

Abstract
Ischemic and excitotoxic events within the brain result in rapid and often unfavorable depletions in neuronal energy levels. Here, we investigated the signaling pathways activated in response to the energetic stress created by transient glutamate excitation in cerebellar granule neurons. We characterized a glucose dependent hyperpolarization of the mitochondrial membrane potential (Δψm) in the majority of neurons after transient glutamate excitation. Expression levels of the primary neuronal glucose transporters (GLUTs) isoforms 1, 3, 4, and 8 were found to be unaltered within a 24 h period after excitation. However, a significant increase only in GLUT3 surface expression was identified 30 min after excitation, with this high surface expression remaining significantly above control levels in many neurons for up to 4 h. Glutamate excitation induced a rapid alteration in the AMP:ATP ratio that was associated with the activation of the AMP-activated protein kinase (AMPK). Interestingly, pharmacological activation of AMPK with AICAR (5-aminoimidazole-4-carboxamide riboside) alone also increased GLUT3 surface expression, with a hyperpolarization of Δψm evident in many neurons. Notably, inhibition of the CaMKK (calmodulin-dependent protein kinase kinase) had little affect on GLUT translocation, whereas the inhibition or knockdown of AMPK (compound C, siRNA) activity prevented GLUT3 translocation to the cell surface after glutamate excitation. Furthermore, gene silencing of GLUT3 eradicated the increase in Δψm associated with transient glutamate excitation and potently sensitized neurons to excitotoxicity. In summary, our data suggest that the activation of AMPK and its regulation of cell surface GLUT3 expression is critical in mediating neuronal tolerance to excitotoxicity.
Introduction
Excessive glutamate release and the overexcitation of glutamate receptors has been identified as a central mediator in the pathophysiology of the neuronal injury associated with ischemic stroke and head trauma (Choi, 1987). The overexcitation of postsynaptic glutamate receptors and the neuronal loading with Na+ and Ca2+ results in the rapid depletion of neuronal energy, with a significant reduction in ATP levels (Tsuji et al., 1994; Atlante et al., 1996) and the activation of energetic signaling pathways (Culmsee et al., 2001; McCullough et al., 2005). Recently, we identified rapid changes in the metabolism of cerebellar granule neurons (CGNs) after transient glutamate excitation, establishing a significant increase in glucose uptake that was associated with a hyperpolarization of the mitochondrial membrane potential (Δψm) (Ward et al., 2007). In previous studies, it is apparent that restrictions to glucose availability increases the sensitivity of primary neurons to glutamate excitation (Delgado-Esteban et al., 2000; Vergun et al., 2003). In neurons, glucose uptake is facilitated by a family of glucose transporters (GLUTs) with the GLUT1 and 3 isoforms believed to be responsible for the majority of glucose uptake within the brain (Duelli and Kuschinsky, 2001). Increased expression of GLUT1 and 3 has been reported in vitro (Maher and Simpson, 1994) and in vivo (Vannucci et al., 1996) after excitotoxic and ischemic events.
The AMP-activated protein kinase (AMPK) has been characterized as a critical regulator of cellular function in response to energy stress within cells (Hardie et al., 2006). AMPK is activated by increased cellular AMP levels (Hawley et al., 2003; Woods et al., 2003) and indirectly by the activation of the Ca2+-sensitive calmodulin-dependent protein kinase kinases (CaMKKs) (Hawley et al., 2005; Woods et al., 2005). The α1/2, β1/2, and γ1 subunits of AMPK are highly expressed in neurons (Turnley et al., 1999), with AMPK activity shown to increase during glucose deprivation, metabolic stress, ischemia, and hypoxia in neurons both in vivo and in vitro (Culmsee et al., 2001; Gadalla et al., 2004; McCullough et al., 2005). Interestingly, AMPK activity plays a central role in increasing cellular glucose uptake, with increased activity facilitating the translocation of the GLUT isoform 4 to the plasma membrane in skeletal and cardiac muscle during exercise (Fujii et al., 2006) and by altering the activation of specific transporters at the plasma membrane (Abbud et al., 2000; Barnes et al., 2002; Cidad et al., 2004).
Here, we investigated the signaling pathways activated in CGNs in response to the energetic stress created by transient glutamate excitation. We have characterized an AMP-mediated activation of AMPK that facilitates the translocation of GLUT3 to the neuronal cell surface after excitation. In addition, a siRNA-mediated knockdown of GLUT3 expression was found to sensitize CGNs to glutamate excitation.
Materials and Methods
Materials.
Fetal calf serum and minimal essential medium were from Invitrogen (Bio Sciences). Glutamate, glycine, and all other reagents were from Sigma (Fluka). Fluo-4 AM and TMRM were purchased from Invitrogen (Bio Sciences). 5-aminoimidazole-4-carboxamide riboside (AICAR) was obtained from Cell Signaling. Compound C and KN93 (water-soluble) obtained from Calbiochem (Merck Biosciences, UK).
Preparation of primary cerebellar granule neurons.
CGNs were prepared as described previously (Ward et al., 2000). Cells were plated on poly-l-lysine-coated glass coverslips, glass Willco dishes, 6-well plates, and 24-well plates at 1 × 106 cells per milliliter, and maintained at 37°C in a humidified atmosphere of 5% CO2/95% air. Neurons were used after 8 d in culture. All animal work was performed with ethics approval from the RCSI and under licenses granted to Weisova and Ward by the Irish Department of health and children.
Determination of neuronal injury: Hoechst staining of nuclear chromatin.
Cells cultured on 24-well plates were stained live with Hoechst 33258 (Sigma) at a final concentration of 1 μg/ml. After incubation for 10 min, nuclear morphology was observed using an Eclipse TE 300 inverted microscope (Nikon) and a 20× dry objective. For each time point and treatment (glutamate/glycine, 100 μm/10 μm; AICAR, 2.5 mm), cells were analyzed for apoptotic morphology in three subfields of each culture (up to 2000 cells per time point). All experiments were performed at least twice with similar results.
Confocal microscopy.
CGNs on Willco dishes were loaded with TMRM (20 nm) only or coloaded with Fluo-4 AM (3 μm) for 30 min at 37°C (in the dark) in experimental buffer containing (in mm): 120 NaCl, 3.5 KCl, 0.4 KH2PO4, 20 HEPES, 5 NaHCO3, 1.2 Na2SO4, 1.2 CaCl2, and 15 glucose, pH 7.4. The Willco dishes with cells were mounted in a nonperfusion (37°C) holder and placed on the stage of an LSM 510 Meta Zeiss confocal microscope. For glutamate-induced apoptosis, neurons were exposed to glutamate and glycine (100 and 10 μm) for 10 min, with (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK-801; 10 μm; Fluka) added to block NMDA receptor activation as required. In separate experiments, neurons were incubated with AICAR (2.5 mm) for up to 24 h. Fluo-4 AM was excited at 488 nm, and the emission was collected through a 505–550 nm barrier filter; TMRM was excited at 543 nm, and the emission was collected through a 560 nm long-pass filter. Images were taken at 60 s intervals, and the resulting fluorescent images were processed using MetaMorph Software version 7.1, release 3 (Molecular Devices).
Gene expression analysis using quantitative real-time RT-PCR analysis.
Total RNA was extracted using the RNeasy mini Kit (Qiagen). First strand cDNA synthesis was performed using 2 μg of total RNA as template and Superscript II reverse transcriptase (Invitrogen) primed with 50 pmol of random hexamers. Quantitative real-time PCR was performed using the LightCycler (Roche Diagnostics) and the QuantiTech SYBR Green PCR kit (Qiagen) as per manufacturer's protocol. Specific primers for each gene analyzed were designed using Primer3 software (http://frodo.wi.mit.edu/). Sense and antisense primers were: AGTATCGTGGCCATCTTTGG and CCCACGATGAAGTTTGAGGT for GLUT1; CGCCTGATTATTGGCATCTT and TCCAAACCAAAGACCTGAGC for GLUT3; TGGCATGATTTCCTCCTTTC and TAGGCACCAACCCTGATGTT for GLUT4; ACTGGTTCATGGCCTTTCTG and CTCGAAATGGGCTGTGATTT for GLUT8; AACAAGCACTTCGGTCATCC and AGAGCAAGAAGGCGACACAT for PGC-1α; CAAAAAGACCTCGGTCAGCA and ATGAGATCACTTCGCCCAAC for mtTFA; CATTATCCACCCCACTA and CATGTTGAGGGTAGGGG for ND5; GGGAAATCGTGCGTGACATT and TGCCACAGGATTCCATACCC for β-actin. Each primer pair was tested with a logarithmic dilution of a cDNA mix to generate a linear standard curve, which was used to calculate the primer pair efficiency. The PCRs were performed in 20 μl volumes with the following parameters: 95°C for 15 min, followed by 40 cycles of 94°C for 20 s, 59°C for 20 s, and 72°C for 20 s. The generation of specific PCR products was confirmed by melting curve analysis and gel electrophoresis. The data were analyzed using the Lightcycler Software 4.0 with all samples normalized to β-actin.
Semiquantitative PCR for neuronal GLUT isoforms.
For semiquantitative PCRs were performed using Platimum Taq polymerase (Invitrogen). PCR amplification was performed in 20 μl volumes with the following parameters: 95°C for 2 min, followed by 40 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30. A 479 bp product of ND5 was amplified with primers 5′-CATTATCCACCCCACTA-3′ and 5′-CATGTTGAGGGTAGGGG-3′ as loading control in combination with the gene of interest. The rat-specific GLUT 1, 3, 4, and GLUT8 primers, as described above, were used to generate fragments of 155 kb for GLUT 1, 155 kb for GLUT 3, 209 bp for GLUT 4, and 176 bp for GLUT 8. Initial experiments were performed to ascertain that PCR was within the linear amplification range for all three investigated genes. PCR products were separated by agarose gel electrophoresis (1.5%) and visualized with 0.1% ethidium bromide under UV transillumination, using a CCD camera-based gel documentation system (Fuji Las-300).
Preparation of whole-cell extracts and Western blotting.
Preparation of cell lysates and Western blotting was performed as described previously (Concannon et al., 2008). The resultant blots were probed with a monoclonal mouse anti-cytochrome c-oxidase subunit IV (COX IV) (Invitrogen A21348, 1:100), a rabbit polyclonal anti-GLUT1 antibody (Millipore Bioscience Research Reagents, 1:1000), a rabbit polyclonal anti-GLUT3 antibody (Millipore Bioscience Research Reagents, 1:1000), a rabbit anti-GLUT4 antibody (Millipore Bioscience Research Reagents, 1:1000), a goat polyclonal anti-GLUT8 antibody (Santa Cruz Biotechnology, 1:1,00), a rabbit polyclonal phospho-(Thr 172)-AMPK (Cell Signaling Technology, 1:1000); a rabbit polyclonal total AMPKα (Cell Signaling Technology, 1:1000), or a mouse, monoclonal anti-α-tubulin antibody (Sigma, clone DM 1A; 1:5000). Membranes were washed with Tris-buffered saline Tween 20 (TBST) three times for 10 min. Immunoreactivity was detected with anti-mouse or anti-rabbit peroxidase-conjugated secondary antibodies (1:5000, Promega) for 2 h at room temperature. Blots were washed and developed using ECL chemiluminescence detection reagent (GE Healthcare), detected using Super-Signal West Pico Chemiluminescent Substrate (Pierce), and imaged using a FujiFilm LAS-3000 imaging system (Fuji).
Immunofluorescence analysis.
For immunofluorescence analysis, cells were fixed on coverslips with 4% paraformaldehyde, washed three times with PBS, and then incubated with blocking solution (PBS with 20% fetal calf serum and 1% BSA) for 1 h at room temperature. GLUT3 was detected using a rabbit polyclonal anti-GLUT3 antibody (Millipore Bioscience Research Reagents). The antibody was used at a concentration of 1:250 in PBS containing 20% horse serum and 1% BSA. After incubation at room temperature for 2 h, cells were washed twice with PBS and incubated with Alexa Fluor 488 goat anti-rabbit IgG (H+L) antibody (Invitrogen), diluted 1:250. The secondary antibody was detected using confocal microscopy as described above.
Flow cytometry: quantification of cell surface GLUT isoform expression.
CGNs were harvested from a 24-well plate using trypsin after appropriate treatments and fixed in 1% paraformaldehyde for 20–25 min at 4°C. After incubation with the GLUT1, 3, 4, or 8 antibody (GLUT1, 3, 4 from Millipore Bioscience Research Reagents, 1:250 and GLUT 8 from Santa-Cruz Biotechnology, 1: 25) diluted in PBS + 0.1% BSA for 1 h; cells were washed and incubated with an Alexa Fluor 488 goat anti-rabbit IgG (H+L) antibody (Invitrogen) diluted 1:250 for 1 h. After washing cells three times with PBS + 0.1% BSA, samples were analyzed immediately by flow cytometry on a Partec CyFlow ML followed by analysis using FloMax software. In all cases, a minimum of 104 events were acquired.
Flow cytometry: quantification of mitochondrial mass.
For the quantification of mitochondrial mass, CGNs were loaded with the mitochondria-specific fluorescent dye MitoTracker Green FM (Invitrogen) in media at a final concentration 150 nm for 30 min at 37°C. After incubation the media was removed, cells gently trypsinated, centrifugated at 6000 rpm for 3 min, and resuspended in PBS. The forward scatter versus side scatter area was used to analyze a homogeneous population of live cells. Samples were analyzed immediately by flow cytometry with cytometric analyses performed on a CyFlow ML (Partec) followed by analysis using FloMax software.
ATP luciferase assays.
CGNs were maintained on poly-d-lysine (5 μg/ml)-coated 24-well plates for 7 d before use. The culturing medium was replaced with an experimental buffer (see above) and the neurons were excited with glutamate for 10 min and lysed at the times indicated. In separate experiments, CGNs were exposed to AICAR (2.5 mm) before being lysed. The neurons were lysed using a hypotonic lysis buffer (Tris-acetate buffer, pH 7.75). Fifty microliters of the sample and 50 μl of the luciferin–luciferase reaction kit (ENLITEN ATP Assay System Bioluminescence Detection kit; Promega) for ATP were reacted to quantify ATP content. The amount of ATP was determined by a concentration standard curve, and ATP content values were normalized according to the protein concentration for each sample (μmol ATP/mg protein).
siRNA silencing of GLUT3 in PC12 cells.
A synthetic small interfering RNA (siRNA) that targeted GLUT3 was designed using the RNA workbench software (Vareková et al., 2008), and GLUT3 siRNA and nontargeting control duplexes were obtained from Sigma-Proligo. PC12 cells were transfected using the Nucleofector (Amaxa Biosystems) as per manufacturer's instructions. Briefly, ∼2 × 106 cells were transfected with 100 nm of either GLUT3 or control siRNA duplexes. Immediately after transfection, the cells were transferred with the recommended plastic pipettes into six-well culture dishes. Two days after the transfection, cells were lysed and the knockdown of GLUT3 was examined by RT-qPCR (see above) and Western blot (see above).
Generation of siRNA expressing vectors and transfection of CGNs.
The GLUT3 and control siRNA were cloned into the pFIV-H1/U6-copGFP siRNA vector (SBI, System Biosciences) as per manufacturer's instructions to allow for the expression of the appropriate siRNA and GFP in parallel. For knockdown of AMPK, a sequence targeting AMPK α1/α2 (Aguilar et al., 2007) was used. Transfection of CGNs was performed according to the protocol for calcium-phosphate-based transfection of primary neurons as previously described (Dahm et al., 2008). Approximately 1 × 106 neurons were maintained on Willco dishes and on day 4 (DIV) were transfected with 3 μg of plasmid. Neurons were used for experiments 3–4 d after transfection
Measurement of intracellular AMP and ATP levels.
Adenine nucleotide levels in CGN lysates were determined by HPLC analysis as previously described with some modifications (Landree et al., 2004). Briefly, each well of a six-well plate was washed with 2 ml of ice-cold PBS, lysed with 70 μl of ice-cold 0.5 m KOH, and scraped. H20 (140 μl) was added to cell lysates and incubated on ice for 5 min, and the pH was then adjusted to 6.5 by addition of 1 m KH2 PO4. Cell lysates were spun through Microcon YM-50 centrifugal filters (Millipore: 10 kDa followed by a 3 kDa) and stored at −80°C for subsequent HPLC analysis. The HPLC used was an Varian ProStar with ProStar 335 Diode Array Detector and equipped with a reverse-phase Gemini column (Phenomenex, 5Å, C18, 4.6 mm deep/250 mm long). Of each sample, 20 μl was injected in the HPLC and a mobile phase consisting of 0.1 m KH2 PO4, pH adjusted to 6 with KOH and 100% acetonitrile, with a flow rate 1 ml/min. Each elution peak was compared with AMP, ADP, and ATP standards (Sigma) to confirm its identity. Additionally, the identity of the AMP peak was compared by spiking method with AMP standard in biological sample. Data are expressed as peak area (mAU.min).
Statistics.
Data are presented as means ± SEM. For statistical comparison, t test or one-way ANOVA and post hoc Tukey's test were used using SPSS software. p values <0.05 were considered to be statistically significant.
Results
Mitochondrial bioenergetics and biosynthesis after transient glutamate excitation
Previous studies have highlighted significant modulations in neuronal metabolism and mitochondrial energetics in response to excitotoxic/ischemic events; with increased glucose uptake, increased NAD(P)H availability and a hyperpolarization of Δψm (Robb-Gaspers et al., 1998; Iijima et al., 2003; Ward et al., 2007). Here, transient glutamate excitation of CGNs resulted in a delayed, caspase-independent apoptotic injury within 52.1 ± 6.5% of the neuronal population within a 24 h period (Fig. 1A) (Ward et al., 2006). Single-cell imaging identified a significant increase (from 101.5 ± 1.5% to 148.1 ± 5.7%) in the whole-cell TMRM fluorescence in >90% of the neurons within a 2 h period after glutamate excitation (Fig. 1B,C). The increase in cellular TMRM fluorescence in response to glutamate excitation has been attributed to an increased accumulation of the probe within the mitochondrial matrix after a hyperpolarization of Δψm within the neurons (Nicholls and Ward, 2000; Ward et al., 2007). However, increased TMRM fluorescence may also be a function of increased mitochondrial biogenesis in response to energy depletion created within the neurons after excitation. Western blot analysis of the mitochondrial protein, COX IV, revealed no significant changes in the expression levels of the protein in the 24 h after glutamate excitation (Fig. 1D). In addition, flow cytometry analysis of CGNs loaded with mitotracker green did not identify an increase in mitochondrial mass over the same period (Fig. 1E). Furthermore, qPCR analysis of the expression levels of two genes, the peroxisome proliferator-activated receptor gamma coactivator 1α (pgc-1α) and the mitochondrial transcription factor A (tfam), believed to play a role in mitochondrial biogenesis, did not display major changes, particularly in the early period (1–2 h) after glutamate excitation (Fig. 1F) in which TMRM fluorescence increased rapidly (Fig. 1B,C). Interestingly, a significant increase in the expression levels of pgc-1α and tfam were detected 4 h (Fig. 1F) after glutamate excitation. Together, this data suggest that the early increase in TMRM fluorescence after glutamate excitation is not attributable to an increase in mitochondrial biogenesis.
Figure 1.

Transient glutamate excitation induces an early hyperpolarization of Δψm that is not a function of increased mitochondrial biogenesis. CGNs were stimulated with glutamate (glutamate/glycine 100 μm/10 μm) for 10 min. A, Nuclei were stained with Hoechst at the indicated time points (0, 4, 16, and 24 h) with uniformly stained nuclei counted as healthy (viable neurons) and condensed nuclei counted as apoptotic (n = 4 experiments in triplicate, *p < 0.01, difference between sham and 4, 8, and 24 h) B, CGNs preloaded with TMRM and Fluo-4 AM were stimulated with glutamate. Traces are representative of the responses in neurons at postnatal day 15 from 10 separate experiments. C, Average TMRM fluorescence in neurons after glutamate excitation are represented as mean ± SEM. Fluorescent images for representative chosen at selected time points (0 and 120 min) after glutamate excitation. *p < 0.001, difference between sham (n = 63) and glutamate-treated neurons (n = 119). D, After glutamate excitation, the expression levels of COX IV were examined by Western blotting. α-Tubulin served as a loading control. Similar responses were observed in samples from three separate cultures. E, CGNs were stimulated with glutamate, loaded with 100 nm MitoTracker Green FM, and mitochondrial mass analyzed using FACs. Data are presented as mean ± SEM. No significant increase was identified. F, CGNs were stimulated with glutamate, and the mRNA expression of the transcription factor tfam and cofactor pgc-1α was analyzed by real-time qPCR. Expression levels were normalized to control cells and data are represented as mean ± SEM from three independent experiments, 2, 4, and 24 h). *, #p < 0.05, difference between sham and glutamate treated neurons at 4 h (n = 3 experiments in triplicate).
Glucose availability dictates neuronal viability and the hyperpolarization of Δψm after transient glutamate excitation
It has previously been established that an inhibition of glycolysis or alterations in glucose availability increased neuronal vulnerability to glutamate excitation (Delgado-Esteban et al., 2000; Vergun et al., 2003). In addition, we have recently described an increased capacity for CGNs to take up glucose after transient glutamate excitation using tritiated 2-deoxyglucose (Ward et al., 2007). Here, we set out to establish how alterations in glucose availability influenced neuronal viability and mitochondrial energetics in response to glutamate excitation in our model. CGNs loaded with TMRM (20 nm) were excited with glutamate in experimental buffer containing normal glucose (15 mm) and low glucose (1 mm). Significantly, neurons incubated in the lower glucose concentration were found to have a more marked reduction in (83.2 ± 6.4%) TMRM fluorescence 60 min after glutamate excitation (Fig. 2A,B) than that (167.2 ± 9.9%) in neurons maintained in normal glucose (15 mm). Previously, we have identified that a hyperpolarization Δψm is associated with cell survival and tolerance (Ward et al., 2007). Interestingly, in neuronal populations maintained in low glucose (1 mm) and excited with glutamate where no hyperpolarization of Δψm was evident, a higher rate of cell death was identified (65.1 ± 7.4% survival in 1 mm glucose and 39.8 ± 6.1% in 15 mm glucose) with an increased number of neurons having pyknotic nuclei within 24 h of excitation (Fig. 2C). This data demonstrate that the increase in Δψm identified in CGNs after transient glutamate excitation is dependent on extracellular glucose availability, and that decreased extracellular glucose sensitizes CGNs to glutamate excitation.
Figure 2.
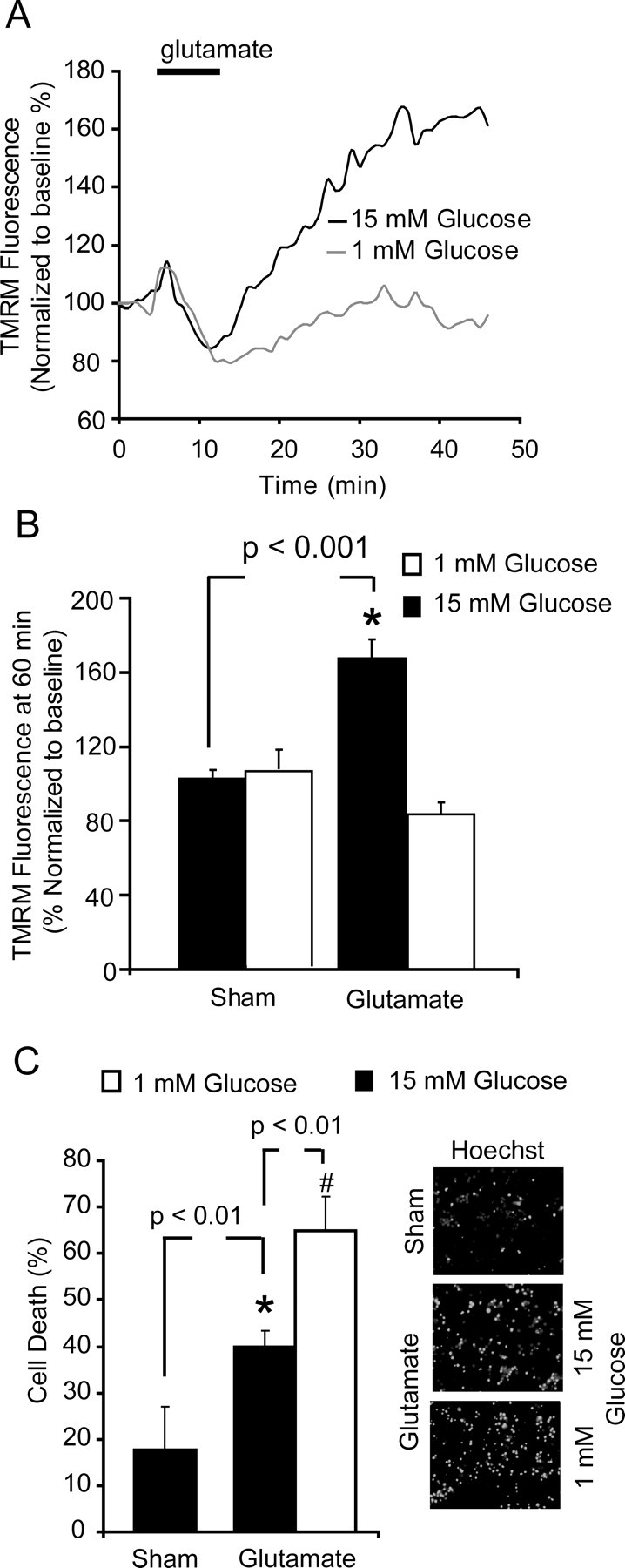
Reduced glucose availability attenuates the increase in Δψm after transient glutamate excitation and increases neuronal sensitivity to excitation. CGNs were stimulated with glutamate/glycine (100 μm/10 μm 10 min) in the presence of normal (15 mm) glucose or low (1 mm) extracellular glucose. A, Representative traces for whole-cell TMRM fluorescence in neurons during transient glutamate excitation in 1 mm (n = 26) or 15 mm (n = 31) extracellular glucose concentration. B, Quantification of the TMRM fluorescence in sham-treated neurons (n = 39) and glutamate-treated neurons with 15 mm (n = 26) or 1 mm glucose (n = 31). Data are presented as mean ± SEM. *p < 0.001, difference between sham-treated and glutamate-treated neurons in 15 mm glucose. C, Neurons were stimulated with glutamate and left to recover in media with different glucose concentrations (1 and 15 mm) for 24 h. Nuclei were stained with Hoechst, with uniformly stained nuclei counted as healthy/viable neurons and condensed nuclei scored as apoptotic. Data were presented as mean ± SEM. *p < 0.01, difference between sham- and glutamate-stimulated neurons in 15 mm glucose. #p < 0.01, difference between glutamate-stimulated neurons recovered in 15 mm glucose and those recovered in 1 mm glucose (n = 3 in triplicate).
Glutamate-induced hyperpolarization of Δψm is coupled with an increase in GLUT3 translocation
Because the changes in mitochondrial energetics appear to be highly dependent on the availability of glucose (Fig. 2A,B) and CGNs have an increased capacity for glucose uptake after transient glutamate excitation (Ward et al., 2007), we investigated the role of GLUTs in facilitating this increased glucose uptake. PCR analysis of glut isoform mRNA expression in cerebellar tissue revealed a high expression level for glut3 and 8, with a much reduced expression of the glut isoforms 1 and 4 (Fig. 3A). Rather surprisingly, no significant increase in the mRNA (Fig. 3B) or protein (Fig. 3C) expression levels were found for the main neuronal GLUT isoforms (1, 3, 4, 8) in the CGNs within a 24 h period after glutamate excitation. However, immunofluorescence analysis of the cell surface expression of the GLUT isoforms identified a significant increase in the expression of only the insulin-insensitive GLUT3 isoform after glutamate excitation (Fig. 4A,B). Because of the inherent problems associated with immunofluorescence (investigator bias, qualitative quantification) and the high variability of GLUT3 cell surface expression in untreated neurons (Fig. 4C, gray shaded area), we used flow cytometry to more accurately determine the GLUT surface expression of the principle neuronal isoforms (1, 3, 4, and 8) in response to transient glutamate excitation in neuronal populations with 104 events per sample (Fig. 4D,E). Similar to the immunofluorescence, flow cytometric analysis detected a significant increase in the cell surface expression of only GLUT3 30 and 60 min after excitation (Fig. 4C,D), with no significant change in surface expression levels of GLUT, 1, 4, and 8 identified (Fig. 4E). Interestingly, GLUT3 is known to have a much higher capacity to transport glucose in neurons at optimal glucose concentrations than other GLUT isoforms (Carruthers, 1990; Maher et al., 1996; Vannucci et al., 1997); therefore, only minor changes in surface expression levels would result in greatly enhanced glucose uptake. Importantly, the increase in GLUT3 cell surface expression identified after glutamate excitation occurred independent of extracellular glucose concentrations (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), indicating that the translocation event is dependent on the activation of intracellular signaling cascades. The current data suggest that the rapid translocation of the GLUT3 to the plasma membrane is central in enabling CGNs to increase glucose uptake in response to the energy deprivation associated with glutamate receptor over activation.
Figure 3.
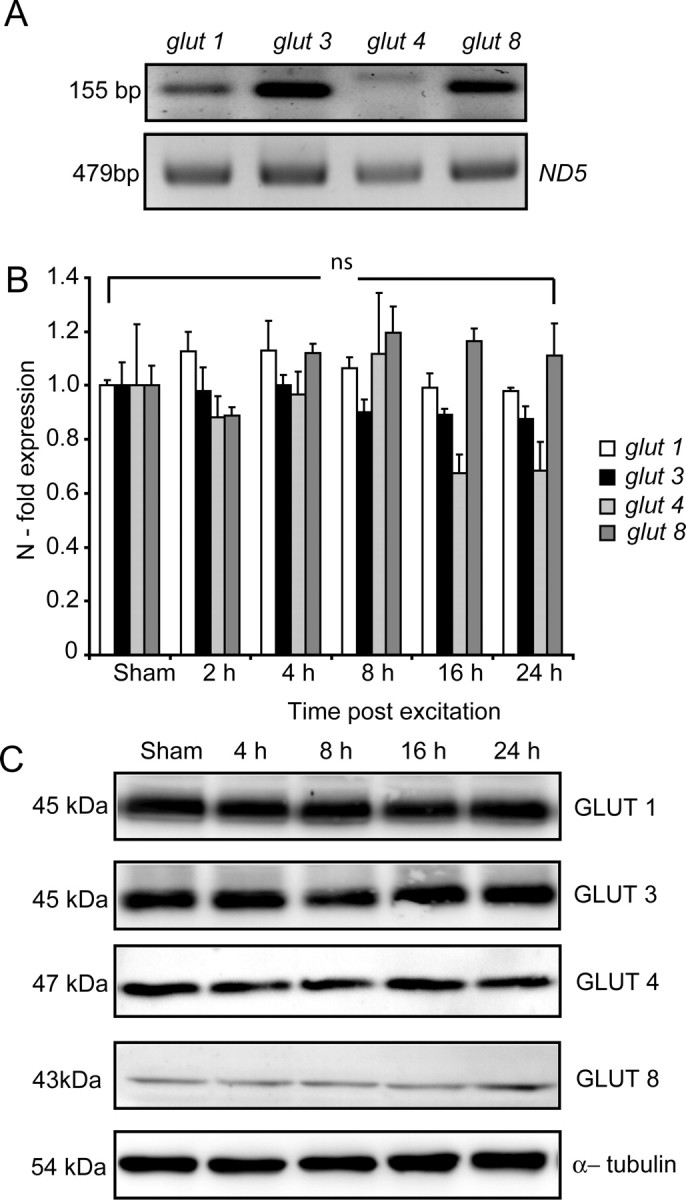
Transient glutamate excitation did not induce a significant change in the expression levels of GLUT1, 3, 4, and 8 over 24 h. A, Representative gel of RT-PCR for semiquantitative assessment of different glucose transporter isoforms localized in the cerebellum. ND5 was used as a control for RNA loading and RT-PCR efficiency. B, CGNs were stimulated with glutamate/glycine (100 μm/10 μm) for 10 min and allowed to recover for indicated time periods. After treatment, mRNA expression of the predominant neuronal glucose transporters (glut1, 3, 4, and 8) were determined by real-time qPCR analysis. Expression levels were normalized to sham-treated neurons and data are represented as SEM from three separate experiments. Data were presented as mean ± SEM. ns, No significance. C, Western blot analysis of GLUT1, 3, 4, and 8 at indicated times of recovery after transient glutamate excitation. α-Tubulin served as a loading control. Similar results were observed in two additional experiments.
Figure 4.
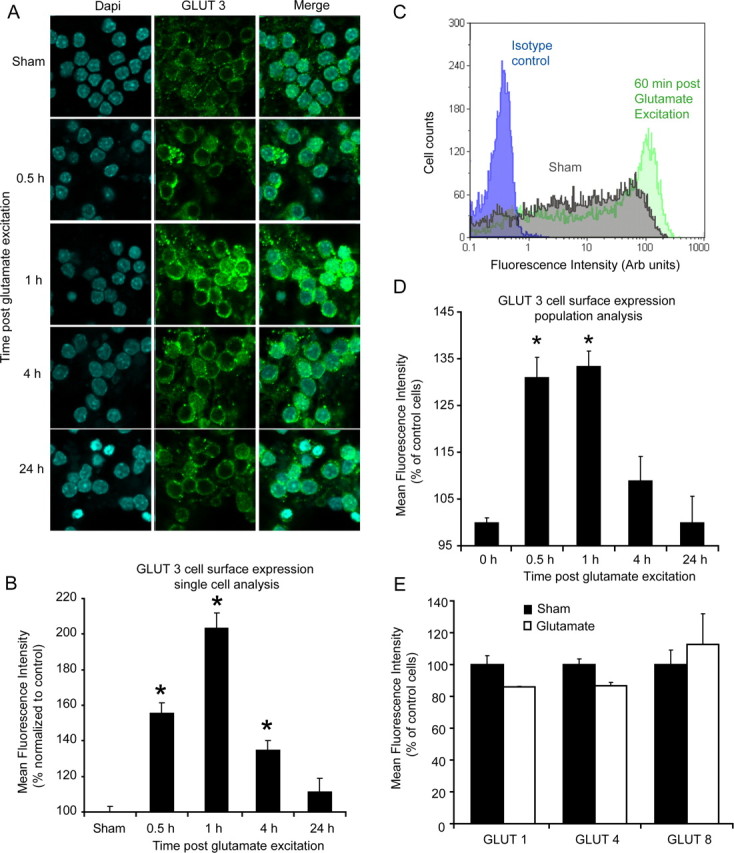
Transient glutamate excitation induces a rapid translocation of GLUT3 to the plasma membrane. A, Immunofluorescence of the cell surface expression of GLUT3 in sham- and glutamate- (glutamate and glycine 100 μm/10 μm, 10 min) treated neurons after 0.5, 1, 4, and 24 h. Nuclei were stained with DAPI (blue) and an Alexa Fluor 488-labeled secondary antibody (green) was used to visualize GLUT3 expression. B, Single-cell evaluation of cell surface GLUT3 expression in sham (1 h) and glutamate-treated neurons over a 24 h period. Data are presented as mean ± SEM. *p < 0.01, difference between sham- (n = 38) and glutamate-excited neurons after 0.5 (n = 43), 1 (n = 34), 4 h (n = 41), and 24 h (n = 39). C, Quantification of relative fluorescence intensity [arbitrary (Arb) units] by flow cytometry for GLUT3 surface expression. Cells were gated according to size. A right shift (gray–green) in distribution indicates an increase in GLUT3 cell surface fluorescence after stimulation with glutamate. Isotype control in blue. D, Population analysis (flow cytometry analysis) of GLUT3 surface expression for sham- and glutamate-stimulated neurons at the indicated time points. Data are presented as mean ± SEM. *p < 0.001, difference between sham-treated neurons and neurons 0.5 and 1 h after glutamate excitation. Experiments were performed in triplicate from three separate cultures. E, Flow cytometry analysis of the plasma membrane expression of other predominant neuronal glucose transporters (GLUT1, 4, and 8) for sham-treated and glutamate-stimulated neurons after 1 h. Results were obtained from three separate cultures and data are presented as mean ± SEM. For flow cytometry experiments a minimum of 104 events were collected per sample.
Activation of AMP-activated protein kinase induces a translocation of GLUT3 to the plasma membrane and a hyperpolarization of Δψm
AMPK has been identified as a highly sensitive energy sensor within cells, and it is believed to play an important part in a cell's response to energy stress (Hardie et al., 2006). Indeed, in muscle tissue there is strong evidence showing that the direct activation of AMPK with AICAR increases glucose transport by an insulin-independent signaling mechanism (Merrill et al., 1997; Hayashi et al., 1998; Bergeron et al., 1999). We therefore investigated the role of AMPK activity after transient glutamate excitation and the potential of AMPK activation to alter GLUT3 cell surface expression in neurons. A rapid increase in the active, phosphorylated state of AMPK (Thr 172) was identified in CGNs 5 min after glutamate excitation (Fig. 5A). Interestingly, cytosolic ATP inversely correlated with the phosphorylated state of AMPK (Thr 172) with a decreased availability of ATP within the neurons (Fig. 5B), mirrored by an increased phosphorylation of AMPK (Fig. 5A). Because AMPK is primarily activated by AMP, we investigated how the changes in ATP levels identified (Fig. 5B) corresponded to alterations in AMP levels within the neurons after excitation. HPLC analysis identified a marked reduction in ATP levels (100 ± 26.3% to 39.6 ± 13.8%) after glutamate excitation that was associated with a significant increase in the AMP levels (100 ± 9.1% to 302.6 ± 13.3%) (Fig. 5C; supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 5.
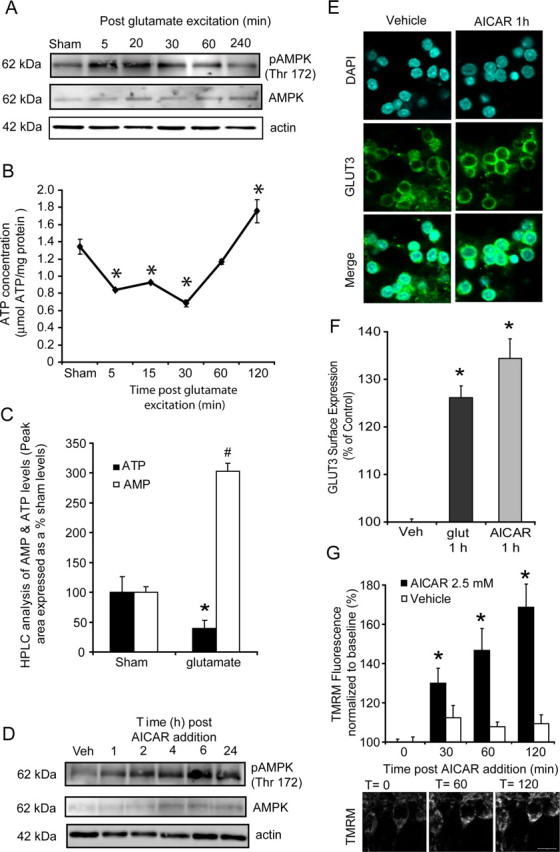
Increased AMPK activity regulates the translocation of GLUT3 to the cell surface in response to both glutamate excitation and activation with AICAR. CGNs were exposed to glutamate/glycine (100 μm/10 μm) for 10 min or continuously with 2.5 mm AICAR. A, Western blot analysis of pAMPK (Thr172) and total AMPK at the times indicated after glutamate excitation. Actin was used as an additional loading control. The observed responses are similar to that obtained in four separate experiments. B, CGNs plated in 24-well plates were exposed to glutamate/glycine for 10 min and their ATP content measured (μmol ATP/mg protein) at the times indicated. (n = 3 experiments in triplicate; *p < 0.01, difference from sham-treated neurons). C, AMP and ATP levels were determined by HPLC for sham and glutamate (10 min glutamate/glycine 100 μm/10 μm and recovered for 5 min) treated neurons. Peak area is expressed as a % of initial sham values. *p < 0.01, difference between ATP levels in sham- and glutamate-treated neurons. #p < 0.001, difference between AMP levels in sham- and glutamate-treated neurons (lysates analyzed from 3 separate experiments). D, Western blot analysis of pAMPK (Thr172) and total AMPK expression in CGNs after incubation with AICAR (2.5 mm). Actin was used as an additional loading control. The observed responses are similar to that obtained in three separate experiments. E, Immunofluorescence of the cell surface expression of GLUT3 in vehicle treated and neurons 1 h after incubation with AICAR (2.5 mm). F, Flow cytometry analysis of GLUT3 surface expression for vehicle-treated neurons and neurons 1 h after glutamate excitation and neurons treated with AICAR for 1 h. (n = 3 in triplicate; data are presented as mean ± SEM). *p < 0.01, difference between vehicle treated and neurons stimulated with AICAR and glutamate. G, Neurons plated on Willco dishes were loaded with TMRM and continuously exposed to AICAR (2.5 mm). Quantification of TMRM fluorescent for neurons was performed in vehicle treated (n = 26) and neurons treated with AICAR (n = 23) at time 0, 30 min, 60 min, and 120 min. Representative images were taken at selected time points (0, 60, and 120 min) during AICAR exposure. Data are presented as mean ± SEM. *p < 0.01, difference from vehicle and AICAR treated neurons.
Through the direct manipulation of AMPK with AICAR, we aimed to determine whether GLUT3 expression and Δψm were altered in a similar manner as that after transient glutamate excitation. Addition of AICAR and its conversion into the AMP mimetic ZMP resulted in a robust increase in the phosphorylated state of AMPK within 1 h (Fig. 5D) in CGNs. This increase in AMPK activity paralleled a significant translocation of GLUT3 to the plasma membrane (Fig. 5E,F) within 1 h of exposure and a marked increase in the whole-cell TMRM fluorescence (hyperpolarization of Δψm, 168.8 ± 15.9%) (Fig. 5G). It must be noted that as well as activating AMPK, AICAR can induce other cellular affects that are not related to AMPK activity (Guigas et al., 2007). Indeed, we found a significant reduction in cellular ATP levels (> 4 h) and a progressive loss of neuronal viability when neurons were incubated with AICAR (2.5 mm) for prolonged periods of time (24–48 h) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Inhibition or knockdown of AMPK activity significantly attenuates the translocation of GLUT3 to the plasma membrane after transient glutamate excitation
To clearly establish whether increased AMPK activity facilitated GLUT3 translocation within our model of glutamate excitation, we inhibited AMPK activity with compound C (Zhou et al., 2001; McCullough et al., 2005) for 30 min before and during the excitation phase. Incubation of CGNs with compound C decreased the phosphorylation of AMPK when compared with neurons excited with glutamate in the absence of an inhibitor (Fig. 6A). On examination of GLUT3 surface expression, glutamate-exposed neurons pretreated with compound C had similar levels of GLUT3 cell surface expression to that of sham-treated neurons (Fig. 6B). Compound C does not exclusively inhibit AMPK and has been reported to inhibit respiration and suppress mitochondrial-generated reactive oxygen species (Emerling et al., 2007). Therefore, we also suppressed AMPK activity by transiently knocking down the expression of the α1 and α2 AMPK subunits with siRNA (Fig. 6C). To knockdown α1 and α2 AMPK subunit expression in CGNs, we constructed a siRNA vector expressing α1/α2 AMPK subunit siRNA or control siRNA in parallel with copGFP to allow the identification of siRNA-expressing neurons. Significantly, those neurons that were transfected with the AMPK siRNA were found to have a much more reduced surface expression level of GLUT3 after glutamate excitation than neurons containing the control siRNA (Fig. 6D).
Figure 6.
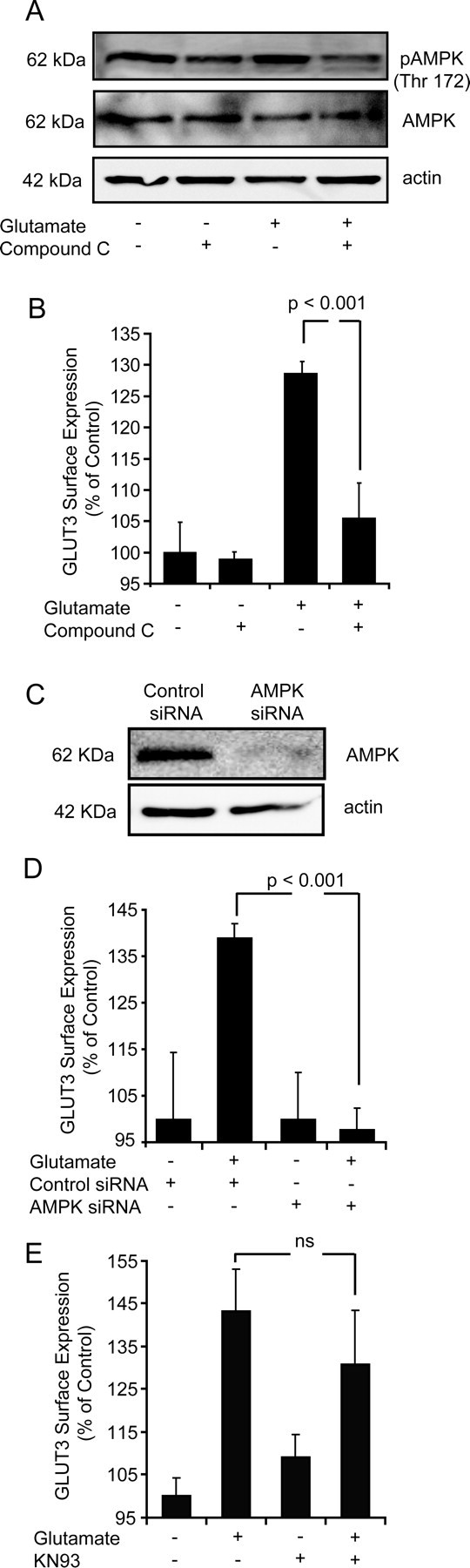
Inhibition of AMPK activity but not CaMKK activity significantly attenuates the translocation of GLUT3 to the cell surface after glutamate excitation. A, Western blot analysis of AMPK activity in sham treated and glutamate excited neurons plus and minus compound C. Total AMPK and actin served as a loading controls. Western blot is representative of three separate blots from three separate experiments. B, Flow cytometry analysis of GLUT3 surface expression in sham, vehicle treated, glutamate treated and neurons treated with glutamate preincubated with compound C (10 μm). Data are presented as mean ± SEM. *p < 0.001, difference between glutamate treated and neurons treated with glutamate in the presence of compound C. (n = 2 in triplicate). C, Western blot analysis of CGNs transfected with siRNA directed against AMPK and a nontargeting siRNA pool which acted as a control (control siRNA) (n = 3). D, Flow cytometry analysis of GLUT3 surface expression in sham treated control siRNA transfected neurons, glutamate treated control siRNA transfected neurons, sham treated AMPK siRNA transfected neurons and glutamate treated AMPK siRNA transfected neurons. *p < 0.001 difference between control siRNA and AMPK siRNA glutamate treated neurons (n = 3 in triplicate). E, Flow cytometry analysis of GLUT3 surface expression in sham and glutamate treated neurons plus or minus incubation with KN93 (10 μm for 16 h). Data are presented as mean ± SEM. No significant difference was identified between vehicle and KN93 treated neurons after glutamate excitation (n = 3 in triplicate).
Because the Ca2+-sensitive CaMKKs are known to play a contributory role to the activation of AMPK (Hawley et al., 2005; Woods et al., 2005; Jensen et al., 2007), and changes in cytosolic Ca2+ are known to play a pivotal role in the progression of excitotoxic injury (Tymianski et al., 1993a,b), we investigated whether CaMKK activity influenced GLUT3 translocation within our model of excitotoxic injury. Interestingly, inhibition of CaMKK activity using KN93 was not found to significantly alter the surface expression levels of GLUT3 after glutamate excitation (Fig. 6E), indicating that the Ca-activated CAMKK pathway may only play a contributory (synergistic) role in the activation of AMPK. Therefore, our data strongly suggest that the AMP-mediated activation of AMPK after transient glutamate excitation facilitates the translocation of GLUT3 to the neuronal cell surface.
siRNA knockdown of GLUT3 expression increases neuronal sensitivity to glutamate excitation
To further explore the relationship between glucose transport, altered mitochondrial energetics, and neuronal viability, we transiently knocked down GLUT3 levels in CGNs before glutamate excitation. Transient transfection of PC12 cells with a GLUT3 siRNA duplex significantly reduced GLUT3 expression at both mRNA (Fig. 7A) and protein level (Fig. 7B), suggesting the functionality of the siRNA sequence. Moreover, the transient reduction in GLUT3 expression did not appear to induce a compensatory upregulation of other GLUT isoforms with no apparent change in the expression levels of GLUT1 (Fig. 7A). To knockdown GLUT3 expression in CGNs, we constructed a siRNA vector expressing GLUT3 siRNA or control siRNA in parallel with copGFP (Fig. 7C). Those neurons transfected with GLUT3 siRNA were found to have a significantly reduced cell surface expression of GLUT3 (Fig. 7D) after glutamate excitation. Interestingly, the decreased expression of GLUT3 in neurons transfected with the GLUT3 siRNA vector was also associated with a significant reduction in TMRM fluorescence after glutamate excitation (Fig. 7E,F). Furthermore, inhibition of GLUT3 expression dramatically increased the vulnerability of CGNs to glutamate excitotoxicity (Fig. 7G). From this data it is evident that the knockdown of GLUT3 expression markedly inhibited the recovery and hyperpolarization of Δψm and significantly increased neuronal sensitivity to glutamate excitation.
Figure 7.
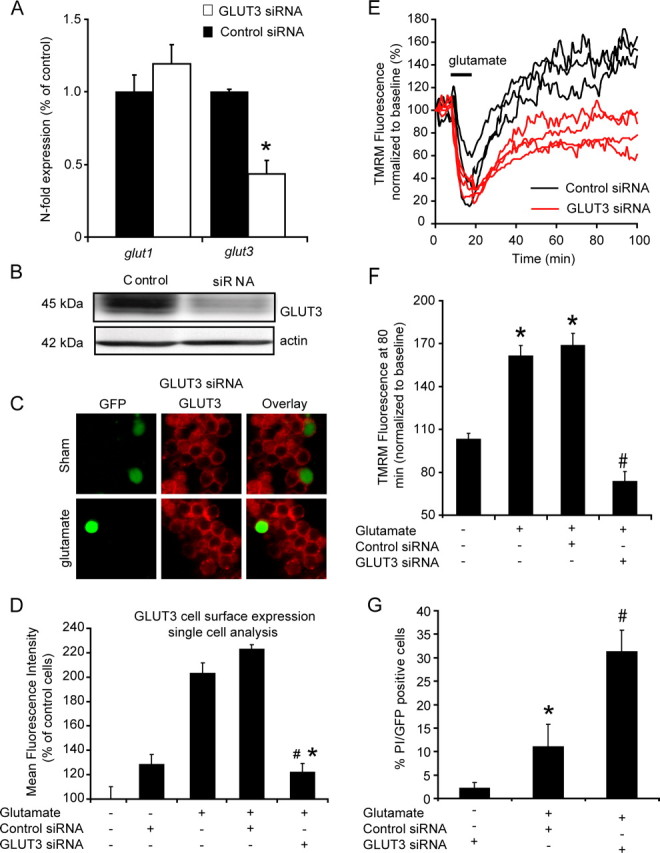
Knockdown of GLUT3 with siRNA increases neuronal sensitivity to glutamate excitation and reduces the recovery and hyperpolarization of Δψm at a single cell level. PC12 cells and CGNs were transfected with siRNA directed against GLUT3 and a nontargeting siRNA pool which acted as a control (control siRNA). A, B, GLUT3 downregulation is shown for PC12 transfected with control siRNA and GLUT3 directed siRNA at a (A) gene (mRNA n = 3) and (B) protein level (Western blot, n = 3). Effect of GLUT3 gene silencing on the gene expression level of GLUT1 isoform is also presented. C, D, Immunofluorescence and quantification of GLUT3 surface expression in neurons transfected with Double-Promoter pFIV-H1/U6 siRNA expression vector containing green fluorescent protein (EGFP) and siRNA against GLUT3 or control siRNA 1 h after glutamate excitation. Data are presented as mean ± SEM. *p < 0.01, difference between nontransfected neurons (n = 34) and neurons transfected with the GLUT siRNA after glutamate excitation (n = 38). #p < 0.01, difference between control siRNA transfected neurons (n = 26) and neurons transfected with the GLUT siRNA after glutamate excitation. Sham (n = 22), control siRNA (n = 17). E, Representative TMRM fluorescence traces for neurons transfected with pFIV-H1/U6-copGFP siRNA vector containing siRNA against GLUT3 and those with control siRNA after glutamate excitation. F, Quantification of the TMRM fluorescence after 80 min for sham treated neurons (n = 36) and glutamate stimulated; nontransfected neurons (n = 42), control siRNA transfected neurons (n = 31) and GLUT3 siRNA transfected neurons (n = 48). Data are presented as mean ± SEM. *p < 0.01, difference between sham, and nontransfected neurons and control siRNA-transfected neurons stimulated with glutamate. #p < 0.001, difference between control siRNA transfected neurons and GLUT3 siRNA-transfected neurons exposed to glutamate. G, Quantification of neuronal viability for sham-treated GLUT3 siRNA-transfected neurons (n = 64), glutamate-stimulated control siRNA-transfected neurons (n = 71), and glutamate-stimulated GLUT3 siRNA-transfected neurons (n = 80). Data are presented as mean ± SEM. *p < 0.01, difference between sham and glutamate treated control siRNA transfected neurons. #p < 0.001, difference between glutamate-treated control siRNA-transfected neurons and glutamate-treated GLUT3 siRNA transfected neurons.
Discussion
Here, we have set out to characterize the energetic signaling pathways activated in response to the energy depletion associated with transient glutamate excitation in CGNs (Ward et al., 2007). We have characterized a hyperpolarization of Δψm in the majority of CGNs after glutamate excitation that was found to be sensitive to both extracellular glucose availability and the expression levels of GLUT3 at the plasma membrane. Significant alterations in the AMP levels were identified after glutamate excitation. The activation of AMPK signaling after glutamate excitation was found to facilitate a translocation of GLUT3 to the plasma membrane with an inhibition or knockdown of AMPK potently blocking the increase in GLUT3 cell surface expression. Finally, we demonstrated that the knockdown of GLUT3 with siRNA significantly reduced the recovery and hyperpolarization of Δψm in CGNs and dramatically increased the sensitivity of CGNs to excitotoxic injury.
Hyperpolarization of Δψm
This study has identified a novel relationship between alterations in glucose transport and mitochondrial bioenergetics after glutamate excitation, with the hyperpolarization of Δψm observed (Fig. 1B,C) dependent on extracellular glucose availability (Fig. 2A,B) and the capacity of neurons to increase glucose transport (Fig. 6E,F). However, it is not clear how Δψm is altered in neurons after an excitotoxic event. Previously, we have established that mitochondria retain Ca2+ within the mitochondrial matrix for an extended period of time after glutamate excitation (Ward et al., 2005), and this Ca2+ has the potential to mediate a protracted increase in mitochondrial dehydrogenase activity (McCormack et al., 1990; Hansford, 1994). Therefore, increased mitochondrial dehydrogenase activity coupled to increased substrate availability could in turn drive proton efflux, increasing Δψm. Interestingly, the activation of AMPK with AICAR alone was sufficient to induce both a translocation of GLUT3 to the plasma membrane and a subsequent hyperpolarization of Δψm (Fig. 5E–G). This would suggest that activation of AMPK signaling and subsequent increased glucose uptake and utilization alter mitochondrial bioenergetics in a Ca2+-independent manner. Indeed, when mouse islet cells were incubated in high extracellular glucose, a Ca2+-independent increase in Δψm was also observed when cellular fuel increased (Heart et al., 2006; Wikstrom et al., 2007).
GLUT3 translocation and neuronal viability
In previous studies, an increase in the total expression levels of GLUT3 in models of neuronal excitation in vitro (Maher and Simpson, 1994) and hypoxia and ischemia in vivo (Vannucci et al., 1996, 1998) have been characterized. In our model of transient glutamate excitation in CGNs, we found no evidence for any significant increase in the expression levels of the main neuronal GLUT isoforms at a gene or protein level; however, in this study we have identified a rapid and significant translocation of the GLUT 3 isoform to the plasma membrane within a relatively short time (30 min) after transient glutamate excitation, although a contribution of other GLUT isoforms cannot be fully excluded. For example, in skeletal muscle the activation of AMPK with AICAR has been shown to induce increased glucose uptake through a translocation of GLUT4 to the plasma membrane (Merrill et al., 1997; Hayashi et al., 1998; Fryer et al., 2000). It is also important to note that increased AMPK activity has been shown to enhance the activity of the glucose transporters at the plasma membrane (Abbud et al., 2000; Barnes et al., 2002; Cidad et al., 2004), independent of translocation events. For instance, in HEK-293T cells transformed to express GLUT3, activation of AMPK enhanced GLUT3-mediated glucose uptake with no apparent increase in the cell surface expression of GLUT3 (Cidad et al., 2004). Therefore, the ability of AMPK to increase the capacity for glucose uptake via GLUT3 and other GLUT isoforms appears to be influenced by the signal- and cell-specific factors.
On further analysis of both immunofluorescence and flow cytometry data, it is evident that GLUT3 surface expression (Fig. 4) has a high cell to cell variability in neurons (Fig. 4C, gray shaded area), with a 10 min glutamate excitation resulting in a significantly increased surface expression level of GLUT3 in the majority of neurons for up to 4 h (Fig. 4A,B). This implies that some neurons require an increased capacity for glucose uptake over an extended period of time (1–4 h) to survive the energy crisis created during glutamate excitation. Interestingly, hyperglycemic conditions during reperfusion after both transient and focal ischemia have been shown to significantly increase infarct size and neuronal injury in vivo (Berger and Hakim, 1986; Nedergaard, 1987). Therefore, it may be the case that many neurons within the penumbra region of an infarct have an increased capacity for glucose uptake for a number of hours after an ischemic event (Vannucci et al., 1996, 1998). If on reperfusion these neurons are then exposed to hyperglycemic conditions, glucose can accumulate to toxic levels within the cell (Tomlinson and Gardiner, 2008), and this glucose-mediated stress coupled to excitotoxic/hypoxic stress signaling pathways may well increase neuronal injury on reperfusion.
Intracellular signaling events leading to AMPK activation and GLUT3 translocation
In a series of comprehensive studies Almeida, Bolanõs and coworkers have established that the inhibition of cytochrome oxidase activity with NO increases cytosolic AMP levels within cells leading to an activation of AMPK, which in turn regulates the glycolytic pathway and glucose transport within neuronal tissue (Almeida et al., 2004; Cidad et al., 2004; Bolaños et al., 2008). In our model of glutamate excitotoxicity, we show that the activation of the NMDA receptor and the subsequent accumulation of Na+ and Ca2+ within the cell results in a rapid depletion of cellular ATP (Fig. 5B), a rapid increase in cellular AMP levels (Fig. 5C), and the activation of AMPK (Fig. 5D). In addition, we show that the activation of AMPK facilitates the translocation of GLUT3 to the cell surface (Figs. 5E,F, 6B,D) and that an inhibition of CaMKK signaling pathway (Fig. 6E) does not significantly influence this. It would then appear that alterations in neuronal AMP levels (Hawley et al., 2003; Woods et al., 2003) is the predominant factor regulating AMPK activity after glutamate excitation. However, NO may also play a contributory role in the regulation of AMPK activity, glucose uptake and glucose utilization during excitotoxic injury (Bolaños et al., 2008).
Role of AMPK in survival and injury signaling
AMPK activity has been shown to be increased in neuronal tissue in response to glucose deprivation, metabolic stress, hypoxia, and ischemia (Culmsee et al., 2001; Gadalla et al., 2004; McCullough et al., 2005). However, further studies may be required to determine whether increased AMPK activity enhances neuronal survival or injury (Culmsee et al., 2001; McCullough et al., 2005). Culmsee and et al. showed that the transient activation of AMPK with low concentrations of AICAR (0.1 mm) protected hippocampal neurons during glucose deprivation, metabolic, excitotoxic, and oxidative stress (Culmsee et al., 2001). As the induction of AMPK activity has been linked to the preconditioning of cells against energetic stress (Peralta et al., 2001; Sukhodub et al., 2007), Culmsee et al. may have inadvertently preconditioned their neurons before stimulation by transiently increasing AMPK activity. In a separate study, McCullough et al. (McCullough et al., 2005) identified that the prolonged exposure of neurons to AICAR and the continuous activation of AMPK increased neuronal injury during ischemia (see also Fig. 5D; supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Together, these data imply that the duration and level of AMPK activity within neurons after an energetic stress is a pivotal factor in the decision between cell death and cell survival. Indeed, this hypothesis is supported by McCullough and coworkers, whereby the AMPK inhibitors, compound C and C75, reduced AMPK activity but enhanced cell survival when applied after a stroke (McCullough et al., 2005).
The activation of the AMPK signaling has been linked to the activation of a member of the FOXO family of Forkhead transcription factors, FOXO3 (Greer et al., 2007a,b). Whereas the activation of FOXO3 is known to promote resistance to oxidative stress, tumor suppression, and longevity in C. elegans (Greer et al., 2007b), in neurons, the activation of FOXO can modulate the expression of molecules such as Bim and Fas, thereby promoting neuronal apoptosis (Gilley et al., 2003; Sunters et al., 2003). In addition, the role of autophagy in cerebral ischemia has been identified as an important modifier of cell death and survival (Adhami et al., 2007; Rami and Kögel, 2008) with AMPK signaling also linked to the stimulation of autophagy (Liang et al., 2007; Matsui et al., 2007). However, the role of autophagic pathways in promoting or decreasing cell survival is still a matter of intensive research and debate.
In conclusion, we have shown that transient glutamate excitation of CGNs induces a rapid increase in neuronal AMP levels and an activation of AMPK. AMPK activation in turn mediates the translocation of GLUT3 to the plasma membrane and a subsequent hyperpolarization of Δψm (Fig. 8). Furthermore, we have demonstrated that the knockdown of GLUT3 expression is sufficient to prevent a hyperpolarization of Δψm and dramatically increases neuronal cell death. This study implicates energetic stress signaling as a key factor in the pathophysiology of neuronal injury after an excitotoxic event.
Figure 8.

Modulations in AMPK activity, glucose transport, and Δψm in response to the energetic stress created by transient glutamate excitation. NMDA receptor activation induces a rapid influx of Na+ and Ca2+ ions. The removal of these ions by membrane ATPases rapidly decrease cellular ATP levels altering the AMP:ATP ratio significantly. This alteration in the AMP:ATP ratio and to a minor degree CAMKK activation lead to a rapid activation of AMPK. The activation of AMPK signaling pathway facilitates the translocation of GLUT3 to the plasma membrane, increasing glucose uptake and its metabolism resulting in an increase in Δψm and ATP production. The ability of neurons to recover an energetic equilibrium (ATP levels) appears to determine whether they will tolerate the excitotoxic event.
Footnotes
This work was supported by grants from Royal College of Surgeons in Ireland (RCSI 839), the Health Research Board (RP/2006/181), and Higher Education Authority Programme for Research in Third Level Institutions (National Biophotonics and Imaging Platform Ireland) to M.W.W., and the Health Research Board (RP/2005/206) and Science Foundation Ireland to J.H.M.P. We would like to thank David A. Kennedy for his help in carrying out the HPLC measurements.
References
- Abbud W, Habinowski S, Zhang JZ, Kendrew J, Elkairi FS, Kemp BE, Witters LA, Ismail-Beigi F. Stimulation of AMP-activated protein kinase (AMPK) is associated with enhancement of Glut1-mediated glucose transport. Arch Biochem Biophys. 2000;380:347–352. doi: 10.1006/abbi.2000.1935. [DOI] [PubMed] [Google Scholar]
- Adhami F, Schloemer A, Kuan CY. The roles of autophagy in cerebral ischemia. Autophagy. 2007;3:42–44. doi: 10.4161/auto.3412. [DOI] [PubMed] [Google Scholar]
- Aguilar V, Alliouachene S, Sotiropoulos A, Sobering A, Athea Y, Djouadi F, Miraux S, Thiaudière E, Foretz M, Viollet B, Diolez P, Bastin J, Benit P, Rustin P, Carling D, Sandri M, Ventura-Clapier R, Pende M. S6 kinase deletion suppresses muscle growth adaptations to nutrient availability by activating AMP kinase. Cell Metab. 2007;5:476–487. doi: 10.1016/j.cmet.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Almeida A, Moncada S, Bolaños JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. 2004;6:45–51. doi: 10.1038/ncb1080. [DOI] [PubMed] [Google Scholar]
- Atlante A, Gagliardi S, Minervini GM, Marra E, Passarella S, Calissano P. Rapid uncoupling of oxidative phosphorylation accompanies glutamate toxicity in rat cerebellar granule cells. Neuroreport. 1996;7:2519–2523. doi: 10.1097/00001756-199611040-00023. [DOI] [PubMed] [Google Scholar]
- Barnes K, Ingram JC, Porras OH, Barros LF, Hudson ER, Fryer LG, Foufelle F, Carling D, Hardie DG, Baldwin SA. Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (AMPK) J Cell Sci. 2002;115:2433–2442. doi: 10.1242/jcs.115.11.2433. [DOI] [PubMed] [Google Scholar]
- Berger L, Hakim AM. The association of hyperglycemia with cerebral edema in stroke. Stroke. 1986;17:865–871. doi: 10.1161/01.str.17.5.865. [DOI] [PubMed] [Google Scholar]
- Bergeron R, Russell RR, 3rd, Young LH, Ren JM, Marcucci M, Lee A, Shulman GI. Effect of AMPK activation on muscle glucose metabolism in conscious rats. Am J Physiol. 1999;276:E938–944. doi: 10.1152/ajpendo.1999.276.5.E938. [DOI] [PubMed] [Google Scholar]
- Bolaños JP, Delgado-Esteban M, Herrero-Mendez A, Fernandez-Fernandez S, Almeida A. Regulation of glycolysis and pentose-phosphate pathway by nitric oxide: impact on neuronal survival. Biochim Biophys Acta. 2008;1777:789–793. doi: 10.1016/j.bbabio.2008.04.011. [DOI] [PubMed] [Google Scholar]
- Carruthers A. Facilitated diffusion of glucose. Physiol Rev. 1990;70:1135–1176. doi: 10.1152/physrev.1990.70.4.1135. [DOI] [PubMed] [Google Scholar]
- Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cidad P, Almeida A, Bolaños JP. Inhibition of mitochondrial respiration by nitric oxide rapidly stimulates cytoprotective GLUT3-mediated glucose uptake through 5′-AMP-activated protein kinase. Biochem J. 2004;384:629–636. doi: 10.1042/BJ20040886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concannon CG, Ward MW, Bonner HP, Kuroki K, Tuffy LP, Bonner CT, Woods I, Engel T, Henshall DC, Prehn JH. NMDA receptor-mediated excitotoxic neuronal apoptosis in vitro and in vivo occurs in an ER stress and PUMA independent manner. J Neurochem. 2008;105:891–903. doi: 10.1111/j.1471-4159.2007.05187.x. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- Dahm R, Zeitelhofer M, Götze B, Kiebler MA, Macchi P. Visualizing mRNA localization and local protein translation in neurons. Methods Cell Biol. 2008;85:293–327. doi: 10.1016/S0091-679X(08)85013-3. [DOI] [PubMed] [Google Scholar]
- Delgado-Esteban M, Almeida A, Bolaños JP. D-Glucose prevents glutathione oxidation and mitochondrial damage after glutamate receptor stimulation in rat cortical primary neurons. J Neurochem. 2000;75:1618–1624. doi: 10.1046/j.1471-4159.2000.0751618.x. [DOI] [PubMed] [Google Scholar]
- Emerling BM, Viollet B, Tormos KV, Chandel NS. Compound C inhibits hypoxic activation of HIF-1 independent of AMPK. FEBS Lett. 2007;581:5727–5731. doi: 10.1016/j.febslet.2007.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer LG, Hajduch E, Rencurel F, Salt IP, Hundal HS, Hardie DG, Carling D. Activation of glucose transport by AMP-activated protein kinase via stimulation of nitric oxide synthase. Diabetes. 2000;49:1978–1985. doi: 10.2337/diabetes.49.12.1978. [DOI] [PubMed] [Google Scholar]
- Fujii N, Jessen N, Goodyear LJ. AMP-activated protein kinase and the regulation of glucose transport. Am J Physiol Endocrinol Metab. 2006;291:E867–E877. doi: 10.1152/ajpendo.00207.2006. [DOI] [PubMed] [Google Scholar]
- Gadalla AE, Pearson T, Currie AJ, Dale N, Hawley SA, Sheehan M, Hirst W, Michel AD, Randall A, Hardie DG, Frenguelli BG. AICA riboside both activates AMP-activated protein kinase and competes with adenosine for the nucleoside transporter in the CA1 region of the rat hippocampus. J Neurochem. 2004;88:1272–1282. doi: 10.1046/j.1471-4159.2003.02253.x. [DOI] [PubMed] [Google Scholar]
- Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol. 2003;162:613–622. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007a;282:30107–30119. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol. 2007b;17:1646–1656. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigas B, Taleux N, Foretz M, Detaille D, Andreelli F, Viollet B, Hue L. AMP-activated protein kinase-independent inhibition of hepatic mitochondrial oxidative phosphorylation by AICA riboside. Biochem J. 2007;404:499–507. doi: 10.1042/BJ20070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansford RG. Physiological role of mitochondrial Ca2+ transport. J Bioenerg Biomembr. 1994;26:495–508. doi: 10.1007/BF00762734. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase–development of the energy sensor concept. J Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Mäkelä TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Hirshman MF, Kurth EJ, Winder WW, Goodyear LJ. Evidence for 5′ AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes. 1998;47:1369–1373. doi: 10.2337/diab.47.8.1369. [DOI] [PubMed] [Google Scholar]
- Heart E, Corkey RF, Wikstrom JD, Shirihai OS, Corkey BE. Glucose-dependent increase in mitochondrial membrane potential, but not cytoplasmic calcium, correlates with insulin secretion in single islet cells. Am J Physiol Endocrinol Metab. 2006;290:E143–E148. doi: 10.1152/ajpendo.00216.2005. [DOI] [PubMed] [Google Scholar]
- Iijima T, Mishima T, Akagawa K, Iwao Y. Mitochondrial hyperpolarization after transient oxygen-glucose deprivation and subsequent apoptosis in cultured rat hippocampal neurons. Brain Res. 2003;993:140–145. doi: 10.1016/j.brainres.2003.09.041. [DOI] [PubMed] [Google Scholar]
- Jensen TE, Rose AJ, Jørgensen SB, Brandt N, Schjerling P, Wojtaszewski JF, Richter EA. Possible CaMKK-dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. Am J Physiol Endocrinol Metab. 2007;292:E1308–E1317. doi: 10.1152/ajpendo.00456.2006. [DOI] [PubMed] [Google Scholar]
- Landree LE, Hanlon AL, Strong DW, Rumbaugh G, Miller IM, Thupari JN, Connolly EC, Huganir RL, Richardson C, Witters LA, Kuhajda FP, Ronnett GV. C75, a fatty acid synthase inhibitor, modulates AMP-activated protein kinase to alter neuronal energy metabolism. J Biol Chem. 2004;279:3817–3827. doi: 10.1074/jbc.M310991200. [DOI] [PubMed] [Google Scholar]
- Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- Maher F, Simpson IA. Modulation of expression of glucose transporters GLUT3 and GLUT1 by potassium and N-methyl-D-aspartate in cultured cerebellar granule neurons. Mol Cell Neurosci. 1994;5:369–375. doi: 10.1006/mcne.1994.1044. [DOI] [PubMed] [Google Scholar]
- Maher F, Davies-Hill TM, Simpson IA. Substrate specificity and kinetic parameters of GLUT3 in rat cerebellar granule neurons. Biochem J. 1996;315:827–831. doi: 10.1042/bj3150827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280:20493–20502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol. 1997;273:E1107–E1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- Nedergaard M. Transient focal ischemia in hyperglycemic rats is associated with increased cerebral infarction. Brain Res. 1987;408:79–85. doi: 10.1016/0006-8993(87)90360-x. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Ward MW. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- Peralta C, Bartrons R, Serafin A, Blazquez C, Blázquez C, Guzmán M, Prats N, Xaus C, Cutillas B, Gelpí E, Roselló-Catafau J. Adenosine monophosphate-activated protein kinase mediates the protective effects of ischemic preconditioning on hepatic ischemia-reperfusion injury in the rat. Hepatology. 2001;34:1164–1173. doi: 10.1053/jhep.2001.29197. [DOI] [PubMed] [Google Scholar]
- Rami A, Kögel D. Apoptosis meets autophagy-like cell death in the ischemic penumbra: two sides of the same coin? Autophagy. 2008;4:422–426. doi: 10.4161/auto.5778. [DOI] [PubMed] [Google Scholar]
- Robb-Gaspers LD, Burnett P, Rutter GA, Denton RM, Rizzuto R, Thomas AP. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 1998;17:4987–5000. doi: 10.1093/emboj/17.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhodub A, Jovanoviæ S, Du Q, Budas G, Clelland AK, Shen M, Sakamoto K, Tian R, Jovanoviæ A. AMP-activated protein kinase mediates preconditioning in cardiomyocytes by regulating activity and trafficking of sarcolemmal ATP-sensitive K(+) channels. J Cell Physiol. 2007;210:224–236. doi: 10.1002/jcp.20862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunters A, Fernández de Mattos S, Stahl M, Brosens JJ, Zoumpoulidou G, Saunders CA, Coffer PJ, Medema RH, Coombes RC, Lam EW. FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J Biol Chem. 2003;278:49795–49805. doi: 10.1074/jbc.M309523200. [DOI] [PubMed] [Google Scholar]
- Tomlinson DR, Gardiner NJ. Glucose neurotoxicity. Nat Rev Neurosci. 2008;9:36–45. doi: 10.1038/nrn2294. [DOI] [PubMed] [Google Scholar]
- Tsuji K, Nakamura Y, Ogata T, Shibata T, Kataoka K. Rapid decrease in ATP content without recovery phase during glutamate-induced cell death in cultured spinal neurons. Brain Res. 1994;662:289–292. doi: 10.1016/0006-8993(94)90828-1. [DOI] [PubMed] [Google Scholar]
- Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–1716. doi: 10.1046/j.1471-4159.1999.721707.x. [DOI] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993a;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Secondary Ca2+ overload indicates early neuronal injury which precedes staining with viability indicators. Brain Res. 1993b;607:319–323. doi: 10.1016/0006-8993(93)91523-u. [DOI] [PubMed] [Google Scholar]
- Vannucci SJ, Seaman LB, Vannucci RC. Effects of hypoxia-ischemia on GLUT1 and GLUT3 glucose transporters in immature rat brain. J Cereb Blood Flow Metab. 1996;16:77–81. doi: 10.1097/00004647-199601000-00009. [DOI] [PubMed] [Google Scholar]
- Vannucci SJ, Maher F, Simpson IA. Glucose transporter proteins in brain: delivery of glucose to neurons and glia. Glia. 1997;21:2–21. doi: 10.1002/(sici)1098-1136(199709)21:1<2::aid-glia2>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Vannucci SJ, Reinhart R, Maher F, Bondy CA, Lee WH, Vannucci RC, Simpson IA. Alterations in GLUT1 and GLUT3 glucose transporter gene expression following unilateral hypoxia-ischemia in the immature rat brain. Brain Res Dev Brain Res. 1998;107:255–264. doi: 10.1016/s0165-3806(98)00021-2. [DOI] [PubMed] [Google Scholar]
- Vareková RS, Bradác I, Plchút M, Skrdla M, Wacenovsky M, Mahr H, Mayer G, Tanner H, Brugger H, Withalm J, Lederer P, Huber H, Gierlinger G, Graf R, Tafer H, Hofacker I, Schuster P, Polcík M. www.rnaworkbench.com: a new program for analyzing RNA interference. Comput Methods Programs Biomed. 2008;90:89–94. doi: 10.1016/j.cmpb.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Vergun O, Han YY, Reynolds IJ. Glucose deprivation produces a prolonged increase in sensitivity to glutamate in cultured rat cortical neurons. Exp Neurol. 2003;183:682–694. doi: 10.1016/s0014-4886(03)00243-7. [DOI] [PubMed] [Google Scholar]
- Ward MW, Rego AC, Frenguelli BG, Nicholls DG. Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 2000;20:7208–7219. doi: 10.1523/JNEUROSCI.20-19-07208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward MW, Kushnareva Y, Greenwood S, Connolly CN. Cellular and subcellular calcium accumulation during glutamate-induced injury in cerebellar granule neurons. J Neurochem. 2005;92:1081–1090. doi: 10.1111/j.1471-4159.2004.02928.x. [DOI] [PubMed] [Google Scholar]
- Ward MW, Rehm M, Duessmann H, Kacmar S, Concannon CG, Prehn JH. Real time single cell analysis of Bid cleavage and Bid translocation during caspase-dependent and neuronal caspase-independent apoptosis. J Biol Chem. 2006;281:5837–5844. doi: 10.1074/jbc.M511562200. [DOI] [PubMed] [Google Scholar]
- Ward MW, Huber HJ, Weisová P, Düssmann H, Nicholls DG, Prehn JH. Mitochondrial and plasma membrane potential of cultured cerebellar neurons during glutamate-induced necrosis, apoptosis, and tolerance. J Neurosci. 2007;27:8238–8249. doi: 10.1523/JNEUROSCI.1984-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikstrom JD, Katzman SM, Mohamed H, Twig G, Graf SA, Heart E, Molina AJ, Corkey BE, de Vargas LM, Danial NN, Collins S, Shirihai OS. beta-Cell mitochondria exhibit membrane potential heterogeneity that can be altered by stimulatory or toxic fuel levels. Diabetes. 2007;56:2569–2578. doi: 10.2337/db06-0757. [DOI] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]