

Abstract
After brain damage such as stroke, topographically organized sensory and motor cortical representations remap onto adjacent surviving tissues. It is conceivable that cortical remapping is accomplished by changes in the temporal precision of sensory processing and regional connectivity in the cortex. To understand how the adult cortex remaps and processes sensory signals during stroke recovery, we performed in vivo imaging of sensory-evoked changes in membrane potential, as well as multiphoton imaging of dendrite structure and tract tracing. In control mice, forelimb stimulation evoked a brief depolarization in forelimb cortex that quickly propagated to, and dissipated within, adjacent motor/hindlimb areas (<100 ms). One week after forelimb cortex stroke, the cortex was virtually unresponsive to tactile forelimb stimulation. After 8 weeks recovery, forelimb-evoked depolarizations reemerged with a characteristic pattern in which responses began within surviving portions of forelimb cortex (<20 ms after stimulation) and then spread horizontally into neighboring peri-infarct motor/hindlimb areas in which depolarization persisted 300–400% longer than controls. These uncharacteristically prolonged responses were not limited to the remapped peri-infarct zone and included distant posteromedial retrosplenial cortex, millimeters from the stroke. Structurally, the remapped peri-infarct area selectively exhibited high levels of dendritic spine turnover, shared more connections with retrosplenial cortex and striatum, and lost inputs from lateral somatosensory cortical regions. Our findings demonstrate that sensory remapping during stroke recovery is accompanied by the development of prolonged sensory responses and new structural circuits in both the peri-infarct zone as well as more distant sites.
Keywords: stroke, plasticity, somatosensory cortex, dendritic spine, motor cortex, membrane potential
Introduction
Recovery from brain injury is dependent on how well surviving functionally homologous brain regions assume the responsibilities of those lost to injury (Corbetta et al., 2005; Ward, 2006). Given the topographic organization of the cerebral cortex, areas situated in close proximity to the site of injury (i.e., peri-infarct regions) are most likely to adopt new functional roles after stroke (Carmichael, 2006; Cramer, 2008). Indeed, there is considerable support for this idea because the peri-infarct zone is heavily enriched with growth-promoting genes, markers of axonal sprouting, new blood vessels, and even new neurons (Stroemer et al., 1995; Carmichael, 2006; Kolb et al., 2007). Furthermore, metabolic or hemodynamic imaging studies have shown that sensorimotor cortical representations can position themselves in surviving peri-infarct regions (Dijkhuizen et al., 2001; Jaillard et al., 2005), which are critical for the restoration of sensorimotor functions (Castro-Alamancos and Borrel, 1995; Werhahn et al., 2003).
Despite considerable progress in our understanding of cortical map plasticity after stroke, many fundamental questions need to be resolved if we hope to maximize functional recovery. For example, we still do not know the spatiotemporal dynamics of sensory information flow in and out of reorganized cortical networks or the temporal precision with which new functional representations process sensory stimuli. This is important because restoration of normal tactile perception after stroke (i.e., to sense contours, texture, and flutter) requires our sensory system to detect and encode tactile stimuli that are separated in both space and time (Blake et al., 1997). One of the primary reasons for the paucity of data on this topic is that previous electrophysiological or imaging studies (Nudo et al., 1996; Xerri et al., 1998; Ward, 2004) could not simultaneously monitor the spatial/temporal spread of neuronal activation across a large area of cortex with millisecond time resolution, appreciably faster than hemodynamic responses (that occur over seconds) that form the basis of most types of functional imaging (Shoham et al., 1999). Second, how and where new structural synaptic circuits form in relation to reorganized sensory maps remains unknown.
To address these issues, we used the photothrombotic method as our model of focal ischemic stroke because it allowed us to precisely target a cortical sensory representation in a relatively noninvasive manner (through the thinned skull or a cranial window). We then used two-photon microscopy and voltage-sensitive dye (VSD) imaging in vivo, to interrogate cortical map structure at the level of individual synapses and function with millisecond temporal resolution (Grinvald and Hildesheim, 2004; Helmchen and Denk, 2005; Ferezou et al., 2006; Kerr and Denk, 2008). Furthermore, given that VSD signals can reflect subthreshold membrane depolarization (Berger et al., 2007; Ferezou et al., 2007), one can visualize latent connections between functionally related areas that previous studies could not detect (Kleinfeld and Waters, 2007). Our data indicate that recovery from focal stroke damage in the adult brain is associated with profound changes in the temporal dynamics of cortical information processing, synaptic structural plasticity, and new patterns of regional connectivity.
Materials and Methods
Animals.
Adult male wild type of yellow fluorescent protein (YFP) transgenic mice (>2 months of age, C57BL/6 background) (Feng et al., 2000) were used. Mice were housed in clear plastic cages, under a 12 h light/dark cycle, and were given ad libitum access to water and standard laboratory diet. All experiments were conducted in accordance with the guidelines set forth by the Canadian Council for Animal Care.
Intrinsic optical signal imaging.
Mice were lightly anesthetized with isoflurane gas (1.5% for induction, 1% for maintenance; mixed in air) while maintaining body temperature at 37°C. For prestroke or sham surgery, intrinsic optical signal (IOS) imaging of forelimb (FL) and hindlimb (HL) representations, a 3 × 3 mm region of skull was carefully thinned to ∼50% of original thickness using a high-speed dental drill and a surgical scalpel (Surgistar #6400). After applying 1.3% low-melt agarose [at 37–38°C; Type 3-A Sigma; A9793, dissolved in a HEPES-buffered artificial CSF (ACSF)] and a coverslip over the skull, the cortical surface was illuminated by red and green sets of light-emitting diodes (LEDs) mounted around the microscope objective driven by a regulated direct current power supply (Circuit Test Electronics). The green LED light source was used for visualizing the surface of the cortex and pattern of vessels. The red LED light source (center at 635 nm) was used to detect sensory-evoked changes in light reflectance in cortical regions in which levels of deoxyhemoglobin and neuronal activity are increased (Frostig et al., 1990). Before functional imaging, the plane of focus was set to 300 μm below the cortical surface to blur the contribution of large surface vessels. Image acquisition was performed using XCAP standard version 2.2 imaging software (EPIX) with a Dalsa M-60 Pantera 12-bit camera mounted on a video macroscope setup that used a short focus front-to-front dual-video lens system (3.8 × 3.8 mm field, 7.5 μm per pixel). Each data collection session consisted of 20–40 trials, each taken 20 s apart. During each trial, 15 image frames were collected over 1.5 s before and after contralateral limb stimulation using a piezoelectric device (100 Hz for 1 s). To generate maps of forelimb and hindlimb areas, trials for each limb were first summed and mean filtered (radius = 3) using NIH Image J software. Responsive areas were then identified by dividing all image frames taken 1.5 s after stimulation by those taken before. Recent work from our laboratory using in vivo single-cell calcium imaging (Winship and Murphy, 2008) has shown that the functional border between forelimb and hindlimb responsive neurons is very sharp. Therefore, we chose to threshold maps at two-thirds of maximal response amplitude that most routinely delineated forelimb and hindlimb responsive areas with minimal overlap. Response maps were then assigned a particular color and merged onto an image of the surface vasculature to create color-coded response maps.
Photothrombosis.
After IOS mapping, unilateral ischemic stroke was induced in the forelimb sensorimotor cortex (outlined with a water-insoluble black marker) using the photothrombotic method (Watson et al., 1985). This model was selected because it is minimally invasive and produces highly localized and reproducible lesions (Maxwell and Dyck, 2005). Briefly, the skull over the forelimb area was illuminated with a collimated beam of green laser light (532 nm, 17 mW; ∼1.5 mm diameter) for 15 min after an injection of 1% Rose Bengal solution (100 mg/kg, i.p., in PBS). Age-matched sham surgery controls were treated in an identical manner with the omission of either laser illumination or Rose Bengal injection. In either case, sham surgery did not produce any signs of damage (i.e., scarring) or structural alterations to neuronal dendrites (Brown et al., 2007). After surgery, mice were returned to their home cages in which they were housed either singly or in pairs and were allowed to recover.
Behavioral testing.
The cylinder task was used to detect changes in the use of each forelimb during recovery from unilateral photothrombotic stroke (Shanina et al., 2006). Animals began testing 1–2 d before the induction of stroke to assess basal levels of forepaw use and/or paw preference and then were tested 1, 2, 3, 4, 6, and 8 weeks after surgery. Each testing session involved placing an individual mouse in a clear plastic cylinder that was 10 cm in diameter and 20 cm high. Forelimb placement onto the walls of the cylinder during rears was recorded for ∼10 min per session with a web camera placed underneath the plastic cylinder. Forelimb use was scored by counting the number of times each individual forelimb or both were used to make the first touch onto the wall of the cylinder during a vertical rear. To quantify the asymmetry in forelimb use after stroke, we first calculated a baseline or preoperative value for each forepaw, which represented the number of times a particular forepaw was used in a rear divided by the total number of rears (rears using both paws were excluded from the total). A baseline score for each paw was used to compensate for the fact that some mice, even before the induction of stroke, appear to show a preference for one paw over the other. This value was then divided by a post-stroke testing score obtained for each forepaw (using same formula described for baseline) and expressed as the percentage change in post-stroke forelimb use relative to baseline.
Longitudinal in vivo imaging of dendritic spines.
In isoflurane-anesthetized mice (1.5% in air), a circular piece of skull (3 mm diameter) overlying the forelimb and hindlimb areas was carefully removed using a high-speed dental drill and fine surgical forceps but the surface vasculature and dura intact were left intact. A 5-mm-round glass coverslip was gently placed over the moistened surface of the exposed brain and permanently fixed into place using cyanoacrylate glue and dental cement. Mice were given a single injection of 2% dexamethasone (0.03 ml, i.m.) to minimize potential brain swelling and fluid accumulation in the trachea. Given that the surgical procedure could potentially affect basal spine turnover levels, all mice were allowed to recover for 4–5 weeks before the first imaging session (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Mice that displayed any signs of brain damage or loss of clarity through the cranial window were not used for imaging (∼60% of mice).
Two-photon imaging of dendritic structure was performed using a modified Olympus BX50W1 upright microscope equipped with titanium:sapphire Coherent Mira 900 mode-locked laser that was tuned to 900 nm for YFP excitation. The laser produced 100 fs pulses at a rate of 76 MHz and was pumped by a 5 W Verdi argon laser. Excitation power measured at the back aperture of the objective was typically between 20 and 35 mW and was adjusted to achieve near identical levels of fluorescence within each imaging session. Images were acquired with 60× Olympus IR-LUMPLanFl water-immersion objective [numerical aperture (NA) 0.9], using custom software routines (IgorPro; Wavemetrics) interfacing with a 12-bit data acquisition card (PCI-6110; National Instruments) in a standard desktop personal computer.
To determine the effect of stroke on spine turnover rates, the apical dendritic tufts of YFP-labeled neurons were imaged for several weeks before and after the induction of stroke (1 week intervals). Just before each imaging session, mice were anesthetized with an injection of ketamine/xylazine (100 and 10 mg/kg, i.p., for ketamine and xylazine, respectively) and fitted into a custom-made frame. This regimen provided effective anesthesia for ∼1 h each week and appeared to be well tolerated by the mice. Using images of the surface vasculature combined with epifluorescence imaging, we were able to identify the same sets of dendrites from week to week or identify the edge of the cortical infarct (see Fig. 7B). In general, dendrites were imaged either relatively close to or far away from the medioposterior edge of the infarct border (“peri-infarct” zone, 0.18 ± 0.01 mm; “distant” cortex, 0.7 ± 0.08 mm). High-resolution image stacks consisted of 70–80 optical sections, taken 1.25 μm apart covering an area of 168 × 168 μm (1024 × 1024 pixels, 0.164 μm per pixel), averaging three images per section. A median filter (radius, 1) was applied to each image to reduce photon or photomultiplier tube noise. We should note that averaging three images per optical section and applying a median filter did not noticeably reduce the clarity of images or our ability to detect spines, given that we found identical numbers of spines using frame averaging or by applying a filter (Brown et al., 2007).
Figure 7.
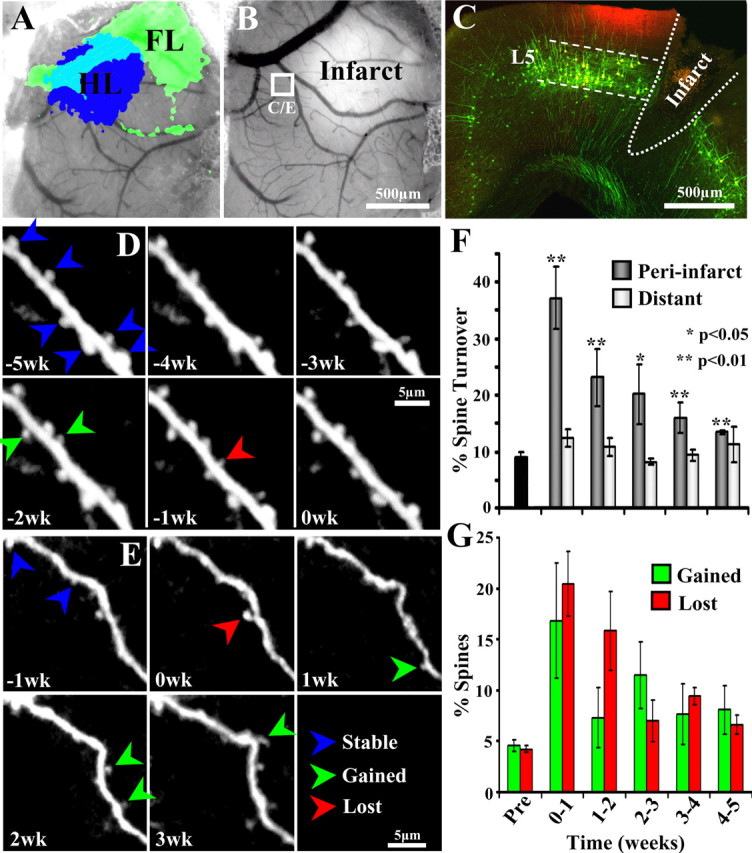
Increased rates of dendritic spine turnover in reorganized forelimb area during stroke recovery. A, IOS mapping of the forelimb and hindlimb representations through the cranial window before the induction of stroke. B, One week after focal stroke in forelimb cortex, tissue in the infarct core appears brighter than adjacent surviving tissue. C, Confocal images showing the approximate location of longitudinal imaging (boxed area in B marked with DiI, appears red) in a coronal section of a YFP transgenic mouse. Note that YFP-labeled dendrites originated almost exclusively from layer 5 of the cortex. D, In vivo time-lapse images of a dendrite imaged for 5 weeks before the induction of stroke. The “0wk” time point represents data taken 1 h before the induction of stroke. E, Time-lapse images of a peri-infarct dendrite (indicated by white box in B) imaged 1 week before and 3 weeks after stroke. Note that more dendritic spines are formed (green arrowheads) and lost (red arrowheads) after stroke. F, Graph shows average weekly rates of dendritic spine turnover (sum of percentage spines gained and lost relative to stable spines) before (Pre) and several weeks after stroke. Spine turnover rates were elevated specifically within the peri-infarct cortex (∼0.18 ± 0.01 mm) but not in more distant cortex (∼0.7 ± 0.08 mm). G, Graph showing percentage spines gained or lost in the peri-infarct region. Both spine formation and elimination rates are similarly affected by stroke. *p < 0.05; **p < 0.01.
To quantify weekly rates of dendritic spine turnover, an observer blind to experimental condition would scroll through and compare three-dimensional images stacks taken 1 week apart using custom-written IgorPro software. For a spine to be considered new or lost, it had to meet several criteria. First, it had to clearly protrude out of the shaft by at least 3 pixels (∼0.5 μm) or had to retract completely into the shaft of the dendrite. Second, it had to be clearly identifiable (i.e., show high signal-to-noise ratio) without any large changes in image quality between sessions. For example, any spines from images that did not appear sharp or crisp, perhaps attributable to sudden movement, were excluded from the analysis. Third, because it is difficult to distinguish dendritic filopodium from thin structures with optical microscopy (Knott et al., 2006), all dendritic protrusions were considered spines. Spine turnover rates were calculated by adding up the total number of spines gained and lost between each imaging session and dividing that by the number of stable spines.
Voltage-sensitive dye imaging.
For in vivo VSD experiments, mice were anesthetized with 15% (w/v) urethane dissolved in distilled water (1.25 g/kg; supplemented at 0.25 g/kg when needed). After carefully removing a 3 × 3 or 6 × 6 mm piece of skull and the underlying dura, the exposed brain was bathed in VSD RH1692 dissolved in HEPES-buffered ACSF for 90 min. After dye incubation, the exposed brain was thoroughly washed and covered with 1.3% low-melt agarose dissolved in a HEPES-buffered ACSF and then sealed with a custom-cut glass coverslip. To further minimize movement artifact, the skull was fastened to a steel plate over a large surface area with cyanoacrylate glue and dental cement, which was then directly screwed onto a metal plate that could be mounted onto the upright microscope.
For VSD data collection, 12-bit image frames were captured every 4 or 5 ms using a Dalsa M-60 Pantera 12-bit camera coupled to an XCAP standard version 2.2 imaging software (EPIX). Voltage-sensitive dyes were excited with red light (Luxeon K2 LED, 627 nm) focused 300 μm below the surface through either an Olympus XLfluor 4× (NA 0.28) or 2× (NA 0.14) objective. Responses in forelimb, hindlimb, motor, and retrosplenial cortex (RS) represent the average of 10–20 stimulation trials. For each trial, images were collected for 205 ms before and 500 ms after a single 5 ms mechanical deflection (∼300 μm amplitude) of the contralateral forelimb or hindlimb delivered through a piezoelectric device. To correct for dye bleaching, images collected during stimulation trials were divided by null stimulation trials. The amplitudes of VSD responses were expressed as the percentage change in VSD signal (ΔF/Fo) by dividing image frames taken after stimulation by the average of those taken before (∼100 ms baseline). Evoked cortical responses were quantified by placing a square region of interest (600 × 600 μm) over the forelimb, hindlimb, motor, and retrosplenial cortex, using NIH ImageJ software. Motor and retrosplenial cortical areas were identified based on stereotaxic coordinates (Paxinos and Franklin, 2001) and their stereotyped position relative to the functionally defined hindlimb area.
Tracer injections, histology, and data analysis.
To map changes in neuronal connectivity in the reorganized forelimb representation, mice were injected with the neuronal tracer Cholera toxin subunit B (1% CtB) (low-salt; List Biological Laboratories) after at least 8 weeks recovery from stroke. Mice were anesthetized with isoflurane gas and fitted into a stereotaxic apparatus, whereupon body temperature was maintained at 37°C. Once the scalp was cut and retracted, the original site of photoactivation was easily identified by the presence of black marker outlining the forelimb area. For tracer injections, a small hole was drilled adjacent to the medial edge of the thinned region of skull overlying the FL cortex, which corresponds to the primary (M1)/secondary (M2) motor cortex. Furthermore, because the photothrombotic method produces exceptionally focal areas of damage with a crisp, well defined border between dead and surviving tissue (Brown et al., 2008), we could be certain that this area of cortex would be close to the edge of the infarction but not ultimately destroyed by it and thus represent “peri-infarct” cortex. Labeling of neuronal projections to and from peri-infarct cortex was achieved by pressure injecting 100 nL of 1% CtB using a 33-gauge Hamilton syringe. Five minutes after the injection, the syringe was withdrawn, the skin was sutured, and animals were returned to their home cages to recover.
Five days after CtB injections, all mice were deeply anesthetized with sodium pentobarbital and were perfused transcardially with 10 ml of PBS followed by 10 ml of 4% paraformaldehyde in PBS. The brain was removed, immersed in 4% paraformaldehyde for 2 d, and then transferred to a 30% sucrose solution in PBS. Coronal brain sections were cut frozen on a microtome at 50 μm. Sections were collected and stored in a series of six wells containing PBS with 0.2% sodium azide.
For CtB immunohistochemistry, all incubation steps were done at room temperature and were followed by washes in PBS (three times for 10 min each), unless otherwise stated. Briefly, sections were pretreated in 0.3% hydrogen peroxide in PBS for 20 min to eliminate endogenous peroxidases that may contribute to background staining. Sections were then incubated overnight in goat anti-CtB primary antisera (1:10,000; List Biological Laboratories), in PBS containing 0.3% Triton X-100 and 2% serum. They were then incubated in biotinylated secondary antibody (anti-goat, 1:1000) followed by streptavidin-HRP (ABC Elite kit; 1:000; Vector Laboratories) in PBS with 0.3% Triton X-100 for 1 h each. Sections were reacted in a chromagen solution containing diaminobenzidine (0.2 mg/ml; Sigma) in PBS with 0.01% hydrogen peroxide and 0.04% nickel chloride. All sections were mounted onto glass slides, dehydrated in an ascending series of ethanols, cleared in xylene, and coverslipped using Permount.
Variability in the exact size and location of CtB injections between animals can yield different patterns of neuronal labeling. Therefore, to ensure that differences in neuronal labeling between stroke and control animals were attributable to stroke-induced reorganization rather than differences in injection size or location, we would process CtB labeling from one set of sections from each stroke animal and then match that to a control with an equivalent injection (both in size and location). Once these pairs had been established, a series of sections from each animal was processed in parallel for CtB immunohistochemistry. After staining, digital photomicrographs of each brain section were taken under identical levels of illumination using a 12-bit camera (Retiga EXi; Q imaging) mounted on a Zeiss Axiophot microscope (2.5× objective) coupled to a standard desktop personal computer running Northern Eclipse software. This was done to facilitate quantitative comparisons between animals, especially for analyses involving optical density measurements of staining intensity.
All analysis of CtB-labeled axons or cell bodies was done using NIH ImageJ software. Axonal labeling in the striatum was quantified using four coronal sections in each animal, spaced 300 μm apart. The striatum was manually outlined in each hemisphere (the ventral border of the striatum was defined at the level of the claustrum), and the mean optical density (using 16-bit grayscale) was calculated for each. To reduce the contribution of nonspecific background staining to these measurements, we measured the optical density of an adjacent area of tissue that displayed no signs of axonal or cellular labeling and subtracted these values from the original measurement. Once an optical density value was established for each animal, we then calculated a ratio for the striatum, using the optical density value of the stroke animal as the numerator and the control value as the denominator.
To quantify retrograde labeling of cell bodies, we first identified regions of the brain in which labeling could typically be found in most animals. These areas could be clearly distinguished by certain cytoarchitectonic features such as the presence of barrels in primary somatosensory cortex or the absence of a prominent layer 4 in motor cortex. An adjacent series of sections was stained for cresyl violet to further aid the delineation of different brain regions based on cytoarchitecture. Similar to our axonal quantification, we were careful to match images of coronal sections (two to three sections from each area) from stroke animals to equivalent sections in controls. Automated counting of cell bodies was performed using a particle analysis macro in NIH Image J. It is important to note that we validated our automated counting approach by comparing cell counts obtained manually from a blind observer (both methods yielded similar estimates).
Statistical analysis.
Statistical comparisons for the effect of time on recovery of forelimb use and spine turnover rates after stroke were made using an ANOVA with post hoc comparisons using Student's t tests. For analysis of CtB labeling, a one-sample t test was used to compare ratios generated in each brain region with a hypothetical value of 1, which reflects the null hypothesis that CtB labeling after stroke is equivalent to controls. All p values ≤0.05 were considered statistically significant, except for multiple post hoc comparisons in which a Bonferroni's (for post hoc VSD comparisons) or sequential Dunn–Sidak correction (for spine turnover comparisons) was used to set the α level. All data are expressed as the mean ± SE.
Results
Functional reorganization during recovery from stroke
To elucidate the functional and structural basis of cortical map reorganization during recovery from stroke, adult male wild-type mice were subjected to a unilateral photothrombotic stroke in the right forelimb sensorimotor cortex and then allowed to recover in their home cages for several weeks (for experimental overview, see Fig. 1A). The photothrombotic method reliably produced a cerebral infarction that extended through all cortical layers (sparing the white matter) and was ∼1 mm in diameter (Fig. 1A). Consistent with previous reports (Gonzalez and Kolb, 2003; Shanina et al., 2006), focal cerebral stroke was sufficient to cause a significant deficit in forelimb function at 1 week after stroke, which gradually recovered to near baseline levels by 8 weeks (Fig. 1B,C) (n = 12, main effect of recovery time: F(5,63) = 2.69, p < 0.05; 8 weeks, −69% vs 1 week, −15%; t(22) = 2.86; p < 0.005).
Figure 1.
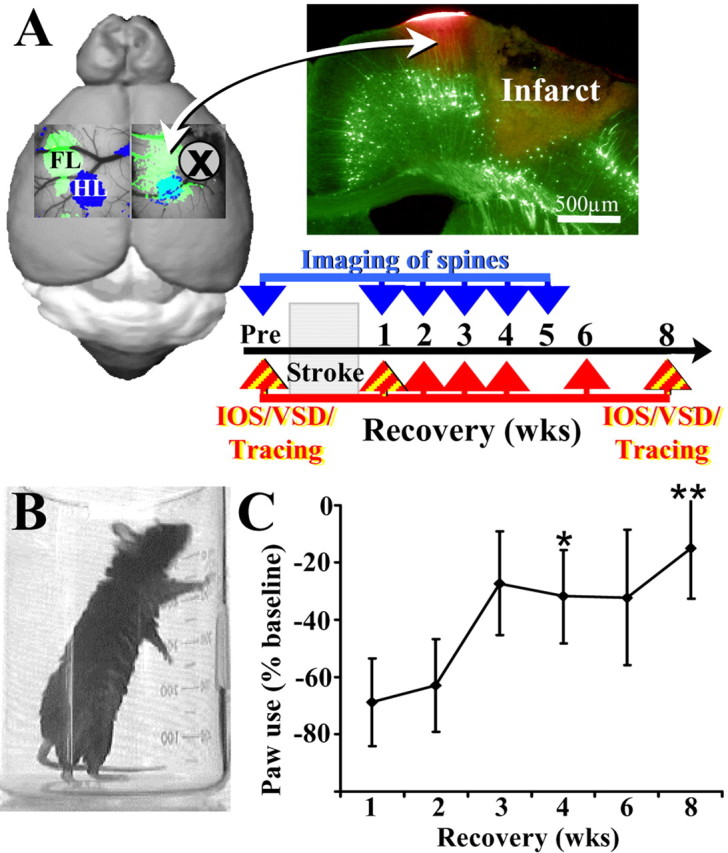
Overview of experiments examining the neurobiological mechanisms of stroke recovery. A, Before and after induction of photothrombotic stroke in the right FL (green area in diagram), we examined functional (using IOS or VSD imaging) and structural changes (using tract tracing or in vivo imaging of dendritic spines) in the peri-infarct cortex over several weeks while the animal was in the process of stroke recovery. Photomicrograph of a coronal section from a YFP-labeled transgenic mouse showing the photothrombotic infarction and the peri-infarct region demarcated in red with DiI. B, Photograph showing a mouse 1 week into recovery from stroke using its un-impaired (right) forelimb for support during upright rears in the cylinder test of forelimb function. C, Graph showing the percentage change in usage of the impaired forelimb (i.e., contralateral to stroke) at different time points during stroke recovery. In the first 1–2 weeks after stroke, mice rarely use the impaired left forelimb for support during vertical rears. Over the ensuing weeks, mice show a significant increase in the usage of the impaired forelimb, particularly by the end of the 8 week testing period relative to 1 week data. *p < 0.01, **p < 0.005.
As a first step toward understanding how the cerebral cortex forms new functional circuits after stroke, we used IOS imaging to map the cortical representation of the forelimb before stroke, 1 week after stroke when behavioral deficits were maximal and then at 8 weeks when the animal had shown significant forelimb recovery. IOS imaging is a useful tool to study cortical map plasticity in vivo because it can be performed repeatedly in a relatively noninvasive manner (i.e., image through the intact skull) in the same animals over several weeks or months. Cortical maps of the forelimb and hindlimb were generated by imaging changes in the reflectance of red light by the brain after mechanically stimulating (100 Hz for 1 s) the contralateral limb (Fig. 2A). Note that certain areas of the cortex appear darker after stimulation (Fig. 2A) attributable to relative increases in deoxyhemoglobin and presumably neuronal activity levels (Frostig et al., 1990). In accordance with previous reports from our laboratory, the forelimb representation in sham-operated controls or prestroke mice was invariably (100% of animals) situated at a 45° angle in the anterolateral direction relative to the hindlimb area (Fig. 2A,B). One and 8 weeks after stroke, forelimb-evoked cortical responses were no longer evident within the original forelimb area (Fig. 2C). However, at 8 weeks recovery, the forelimb representation reemerged within medial and posterior regions of peri-infarct cortex, corresponding to primary motor and hindlimb cortex, respectively (Fig. 2B, raw forelimb response maps are shown in insets). To summarize these changes quantitatively across animals (n = 6 per group), IOS map amplitudes were measured in forelimb, hindlimb, and motor cortical areas before (light bars) and 1 week (striped bars) and 8 weeks (dark bars) after stroke. As shown in Figure 2C, forelimb-evoked responses were robust in the original forelimb area before stroke but were virtually absent 1 or 8 weeks afterward [FL control, −0.038 ± 0.013% vs 1week recovery, −0.0005 ± 0.001% (t(10) = 2.89; p = 0.007) vs 8 weeks recovery, 0.001 ± 0.005% (t(10) = 2.63; p = 0.012)]. In contrast, at 8 weeks recovery, motor and hindlimb cortical areas became uncharacteristically responsive to forelimb stimulation (Fig. 2B,C), indicated by significantly more negative light reflectance values 8 weeks after stroke relative to controls [M1 control, −0.004 ± 0.004% vs 8 weeks recovery, −0.024 ± 0.004% (t(10) = 3.98; p = 0.0013); HL control, −0.003 ± 0.001% vs 8 weeks recovery, −0.019 ± 0.003 (t(10) = 4.37; p = 0.0007)].
Figure 2.
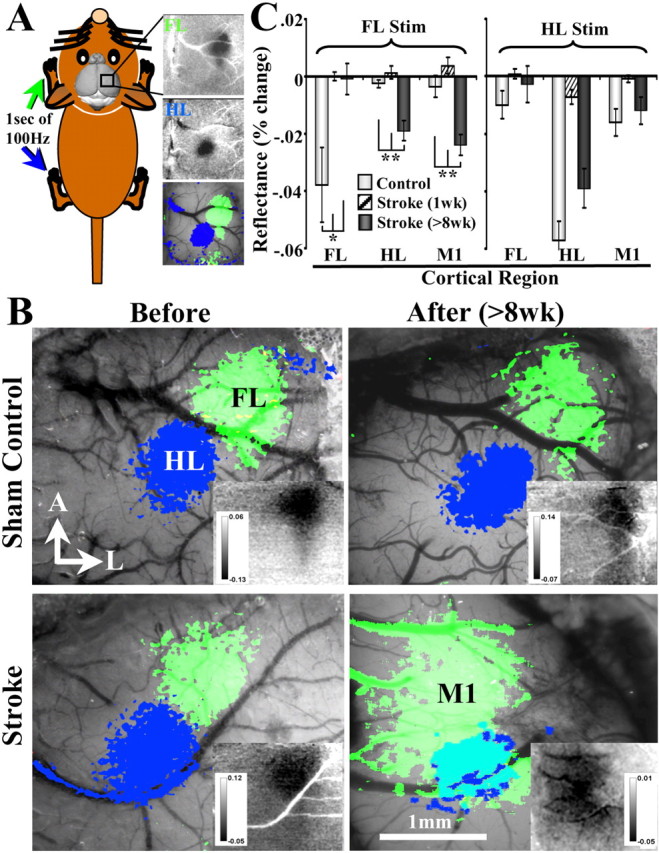
In vivo IOS imaging reveals that behavioral recovery is associated with remapping of the forelimb representation into adjacent peri-infarct motor and hindlimb cortex. A, Diagram showing how IOS maps were generated. Maps were generated by recording the change in the reflectance of red light off the surface of the brain after mechanical stimulation of the contralateral forelimb or hindlimb (green and blue areas, respectively). Note that distinct parts of the cerebral cortex appear darker after stimulation, which corresponds with increased levels of deoxyhemoglobin. B, Images showing the functional representation of the forelimb and hindlimb before and 8 weeks after sham surgery (top) or photothrombotic stroke (bottom). Before stroke, IOS-derived maps were generated using a transcranial imaging preparation, whereas those taken at 8 weeks recovery were performed with the bone removed. The areal extent of each functional map was determined by thresholding images at two-thirds of maximal intrinsic signal amplitude. Insets in each image show raw percentage change in reflectance after forelimb stimulation. Notice that, in sham controls and prestroke mice, the functional representation of the forelimb was reliably located at a 45° angle from the hindlimb, which did not change 8 weeks after sham surgery. Eight weeks after stroke, the functional representation of the forelimb shifted into the peri-infarct M1 and HL. C, Graphs summarizing quantitative changes in cortical responsiveness (as indicated by more negative reflectance values) to stimulation of forelimb or hindlimb in controls, 1 and 8 weeks after stroke. In controls, forelimb and hindlimb areas responded selectively to stimulation of their respective limbs. One week after stroke, forelimb responses were virtually absent, although a small response to hindlimb stimulation could be detected in the hindlimb cortex. Eight weeks after stroke, motor and hindlimb cortical areas showed a significant hemodynamic response to forelimb stimulation. Cortical responses to hindlimb stimulation dropped significantly at 1 week but were not significantly different from control levels at 8 weeks recovery. *p < 0.05, **p < 0.005.
Data generated from IOS imaging suggested that peri-infarct primary motor and hindlimb regions became progressively more involved in processing forelimb-driven sensory information after stroke. However, IOS imaging is limited by virtue of the fact that it makes indirect inferences about neuronal activity based on hemodynamic responses (Frostig et al., 1990), which could be confounded in the ischemic brain, particularly in the first week after stroke (note very small IOS signals at 1 week recovery in Fig. 2C). More importantly, IOS imaging does not provide any information about the temporal dynamics of cortical sensory information processing. To address these issues, we used in vivo VSD imaging that allows one to directly assess changes in membrane potential on the order of milliseconds, thereby enabling real-time visualization of the spatiotemporal dynamics of cortical sensory processing (Shoham et al., 1999). To do this, VSD RH-1692 was topically applied to the surface of the brain after removing the skull and dura from acutely anesthetized controls (n = 9) or mice that had recovered for 1 week (n = 6) or 8 weeks (n = 8) after stroke (note that VSD imaging was done only at single time points). Previous work has shown that the source of VSD signals in vivo originates from superficial cortical layer 2/3 cells and that changes in VSD fluorescence are linearly related to membrane potential (Petersen et al., 2003; Grinvald and Hildesheim, 2004; Berger et al., 2007).
Just before VSD imaging, we attempted to visualize the forelimb and hindlimb representation in all groups using IOS imaging. Although we were able to clearly generate forelimb and hindlimb sensory maps in controls and at 8 weeks recovery (Fig. 3A,G), IOS responses at 1 week recovery was generally weak and unreliable (therefore, in Fig. 3D, an IOS map was not superimposed onto the brain surface). As shown in Figure 3, A and G, the putative center of forelimb responsive cortex visualized with IOS imaging in both controls and stroke-recovered mice (>8 weeks) were very similar to those generated with VSD imaging. The fact that these two different types of functional imaging techniques yielded almost identical forelimb maps substantiated our previous claims using IOS imaging that the forelimb representation reorganized onto adjacent motor and hindlimb areas. However, because of the exquisite temporal resolution of VSD imaging, we were now able to determine whether the temporal dynamics of forelimb sensory processing were different between controls and stroke-recovered mice. In control mice, a brief (5 ms) tap of the forelimb was accompanied by a robust cortical depolarization in the putative center of the forelimb representation of the contralateral hemisphere that typically appeared within 20 ms (Fig. 3B), spread to adjacent motor and hindlimb areas, and decayed to baseline levels within 100 ms (Fig. 3C) (supplemental Movie 1, available at www.jneurosci.org as supplemental material). At 1 week recovery from stroke, forelimb-evoked responses were virtually absent in the cortex (Fig. 3F), with the exception of a small depolarization in the remaining piece of the original forelimb area posterior to the infarct (Fig. 3E, white arrow). However, we should point out that this was not attributable exclusively to a general loss of peri-infarct responsiveness at 1 week given that hindlimb stimulation could readily drive neuronal responses in peri-infarct hindlimb cortex (Fig. 4B). After 8 weeks recovery from stroke, the amplitude and kinetics of the cortical response to forelimb stimulation changed significantly. Here, forelimb stimulation elicited a robust, short-latency depolarization in the remaining piece of the original forelimb area (<20 ms) (Fig. 3H, white arrows) that spread horizontally around the infarct to adjacent motor and hindlimb cortical areas and persisted for 300 ms (note the green crescent of activation in Fig. 3I) (supplemental Movie 2, available at www.jneurosci.org as supplemental material). Quantitatively (Fig. 3J–N), the amplitude of forelimb-evoked cortical depolarization dropped significantly in all three cortical regions 1 week after stroke (Fig. 3L) (peak ΔF/Fo in controls vs 1 week recovery, p < 0.001 in FL, M1, and HL areas), which then recovered to prestroke levels in motor and hindlimb areas by 8 weeks (Fig. 3L) [M1 peak ΔF/Fo in controls vs 8 weeks recovery, 0.232 ± 0.03 vs 0.167 ± 0.02 (t(15) = 1.56; p = 0.07); HL peak ΔF/Fo in controls vs 8 weeks recovery, 0.195 ± 0.03 vs 0.154 ± 0.03 (t(15) = 1.13; p = 0.13)] but not in remaining forelimb cortex. With respect to timing, forelimb-evoked depolarization in motor and hindlimb areas at 8 weeks recovery peaked later in time than controls (Fig. 3M) [M1 cortex time-to-peak in controls vs 8 weeks recovery: 51 ± 12 vs 128 ± 17 ms (t(15) = 3.73; p = 0.001); HL cortex time-to-peak in controls vs 8 weeks recovery, 50 ± 12 vs 143 ± 13 ms (t(15) = 5.25; p = 0.00005)]. Furthermore, forelimb-evoked responses in motor and hindlimb areas at 8 weeks recovery persisted significantly longer than controls (Fig. 3N) [M1 cortex half-width in controls vs 8 weeks recovery, 54 ± 13 vs 201 ± 26 ms (t(15) = 5.26; p = 0.00005); HL cortex half-width in controls vs 8 weeks recovery, 52 ± 11 vs 154 ± 17 ms (t(15) = 5.13; p = 0.00006)]. Of note, these changes in forelimb-evoked cortical activation patterns were not induced when the stroke occurred anterior of the forelimb area (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Collectively, these findings indicate that remapping of the forelimb representation onto motor and hindlimb areas is specific to forelimb cortex damage. Furthermore, forelimb remapping is accompanied by a progressive recovery in peri-infarct cortical responsiveness and an increase in the amount of time these areas spend processing forelimb-evoked sensory information.
Figure 3.
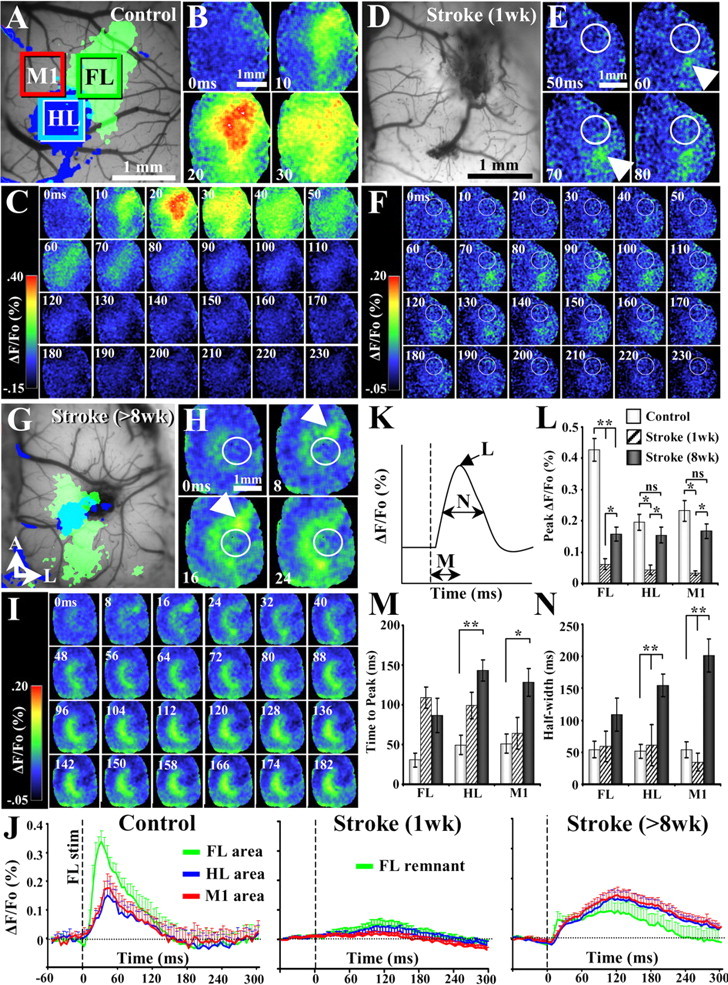
VSD imaging reveals progressive changes in the spatiotemporal dynamics of forelimb-evoked cortical responses after stroke. A, D, G, Before VSD imaging, IOS imaging was used to visualize the functional representation of the forelimb and hindlimb in controls (A) and after 1 week (D) and 8 weeks (G) recovery from stroke. However, because of the fact that forelimb-evoked IOS signals at 1 week recovery were exceedingly difficult to obtain, only the surface of the brain is shown at this time point. B, C, VSD imaging of cortical responses to forelimb stimulation (one 5 ms tap per trial) in a control mouse. The earliest VSD response originates in the center of the forelimb and quickly spreads to adjacent cortical areas. Note the close correspondence between IOS (A) and VSD-derived maps of the forelimb. E, F, At 1 week recovery, the cortex was mostly unresponsive to forelimb stimulation except in the surviving portion of forelimb cortex posterior to the infarct (infarct denoted by white circle). H, I, After at least 8 weeks recovery from stroke, the earliest VSD signal arises from the remaining piece of the forelimb cortex (indicated by white arrow in H), which then spreads horizontally to motor and hindlimb cortical areas, which are medial and posterior to the infarct (infarct denoted by white circle). Note that, after stroke, motor and hindlimb areas show very prolonged responses to forelimb stimulation. J, Plots summarizing average forelimb, hindlimb, and motor cortical responses (plots derived from green, blue, and red boxes shown in A) to stimulation of the forelimb in controls and stroke-recovered mice at 1 and 8 weeks. Note that, in controls (n = 9 mice), responses in all three areas were relatively brief, peaking within the first 50 ms and rapidly decayed. One week after stroke (n = 6 mice), cortical responses were barely detectable. However, after 8 weeks recovery (n = 6 mice), the amplitude of motor and hindlimb area responses returned to near control levels. Furthermore, peri-infarct motor and hindlimb responses to forelimb stimulation persisted for considerably longer periods of time relative to controls. K–N, Quantification of VSD signal response amplitude (L), time-to-peak (M), and duration (N; time spent at half-maximal response amplitude) in controls and stroke-recovered mice at 1 and 8 weeks. *p < 0.01, **p < 0.001.
Figure 4.
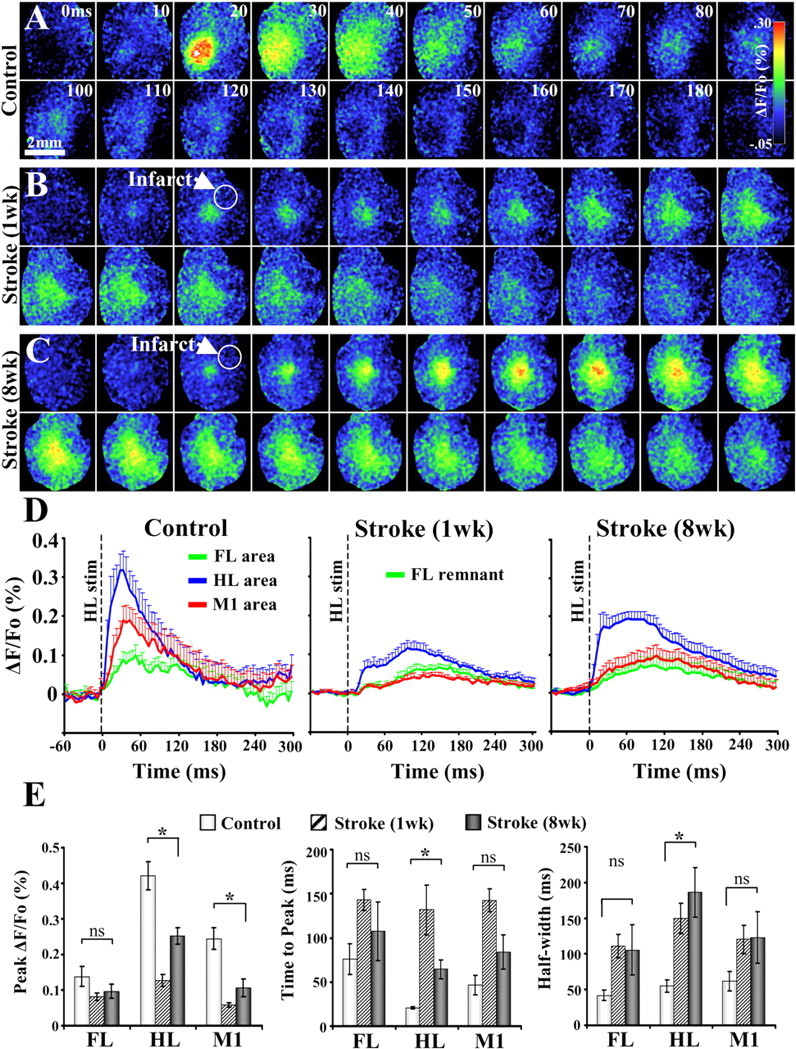
VSD imaging of cortical responses to hindlimb stimulation. A–C, Montages showing the spatiotemporal dynamics of cortical responses to hindlimb stimulation (average of 10–20 trials, 1 5 ms tap delivered per trial) in controls (A) and 1 week (B) and 8 weeks (C) after stroke. Scale bar, 2 mm. D, Average forelimb (green), hindlimb (blue), and motor (red) cortical responses to hindlimb stimulation in controls (n = 9 mice) and 1 week (n = 6 mice) and 8 weeks (n = 6 mice) after stroke. E, Quantification of hindlimb-evoked VSD cortical responses in controls and after stroke. Our analysis showed that stroke in the forelimb cortex caused significant changes in the peak amplitude, time-to-peak, and duration (i.e., half-width) of responses in hindlimb cortex. *p < 0.01.
Although hindlimb cortex escaped obvious structural damage, the magnitude and temporal dynamics of hindlimb-evoked sensory responses in this area were not entirely normal. In control animals, hindlimb stimulation elicited readily detectable but relatively short-lived responses in the putative center of hindlimb cortex (Fig. 4A,D). One week after forelimb cortex stroke, hindlimb stimulation could still drive responses in the native hindlimb cortical region albeit to a lower amplitude than normal animals (Fig. 4B,D). At 8 weeks recovery, the strength of hindlimb-evoked cortical responses improved relative to 1 week recovery (Fig. 4C,D). Analysis of the peak amplitudes revealed that hindlimb-evoked cortical responses in hindlimb and motor cortex were significantly reduced after stroke (Fig. 4E) [HL peak ΔF/Fo in controls vs 8 weeks recovery, 0.42 ± 0.04 vs 0.25 ± 0.02 (t(13) = 3.23; p = 0.003); M1 peak ΔF/Fo in controls vs 8 weeks recovery, 0.24 ± 0.03 vs 0.11 ± 0.02 (t(13) = 3.24; p = 0.003)]. Our temporal analysis indicated that hindlimb-evoked cortical responses were only significantly delayed and prolonged in the hindlimb cortex (Fig. 4E) [HL cortex time-to-peak in controls vs 8 weeks recovery, 20.6 ± 1 vs 64.5 ± 11 ms (t(13) = 5.0; p = 0.0001); HL cortex half-width in controls vs 8 weeks recovery, 55 ± 8 vs 186 ± 34 ms (t(13) = 3.67; p = 0.005)] but not in forelimb or motor cortex (all p values >0.05). These results show that forelimb cortex stroke alters the spatiotemporal dynamics of hindlimb-evoked cortical responses but to a lesser degree than that observed with forelimb stimulation.
New patterns of neuronal connectivity in reorganized forelimb area
Because the functional representation of the forelimb remapped onto adjacent peri-infarct motor cortical areas at 8 weeks recovery, we then asked whether this functional rewiring was accompanied by changes in neuronal connectivity (schematic in Fig. 5A). To address this issue, sham-operated or stroke-recovered mice (>8 weeks recovery) were given a unilateral microinjection (0.2 μl) of the neuronal tracer CtB into the reorganized forelimb area (medial to the infarct zone), which corresponds stereotaxically with the M1 and M2 (Paxinos and Franklin, 2001). For all animals analyzed (n = 14), the injection site (indicated by dense CtB immunoreactivity) spanned all cortical layers and spread ∼1 mm in diameter (Fig. 5C). To ensure that CtB injections in stroke and control groups were identical in both size and uptake, we counted all labeled cells in the contralateral hemisphere and found equivalent levels of cellular labeling (average cell count in stroke vs control groups, 2591 ± 377 vs 2702 ± 214 cells; p = 0.40). Consistent with previous studies (Alloway et al., 2006), CtB was transported in both anterograde and retrograde directions. CtB immunoreactivity was detected in axons (Fig. 5C, inset) as well as cell bodies (Fig. 5E, inset shows layer 5 neurons). For the most part, retrograde labeling of cell bodies in local and distant brain regions was associated with reciprocal axonal labeling, with the one exception being the caudate–putamen (CPu), which showed only axonal labeling (Fig. 5C).
Figure 5.
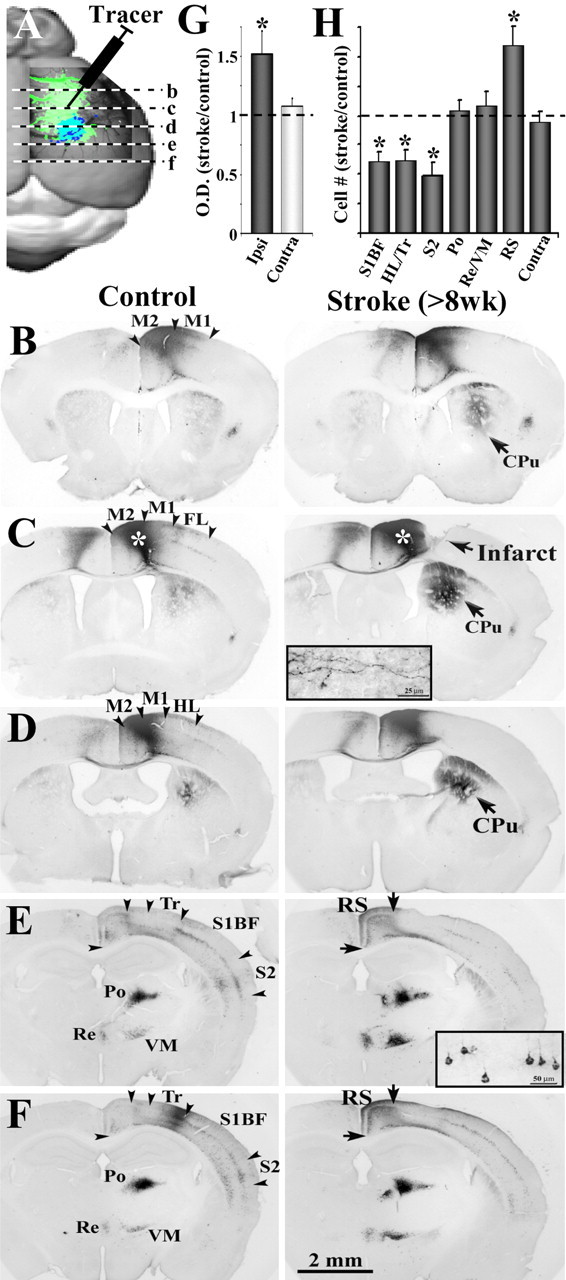
Tracing new patterns of neuronal connectivity in the functionally reorganized forelimb representation. A, Diagram showing the injection site of the neuronal tracer CtB and the relative anteroposterior position of coronal sections shown in B–F. B–F, Light photomicrographs showing the distribution of labeled axons and cell bodies in coronal sections, after an injection of CtB into the motor cortex of controls and after 8 weeks recovery (see white asterisks over darkly stained area in C). Insets show higher-magnification images of anterograde labeling of axons in the striatum (C) and retrograde labeling of cell bodies in the cortex (E). In general, both control and stroke recovered mice showed CtB-labeled axons and somata in the ipsilateral and contralateral M1 and M2, the primary somatosensory regions of the HL, Tr and whiskers (S1BF), S2, RS, and the Po, VM, and Re nucleus of the thalamus. Note that, 8 weeks after stroke, there is much greater CtB labeling in the ipsilateral striatum (CPu) and RS cortex, whereas labeling is reduced in more lateral cortical areas such as the HL/Tr, S1BF, and S2. G, Relative to controls, the optical density of axonal labeling in the striatum was significantly greater in the ipsilateral but not contralateral hemisphere after stroke. H, Quantification of retrogradely labeled cell bodies in stroke-recovered mice relative to controls. After stroke, there were significantly fewer labeled cell bodies in the HL/Tr, S1BF, and S2 but more labeled somata in the RS cortex. No change in cell labeling was found in the thalamic nuclei or homotopic regions of the contralateral cortex. *p < 0.05.
In agreement with a previous report (Donoghue and Parham, 1983), CtB labeling was evident within the homotopic region of the contralateral cerebral cortex, the ipsilateral caudate–putamen, thalamus, retrosplenial, primary, and secondary somatosensory cortical areas. Although the regional pattern of connections was similar between stroke-affected and control groups, the relative abundance of connections to and from motor cortex differed significantly. These changes in CtB labeling are illustrated through photomicrographs of a representative control and stroke-recovered mouse shown in Figure 5B–F. We substantiated our qualitative observations by comparing levels of CtB labeling between pairs of stroke-recovered and control animals (n = 14, 7 pairs). Our analysis indicated that stroke (>8 weeks recovery) was accompanied by a loss of peri-infarct motor cortex connections with lateral somatosensory cortical regions (Fig. 5D–F,H) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), given that we found much fewer CtB-labeled cells in primary somatosensory regions of the hindlimb/trunk (HL/Tr) (HL/Tr stroke/control ratio, 0.62 ± 0.09; t(6) = 4.50; p = 0.004), whiskers (S1BF, 0.61 ± 0.08; t(6) = 4.47; p = 0.004), and secondary somatosensory cortex (S2) (0.49 ± 0.11; t(6) = 5.05; p = 0.002). Conversely, stroke-recovered mice showed increased CtB labeling of axons in the ipsilateral CPu (1.52 ± 0.18; t(6) = 2.76; p = 0.03) (Fig. 5B–D,G) and cell bodies in the RS (1.60 ± 0.16; t(6) = 3.69; p = 0.01) (Fig. 5E,F,H). It is important to note that neuronal connectivity was not affected in all regions examined, such as the caudate–putamen (1.08 ± 0.11; t(6) = 0.93; p = 0.38) (Fig. 5B–D,G) or homotopic cortical areas of the undamaged hemisphere (0.94 ± 0.09; t(6) = 0.62; p = 0.55) (Fig. 5B–D,G). Furthermore, there were no significant changes in CtB labeling in the posterior (Po) (1.04 ± 0.09; t(6) = 0.46; p = 0.65) and ventromedial/reunions (VM/Re) (1.08 ± 0.12; t(6) = 0.73; p = 0.49) nuclei of the thalamus (Fig. 5E,F,H). Collectively, these data indicate that the functionally rewired forelimb representation loses connections with lateral somatosensory areas but gains inputs from the retrosplenial cortex and sends more output projections to the ipsilateral striatum.
New patterns of brain activation in more remote cortical domains
Using the knowledge gained from the neuronal tract tracing experiments, we examined whether the enhanced connectivity of the reorganized forelimb with the retrosplenial cortex was accompanied by changes in circuit function. Therefore, we removed a large portion of the skull (∼6 × 6 mm window) (Ferezou et al., 2007) and performed VSD imaging of forelimb-evoked cortical responses in controls (n = 5) and at 1 week (n = 6) and 8 weeks (n = 5) recovery from stroke. Similar to our previous descriptions, sensory-evoked depolarization in control mice emerged within 20 ms of forelimb stimulation, in the putative center of the forelimb cortex (Fig. 6A, anterolateral to hindlimb area indicated by red circle) (supplemental Movie 3, available at www.jneurosci.org as supplemental material), which briefly spread to distant cortical regions such as the homotopic region of the contralateral hemisphere and the ipsilateral RS (Fig. 6A, yellow arrow). At 1 week recovery, very little cortical activation was observed in the retrosplenial cortex after forelimb stimulation (for response profile, see Fig. 6C). In mice that recovered for 8 weeks (Fig. 6B) (supplemental Movie 4, available at www.jneurosci.org as supplemental material), forelimb stimulation evoked prolonged cortical responses in motor and hindlimb areas (see crescent-shaped pattern of activation in and around HL area), which was followed by considerably long-lived activation in posteromedial retrosplenial cortex several millimeters away (yellow arrows). We should note that we verified the position of the retrosplenial cortex using histological specimens and stereotaxic coordinates. Quantification of forelimb-evoked VSD responses in the retrosplenial area (Fig. 6C,D) revealed a significant shift in the timing (time-to-peak in controls vs 8 weeks recovery, 76 ± 37 vs 248 ± 55 ms; t(8) = 2.58; p = 0.016) and duration of depolarization (half-width in controls vs 8 weeks recovery, 57 ± 7 vs 167 ± 48 ms; t(8) = 2.27; p = 0.026) at 8 weeks recovery relative to controls. In contrast, peak depolarization in the retrosplenial cortex was reduced at 1 week (but did not reach statistical significance; peak ΔF/Fo in controls vs 1 week recovery, 0.045 ± 0.009 vs 0.018 ± 0.001; t(9) = 1.61; p = 0.07) and returned to control levels by 8 weeks recovery (peak ΔF/Fo in controls vs 8 weeks recovery, 0.045 ± 0.009 vs 0.051 ± 0.006; t(8) = 0.56; p = 0.29).
Figure 6.
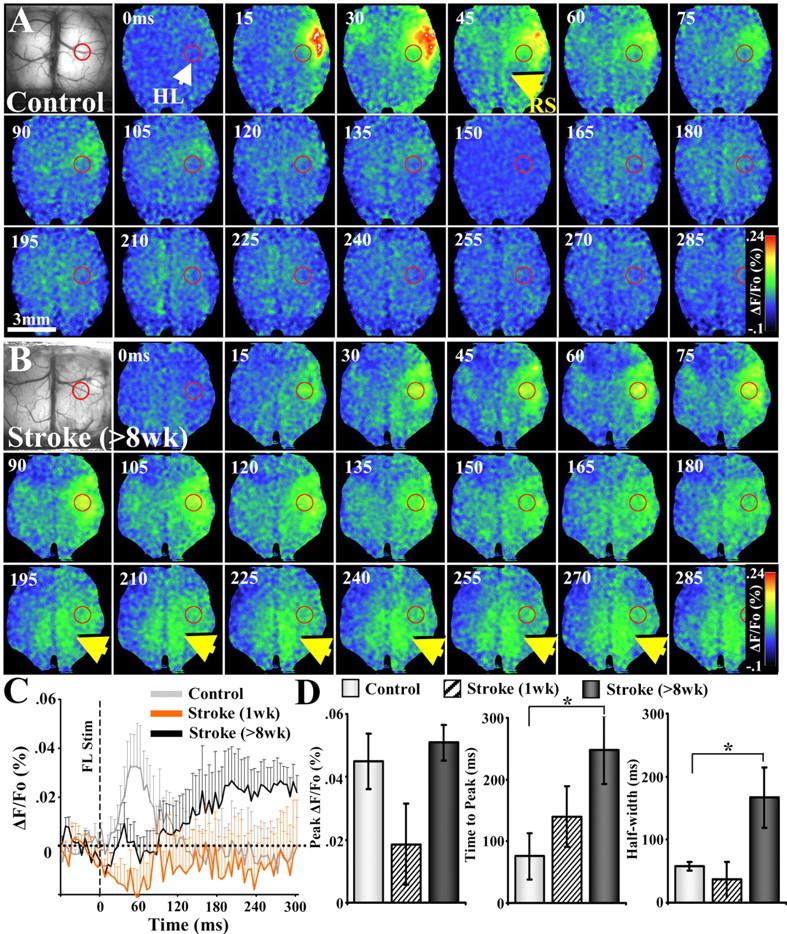
Bihemispheric VSD imaging of forelimb-evoked responses in control and stroke-recovered mice. A, Montage shows the timing and spread of VSD signals in a control mouse after stimulation of the left forelimb. Note the robust responses in right forelimb cortex (anterolateral to hindlimb cortex shown by a red circle) that peaks within 15–45 ms after stimulation and quickly dissipates. B, In mice that had recovered for 8 weeks, forelimb-evoked responses were especially prominent in peri-infarct motor and hindlimb areas during the first 200 ms, which was then followed by prolonged activation in the posteromedial retrosplenial cortex (see yellow arrows). C, Average forelimb-evoked VSD signals in the retrosplenial cortex of controls (n = 5) and stroke-recovered mice at 1 week (n = 6) and 8 weeks (n = 5). D, Forelimb-evoked VSD signals in the retrosplenial cortex of mice that had recovered for at least 8 weeks after stroke (>8 weeks) were shifted significantly in time and duration but not amplitude. *p < 0.05.
Chronic in vivo imaging of excitatory synapse turnover during stroke recovery
Dendritic spines are the postsynaptic targets of most cortical projections in the cortex (Harris and Kater, 1994). Therefore, documenting how they form and retract after stroke provides a measure of excitatory synapse rewiring that is necessary for large-scale changes in functional cortical maps. We have shown previously in acute in vivo imaging experiments that dendritic spine formation is elevated in the apical dendrites of layer 5 neurons in peri-infarct cortical regions (Brown et al., 2007). However, these changes in dendritic structure were tracked for only 6 h in an anesthetized animal, which would preclude an accurate estimate of spine turnover rates (over weeks) that are likely nonlinear in nature (Holtmaat et al., 2005). Therefore, in the present study, we used the chronic cranial window preparation (Trachtenberg et al., 2002; Majewska et al., 2006) in transgenic mice that express YFP/green fluorescent protein (GFP) within predominately layer 5 cortical neurons (Feng et al., 2000), to track longitudinal changes in dendritic structure over several weeks time, while the animal was in the process of recovery.
Although chronic imaging has obvious advantages, previous work from our laboratory and others has suggested that basal levels of dendritic spine turnover in the brain can be influenced by injury (Brown et al., 2007). A recent study by Xu et al. (2007) has suggested that craniotomies can produce reactive gliosis and unusually high levels of dendritic spine plasticity. In the present study, we tried to mitigate any methodological/surgical artifacts by allowing mice to recover for at least 4–5 weeks after the implantation of a glass window over the exposed brain. In our hands, we found virtually no signs of significant tissue damage 4–5 weeks after surgery, as indicated by the absence of GFAP-positive astrocytes in the cortex (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) or blebbed dendritic arbors. Furthermore, functional mapping of forelimb and hindlimb representations through the cranial window appeared normal (Fig. 7A) and were indistinguishable from those generated using a thinned skull preparation.
To provide an estimate of dendritic spine turnover, we imaged YFP- or GFP-labeled dendrites at weekly intervals before (∼2–5 weeks) and after the induction of stroke. Because of the fact that stroke induces progressive warping of dendritic structure and angiogenesis in the peri-infarct region (Wei et al., 2001; Brown et al., 2007), obtaining exceptionally clear images of the same segment of dendrite for 5 weeks of time after stroke was exceedingly difficult. Therefore, in the present study, estimates of spine formation and elimination were made by comparing two imaging sessions (1 week apart) rather than over multiple imaging sessions and weeks time, which has been used previously to estimate spine stability in unlesioned mice (Holtmaat et al., 2006). Consistent with the notion that our imaging preparation did not influence basal dendritic plasticity, spine turnover was relatively low and constant throughout the prestroke imaging period (Fig. 7D) (weekly turnover rate, 8.8 ± 0.8%; gained, 4.6 ± 0.5%; lost, 4.2 ± 0.3%; n = 5). After this period, focal ischemic stroke was induced in the forelimb representation using the photothrombotic method (n = 4). The infarct zone could be easily verified by virtue of the fact that it lacked fluorescently labeled dendrites (see histological specimen in Fig. 7C) and reflected light more strongly than healthy surviving tissue (see brighter area in Fig. 7B). Our analysis indicated that rates of spine turnover were significantly elevated within the peri-infarct zone after stroke (F(5,16) = 8.48; p < 0.0005). Relative to prestroke levels, spine turnover rates in peri-infarct cortex peaked in the first week after stroke (Fig. 7F,G) (0–1 week turnover, 37.2 ± 5.4%; t(6) = 6.96; p < 0.001) and remained significantly above prestroke levels for 5 weeks after stroke (1–2 weeks, 23.1 ± 5.1%, t(6) = 3.69, p = 0.005; 2–3 weeks, 20.2 ± 5.2%, t(7) = 2.83, p = 0.01; 3–4 weeks, 16 ± 2.7%, t(7) = 3.42, p = 0.005; 4–5 weeks, 13.4 ± 0.3%, t(6) = 4.41, p = 0.002). However, the effect of stroke on spine turnover was spatially limited given that spine turnover rates in cortical territory more distant from the infarct (0.7 ± 0.1 mm away from stroke border) was not significantly different from prestroke levels (Fig. 7F) (F(5,16) = 1.09, p = 0.40; 0–1 week, 12.3 ± 1.5%; 1–2 weeks, 10.7 ± 1.6%; 2–3 weeks, 8.2 ± 0.5%; 3–4 weeks, 9.4 ± 1.0%; 4–5 weeks, 11.2 ± 3.2%).
Discussion
Imaging new routes of sensory information processing during stroke recovery
It is well known that damage to the nervous system can remodel the topographic organization of sensory and motor cortical maps (Nudo and Milliken, 1996; Xerri et al., 1998; Dijkhuizen et al., 2001; Conner et al., 2005; Keck et al., 2008; Winship and Murphy, 2008). The transfer of sensory or motor functions to surviving tissues appears to represent an adaptive response by the injured brain given that secondary lesions or neuronal inactivation of the remaining sensorimotor system disrupts recovery from injury (Castro-Alamancos and Borrel, 1995; Frost et al., 2000; Werhahn et al., 2003; Biernaskie et al., 2005). However, what is not known is precisely how sensory information propagates to and from these reorganized zones in real time and whether these areas process information with the same temporal fidelity as the original sensory representation. The reason these issues remain outstanding is because (1) metabolic and hemodynamic imaging techniques [functional magnetic resonance imaging (fMRI), positron emission tomography (PET), and IOS] cannot provide sufficient time resolution to capture sensory information processing that occurs within just milliseconds and (2) intracortical microstimulation or single-cell optical or electrical recordings lack the ability to view the spread of neuronal activity across a large area. To overcome these issues, we used large-scale in vivo imaging of neuronal membrane potentials that allows one to visualize, with millisecond time resolution, the dynamics of cortical sensory information processing (Shoham et al., 1999). Our data show that new sensory map formation after stroke is accomplished by the progressive recovery of neuronal responsiveness to the paretic limb, presumably within the surviving portion of forelimb cortex that escaped damage, which propagates to and reverberates (three to four times longer than controls) within the peri-infarct motor and hindlimb representations. To our knowledge, this is the first study to directly visualize the sequential activation of forelimb-related circuits after stroke and to show that stroke induces profound changes in the temporal precision of sensory information processing. We should note, however, that the prolonged activation of peri-infarct motor and hindlimb regions is reconcilable with previous descriptions showing increased activity in peri-infarct cortex using IOS (Wei et al., 2001; Zepeda et al., 2004), fMRI (Dijkhuizen et al., 2001; Jaillard et al., 2005), or PET (Chollet et al., 1991) studies, which image the integrated hemodynamic or metabolic responses of cortical areas over several hundred or thousands of milliseconds.
Nature of sensory-evoked membrane potential changes after stroke: suprathreshold versus subthreshold responses
The fact that VSD imaging is sensitive enough to detect subthreshold neuronal activity in the cortex (Berger et al., 2007) begs the question of whether altered VSD responses in reorganized cortex represent subthreshold or suprathreshold spiking activation. Suprathreshold forelimb-evoked activity in the hindlimb cortex has been investigated previously in vivo after stroke (Winship et al., 2007; Winship and Murphy, 2008), using AM calcium dyes that detect action potential-mediated increases in intracellular calcium levels (Stosiek et al., 2003; Berger et al., 2007; Sato et al., 2007). Winship and Murphy (2008) demonstrated a 10-fold increase in the incidence but not amplitude of forelimb-evoked somatic calcium transients in hindlimb cortex after stroke. However, the incidence of calcium responses comprised a relatively small percentage of cells, with only 1.3% of neurons responding in controls versus 11.2% of neurons 1 month after stroke. Based on these studies, we suggest that forelimb-evoked VSD activation in motor and hindlimb cortex after stroke reflect both subthreshold and suprathreshold activity. Whether stroke-induced changes in VSD signals in the retrosplenial cortex represent mostly subthreshold versus suprathreshold activation is unknown and warrants additional investigation.
Mechanisms and functional significance of prolonged sensory responses
Although a switch to a persistent mode of activity was clear, the mechanisms behind prolonged neuronal responses in the stroke-recovered brain are unclear. One well documented side effect of stroke is the loss of GABAergic inhibitory neurotransmission in the peri-infarct zone (Neumann-Haefelin et al., 1995; Redecker et al., 2002). In vitro work from Buchkremer-Ratzmann et al. (1996, 1998) has shown that cortical stroke increased the frequency of electrically evoked cortical discharges as well as the amplitude of pair-pulse excitatory responses. Therefore, prolonged sensory responses after stroke may reflect a loss of inhibition within the cortex or feedback circuits in the thalamus, which are normally required to terminate cortical responses to sensory stimuli. Somewhat unexpectedly, longer cortical responses after stroke were not accompanied by a significant increase in the amplitude of evoked VSD responses (Fig. 3), as one might predict from a generalized loss of cortical inhibition. These data would suggest that stroke-induced changes in responsiveness may be more nuanced, perhaps akin to the developing brain in which cortical responses to tactile whisker stimulation are typically slower and longer lived relative to older animals (Stern et al., 2001; Borgdorff et al., 2007). Alternatively, stroke may alter the composition of excitatory receptor subtypes in the peri-infarct zone (Liu et al., 2007). Single-unit recordings have shown previously that much of the longer-latency cortical responses (10–100 ms) to tactile stimulation are sensitive to NMDA receptor blockers (Armstrong-James et al., 1993). Long-lived sensory responses after stroke could therefore be mediated by a selective increase in the expression of NMDA receptors with longer decay kinetics, such as those containing NR2B subunits.
For the somatosensory system to accurately sense textures, vibrations, and contours, the cortex must receive and interpret sensory signals that are separated in both space and time. Although our behavioral data indicated that mice progressively recover the usage of the impaired forelimb (Fig. 1C), the fact that the new forelimb representation spent significantly more time responding to forelimb stimulation suggests that there may be more subtle disturbances to sensory function. Indeed, clinical data has shown that 50–85% of human stroke patients display deficits in the ability to accurate sense and discriminate tactile stimuli (Kim and Choi-Kwon, 1996; Blennerhassett et al., 2007). Our data provide a neurophysiological explanation for these deficits and a model system that future therapeutic interventions can use to help sharpen the temporal precision of cortical circuits after stroke.
New patterns of regional connectivity
Supporting new patterns of functional brain activation are changes in neuronal connectivity to and from reorganized forelimb cortex. These findings extend previous data showing that focal cerebral stroke can remodel short-range connections within somatosensory cortex (Carmichael et al., 2001) and promote axonal plasticity in cortical regions distant from the infarct (Dancause et al., 2005). Although the significance of such changes in neuronal connectivity are unclear, it is interesting to note that, although the peri-infarct motor cortex lost connections with lateral somatosensory cortical domains, it became more connected with the medioposterior retrosplenial cortex. The retrosplenial cortex appears to play a role in visuomotor functions given that excitotoxic lesions of the retrosplenial cortex disrupt spatial navigation. Furthermore, cells within the retrosplenial cortex receive inputs from visual and somatosensory cortical areas that encode various aspects of movement, location, or head direction of the animal (van Groen and Wyss, 1992; Cho and Sharp, 2001; Vann and Aggleton, 2004). Indeed, if the motor cortex loses information from somatosensory regions, one might expect that it may rely more heavily on other cortical areas such as the retrosplenial cortex that process and integrate visual/somatosensory information during movement. Alternatively, the enhanced connectivity and functional responsiveness of the retrosplenial cortex may reflect more general changes in movement induced by cortical stroke, such as changes in gait, posture, or head/neck movements (Whishaw et al., 2008).
Spine dynamics during new sensory map formation
We used time-lapse imaging of layer 5 dendrites over several weeks to determine whether the establishment of new functional circuits and long-range connections in peri-infarct regions was accompanied by changes in fine synaptic structure. Before stroke, 9% of spines were gained or lost in the mature cortex from week to week, which approximates previous estimates in fully mature mice (Zuo et al., 2005; Majewska et al., 2006; Keck et al., 2008). After stroke, spine turnover increased substantially in the peri-infarct zone, which persisted, albeit to a lesser extent for up to 5 weeks after stroke. This finding agrees with previous imaging studies showing that dendritic spine stability can be augmented by changes in sensory experience (Trachtenberg et al., 2002; Holtmaat et al., 2006), neuronal activity, or disease (Kirov and Harris, 1999; Rensing et al., 2005; Fuhrmann et al., 2007). Farther away from the infarct border (∼700–800 μm), spine turnover rates were similar to unlesioned animals. These results agree with recent work showing that stroke-induced spine loss or reorientation of dendritic arbors is restricted to within a couple hundred micrometers of the infarct border (Brown et al., 2007, 2008), and support the idea that stroke-related structural plasticity is especially prominent in the peri-infarct zone (Stroemer et al., 1995; Carmichael et al., 2005). We should note that our spine imaging data does not identify the source of the presynaptic partners for the spines that turn over but does provide an indication of the degree to which spiny synapses turnover and in what brain regions this occurs (i.e., peri-infarct zone). Future experiments combining long-term imaging of fluorescently labeled axons from select populations of neurons will be necessary to resolve these issues (Portera-Cailliau et al., 2005).
In conclusion, we have used a number of experimental approaches (results summarized in Fig. 8) to show how sensory cortex maps are rebuilt during stroke recovery. Our data indicate that stroke causes (1) an initial loss of cortical responsiveness to the impaired limb that is gradually restored and remerges predominately within the peri-infarct motor and hindlimb regions, (2) the development of prolonged cortical sensory responses in local and distant regions, (3) large-scale changes in neuronal connectivity with the retrosplenial cortex, primary and secondary somatosensory cortical regions, and the striatum, and (4) enhanced turnover of the presumptive postsynaptic targets of cortical connections in the reorganized forelimb area (i.e., dendritic spines) for several weeks after stroke. Collectively, these findings provide a new, comprehensive circuit model for understanding how the adult cerebral cortex adapts to focal brain damage.
Figure 8.

Summary of new functional and structural circuits, viewed from the dorsal (A) or coronal (B) plane. Several weeks after focal ischemic stroke (gray area in right hemisphere of A), forelimb-evoked responses reemerge in adjacent peri-infarct motor and hindlimb cortical areas (note the shaded green area overlapping with blue and red zones that correspond with hindlimb and motor areas, respectively). Longitudinal two-photon imaging indicated that layer 5 apical dendrites in the reorganized zone were sites of heightened dendritic spine turnover (see inset outlined in red box in B). Tracing of neuronal connectivity in the reorganized forelimb area revealed that it had lost inputs from more lateral primary and secondary somatosensory cortical regions (see dotted arrows from orange and purple zones) but gained connections from the retrosplenial cortex (thick arrow from yellow zone in A and B) and sent more output projections to the ipsilateral striatum (thick arrow entering CPu). In contrast, connectivity with the contralateral cerebral cortex/striatum, as well as the ipsilateral thalamus did not change significantly (see thin black arrows).
Footnotes
This work was supported by the Michael Smith Foundation for Health Research, the Natural Sciences and Engineering Research Council of Canada, and Canadian Institutes of Health Research (CIHR) postdoctoral fellowships (C.E.B.), CIHR Operating Grant MOP49586 (T.H.M.), and funds from the Canadian Stroke Network (T.H.M.). We thank Charles Wong for assisting in data analysis, PuMin Wang for surgical expertise, and Albrecht Sigler for assistance with computer programming. We also thank Nicholas Swindale, Jamie Boyd, and Majid Mohajerani for providing helpful comments on this manuscript.
References
- Alloway KD, Lou L, Nwabueze-Ogbo F, Chakrabarti S. Topography of cortical projections to the dorsolateral neostriatum in rats: multiple overlapping sensorimotor pathways. J Comp Neurol. 2006;499:33–48. doi: 10.1002/cne.21039. [DOI] [PubMed] [Google Scholar]
- Armstrong-James M, Welker E, Callahan CA. The contribution of NMDA and non-NMDA receptors to fast and slow transmission of sensory information in the rat SI barrel cortex. J Neurosci. 1993;13:2149–2160. doi: 10.1523/JNEUROSCI.13-05-02149.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger T, Borgdorff A, Crochet S, Neubauer FB, Lefort S, Fauvet B, Ferezou I, Carleton A, Lüscher HR, Petersen CC. Combined voltage and calcium epifluorescence imaging in vitro and in vivo reveals subthreshold and suprathreshold dynamics of mouse barrel cortex. J Neurophysiol. 2007;97:3751–3762. doi: 10.1152/jn.01178.2006. [DOI] [PubMed] [Google Scholar]
- Biernaskie J, Szymanska A, Windle V, Corbett D. Bi-hemispheric contribution to functional motor recovery of the affected forelimb following focal ischemic brain injury in rats. Eur J Neurosci. 2005;21:989–999. doi: 10.1111/j.1460-9568.2005.03899.x. [DOI] [PubMed] [Google Scholar]
- Blake DT, Hsiao SS, Johnson KO. Neural coding mechanisms in tactile pattern recognition: the relative contributions of slowly and rapidly adapting mechanoreceptors to perceived roughness. J Neurosci. 1997;17:7480–7489. doi: 10.1523/JNEUROSCI.17-19-07480.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennerhassett JM, Matyas TA, Carey LM. Impaired discrimination of surface friction contributes to pinch grip deficit after stroke. Neurorehabil Neural Repair. 2007;21:263–272. doi: 10.1177/1545968306295560. [DOI] [PubMed] [Google Scholar]
- Borgdorff AJ, Poulet JF, Petersen CC. Facilitating sensory responses in developing mouse somatosensory barrel cortex. J Neurophysiol. 2007;97:2992–3003. doi: 10.1152/jn.00013.2007. [DOI] [PubMed] [Google Scholar]
- Brown CE, Li P, Boyd JD, Delaney KR, Murphy TH. Extensive turnover of dendritic spines and vascular remodeling in cortical tissues recovering from stroke. J Neurosci. 2007;27:4101–4109. doi: 10.1523/JNEUROSCI.4295-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Wong C, Murphy TH. Rapid morphologic plasticity of peri-infarct dendritic spines after focal ischemic stroke. Stroke. 2008;39:1286–1291. doi: 10.1161/STROKEAHA.107.498238. [DOI] [PubMed] [Google Scholar]
- Buchkremer-Ratzmann I, August M, Hagemann G, Witte OW. Electrophysiological transcortical diaschisis after cortical photothrombosis in rat brain. Stroke. 1996;27:1105–1109. doi: 10.1161/01.str.27.6.1105. discussion 1109–1111. [DOI] [PubMed] [Google Scholar]
- Buchkremer-Ratzmann I, August M, Hagemann G, Witte OW. Epileptiform discharges to extracellular stimuli in rat neocortical slices after photothrombotic infarction. J Neurol Sci. 1998;156:133–137. doi: 10.1016/s0022-510x(98)00034-3. [DOI] [PubMed] [Google Scholar]
- Carmichael ST. Cellular and molecular mechanisms of neural repair after stroke: making waves. Ann Neurol. 2006;59:735–742. doi: 10.1002/ana.20845. [DOI] [PubMed] [Google Scholar]
- Carmichael ST, Wei L, Rovainen CM, Woolsey TA. New patterns of intracortical projections after focal cortical stroke. Neurobiol Dis. 2001;8:910–922. doi: 10.1006/nbdi.2001.0425. [DOI] [PubMed] [Google Scholar]
- Carmichael ST, Archibeque I, Luke L, Nolan T, Momiy J, Li S. Growth-associated gene expression after stroke: evidence for a growth-promoting region in peri-infarct cortex. Exp Neurol. 2005;193:291–311. doi: 10.1016/j.expneurol.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Borrel J. Functional recovery of forelimb response capacity after forelimb primary motor cortex damage in the rat is due to the reorganization of adjacent areas of cortex. Neuroscience. 1995;68:793–805. doi: 10.1016/0306-4522(95)00178-l. [DOI] [PubMed] [Google Scholar]
- Cho J, Sharp PE. Head direction, place, and movement correlates for cells in the rat retrosplenial cortex. Behav Neurosci. 2001;115:3–25. doi: 10.1037/0735-7044.115.1.3. [DOI] [PubMed] [Google Scholar]
- Chollet F, DiPiero V, Wise RJ, Brooks DJ, Dolan RJ, Frackowiak RS. The functional anatomy of motor recovery after stroke in humans: a study with positron emission tomography. Ann Neurol. 1991;29:63–71. doi: 10.1002/ana.410290112. [DOI] [PubMed] [Google Scholar]
- Conner JM, Chiba AA, Tuszynski MH. The basal forebrain cholinergic system is essential for cortical plasticity and functional recovery following brain injury. Neuron. 2005;46:173–179. doi: 10.1016/j.neuron.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Corbetta M, Kincade MJ, Lewis C, Snyder AZ, Sapir A. Neural basis and recovery of spatial attention deficits in spatial neglect. Nat Neurosci. 2005;8:1603–1610. doi: 10.1038/nn1574. [DOI] [PubMed] [Google Scholar]
- Cramer SC. Repairing the human brain after stroke. I. Mechanisms of spontaneous recovery. Ann Neurol. 2008;63:272–287. doi: 10.1002/ana.21393. [DOI] [PubMed] [Google Scholar]
- Dancause N, Barbay S, Frost SB, Plautz EJ, Chen D, Zoubina EV, Stowe AM, Nudo RJ. Extensive cortical rewiring after brain injury. J Neurosci. 2005;25:10167–10179. doi: 10.1523/JNEUROSCI.3256-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkhuizen RM, Ren J, Mandeville JB, Wu O, Ozdag FM, Moskowitz MA, Rosen BR, Finklestein SP. Functional magnetic resonance imaging of reorganization in rat brain after stroke. Proc Natl Acad Sci U S A. 2001;98:12766–12771. doi: 10.1073/pnas.231235598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoghue JP, Parham C. Afferent connections of the lateral agranular field of the rat motor cortex. J Comp Neurol. 1983;217:390–404. doi: 10.1002/cne.902170404. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Ferezou I, Bolea S, Petersen CC. Visualizing the cortical representation of whisker touch: voltage-sensitive dye imaging in freely moving mice. Neuron. 2006;50:617–629. doi: 10.1016/j.neuron.2006.03.043. [DOI] [PubMed] [Google Scholar]
- Ferezou I, Haiss F, Gentet LJ, Aronoff R, Weber B, Petersen CC. Spatiotemporal dynamics of cortical sensorimotor integration in behaving mice. Neuron. 2007;56:907–923. doi: 10.1016/j.neuron.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Frost DO, Boire D, Gingras G, Ptito M. Surgically created neural pathways mediate visual pattern discrimination. Proc Natl Acad Sci U S A. 2000;97:11068–11073. doi: 10.1073/pnas.190179997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frostig RD, Lieke EE, Ts'o DY, Grinvald A. Cortical functional architecture and local coupling between neuronal activity and the microcirculation revealed by in vivo high-resolution optical imaging of intrinsic signals. Proc Natl Acad Sci U S A. 1990;87:6082–6086. doi: 10.1073/pnas.87.16.6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann M, Mitteregger G, Kretzschmar H, Herms J. Dendritic pathology in prion disease starts at the synaptic spine. J Neurosci. 2007;27:6224–6233. doi: 10.1523/JNEUROSCI.5062-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez CL, Kolb B. A comparison of different models of stroke on behaviour and brain morphology. Eur J Neurosci. 2003;18:1950–1962. doi: 10.1046/j.1460-9568.2003.02928.x. [DOI] [PubMed] [Google Scholar]
- Grinvald A, Hildesheim R. VSDI: a new era in functional imaging of cortical dynamics. Nat Rev Neurosci. 2004;5:874–885. doi: 10.1038/nrn1536. [DOI] [PubMed] [Google Scholar]
- Harris KM, Kater SB. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci. 1994;17:341–371. doi: 10.1146/annurev.ne.17.030194.002013. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat Methods. 2005;2:932–940. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- Holtmaat AJ, Trachtenberg JT, Wilbrecht L, Shepherd GM, Zhang X, Knott GW, Svoboda K. Transient and persistent dendritic spines in the neocortex in vivo. Neuron. 2005;45:279–291. doi: 10.1016/j.neuron.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Wilbrecht L, Knott GW, Welker E, Svoboda K. Experience-dependent and cell-type-specific spine growth in the neocortex. Nature. 2006;441:979–983. doi: 10.1038/nature04783. [DOI] [PubMed] [Google Scholar]
- Jaillard A, Martin CD, Garambois K, Lebas JF, Hommel M. Vicarious function within the human primary motor cortex? A longitudinal fMRI stroke study. Brain. 2005;128:1122–1138. doi: 10.1093/brain/awh456. [DOI] [PubMed] [Google Scholar]
- Keck T, Mrsic-Flogel TD, Vaz Afonso M, Eysel UT, Bonhoeffer T, Hübener M. Massive restructuring of neuronal circuits during functional reorganization of adult visual cortex. Nat Neurosci. 2008;11:1162–1167. doi: 10.1038/nn.2181. [DOI] [PubMed] [Google Scholar]
- Kerr JN, Denk W. Imaging in vivo: watching the brain in action. Nat Rev Neurosci. 2008;9:195–205. doi: 10.1038/nrn2338. [DOI] [PubMed] [Google Scholar]
- Kim JS, Choi-Kwon S. Discriminative sensory dysfunction after unilateral stroke. Stroke. 1996;27:677–682. doi: 10.1161/01.str.27.4.677. [DOI] [PubMed] [Google Scholar]
- Kirov SA, Harris KM. Dendrites are more spiny on mature hippocampal neurons when synapses are inactivated. Nat Neurosci. 1999;2:878–883. doi: 10.1038/13178. [DOI] [PubMed] [Google Scholar]
- Kleinfeld D, Waters J. Wilder Penfield in the age of YouTube: visualizing the sequential activation of sensorimotor areas across neocortex. Neuron. 2007;56:760–762. doi: 10.1016/j.neuron.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Knott GW, Holtmaat A, Wilbrecht L, Welker E, Svoboda K. Spine growth precedes synapse formation in the adult neocortex in vivo. Nat Neurosci. 2006;9:1117–1124. doi: 10.1038/nn1747. [DOI] [PubMed] [Google Scholar]
- Kolb B, Morshead C, Gonzalez C, Kim M, Gregg C, Shingo T, Weiss S. Growth factor-stimulated generation of new cortical tissue and functional recovery after stroke damage to the motor cortex of rats. J Cereb Blood Flow Metab. 2007;27:983–997. doi: 10.1038/sj.jcbfm.9600402. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska AK, Newton JR, Sur M. Remodeling of synaptic structure in sensory cortical areas in vivo. J Neurosci. 2006;26:3021–3029. doi: 10.1523/JNEUROSCI.4454-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell KA, Dyck RH. Induction of reproducible focal ischemic lesions in neonatal mice by photothrombosis. Dev Neurosci. 2005;27:121–126. doi: 10.1159/000085983. [DOI] [PubMed] [Google Scholar]
- Neumann-Haefelin T, Hagemann G, Witte OW. Cellular correlates of neuronal hyperexcitability in the vicinity of photochemically induced cortical infarcts in rats in vitro. Neurosci Lett. 1995;193:101–104. doi: 10.1016/0304-3940(95)11677-o. [DOI] [PubMed] [Google Scholar]
- Nudo RJ, Milliken GW. Reorganization of movement representations in primary motor cortex following focal ischemic infarcts in adult squirrel monkeys. J Neurophysiol. 1996;75:2144–2149. doi: 10.1152/jn.1996.75.5.2144. [DOI] [PubMed] [Google Scholar]
- Nudo RJ, Wise BM, SiFuentes F, Milliken GW. Neural substrates for the effects of rehabilitative training on motor recovery after ischemic infarct. Science. 1996;272:1791–1794. doi: 10.1126/science.272.5269.1791. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KB. The mouse brain in stereotaxic coordinates. New York: Academic; 2001. [Google Scholar]
- Petersen CC, Grinvald A, Sakmann B. Spatiotemporal dynamics of sensory responses in layer 2/3 of rat barrel cortex measured in vivo by voltage-sensitive dye imaging combined with whole-cell voltage recordings and neuron reconstructions. J Neurosci. 2003;23:1298–1309. doi: 10.1523/JNEUROSCI.23-04-01298.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portera-Cailliau C, Weimer RM, De Paola V, Caroni P, Svoboda K. Diverse modes of axon elaboration in the developing neocortex. PLoS Biol. 2005;3:e272. doi: 10.1371/journal.pbio.0030272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redecker C, Wang W, Fritschy JM, Witte OW. Widespread and long-lasting alterations in GABA(A)-receptor subtypes after focal cortical infarcts in rats: mediation by NMDA-dependent processes. J Cereb Blood Flow Metab. 2002;22:1463–1475. doi: 10.1097/01.WCB.0000034149.72481.BD. [DOI] [PubMed] [Google Scholar]
- Rensing N, Ouyang Y, Yang XF, Yamada KA, Rothman SM, Wong M. In vivo imaging of dendritic spines during electrographic seizures. Ann Neurol. 2005;58:888–898. doi: 10.1002/ana.20658. [DOI] [PubMed] [Google Scholar]
- Sato TR, Gray NW, Mainen ZF, Svoboda K. The functional microarchitecture of the mouse barrel cortex. PLoS Biol. 2007;5:e189. doi: 10.1371/journal.pbio.0050189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanina EV, Schallert T, Witte OW, Redecker C. Behavioral recovery from unilateral photothrombotic infarcts of the forelimb sensorimotor cortex in rats: role of the contralateral cortex. Neuroscience. 2006;139:1495–1506. doi: 10.1016/j.neuroscience.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Shoham D, Glaser DE, Arieli A, Kenet T, Wijnbergen C, Toledo Y, Hildesheim R, Grinvald A. Imaging cortical dynamics at high spatial and temporal resolution with novel blue voltage-sensitive dyes. Neuron. 1999;24:791–802. doi: 10.1016/s0896-6273(00)81027-2. [DOI] [PubMed] [Google Scholar]
- Stern EA, Maravall M, Svoboda K. Rapid development and plasticity of layer 2/3 maps in rat barrel cortex in vivo. Neuron. 2001;31:305–315. doi: 10.1016/s0896-6273(01)00360-9. [DOI] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, Konnerth A. In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci U S A. 2003;100:7319–7324. doi: 10.1073/pnas.1232232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroemer RP, Kent TA, Hulsebosch CE. Neocortical neural sprouting, synaptogenesis, and behavioral recovery after neocortical infarction in rats. Stroke. 1995;26:2135–2144. doi: 10.1161/01.str.26.11.2135. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- van Groen T, Wyss JM. Connections of the retrosplenial dysgranular cortex in the rat. J Comp Neurol. 1992;315:200–216. doi: 10.1002/cne.903150207. [DOI] [PubMed] [Google Scholar]
- Vann SD, Aggleton JP. Testing the importance of the retrosplenial guidance system: effects of different sized retrosplenial cortex lesions on heading direction and spatial working memory. Behav Brain Res. 2004;155:97–108. doi: 10.1016/j.bbr.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Ward NS. Functional reorganization of the cerebral motor system after stroke. Curr Opin Neurol. 2004;17:725–730. doi: 10.1097/00019052-200412000-00013. [DOI] [PubMed] [Google Scholar]
- Ward NS. The neural substrates of motor recovery after focal damage to the central nervous system. Arch Phys Med Rehabil. 2006;87:S30–S35. doi: 10.1016/j.apmr.2006.08.334. [DOI] [PubMed] [Google Scholar]
- Watson BD, Dietrich WD, Busto R, Wachtel MS, Ginsberg MD. Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann Neurol. 1985;17:497–504. doi: 10.1002/ana.410170513. [DOI] [PubMed] [Google Scholar]
- Wei L, Erinjeri JP, Rovainen CM, Woolsey TA. Collateral growth and angiogenesis around cortical stroke. Stroke. 2001;32:2179–2184. doi: 10.1161/hs0901.094282. [DOI] [PubMed] [Google Scholar]
- Werhahn KJ, Conforto AB, Kadom N, Hallett M, Cohen LG. Contribution of the ipsilateral motor cortex to recovery after chronic stroke. Ann Neurol. 2003;54:464–472. doi: 10.1002/ana.10686. [DOI] [PubMed] [Google Scholar]
- Whishaw IQ, Alaverdashvili M, Kolb B. The problem of relating plasticity and skilled reaching after motor cortex stroke in the rat. Behav Brain Res. 2008;192:124–136. doi: 10.1016/j.bbr.2007.12.026. [DOI] [PubMed] [Google Scholar]
- Winship IR, Murphy TH. In vivo calcium imaging reveals functional rewiring of single somatosensory neurons after stroke. J Neurosci. 2008;28:6592–6606. doi: 10.1523/JNEUROSCI.0622-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winship IR, Plaa N, Murphy TH. Rapid astrocyte calcium signals correlate with neuronal activity and onset of the hemodynamic response in vivo. J Neurosci. 2007;27:6268–6272. doi: 10.1523/JNEUROSCI.4801-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xerri C, Merzenich MM, Peterson BE, Jenkins W. Plasticity of primary somatosensory cortex paralleling sensorimotor skill recovery from stroke in adult monkeys. J Neurophysiol. 1998;79:2119–2148. doi: 10.1152/jn.1998.79.4.2119. [DOI] [PubMed] [Google Scholar]
- Xu HT, Pan F, Yang G, Gan WB. Choice of cranial window type for in vivo imaging affects dendritic spine turnover in the cortex. Nat Neurosci. 2007;10:549–551. doi: 10.1038/nn1883. [DOI] [PubMed] [Google Scholar]
- Zepeda A, Sengpiel F, Guagnelli MA, Vaca L, Arias C. Functional reorganization of visual cortex maps after ischemic lesions is accompanied by changes in expression of cytoskeletal proteins and NMDA and GABAA receptor subunits. J Neurosci. 2004;24:1812–1821. doi: 10.1523/JNEUROSCI.3213-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Lin A, Chang P, Gan WB. Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron. 2005;46:181–189. doi: 10.1016/j.neuron.2005.04.001. [DOI] [PubMed] [Google Scholar]