

Abstract
Glucose sensing by hypothalamic neurons triggers adaptive metabolic and behavioral responses. In orexin neurons, extracellular glucose activates a leak K+ current promoting electrical activity inhibition. Sensitivity to external acidification and halothane, and resistance to ruthenium red designated the tandem-pore K+ (K2P) channel subunit TASK3 as part of the glucose-induced channel. Here, we show that glucose inhibition and its pH sensitivity persist in mice lacking TASK3 or TASK1, or both subunits. We also tested the implication of another class of K2P channels activated by halothane. In the corresponding TREK1/2/TRAAK triple knock-out mice, glucose inhibition persisted in hypothalamic neurons ruling out a major contribution of these subunits to the glucose-activated K+ conductance. Finally, block of this glucose-induced hyperpolarizing current by low Ba2+ concentrations was consistent with the conclusion that K2P channels are not required for glucosensing in hypothalamic neurons.
Keywords: orexin neuron, leak potassium channel, resting potential, gene inactivation, neuropeptide, secretion
Introduction
Modulation of neuronal activity by glucose is pivotal in coordinating energy balance and behavior. Changing levels of extracellular glucose can lead to either excitation or inhibition of glucose-sensitive hypothalamic neurons. Neurons excited by rising glucose behave similarly to the β cells of the endocrine pancreas (Ibrahim et al., 2003). In these cells, an increase in glucose metabolism gives rise to elevated ATP levels that promote closure of ATP-sensitive K+ channels (KATP) and depolarization. Other hypothalamic neurons, such as neurons producing orexins/hypocretins, are inhibited by rising glucose (Yamanaka et al., 2003; Burdakov et al., 2005). They project to all major brain areas, with prominent innervation of arousal and autonomic regions where orexins are released and act on specific G-protein-coupled receptors. Defect in orexin signaling results in narcolepsy (Chemelli et al., 1999), although orexin neurons also regulate appetite and metabolism (Hara et al., 2001).
Recently, Burdakov et al. (2006) provided a careful description of the glucosensing mechanism in orexin neurons. They showed that the glucose-activated K+ conductance displays rapid activation and minimal time-dependent decay, as well as a current–potential relationship exhibiting Goldman–Hodgkin–Katz (GHK) outward rectification in physiological K+ conditions. Burdakov et al. (2006) next tested the sensitivity of the glucosensing mechanism to external pH and ruthenium red, and measured the unitary conductance of the glucose-induced channel. Based on these characteristics, they concluded that the K2P Twik1-related acid-sensitive K+ channel subunit TASK3 (Kcnk9, K2P9.1) was a major constituent of the glucose-induced channel.
To study the role of this neuronal nutrient sensor on behavior and metabolism of living animals, we developed TASK3 knock-out (KO) mice. Surprisingly, the glucose-induced hyperpolarization of orexin neurons was unaffected in these animals or even in TASK1 KO (Kcnk3, K2P3.1) and double TASK1/TASK3 KO mice, ruling out any functional compensation between the two TASK channel subunits. Gene inactivation of another class of halothane-activated K2P channels was also without effect on glucosensing. Finally, the dose-dependent inhibition of the glucose response by low concentrations of Ba2+ in WT mice confirmed the absence of implication of K2P channels in glucosensing by orexin neurons of the lateral hypothalamus.
Materials and Methods
Knock-out mice.
We targeted TASK3 gene locus for homologous recombination in 129/Sv embryonic stem (ES) cells. The vector was designed to allow cre-mediated deletion of exon 2, which encodes pore domains P1 and P2, transmembrane domains M2 to M4 as well as the cytoplasmic C-ter of TASK3. Primers were designed to amplify three contiguous DNA fragments of 6.6 kb (long arm), 1 kb (containing exon 2), and 2.9 kb (short arm) from 129/Sv genomic DNA. PCR products were subcloned into a modified pBluescript (Stratagene) containing a neomycin resistance cassette for positive selection, and thymidine kinase (TK) and diphtheria toxin (DT) genes to select against random incorporations. LoxP sites were added flanking the second exon and the neomycin resistance cassette for further gene inactivation by excision using the cre recombinase. Relative positions of these different elements are shown in Figure 1A. This construct was verified by sequencing TASK3 coding sequence, and 5′ and 3′ ends of long and short arms, as well as the loxP sites and the regions flanking the neo cassette. After linearization, the targeting vector was transfected by electroporation in ES cells by Genoway. DNA extracted from G418-resistant colonies was analyzed by PCR using a 5′ primer overlapping the sequence junction between the loxP sequence and the 5′ end of the short arm (P1, 5′-CCTGCAGGCGCGCCGATGTAA-3′) and a 3′ primer external to the targeted sequence (P2, 5′-GAGACACAATAAAGTCCAGTAGTAGG-3′). A positive clone was further characterized by Southern blot analysis using probes corresponding to 5′ and 3′ sequences flanking the targeted regions to ensure integrity of the targeted locus (data not shown). Genoway performed blastocyst injection. Chimeric animals were identified by coat color and crossed to C57BL/6J mice. Tail DNA was analyzed by PCR to select progeny bearing the floxed allele, which was then crossed to a mouse strain expressing the cre recombinase under the CMV promoter (CMV-cre). Exon 2 excision in offspring was assessed by PCR using primers flanking this region (P3, 5′-TGCGAGCTTCAGAGAGAGGATG-3′ and P4, 5′-ATGCTCTAATCTCCAGTCTG-3′). TASK−/− mice were generated by crossing TASK3−/− and TASK1−/− mice (Aller et al., 2005). Resulting TASK3+/−TASK1+/− animals were further crossed, and double KO progeny (TASK−/−) was identified by analyzing tail DNA.
Figure 1.
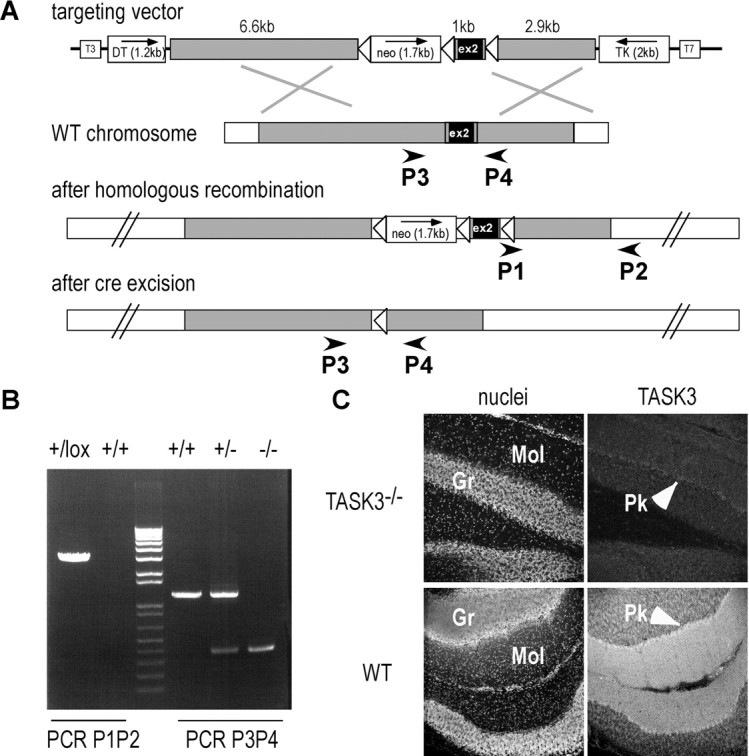
TASK3 gene inactivation. A, Inactivation strategy. Open triangles represent loxP sequences. Exon 2 is deleted in KO mice. B, PCR analysis of genomic DNA. Primer positions are shown in A. C, TASK3 immunodetection in adult mouse cerebellum. TASK3 expression observed in the molecular layer (Mol) of wild-type (WT) mouse was lacking in KO mouse. Gr, Granule cell layer; Pk, Purkinje cells.
TREK2 was inactivated by gene gap repair (Zheng et al., 1999). A 7.8 kb DNA fragment containing the 335 bp exon 2 of TREK2 was amplified by PCR from 129/Sv DNA and subcloned into a pBluescript (Stratagene) containing a neomycin resistance cassette. Relative positions of these different elements are shown in Figure 4A. An internal fragment of 1.5 kb was removed by NcoI digestion, and linearized plasmid was transfected by electroporation in 129/Sv ES cells (Genoway). Genomic DNA from G418-resistant colonies was analyzed by PCR using a 5′ primer absent in the targeting construct (P3, 5′-GGTCAGATGTCAGTGTTAGGTATGTC-3′) and a 3′ primer internal to pBS (P4, 5′-TTAACCCTCACTAAAGGGAACAAAAG-3′). A positive colony giving the expected PCR product of 3 kb was next characterized by Southern blot analysis to ensure integrity of targeted and flanking regions (data not shown). Blastocyst injection was performed by Genoway. Chimeric animals were identified by coat color and crossed to C57BL/6J mice. Tail DNA was analyzed by PCR using P3 and P4 primers to identify the recombinant allele, and P1 (5′-CACTTCTAGCTGCTGAAATGTG-3′) and P2 (5′-TCTTGGTCTGGCTCACTTGC-3′) primers external to the targeted locus to identify the 8 kb wild-type allele. In the recombinant allele, P1P2 fragment is 16 kb long and was no longer amplified by PCR (see Fig. 4B). PCR was performed using Hi-Fi polymerase mix from Fermentas according to the manufacturer‘s specifications. Amplification conditions were 95°C/1 min; [95°C/10 s; 62°C/30 s (+1°C/cycle); 68°C/3 min] 5×; [95°C/10 s; 57°C/30 s; 68°C/3 min (+5 s/cycle)] 32×; 68°C/7 min. Mouse brain cDNAs were analyzed by PCR using primers immediately flanking exon 2 (5′ primer: 5′-ATGAAATTTCCAATCGAGACGCC-3′ and 3′ primer: 5′-GCAATGCTTTTGCCAAAGATGGTT-3′). Multiple PCR products of 644, 979, and 1314 bp were amplified from −/− animals that correspond to alternative hybridizations from duplicated exon 2 sequences. Brain proteins were analyzed by Western blot using anti-TREK2 affinity-purified antibodies raised in rabbits against the last 60 C-ter residues of TREK2. To get TREK−/− triple KO mice, we first prepared TREK1−/−TREK2−/− and TREK1−/−TRAAK−/− mice and then we crossed them. Resulting TREK1+/−TREK2+/−TRAAK+/− were further crossed and triple KO progeny (TREK−/−) was identified by analyzing tail DNA.
Figure 4.

TREK2 gene inactivation. A, Inactivation strategy. Exon 2 is duplicated in KO mice introducing a frameshift resulting in a premature stop codon. B, PCR analysis of genomic DNA from mutant mice. Primer positions are shown in A. C, RT-PCR analysis of brain cDNAs. Primer positions are shown in A. Multiple PCR products are amplified from KO animals that correspond to alternative hybridizations from the exon 2 repeat. D, Western analysis of adult mouse brain proteins using anti-TREK2 antibodies.
Male C57BL/6J and mutant mice were bred in the local animal facility and maintained on a 12 h dark/light cycle (7 A.M./7 P.M.) with food and water ad libitum. All the experiments were performed in accordance with French standard ethical guidelines for laboratory animals (Agreement number 75-178, 05/16/2000).
Immunohistochemistry.
Immunostaining experiments were performed on C57BL/6J and TASK3−/− adult mice mouse. They were anesthetized with pentobarbital (50 mg/kg) and perfused transcardially with 4% paraformaldehyde in 0.1 m phosphate buffer. Brains were removed and postfixed overnight in the perfusion solution. Sixty micrometer sections were obtained on a vibratome (Leica). Free-floating slices were permeabilized for 10 min in PBS containing 1% Triton X-100 and then incubated for 30 min in the blocking solution (PBS, 5% horse serum, 0.5% Triton X-100). Slices were incubated with affinity-purified anti-TASK3 directed against its C-ter (Zanzouri et al., 2006) in the blocking solution overnight at 4°C (dilution 1:1000). After washing, slices were incubated with secondary antibodies (Alexa-488 anti-rabbit at 1:1000) and Hoechst 33342 (5 mg · ml−1) in the blocking solution for 1 h at room temperature. After washing, slices were mounted on glass coverslips and viewed on a confocal microscope (SP5, Leica).
Brain slices for electrophysiology.
Twelve- to twenty-seven-day-old mice were anesthetized with 1% halothane. Following decapitation, brains were rapidly removed and placed in cold phosphate/bicarbonate-buffered solution (PBBS, 4°C) composed of (in mm) 125 NaCl, 2.5 KCl, 0.4 CaCl2, 1 MgCl2, 25 glucose, 1.25 NaH2PO4, 26 NaHCO3, pH 7.4 when bubbled with 95% O2/5% CO2. Transversal 250-μm-thick SN slices cut with a vibrating microtome (Microm) were then transferred to an incubating chamber maintained at 34°C in oxygenated PBBS. After 1 h, slices were transferred to another incubating chamber at room temperature (22–25°C) filled with PBBS containing additional CaCl2 (final concentration: 2 mm). Before recording, slices were transferred to another incubating chamber filled with ACSF [glucose] containing (in mm) 125 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 0.2 glucose, 1.25 NaH2PO4, 26 NaHCO3, 10 HEPES, pH 7.3 when bubbled with 95% O2/5% CO2. The modest osmolarity changes associated with changing [glucose] in ACSF were left uncompensated. When indicated, ACSF pH was modified by addition of NaOH or HCl. All the chemicals were from Sigma.
Whole-cell patch-clamp recording and data analysis.
Brain slices were placed under a Nomarski microscope (Zeiss) equipped with infrared video camera (Axiocam, Zeiss) in a recording chamber superfused at a flow rate of 1 ml · min−1 with oxygenated ACSF [glucose]. Pictures were taken by using a digital camera (Axiocam, Zeiss) connected to image-acquisition software (Axiovision). Recordings were made at room temperature (25 ± 2°C) using an Axopatch 200B (Axon Instruments). Patch-clamp pipettes made from borosilicate glass capillary (Hilgenberg) had a resistance of 4–10 MΩ when filled with the internal solution containing (mm) 130 KCl, 2 MgCl2, 0.1 EGTA, 5 Na2ATP, 10 HEPES (pH adjusted to 7.3 with KOH). Values of access resistance ranged from 12 to 20 MΩ and were not compensated. Measurements were made 2–3 min after obtaining the whole cell to ensure dialysis. Cell capacitance and resistance were measured in voltage clamp using the pClamp Clampex software by applying 5 mV voltage steps. Changes of extracellular solution were obtained by a fast multibarrel delivery system positioned close to the cell tested. All chemicals were from Sigma. Current-clamp recordings were made in the IClamp fast mode of the Axopatch 200B. The cytoplasm of the neuron was sucked at the end of the recording when possible.
Statistical significance between groups (average data expressed as mean ± SEM, n = number of neurons) was tested using either the Student's t test or the ANOVA followed by t test, and were considered significant at p < 0.05. Statistical analysis was done using SigmaPlot (Jandel) and Origin (Microcal) software.
Results
TASK3 gene targeting and knock-out
Animals bearing floxed alleles of the TASK3 second exon were generated by standard procedures (Fig. 1A). Resulting mice were crossed with a CMV-cre mouse strain. Successful recombination and germline transmission were confirmed by PCR (Fig. 1B) and Southern blot analysis (data not shown). Deletion of the sequence encoding the second exon in TASK3 mRNA was verified by RT-PCR analysis from adult brain cDNAs (data not shown). In adult mouse, TASK3 is expressed at a high level in the cerebellum (Aller et al., 2005). As expected, a strong TASK3 immunoreactivity was detected in the molecular layer of the cerebellum. This labeling was lacking in TASK3−/− mice (Fig. 1C). In hypothalamic neurons, anti-TASK3 antibodies gave only a faint background labeling with no significant differences between WT and KO mice indicating that TASK3 expression is too modest in these cells to be detected by immunohistochemistry in our conditions (data not shown). TASK3−/− mice were viable and showed no obvious change in weight or size. TASK3−/− and the other mutant mice used in this study were back crossed with C57BL/6J mice for 10 generations to get mice with homogenous genetic backgrounds.
Orexin neuron identification
To study electrical activity of orexin neurons, Burdakov et al. (2006) made transgenic mice in which the expression of eGFP is driven by the human pre-pro-orexin promoter (orexin-eGFP mice). Eighty percent of orexin-containing neurons in brain slices were labeled with eGFP and fluorescent and were used for electrophysiology. Here, we identified orexin neurons in acutely isolated brain slices of the hypothalamus according to electrophysiological criteria, i.e., by the presence of a hyperpolarization-activated cation current (Ih) and a very low adaptation in action potential discharge in response to a depolarizing pulse when recording in the current-clamp mode (Fig. 2A,B). When it was possible, the cytoplasm was sucked at the end of recording for single-cell RT-PCR. Orexin mRNA was amplified in all presumed orexin neurons (n = 14) (Fig. 2C). Mean input resistance of the whole cells was 620 ± 93 MΩ (n = 36), and mean input capacitance was 55 ± 6 pF (n = 36). Orexin neurons usually displayed a slow hyperpolarization when glucose concentration was elevated from 0.2 mm to 4.5 mm, as described previously (Burdakov et al., 2005). This hyperpolarization was not significantly different in WT C57BL/6J mice (n = 6) and knock-in (KI) MCH-GFP mice (n = 18). In MCH-GFP mice, GFP expression is under the control of the MCH promoter. In the lateral hypothalamus, orexin neurons neighbor MCH neurons. In these mice, neurons expressing MCH are fluorescent and adjacent neurons, which are not fluorescent, are mainly orexin neurons (Guyon and Nahon, 2007). The properties of orexin neurons recorded in these KI mice and in C57BL/6J mice were similar thus results obtained in both strains of mice were pooled (mean amplitude of the glucose-induced hyperpolarization = −12.5 ± 1.16 mV, n = 24).
Figure 2.

Responses to glucose of orexin neurons in K2P channel KO mice. A, Typical current response of a orexin neuron recorded in the voltage-clamp mode in response to a 500 ms hyperpolarizing pulse to −100 mV from a holding potential of −60 mV. Notice the presence of a slowly activating inward current (Ih). B, Superimposed typical responses to depolarizing and hyperpolarizing pulses delivered from the resting potential of a orexin neuron recorded in the current-clamp mode. Notice that the discharge of action potential shows very few adaptation. C, Examples of typical pattern obtained in single cell multiplex RT-PCR from one presumed orexin neuron. Multiplex RT-PCR was performed as described by Guyon et al. (2005). A negative control without reverse transcriptase (NRT) was performed. D, Current-clamp recordings showing the effect of increases in glucose concentration from 0.2 to 4.5 mm in double TASK1−/−TASK3−/− (TASK−/−) and triple TREK1−/−TREK2−/−TRAAK−/− (TREK−/−) orexin neurons. E, Mean maximum amplitude (+SEM) of the hyperpolarization recorded in control and KO mice (number of neurons tested in parenthesis).
Glucose inhibition in orexin neurons from TASK−/− mice
Five criteria were used to identify TASK channels as part of the glucose-sensing mechanism: a GHK current–voltage relationship, a single-channel conductance of 40 pS, channel activation by halothane and external acidification, and resistance to ruthenium red (Burdakov et al., 2006). Burdakov et al. (2006) reported that 50% of the glucose-induced hyperpolarization is blocked at pH 6.9, a value closer to those of TASK3 homodimers (IC50 = 6.7) and TASK1/TASK3 heterodimers than TASK1 homodimers (IC50 = 7.3). Furthermore, resistance to a ruthenium red dose that blocks TASK3 homomers as well as the unitary conductance pointed out TASK1/TASK3 heterodimers as the most plausible molecular substrates for glucosensing in orexin neurons. TASK2 is not a good candidate because in mouse brain its expression cannot be detected by Northern blot (Warth et al., 2004) or in situ hybridization (Aller and Wisden, 2008). Moreover, its single-channel conductance of 59.9 pS in high-K+ conditions does not fit the unitary conductance of the glucose-sensitive channels in hypothalamic neurons (Reyes et al., 1998). In orexin neurons of TASK3−/− mice, we could still observe hyperpolarizations in response to glucose (−20 ± 2.9 mV, n = 3, data not shown). This response was not significantly different from that of control or TASK1−/− mice. However, because the absence of TASK3 in KO mice could be compensated by TASK1, we also tested glucosensing in TASK1−/− mice (Aller et al., 2005). We also crossed TASK3−/− and TASK1−/− mice to obtain double TASK KO mice (hereafter termed TASK−/− mice). In TASK1−/− and TASK−/− mice, the maximum amplitude of the effect as well as the kinetics of the response were not significantly different from that of responses recorded in orexin neurons from control mice (Fig. 2D,E).
pH sensitivity of the glucose response in TASK−/− mice
As illustrated in Figure 3A, extracellular acid (pH = 6) reversed the effects of glucose on membrane potential in control C57BL/6J mice (n = 4) similar to what was observed by Burdakov et al. (2006) in orexin-eGFP mice. Same effect of acidification was observed in TASK−/− mice (n = 4) (Fig. 3B). Acid-induced depolarization was always slow with kinetics comparable to those of the glucose-induced hyperpolarization. Although the brain slice tissue has powerful endogenous pH-buffering mechanisms that could distort the response onset, this does not seem in accordance with the very rapid effect of acidification on cloned TASK1 and TASK3 channels (Duprat et al., 1997; Rajan et al., 2000). Inhibition of TASK channels is fast because it relies on protonation of a histidine residue lining the external mouth of the conduction pathway (Rajan et al., 2000). The slow kinetics of inhibition suggests that extracellular acidification has an indirect action on the intracellular pH or on the cascade linking glucose elevation to K+ channel activation rather than a direct effect on the K+ channels opened by glucose.
Figure 3.

Pharmacology of the glucose-response in orexin neurons. A, B, Current-clamp recordings showing the effect of external pH (pHe) on glucose-induced hyperpolarization in WT (A) and TASK−/− (B) neurons. C, D, Responses to glucose are reversed by Ba2+. Examples of the effects of 200 μm Ba2+ (C) or increasing Ba2+ concentrations (D) are shown. Recordings were obtained in control C57BL/6J mice.
Glucose-induced inhibition in orexin neurons from TREK−/− KO mice
Beside pH sensitivity, activation by halothane was another strong discriminating criterion. Among K2P channels, only members of TASK and TREK subclasses are activated by the volatile anesthetic halothane at clinical doses (Patel et al., 1999; Lesage et al., 2000). TREK1 (Kcnk2, K2P2.1), TRAAK (Kcnk4, K2P4.1), and TREK2 (Kcnk10, K2P10.1) form a subfamily of mechanosensitive and polyunsaturated fatty acid-activated background K+ channels. We prepared TREK2−/− mice by gene gap repair (Fig. 4A–C) and verified that TREK2 protein was not longer detectable in KO mice (Fig. 4D). We next crossed TREK2−/− mice with TREK1−/− and TRAAK−/− mice (Heurteaux et al., 2004) to get triple KO mice, termed TREK−/− mice. In TREK−/− mice, glucose-induced hyperpolarization persisted in orexin neurons (n = 3) (Fig. 2D,E), ruling out a major contribution of these halothane-activated K2P channels in glucosensing.
Inhibition of the glucose response by Ba2+
K2P are not sensitive to the classical blockers of K+ channels such as TEA, 4-AP, or Cs+ (Lesage and Lazdunski, 2000; Lesage, 2003). They are only moderately inhibited by Ba2+, the most sensitive being TWIK1 (Kcnk1, K2P1.1) and TWIK2 (KCNK6, K2P6.1), with Ba2+ concentrations inhibiting 50% of the max currents (IC50) around 100 μm (Lesage et al., 1996; Patel et al., 2000). Ba2+ IC50 for TASK1 and TASK3 are much more important and comprised between 300 μm and 3 mm (for review, see Goldstein et al., 2005). We tested the effects of low concentrations of Ba2+ (10–200 μm) on glucose-induced hyperpolarization. Ba2+ alone had a modest depolarizing effect (2.00 ± 0.63 mV, n = 8), suggesting that a few Ba2+-sensitive K+ channels are spontaneously active in orexin neurons. In neurons in which the shift from 0.2 to 4.5 mm glucose induced a hyperpolarization of 11.29 ± 1.64 mV (n = 17), Ba2+ reversed glucose-induced hyperpolarization by 100% in 15 cells (Fig. 3C) and by 50% in two other neurons. Ba2+ (50 μm) was sufficient to fully reverse the glucose-induced hyperpolarization (n = 3) (Fig. 3D). In the presence of Ba2+, the effect of acidification on the glucose response was prevented (n = 4, data not shown), confirming that both treatments probably affected the same conductance. The kinetics of the Ba2+ block are slow. Although it is difficult to infer from kinetics of response onset when using applications in brain slice tissue, such slow response is consistent with a voltage-dependent block as previously shown for inwardly rectifying K+ channels (Kir). In these channels, Ba2+ binds to a site located at half the membrane span (Klein et al., 1999).
Discussion
It has been suggested that hyperpolarizing response to glucose of hypothalamic neurons is due to the opening of TASK3-containing K+ channels, mainly based on their sensitivity to pH and halothane (Burdakov et al., 2006). Here, we have demonstrated that the hyperpolarizing response to glucose persists in orexin neurons of the lateral hypothalamus of TASK3−/− mice. A similar response to glucose in double TASK−/− mice ruled out the eventuality of functional compensations between TASK1 and TASK3. Also, in orexin neurons of TASK−/− mice prepared independently, voltage-clamp studies have shown that glucose-sensitive currents were not changed (D. Burdakov, personal communication). Finally, inhibition of glucose response by low Ba2+ in WT mice definitely demonstrated that TASK channels are not involved in glucosensing by orexin neurons. This study and another recent one (Mulkey et al., 2007) using KO mice prove that it is still necessary to be cautious about the speculated roles of these channels regarding chemosensing and nutrient sensing.
We also have shown that halothane-activated TREK channels are not required for glucosensing. What could be the molecular nature of these glucose-sensitive K+ channels? Ba2+ sensitivity and kinetics of the Ba2+ block point out Kir channels as very attractive candidates. However, the GHK current–voltage relationship of the glucose-induced conductance is not in favor of this hypothesis. K+ channels form the largest family of ion channels with 77 genes encoding pore-forming subunits (Goldstein et al., 2005; Gutman et al., 2005). Often, different subunits form heteromultimers with novel properties. Heteromultimers and homomultimers can also interact with auxiliary and regulatory subunits that themselves modify the electrophysiological and/or pharmacological characteristics. K2P channels are not the only subunits to produce background channels. For example SK channels, KCNQ subunits associated with KCNE subunits, weakly rectifying Kir channels can all produce background conductances. The glucose-induced conductance may be produced by one of these channels or by a yet uncharacterized combination of subunits and regulatory proteins. Further works, in particular a better pharmacological characterization, should help to decipher the molecular nature of the glucosensing mechanism in orexin neurons.
Footnotes
This work was supported partly by the Centre National de la Recherche Scientifique (program Origine de l'Homme, du Langage, et des Langues) and Agence Nationale pour le Recherche (ANR-08-MNP-018), the Convention Hospital Laval (05168), the Association pour la Recherche contre le Cancer (ARC Grant), and the Fondation pour la Recherche Médicale. F. Lesage is the recipient of a “contrat d'interface” Inserm/Centre Hospitalier Universitaire, Service de Neurologie, Hôpital Pasteur, Nice. MCH-GFP mice were kindly provided by Prof. J. M. Friedman (The Rockefeller University, New York, NY) and TASK1−/− mice by Prof. W. Wisden (University of Aberdeen, Aberdeen, UK).
References
- Aller MI, Wisden W. Changes in expression of some two-pore domain potassium channel genes (KCNK) in selected brain regions of developing mice. Neuroscience. 2008;151:1154–1172. doi: 10.1016/j.neuroscience.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Aller MI, Veale EL, Linden AM, Sandu C, Schwaninger M, Evans LJ, Korpi ER, Mathie A, Wisden W, Brickley SG. Modifying the subunit composition of TASK channels alters the modulation of a leak conductance in cerebellar granule neurons. J Neurosci. 2005;25:11455–11467. doi: 10.1523/JNEUROSCI.3153-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdakov D, Gerasimenko O, Verkhratsky A. Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J Neurosci. 2005;25:2429–2433. doi: 10.1523/JNEUROSCI.4925-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdakov D, Jensen LT, Alexopoulos H, Williams RH, Fearon IM, O'Kelly I, Gerasimenko O, Fugger L, Verkhratsky A. Tandem-pore K+ channels mediate inhibition of orexin neurons by glucose. Neuron. 2006;50:711–722. doi: 10.1016/j.neuron.2006.04.032. [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Duprat F, Lesage F, Fink M, Reyes R, Heurteaux C, Lazdunski M. TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J. 1997;16:5464–5471. doi: 10.1093/emboj/16.17.5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein SA, Bayliss DA, Kim D, Lesage F, Plant LD, Rajan S. International Union of Pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol Rev. 2005;57:527–540. doi: 10.1124/pr.57.4.12. [DOI] [PubMed] [Google Scholar]
- Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stühmer W, Wang X. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev. 2005;57:473–508. doi: 10.1124/pr.57.4.10. [DOI] [PubMed] [Google Scholar]
- Guyon A, Nahon JL. Multiple actions of the chemokine stromal cell-derived factor-1alpha on neuronal activity. J Mol Endocrinol. 2007;38:365–376. doi: 10.1677/JME-06-0013. [DOI] [PubMed] [Google Scholar]
- Guyon A, Banisadr G, Rovère C, Cervantes A, Kitabgi P, Melik-Parsadaniantz S, Nahon JL. Complex effects of stromal cell-derived factor-1 alpha on melanin-concentrating hormone neuron excitability. Eur J Neurosci. 2005;21:701–710. doi: 10.1111/j.1460-9568.2005.03890.x. [DOI] [PubMed] [Google Scholar]
- Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, Sugiyama F, Yagami K, Goto K, Yanagisawa M, Sakurai T. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–354. doi: 10.1016/s0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- Heurteaux C, Guy N, Laigle C, Blondeau N, Duprat F, Mazzuca M, Lang-Lazdunski L, Widmann C, Zanzouri M, Romey G, Lazdunski M. TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J. 2004;23:2684–2695. doi: 10.1038/sj.emboj.7600234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim N, Bosch MA, Smart JL, Qiu J, Rubinstein M, Rønnekleiv OK, Low MJ, Kelly MJ. Hypothalamic proopiomelanocortin neurons are glucose responsive and express KATP channels. Endocrinology. 2003;144:1331–1340. doi: 10.1210/en.2002-221033. [DOI] [PubMed] [Google Scholar]
- Klein H, Garneau L, Coady M, Lemay G, Lapointe JY, Sauvé R. Molecular characterization of an inwardly rectifying K+ channel from HeLa cells. J Membr Biol. 1999;167:43–52. doi: 10.1007/s002329900470. [DOI] [PubMed] [Google Scholar]
- Lesage F. Pharmacology of neuronal background potassium channels. Neuropharmacology. 2003;44:1–7. doi: 10.1016/s0028-3908(02)00339-8. [DOI] [PubMed] [Google Scholar]
- Lesage F, Lazdunski M. Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol. 2000;279:F793–F801. doi: 10.1152/ajprenal.2000.279.5.F793. [DOI] [PubMed] [Google Scholar]
- Lesage F, Guillemare E, Fink M, Duprat F, Lazdunski M, Romey G, Barhanin J. TWIK-1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J. 1996;15:1004–1011. [PMC free article] [PubMed] [Google Scholar]
- Lesage F, Terrenoire C, Romey G, Lazdunski M. Human TREK2, a 2P domain mechano-sensitive K+ channel with multiple regulations by polyunsaturated fatty acids, lysophospholipids, and Gs, Gi, and Gq protein-coupled receptors. J Biol Chem. 2000;275:28398–28405. doi: 10.1074/jbc.M002822200. [DOI] [PubMed] [Google Scholar]
- Mulkey DK, Talley EM, Stornetta RL, Siegel AR, West GH, Chen X, Sen N, Mistry AM, Guyenet PG, Bayliss DA. TASK channels determine pH sensitivity in select respiratory neurons but do not contribute to central respiratory chemosensitivity. J Neurosci. 2007;27:14049–14058. doi: 10.1523/JNEUROSCI.4254-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AJ, Honoré E, Lesage F, Fink M, Romey G, Lazdunski M. Inhalational anesthetics activate two-pore-domain background K+ channels. Nat Neurosci. 1999;2:422–426. doi: 10.1038/8084. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Maingret F, Magnone V, Fosset M, Lazdunski M, Honoré E. TWIK-2, an inactivating 2P domain K+ channel. J Biol Chem. 2000;275:28722–28730. doi: 10.1074/jbc.M003755200. [DOI] [PubMed] [Google Scholar]
- Rajan S, Wischmeyer E, Xin Liu G, Preisig-Müller R, Daut J, Karschin A, Derst C. TASK-3, a novel tandem pore domain acid-sensitive K+ channel. An extracellular histidine as pH sensor. J Biol Chem. 2000;275:16650–16657. doi: 10.1074/jbc.M000030200. [DOI] [PubMed] [Google Scholar]
- Reyes R, Duprat F, Lesage F, Fink M, Salinas M, Farman N, Lazdunski M. Cloning and expression of a novel pH-sensitive two pore domain K+ channel from human kidney. J Biol Chem. 1998;273:30863–30869. doi: 10.1074/jbc.273.47.30863. [DOI] [PubMed] [Google Scholar]
- Warth R, Barrière H, Meneton P, Bloch M, Thomas J, Tauc M, Heitzmann D, Romeo E, Verrey F, Mengual R, Guy N, Bendahhou S, Lesage F, Poujeol P, Barhanin J. Proximal renal tubular acidosis in TASK2 K+ channel-deficient mice reveals a mechanism for stabilizing bicarbonate transport. Proc Natl Acad Sci U S A. 2004;101:8215–8220. doi: 10.1073/pnas.0400081101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami K, Sugiyama F, Goto K, Yanagisawa M, Sakurai T. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron. 2003;38:701–713. doi: 10.1016/s0896-6273(03)00331-3. [DOI] [PubMed] [Google Scholar]
- Zanzouri M, Lauritzen I, Duprat F, Mazzuca M, Lesage F, Lazdunski M, Patel A. Membrane potential-regulated transcription of the resting K+ conductance TASK-3 via the calcineurin pathway. J Biol Chem. 2006;281:28910–28918. doi: 10.1074/jbc.M606092200. [DOI] [PubMed] [Google Scholar]
- Zheng B, Mills AA, Bradley A. A system for rapid generation of coat color-tagged knockouts and defined chromosomal rearrangements in mice. Nucleic Acids Res. 1999;27:2354–2360. doi: 10.1093/nar/27.11.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]