially, a function known to depend on L1–ankyrin association (Buhusi et al., 2008).
Figure 6.

Axon branching and positioning in the SC of WT and ALCAM null mice. A, Diagram of directed branch orientation of VT axons along the mediolateral SC axis at P2. The SC was divided into three bins: lateral (L), TZ, and medial (M), separated by dashed lines in relation to the forming TZ (shaded circle). The orientation of each branch was scored and graphed by bin. T, Temporal retina; V, ventral retina. B, Distribution of branches in P2–P3 WT and mutant mice after VT retinal injections with DiI. Arrows depict the direction of branch preference in each region. Results were expressed as a branch DC as described previously (Hindges et al., 2002). The DC was calculated in each bin per mouse as the difference in number of medially oriented branches minus laterally oriented branches, divided by the total number of branches. C, Diagram of RGC axon positions in the SC in WT and ALCAM null mutant mice. The SC was divided into 10 regions along the mediolateral axis, and labeled axons in each bin were counted and represented relative to the position of the developing TZ for each injection. D, Distribution of labeled VT axons along the mediolateral SC axis, expressed as percentage of total axons. * Significant differences in means by a mixed ANOVA, with p values indicated in Results. KO, Knock-out.
To evaluate whether disruption of ALCAM resulted in altered patterns of axonal location in the anterior SC that might reflect axon entry, the distribution of DiI-labeled VT axons entering the SC was evaluated at P2–P3, after retinal injections at P1. For each mouse, the entrance zone of the SC was divided into 10 equal bins: four bins located medial and six bins lateral to normally positioned TZs (Fig. 6C). Axons were scored in each bin of WT mice (n = 9 mice; 457 axons) and ALCAM null mice (n = 7 mice; 425 axons), percentages were calculated, and means were analyzed by a mixed ANOVA. There was a significant difference in the distribution of axon entrance between WT and ALCAM null mice when all bins were analyzed (F(9,126) = 83.44, p < 0.001). Axons of WT mice preferentially entered the SC in the vicinity of the normal ventromedial TZ site (F(9,72) = 8.39, p < 0.001), whereas axons of ALCAM null mice did not show a statistically significant preferential location (F(9,54) = 0.76, p = 0.66) (Fig. 6D). Mann–Whitney tests on individual bins showed significant differences between genotypes in the two bins closest to the normal TZ location (p < 0.01) and in the extreme lateral bin (p < 0.05) but not in the other three bins. This may indicate an effect of ALCAM on the distribution of SC axon entry or may simply reflect the altered branch orientation that otherwise would start to correct the position of axons soon after entry into the SC.
Normal pretarget sorting of dorsal and ventral retinal axons in ALCAM mutants
Pretarget RGC axon order in the optic tract contributes to the development of the retinocollicular map (Simon and O'Leary, 1992; Hindges et al., 2002; Plas et al., 2005). Axons from the dorsal and ventral retina are initially segregated at the optic nerve head, but this order is lost in the optic nerve and chiasm, reemerging in the optic tract as axons approach and enter the brachium (entry zone) of the SC (Plas et al., 2005). Axons from the dorsal and ventral retina become segregated in the optic tract of the mouse by P1, whereas axons from the nasal and temporal retina show no evidence of pretarget sorting at any stage (Plas et al., 2005). Although the nearly complete downregulation of ALCAM on RGC axons by the time they reach the chiasm (Fig. 1) suggested that mapping defects in ALCAM null mutants were unlikely to be because of defects in pretarget axon sorting, we confirmed this directly. Dorsal and ventral retinal quadrants of ALCAM mutant and WT littermates at P0–P1 were injected with DiA and DiI, respectively, and the distribution of labeled axons was analyzed in the optic tract and brachium of the SC after 48 h. In four of five ALCAM mutant mice, axons from the dorsal and ventral retina were segregated within the optic tract similar to WT axons (6 of 6). In the optic tract between the chiasm and the delta (near the LGN), DiA-labeled dorsal axons (green) were localized medially, whereas DiI-labeled ventral axons (red) were localized laterally (Fig. 7). At the brachium of the SC, dorsoventral axon order switches as the optic tract twists (Plas et al., 2005). In both WT mice (4 of 4) and ALCAM mutant mice (4 of 4), dorsal axons were localized laterally, and ventral axons localized medially at the brachium (data not shown). In one ALCAM mutant, dorsal and ventral order was reversed before the delta, but, because the order was normal at the brachium, this anomaly most likely reflected a slight delay in reemergence of order after the chiasm. In adult ALCAM mutants (P28), dorsal and ventral RGC axons were also normally segregated in the optic tract, with dorsal axons positioned medially and ventral axons positioned laterally (data not shown). Furthermore, in ALCAM null mutants, the optic chiasm was of normal size and morphology and displayed a prominent contralateral projection and small ipsilateral projection, typical of WT. No obvious defasciculation was observed in the mutant optic nerve or tract. It should be noted that photoreceptor ectopias, a phenotype initially observed in ALCAM null mutants with incomplete penetration (Weiner et al., 2004), were not observed in ALCAM null mutant mice after extensive intercrossing onto the C57BL/6 background.
Figure 7.
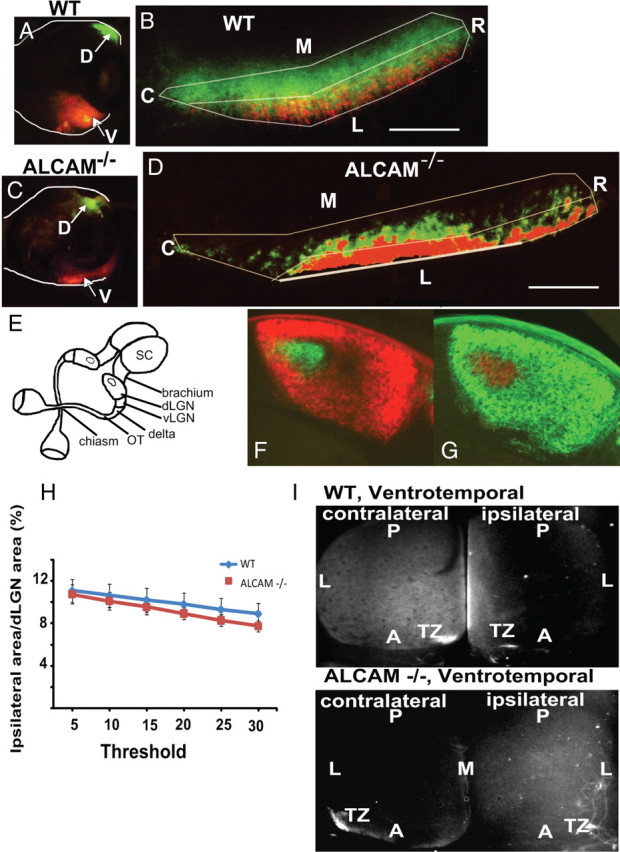
Normal pretarget sorting of dorsal and ventral retinal axons in the optic tract and ipsilateral projections in ALCAM null mutants. A, C, Location of paired injections of DiI and DiA into ventral (V; DiI, red) and dorsal (D; DiA, green) retinal quadrants of WT and ALCAM mutant mice at P0, as shown in flat mounts. B, D, Horizontal sections through the optic tract located between the optic chiasm and delta of WT and ALCAM mutant mice at P2 showed spatial segregation of DiI and DiA, indicating normal sorting of dorsal and ventral retinal axons in ALCAM null mice. The optic chiasm would be located rostrally and the delta located caudally with respect to the sections shown in B and D (refer to diagram E). D, Dorsal; V, ventral; M, medial; L, lateral; R, rostral; C, caudal. Scale bar, 125 μm. E, Diagram of anatomical features of the mouse retinal axon projection. OT, Optic tract; vLGN and dLGN, ventral and dorsal lateral geniculate nucleus of thalamus. F, G, Overlaid images of retinogeniculate projections in WT (F) and ALCAM−/− (G) mice at P12, displaying similar phenotypes for mapping of ipsilateral (F, green; G, red) and contralateral (F, red; G, green) axons in the dLGN. H, Quantification of the percentage of dLGN area occupied by the ipsilateral patch in ALCAM−/− versus WT mice at P12. Data are represented as mean ± SEM. There were no statistically significant differences (p > 0.05, two-tailed t test) between ALCAM null (n = 5) and WT (n = 5) mice for all threshold levels. I, Defective mediolateral targeting of ipsilateral RGC axon projections in ALCAM null mutants. Top, DiI injections into the ventrotemporal retina of WT mice at P8 labeled a single TZ in the anteromedial corner of the contralateral SC and a diffuse zone in the medial SC when analyzed at P10. In contrast, ventrotemporal DiI injections of ALCAM null mutant mice labeled TZs that were laterally shifted in both the contralateral SC and ipsilateral SC. Representative images are shown for WT (n = 4) and ALCAM null mutant (n = 4) mice.
Ipsilateral projections in ALCAM mutant mice
Ipsilateral RGC axons project from the ventrotemporal retina to the dLGN, in which they form an ipsilateral patch surrounded by contralateral axon terminations, with the ipsilateral projection occurring at peak stages of E14–E16 and to a lesser extent at later stages E17 to P0 (Williams et al., 2006). To visualize final retinogeniculate targeting and eye-specific segregation, RGC axons were labeled by injection of cholera toxin B conjugated to Alexa Fluors (Alexa 488, green; Alexa 594, red) into right or left eyes of WT and ALCAM null mutant mice at P10 and analyzed at P12 (Fig. 7F–H). Most mouse ipsilateral projections segregate into a patch in the dorsomedial dLGN by this stage. The pattern of ipsilateral projections to the dLGN of ALCAM mutants appeared similar to WT, with little evident overlap with contralateral projections (Fig. 7F,G). Quantification of the area of the ipsilateral patch relative to the dLGN area, as well as the area of overlap between the ipsilateral and contralateral areas, was made by measurement of pixels within designated areas of images using a multi-threshold protocol (Torborg and Feller, 2004; Torborg et al., 2005; Bjartmar et al., 2006). There was no difference in the relative area of the ipsilateral patch between WT and ALCAM mutant mice at any threshold level, each of which was compared by the t test. Furthermore, the overlap area relative to the dLGN area was very small (0.5–2%) for both genotypes, and there was no significant difference between them at any threshold level.
DiI injections into the ventrotemporal retina of WT mice at P8 (n = 4) and analysis at P10 showed labeling of a single TZ in the anteromedial corner of the contralateral SC and an elongated zone in the medial SC (Fig. 7I), as expected (Plas et al., 2005). In contrast, ventrotemporal DiI injections in ALCAM null mutant mice (n = 4) labeled TZs that were laterally shifted in the contralateral SC as well as the ipsilateral SC (Fig. 7I). Thus, ipsilaterally projecting RGC axons to the SC exhibited a similar mediolateral mapping defect to contralaterally projecting axons in the absence of ALCAM.
ALCAM as substrate increases L1-dependent cell adhesion
The similarity between the phenotype of L1–ankyrin-deficient mutants (Buhusi et al., 2008) and that of ALCAM null mutant mice suggested that mistargeting of retinal axons could be because of altered L1-dependent adhesion to ALCAM as a substrate in the SC, resulting in impaired medial attraction of laterally displaced axonal branches. ALCAM is known to participate not only in homophilic (Nelissen et al., 2000; Neiiendam et al., 2004; Zimmerman et al., 2004) but also heterophilic adhesion, for example, to T-cell antigen CD6 (Bowen et al., 2000; Te Riet et al., 2007), and in chick, possibly L1 (DeBernardo and Chang, 1996). Attractive responses of retinal axons to mediolateral guidance cues are difficult to study in vivo, unlike repellent axon guidance, as discussed previously (Hindges et al., 2002). However, assays of adhesion of cells to defined substrates in culture have been exploited successfully to identify L1- and EphrinB-dependent cellular mechanisms (Holash and Pasquale, 1995; Buhusi et al., 2008).
To ask whether ALCAM was capable of promoting L1-dependent cell adhesion as a substrate-bound ligand, we studied the adhesion of the uveal melanoma cell line MUM2C, derived from the choroid (Seftor et al., 2002) to purified ALCAM–Fc fusion protein in vitro. Western blotting showed that MUM2C cells express L1 (220/200 kDa doublet) but not ALCAM, at both the protein (Fig. 8A) and transcript (assessed by reverse transcription-PCR; data not shown) levels. As a control for ALCAM blotting, we included a distinct cell line derived from the same metastasis as MUM2C (MUM2B), which shows strong expression of ALCAM but not L1. The L1-positive/ALCAM-negative MUM2C cells were allowed to adhere to wells coated sequentially with the Fab′2 fragment of anti-human Fc-specific antibody and with purified ALCAM–Fc fusion protein, and, after 2 h, adherent cells were scored. MUM2C cell adhesion was significantly increased on the ALCAM–Fc substrate compared with the control, anti-Fc-only substrate (Fig. 8B). To determine whether enhanced adhesion to the ALCAM substrate was dependent on cell surface L1, MUM2C cells were preincubated with a function-blocking monoclonal antibody (Neuro4) that recognizes a determinant in the extracellular region of L1 required for cell–cell interactions (Thelen et al., 2002) or with normal IgG as a control. L1 antibodies reduced cell adhesion on ALCAM–Fc to levels on control anti-Fc substrate. Addition of normal IgG had no effect on adhesion to ALCAM–Fc or anti-Fc. Thus, ALCAM presented as a substrate increased L1-dependent adhesion of the ALCAM-negative MUM2C melanoma cell line.
Figure 8.

ALCAM as substrate increases L1-dependent adhesion of MUM2C and retinal cells in culture. A, Western blotting of MUM2B and MUM2C cell extracts (30 μg) with antibodies to L1 and ALCAM and β-tubulin as a loading control. B, MUM2C cell adhesion onto anti-human Fc-(Fab′)2 with or without ALCAM–Fc substrates was quantified by determining the mean number of cells per field from three or more assays. Adhesion of MUM2C cells to ALCAM–Fc substrate (with anti-Fc) was significantly greater than adhesion to anti-Fc alone. Adhesion of MUM2C cells to control anti-Fc or ALCAM–Fc (with anti-Fc) substrates was unaffected in the presence of normal IgG (NIgG; 20 μg/ml). MUM2C adhesion to ALCAM–Fc (with anti-Fc) was significantly decreased in the presence of L1 function-blocking antibodies (Neuro4 mAb, 20 μg/ml) but not normal IgG. *p < 0.05, statistical significance by Student's two-tailed t test; SEM. C, Adhesion of dissociated retinal cells from ventral (black bars) or dorsal (gray bars) hemiretinae of WT P3 mice was increased on an ALCAM–Fc substrate (with anti-Fc and fibronectin) compared with a control substrate (anti-Fc with fibronectin). Preclustered EphrinB1–Fc increased adhesion of ventral retinal cells to ALCAM–Fc compared with cells treated with normal IgG (NIgG). EphrinB1–Fc partially decreased adhesion of dorsal retinal cells to ALCAM–Fc. *p < 0.05, statistical significance by Student's two-tailed t test.
To determine whether retinal cells displayed increased adhesion to ALCAM–Fc, cells from the ventral or dorsal halves of WT retinas (P3) were plated on ALCAM–Fc or control anti-Fc together with fibronectin, a substrate present on radial glia in the tectum/SC (Stettler and Galileo, 2004), and assayed for adhesion after 24 h. Adhesion of both ventral and dorsal retinal cells was significantly increased on the ALCAM–Fc substrate compared with the control substrate (Fig. 8C). To determine whether EphrinB1 altered adhesion, preclustered EphrinB1–Fc (or control nonimmune IgG) was added to cells before plating on the ALCAM–Fc substrate. EphrinB1–Fc increased adhesion of ventral retinal cells, which express higher levels of EphB receptors than do dorsal retinal cells (Fig. 8C). EphrinB1 did not increase adhesion of dorsal retinal cells to ALCAM–Fc but instead decreased their adhesion. EphrinB1-induced decrease in adhesion of dorsal retinal cells to ALCAM might reflect changes in intracellular signaling because of different levels of EphB receptors, because it has been shown that EphB signaling increases or decreases cell adhesion depending on the state of receptor clustering (Stein et al., 1998; Huynh-Do et al., 1999; Pasquale, 2005). A caveat is that, unlike RGC growth cones within the SC or MUM2C cells, dissociated retinal cells express ALCAM on the surface, which can mediate homophilic adhesion to the ALCAM substrate. Furthermore, reverse signaling from EphrinB1, which is likely to occur in SC cells, would not be measured in this assay. In any case, these in vitro results suggest that ALCAM can mediate heterophilic adhesion via L1, increasing the adhesion of retinal cells, and that EphrinB1 preferentially enhances ALCAM-dependent adhesion of ventral retinal cells.
Discussion
Analysis of ALCAM null mice has identified this IgSF recognition molecule as a new mediator of retinotopic mapping important for mediolateral targeting of RGC axon subpopulations in the SC. Loss of ALCAM caused RGC axons from the temporal retinal quadrant to terminate abnormally at laterally displaced ectopic TZs in the SC and disrupted the orientation of interstitial RGC axon branches required for correct mediolateral mapping. During development of the retinocollicular projection, ALCAM was expressed in cells of the SC and on RGC axons proximal to the eye but not on the distal part of L1-expressing retinal axons entering the SC. The expression pattern, ability of ALCAM to promote cell adhesion through L1 in cell culture, and similarity of retinocollicular targeting defects in ALCAM null, EphB, and L1-mutant mice support a mechanism in which ALCAM on the surface of SC cells functionally interacts with L1 on retinal axons to promote directional axon branch orientation toward the medial-high EphrinB1 gradient in the SC.
The ALCAM null retinocollicular map was remarkably similar to that of L1(Y1229H) (Buhusi et al., 2008), EphB2/B3 null, and EphB2ki/ki (kinase inactive) forward-signaling mutant mice (Hindges et al., 2002). In each of these mutants, axons from the temporal retina terminate in prominent laterally shifted TZs in the SC (with smaller posterior shifts) and exhibit a decreased preference in medial orientation of interstitial axon branches during mapping. Attraction of interstitial RGC axon branches toward EphrinB1 in the medial SC requires tyrosine kinase signaling from EphB receptors (Hindges et al., 2002; McLaughlin et al., 2003), as well as L1 linkage to the actin cytoskeleton through ankyrin (Buhusi et al., 2008). ALCAM as a substrate in the SC might engage L1 on RGC axons directly or indirectly to promote transient adhesive contacts in growth cones necessary for medial attraction of axonal branches. In accord with this possibility, purified ALCAM–Fc fusion protein potentiated adhesion of retinal cells in culture and increased adhesion of an L1-positive, ALCAM-negative uveal melanoma cell line in an L1-dependent manner. Because the formation of laterally directed interstitial branches was unaffected in ALCAM mutants, ALCAM does not appear to be involved in the repellent counterforce mediated by Wnt3/Ryk (Schmitt et al., 2006).
A functional L1–ALCAM interaction mediating retinal branch extension is in accord with in vitro studies showing that the chick ortholog of ALCAM (DM-GRASP/BEN) promotes retinal axon growth dependent on the ortholog of L1 [NgCAM (neuron–glia cell adhesion molecule)] (DeBernardo and Chang, 1996; Avci et al., 2004). ALCAM in the SC might interact with L1 on RGC axons directly or indirectly through a distinct coreceptor on retinal axons. However, such a receptor is unlikely to be CD6, because this molecule is not detectable in the retina or SC (Bowen et al., 2000) (J.A.W., unpublished data). Although ALCAM did not coimmunoprecipitate with L1 from lysates (1% NP-40) of neonatal mouse retina or brain (data not shown), an interaction between these molecules could be transient, low in affinity, or simply difficult to demonstrate in the absence of normal membrane topology. Eye enucleation experiments suggested that L1 was not present in the retino-recipient layer of the hamster SC (Lyckman et al., 2000), but we did detect L1 transcripts in a nongraded pattern in mouse SC, although at levels much lower than those observed in RGCs. Thus, L1 on RGC axons might bind ALCAM within the SC in trans or homophilically bind to L1 in a cis-interacting complex with ALCAM on the SC cell surface.
Mistargeting of retinal axons in ALCAM mutant mice did not appear to result from an impairment of intra-retinal pathfinding. ALCAM null mutants displayed a few misrouted axons within the retina and showed subtle effects of defasciculation, but most axons grew normally to the optic disc (Weiner et al., 2004). In this regard, mammalian ALCAM may function differently from Neurolin, the fish homolog of ALCAM, which mediates intra-retinal RGC axon pathfinding (Ott et al., 1998) and guidance in the optic tract (Diekmann and Stuermer, 2009). Alternatively, there may simply be higher redundancy of IgSF members in mammalian RGC axon pathfinding. In any case, ALCAM was not essential for retinal axon outgrowth in mice per se, because the optic nerve and tract of null mutants was normal in size, and axons entered the SC at an appropriate stage. In addition, ipsilateral RGC axon guidance at the optic chiasm, which depends on EphrinB2/EphB1 interactions (Williams et al., 2006), did not seem to require ALCAM, because axons projected ipsilaterally at the optic chiasm and normally targeted the dorsal lateral geniculate nucleus, in which they segregated from contralateral axons. ALCAM restriction to RGC axon segments before but not after the optic chiasm may be because of downregulation as a result of defasciculation, which occurs at the chiasm and as axons enter the SC (Plas et al., 2005). Similarly, zebrafish ALCAM (known as DM-GRASP), which is expressed on fasciculating segments of motor axons, is downregulated on defasciculation during innervation of the muscle target (Fashena and Westerfield, 1999).
Our studies also suggest that ALCAM does not play a critical role in pretarget sorting of dorsal and ventral axons in the optic tract and exerts only a minor effect on axon entry or early distribution in the anterior SC. This is in accord with the decreased expression of ALCAM on distal RGC axons, although it was present on meninges surrounding the optic nerve and the surface of the SC. Pretarget sorting of RGC axons is known to be restricted to the dorsoventral axis of the retina (Plas et al., 2005) and is relatively independent of EphB2/EphB3 (Plas et al., 2008). Topographic mapping of temporal and ventrotemporal RGC axons along the mediolateral SC axis appeared to be more strongly influenced by axon–target interactions involving ALCAM than was the mapping of ventral or dorsal axons. Although ventral retinal axons express high levels of EphB, mediolateral targeting of ventral retinal axons is chiefly determined by pretarget sorting (Plas et al., 2008). Thus, the observed normal mapping of ventral axons in the absence of ALCAM would be consistent with a preferential mechanism of pretarget sorting that is relatively independent of both ALCAM and EphB. Alternatively, EphB signaling in this subpopulation of retinal neurons may be qualitatively different from that in ventrotemporal or temporal retinal neurons, which express lower levels of EphB, and thus may not influence ALCAM-dependent adhesion detectably.
Our studies suggest that ALCAM interaction with L1 may regulate adhesion in response to EphrinB/EphB signaling to promote directional branch extension of RGC axons from the temporal retina, necessary for topographic mapping along the mediolateral SC axis. Signaling through EphB2/EphB3 in retinal axons might modulate L1 phosphorylation on Tyr1229, altering ankyrin binding, cytoskeletal engagement, and adhesion to ALCAM. Because ALCAM is prominently expressed not only in the visual system but also in other axonal projections, such as spinal cord pathways (Fashena and Westerfield, 1999; Arancio et al., 2001; Dillon et al., 2005), ALCAM may have a broader role in L1-dependent synaptic targeting, vital to establishment of connectivity in multiple regions of the developing nervous system. Future studies will be aimed at exploring ALCAM–L1 genetic and physical interactions in other axonal projections during brain development.
Footnotes
This work was supported by National Science Foundation Grant 0618176 (P.F.M.), an E. Matilda Ziegler Foundation for the Blind grant (J.A.W.), and National Science Foundation Research Experiences for Undergraduates Site Award DBI 0755219. Blaine Nesbitt and Leah Fuller are gratefully acknowledged for breeding and genotyping of mice. Beth Stevens is acknowledged for methods of retinogeniculate analysis.
References
- Arancio O, Antonova I, Gambaryan S, Lohmann SM, Wood JS, Lawrence DS, Hawkins RD. Presynaptic role of cGMP-dependent protein kinase during long-lasting potentiation. J Neurosci. 2001;21:143–149. doi: 10.1523/JNEUROSCI.21-01-00143.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avci HX, Zelina P, Thelen K, Pollerberg GE. Role of cell adhesion molecule DM-GRASP in growth and orientation of retinal ganglion cell axons. Dev Biol. 2004;271:291–305. doi: 10.1016/j.ydbio.2004.03.035. [DOI] [PubMed] [Google Scholar]
- Bartsch U, Faissner A, Trotter J, Dörries U, Bartsch S, Mohajeri H, Schachner M. Tenascin demarcates the boundary between the myelinated and nonmyelinated part of retinal ganglion cell axons in the developing and adult mouse. J Neurosci. 1994;14:4756–4768. doi: 10.1523/JNEUROSCI.14-08-04756.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjartmar L, Huberman AD, Ullian EM, Rentería RC, Liu X, Xu W, Prezioso J, Susman MW, Stellwagen D, Stokes CC, Cho R, Worley P, Malenka RC, Ball S, Peachey NS, Copenhagen D, Chapman B, Nakamoto M, Barres BA, Perin MS. Neuronal pentraxins mediate synaptic refinement in the developing visual system. J Neurosci. 2006;26:6269–6281. doi: 10.1523/JNEUROSCI.4212-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen MA, Aruffo AA, Bajorath J. Cell surface receptors and their ligands: in vitro analysis of CD6-CD166 interactions. Proteins. 2000;40:420–428. [PubMed] [Google Scholar]
- Braisted JE, McLaughlin T, Wang HU, Friedman GC, Anderson DJ, O'Leary DD. Graded and lamina-specific distributions of ligands of EphB receptor tyrosine kinases in the developing retinotectal system. Dev Biol. 1997;191:14–28. doi: 10.1006/dbio.1997.8706. [DOI] [PubMed] [Google Scholar]
- Buhusi M, Schlatter MC, Demyanenko GP, Thresher R, Maness PF. L1 interaction with ankyrin regulates mediolateral topography in the retinocollicular projection. J Neurosci. 2008;28:177–188. doi: 10.1523/JNEUROSCI.3573-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBernardo AP, Chang S. Heterophilic interactions of DM-GRASP: GRASP-NgCAM interactions involved in neurite extension. J Cell Biol. 1996;133:657–666. doi: 10.1083/jcb.133.3.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demyanenko GP, Maness PF. The L1 cell adhesion molecule is essential for topographic mapping of retinal axons. J Neurosci. 2003;23:530–538. doi: 10.1523/JNEUROSCI.23-02-00530.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekmann H, Stuermer CA. Zebrafish neurolin-a and -b, orthologs of ALCAM, are involved in retinal ganglion cell differentiation and retinal axon pathfinding. J Comp Neurol. 2009;513:38–50. doi: 10.1002/cne.21928. [DOI] [PubMed] [Google Scholar]
- Dillon AK, Fujita SC, Matise MP, Jarjour AA, Kennedy TE, Kollmus H, Arnold HH, Weiner JA, Sanes JR, Kaprielian Z. Molecular control of spinal accessory motor neuron/axon development in the mouse spinal cord. J Neurosci. 2005;25:10119–10130. doi: 10.1523/JNEUROSCI.3455-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fashena D, Westerfield M. Secondary motoneuron axons localize DM-GRASP on their fasciculated segments. J Comp Neurol. 1999;406:415–424. doi: 10.1002/(sici)1096-9861(19990412)406:3<415::aid-cne9>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Feldheim DA, Kim YI, Bergemann AD, Frisén J, Barbacid M, Flanagan JG. Genetic analysis of ephrin-A2 and ephrin-A5 shows their requirement in multiple aspects of retinocollicular mapping. Neuron. 2000;25:563–574. doi: 10.1016/s0896-6273(00)81060-0. [DOI] [PubMed] [Google Scholar]
- Garrett AM, Weiner JA. Control of CNS synapse development by γ-protocadherin-mediated astrocyte–neuron contact. J Neurosci. 2009;29:11723–11731. doi: 10.1523/JNEUROSCI.2818-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garver TD, Ren Q, Tuvia S, Bennett V. Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J Cell Biol. 1997;137:703–714. doi: 10.1083/jcb.137.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen MJ, Dallal GE, Flanagan JG. Retinal axon response to ephrin-as shows a graded, concentration-dependent transition from growth promotion to inhibition. Neuron. 2004;42:717–730. doi: 10.1016/j.neuron.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Hindges R, McLaughlin T, Genoud N, Henkemeyer M, O'Leary DD. EphB forward signaling controls directional branch extension and arborization required for dorsal-ventral retinotopic mapping. Neuron. 2002;35:475–487. doi: 10.1016/s0896-6273(02)00799-7. [DOI] [PubMed] [Google Scholar]
- Holash JA, Pasquale EB. Polarized expression of the receptor protein tyrosine kinase Cek5 in the developing avian visual system. Dev Biol. 1995;172:683–693. doi: 10.1006/dbio.1995.8039. [DOI] [PubMed] [Google Scholar]
- Huynh-Do U, Stein E, Lane AA, Liu H, Cerretti DP, Daniel TO. Surface densities of ephrin-B1 determine EphB1-coupled activation of cell attachment through alphavbeta3 and alpha5beta1 integrins. EMBO J. 1999;18:2165–2173. doi: 10.1093/emboj/18.8.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenwrick S, Watkins A, De Angelis E. Neural cell recognition molecule L1: relating biological complexity to human disease mutations. Hum Mol Genet. 2000;9:879–886. doi: 10.1093/hmg/9.6.879. [DOI] [PubMed] [Google Scholar]
- Lyckman AW, Moya KL, Confaloni A, Jhaveri S. Early postnatal expression of L1 by retinal fibers in the optic tract and synaptic targets of the Syrian hamster. J Comp Neurol. 2000;423:40–51. [PubMed] [Google Scholar]
- Macias M, Fehr S, Dwornik A, Sulejczak D, Wiater M, Czarkowska-Bauch J, Skup M, Schachner M. Exercise increases mRNA levels for adhesion molecules N-CAM and L1 correlating with BDNF response. Neuroreport. 2002;13:2527–2530. doi: 10.1097/00001756-200212200-00029. [DOI] [PubMed] [Google Scholar]
- Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10:19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- Mann F, Ray S, Harris W, Holt C. Topographic mapping in dorsoventral axis of the Xenopus retinotectal system depends on signaling through ephrin-B ligands. Neuron. 2002;35:461–473. doi: 10.1016/s0896-6273(02)00786-9. [DOI] [PubMed] [Google Scholar]
- McLaughlin T, O'Leary DD. Molecular gradients and development of retinotopic maps. Annu Rev Neurosci. 2005;28:327–355. doi: 10.1146/annurev.neuro.28.061604.135714. [DOI] [PubMed] [Google Scholar]
- McLaughlin T, Hindges R, Yates PA, O'Leary DD. Bifunctional action of ephrin-B1 as a repellent and attractant to control bidirectional branch extension in dorsal-ventral retinotopic mapping. Development. 2003;130:2407–2418. doi: 10.1242/dev.00467. [DOI] [PubMed] [Google Scholar]
- Neiiendam JL, Køhler LB, Christensen C, Li S, Pedersen MV, Ditlevsen DK, Kornum MK, Kiselyov VV, Berezin V, Bock E. An NCAM-derived FGF-receptor agonist, the FGL-peptide, induces neurite outgrowth and neuronal survival in primary rat neurons. J Neurochem. 2004;91:920–935. doi: 10.1111/j.1471-4159.2004.02779.x. [DOI] [PubMed] [Google Scholar]
- Nelissen JM, Peters IM, de Grooth BG, van Kooyk Y, Figdor CG. Dynamic regulation of activated leukocyte cell adhesion molecule-mediated homotypic cell adhesion through the actin cytoskeleton. Mol Biol Cell. 2000;11:2057–2068. doi: 10.1091/mbc.11.6.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott H, Bastmeyer M, Stuermer CA. Neurolin, the goldfish homolog of DM-GRASP, is involved in retinal axon pathfinding to the optic disk. J Neurosci. 1998;18:3363–3372. doi: 10.1523/JNEUROSCI.18-09-03363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquale EB. Eph receptor signalling casts a wide net on cell behaviour. Nat Rev Mol Cell Biol. 2005;6:462–475. doi: 10.1038/nrm1662. [DOI] [PubMed] [Google Scholar]
- Plas DT, Lopez JE, Crair MC. Pretarget sorting of retinocollicular axons in the mouse. J Comp Neurol. 2005;491:305–319. doi: 10.1002/cne.20694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plas DT, Dhande OS, Lopez JE, Murali D, Thaller C, Henkemeyer M, Furuta Y, Overbeek P, Crair MC. Bone morphogenetic proteins, eye patterning, and retinocollicular map formation in the mouse. J Neurosci. 2008;28:7057–7067. doi: 10.1523/JNEUROSCI.3598-06.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AM, Shi J, Wolf AM, Lu CC, King LA, Zou Y. Wnt-Ryk signalling mediates medial-lateral retinotectal topographic mapping. Nature. 2006;439:31–37. doi: 10.1038/nature04334. [DOI] [PubMed] [Google Scholar]
- Seftor EA, Meltzer PS, Kirschmann DA, Pe'er J, Maniotis AJ, Trent JM, Folberg R, Hendrix MJ. Molecular determinants of human uveal melanoma invasion and metastasis. Clin Exp Metastasis. 2002;19:233–246. doi: 10.1023/a:1015591624171. [DOI] [PubMed] [Google Scholar]
- Simon DK, O'Leary DD. Influence of position along the medial-lateral axis of the superior colliculus on the topographic targeting and survival of retinal axons. Brain Res Dev Brain Res. 1992;69:167–172. doi: 10.1016/0165-3806(92)90155-p. [DOI] [PubMed] [Google Scholar]
- Stein E, Lane AA, Cerretti DP, Schoecklmann HO, Schroff AD, Van Etten RL, Daniel TO. Eph receptors discriminate specific ligand oligomers to determine alternative signaling complexes, attachment, and assembly responses. Genes Dev. 1998;12:667–678. doi: 10.1101/gad.12.5.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stettler EM, Galileo DS. Radial glia produce and align the ligand fibronectin during neuronal migration in the developing chick brain. J Comp Neurol. 2004;468:441–451. doi: 10.1002/cne.10987. [DOI] [PubMed] [Google Scholar]
- Te Riet J, Zimmerman AW, Cambi A, Joosten B, Speller S, Torensma R, van Leeuwen FN, Figdor CG, de Lange F. Distinct kinetic and mechanical properties govern ALCAM-mediated interactions as shown by single-molecule force spectroscopy. J Cell Sci. 2007;120:3965–3976. doi: 10.1242/jcs.004010. [DOI] [PubMed] [Google Scholar]
- Thelen K, Kedar V, Panicker AK, Schmid RS, Midkiff BR, Maness PF. The neural cell adhesion molecule L1 potentiates integrin-dependent cell migration to extracellular matrix proteins. J Neurosci. 2002;22:4918–4931. doi: 10.1523/JNEUROSCI.22-12-04918.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torborg CL, Feller MB. Unbiased analysis of bulk axonal segregation patterns. J Neurosci Methods. 2004;135:17–26. doi: 10.1016/j.jneumeth.2003.11.019. [DOI] [PubMed] [Google Scholar]
- Torborg CL, Hansen KA, Feller MB. High frequency, synchronized bursting drives eye-specific segregation of retinogeniculate projections. Nat Neurosci. 2005;8:72–78. doi: 10.1038/nn1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kilsdonk JW, Wilting RH, Bergers M, van Muijen GN, Schalkwijk J, van Kempen LC, Swart GW. Attenuation of melanoma invasion by a secreted variant of activated leukocyte cell adhesion molecule. Cancer Res. 2008;68:3671–3679. doi: 10.1158/0008-5472.CAN-07-5767. [DOI] [PubMed] [Google Scholar]
- Weiner JA, Koo SJ, Nicolas S, Fraboulet S, Pfaff SL, Pourquié O, Sanes JR. Axon fasciculation defects and retinal dysplasias in mice lacking the immunoglobulin superfamily adhesion molecule BEN/ALCAM/SC1. Mol Cell Neurosci. 2004;27:59–69. doi: 10.1016/j.mcn.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Williams SE, Grumet M, Colman DR, Henkemeyer M, Mason CA, Sakurai T. A role for Nr-CAM in the patterning of binocular visual pathways. Neuron. 2006;50:535–547. doi: 10.1016/j.neuron.2006.03.037. [DOI] [PubMed] [Google Scholar]
- Yates PA, Roskies AL, McLaughlin T, O'Leary DD. Topographic-specific axon branching controlled by ephrin-As is the critical event in retinotectal map development. J Neurosci. 2001;21:8548–8563. doi: 10.1523/JNEUROSCI.21-21-08548.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman AW, Nelissen JM, van Emst-de Vries SE, Willems PH, de Lange F, Collard JG, van Leeuwen FN, Figdor CG. Cytoskeletal restraints regulate homotypic ALCAM-mediated adhesion through PKCalpha independently of Rho-like GTPases. J Cell Sci. 2004;117:2841–2852. doi: 10.1242/jcs.01139. [DOI] [PubMed] [Google Scholar]