

Abstract
SUMMARY
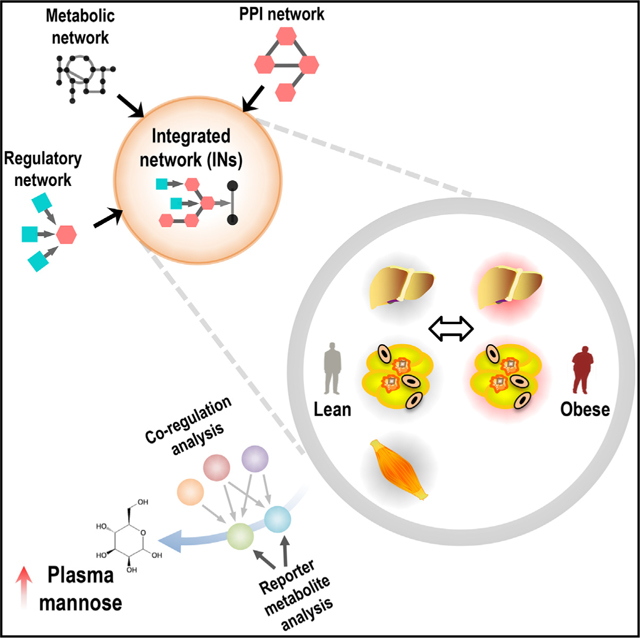
To investigate the biological processes that are altered in obese subjects, we generated cell-specific integrated networks (INs) by merging genome-scale metabolic, transcriptional regulatory and protein-protein interaction networks. We performed genome-wide transcriptomics analysis to determine the global gene expression changes in the liver and three adipose tissues from obese subjects undergoing bariatric surgery and integrated these data into the cell-specific INs. We found dysregulations in mannose metabolism in obese subjects and validated our predictions by detecting mannose levels in the plasma of the lean and obese subjects. We observed significant correlations between plasma mannose levels, BMI, and insulin resistance (IR). We also measured plasma mannose levels of the subjects in two additional different cohorts and observed that an increased plasma mannose level was associated with IR and insulin secretion. We finally identified mannose as one of the best plasma metabolites in explaining the variance in obesity-independent IR.
In Brief
Lee et al. merged genome-scale metabolic models (GEMs), transcriptional regulatory networks (TRNs), and protein-protein interaction networks (PPINs) to generate cell-specific integrated networks for hepatocytes, myocytes, and adipocytes of lean and obese subjects undergoing bariatric surgery. They identified, and independently validated, plasma mannose as highly associated with insulin resistance, independent of BMI.
Graphical Abstract
INTRODUCTION
Obesity is associated with an increased risk for a wide range of morbidities, including insulin resistance (IR), type 2 diabetes (T2D), non-alcoholic fatty liver disease (NAFLD), and cardiovascular disease (CVD). Although the prevalence of obesity continues to dramatically increase worldwide, a clear understanding of the underlying molecular mechanisms involved in the progression of associated disorders is still lacking. Several attempts have been made to reveal the metabolic processes that are altered in obesity (Mardinoglu et al., 2013, 2015a, 2014b), NAFLD (Hyötyläinen et al., 2016; Mardinoglu et al., 2014a), and T2D (Väremo et al., 2015) through the use of genome-scale metabolic models (GEMs). GEMs are collections of biochemical reactions and their catalyzing protein-coding genes and they have been widely used in the identification of biomarkers as well as drug targets for the development of effective treatment strategies for metabolism-related disorders (Björnson et al., 2015; Bordbar et al., 2014; Mardinoglu and Nielsen, 2012, 2015; Mardinoglu et al., 2015b; O’Brien et al., 2015; Shoaie et al., 2015; Uhlén et al., 2016; Yizhak et al., 2013, 2014a, 2014b; Zhang et al., 2015).
Metabolism is precisely regulated in response to internal and external stimuli, and the expression of metabolic enzymes is uniquely controlled by transcriptional regulation in each cell/tissue. Hence, it is necessary to integrate GEMs with transcriptional regulatory networks (TRNs) that control the transcription state of the genome to increase their predictive ability. TRNs include information on the interactions between transcription factors (TFs) and provide insights into network hubs, hierarchical organization, and network motifs. The architecture of TRNs derived from ENCODE data was studied by determining the genomic binding information of 119TFs (Gerstein et al., 2012). Moreover, an additional TRN that includes the associations between 475 sequence-specific TFs has been presented and its dynamic properties have been studied in different cell/tissue types (Neph et al., 2012b). The robustness of the gene expression phenotypes of these highly cell- and tissue-specific TRNs has been extensively studied (Pechenick et al., 2014), and the integration of TRNs and GEMs may lead to improved reconstructions of human metabolism and its regulation.
Interactions between proteins are central to all biological functions, and protein-protein interaction networks (PPINs) provide further insights into the functional organization of individual proteins because proteins rarely act alone (Li et al., 2015). PPINs have been widely used to identify potential drug targets and study the effects of already known targets through the mapping of protein expression data (Snider et al., 2015). Consistent PPINs are currently available, and these networks are presented in a number of databases constructed using community standards (De Las Rivas and Fontanillo, 2010; Orchard et al., 2012; Szklarczyk et al., 2015). The size of the PPINs continues to increase substantially with the development of high-throughput experimental technologies, manual evaluations of relevant literature and predictions using computational methods (Szklarczyk et al., 2015). Recently, Rolland et al. (2014) presented a systematic map of 14,000 human binary protein-protein interactions, and this high-quality reference PPIN was used to generate the cell-specific PPINs used in our study.
While GEMs, TRNs, and PPINs can each provide new insights as to the metabolic processes that become dysfunctional in obesity, the integration of these networks can likely provide a more complete and multifaceted understanding of these biological processes. Such integration has not previously been attempted. Here, we integrated functional GEMs for hepatocytes (iHepatocytes2322) (Mardinoglu et al., 2014a), myocytes (iMyocytes2419) (Väremo et al., 2015), and adipocytes (iAdipocytes1850) (Mardinoglu et al., 2013, 2014b) with TRNs and PPINs to generate cell-specific integrated networks (INs). We employed these INs to explore the biological processes that are altered in obesity in the context of whole-body functions (Figure 1A).
Figure 1. Generation of Cell-Specific Regulatory Networks.
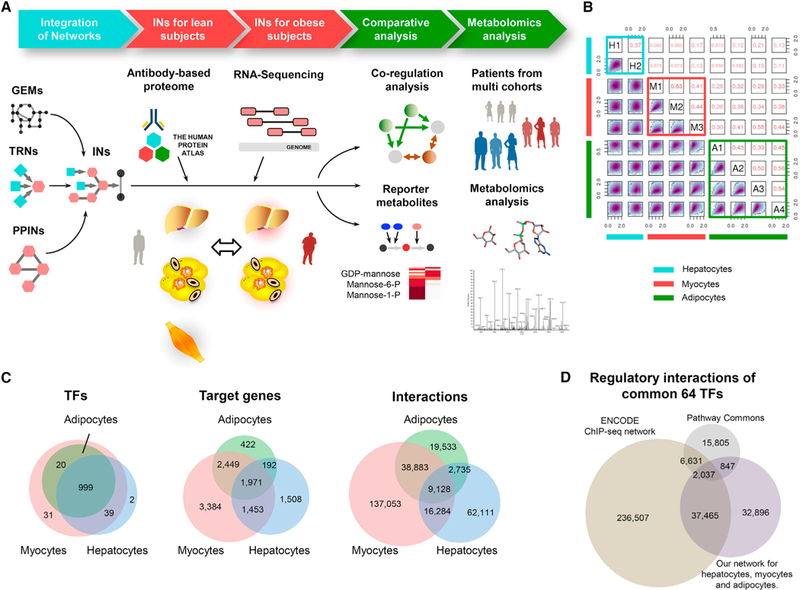
(A) To create integrated networks (INs) for hepatocytes, myocytes, and adipocytes, we generated cell-specific regulatory networks (RNs) using DNase-seq data and integrated these networks with cell-specific protein-protein interaction networks (PPINs) and genome-scale metabolic models (GEMs). We refined the INs for the healthy subjects based on the cell-specific antibody-based proteomic data and tissue-specific transcriptomic data in the Human Protein Atlas. We also generated RNA sequencing data from liver tissue and three different adipose tissues of the obese subjects and used these data in the generation of the INs for the obese subjects. We performed a comparative analysis between the INs of the lean and obese subjects and identified metabolic pathways with decreased or increased co-regulation. Finally, we validated our network-based predictions in three different independent cohorts with varying BMI values, insulin resistance, and insulin secretion values.
(B) We compared replicates of the DNase-seq samples based on their footprint occupancy scores (FOS) in a pairwise manner and found high correlations between the cell-type replicates.
(C) After selecting potential TF-binding sites with high FOS scores over all of the replicates of a given cell type, we generated cell-specific TRNs based on the irreproducible discovery rate (IDR). We compared the TFs, target genes and interactions in each cell-specific TRN.
(D) We evaluated our cell-specific TRNs with other TRNs generated based on ENCODE ChIP-seq data and manually curated knowledge from the literature in the Pathway Commons database. We compared the corresponding regulatory interactions of 64 TFs that were consistently present in all three TRNs with other target genes. We found that our TRN had a high overlap with the ENCODE ChIP-seq data (53.9%).
See also Figures S1 and S7, Tables S1–S3, and Data S1.
RESULTS
Human Cell-Specific Regulatory Networks
The generation of INs by integrating TRNs, PPINs, and GEMs is necessary for a more comprehensive understanding of the mechanisms involved in the progression of chronic diseases. To generate human cell-specific TRNs, we retrieved DNase sequencing (DNase-seq) data from metabolically active cell types, including hepatocytes, myocytes, and adipocytes, from the ENCODE repository (ENCODE Project Consortium, 2012) (Table S1). DNase-seq identifies regions of open (and hence actively regulated) chromatin based on accessibility by the DNase I endonuclease (Crawford et al., 2006). We also retrieved DNase-seq data from hepatic stellate cells, skeletal myoblasts, and myotubes to evaluate our cell-specific TRNs from hepatocytes and myocytes. We first checked the quality of the DNase-seq samples and analyzed the DNase-seq data following the pipeline introduced by the ENCODE consortium (Neph et al., 2012c) (Figure S1A).
We compared the footprint occupancy scores (FOS) of potential TF-binding sites among the samples in a pairwise manner and found that the samples with the same tissue of origin had overall higher correlations (Figure 1B). After generating cell-specific reference TRNs for hepatocytes, myocytes, and adipocytes (Figure S1; Table S1), we compared the contents of the TRNs (Figure 1C) and observed that the interactions between the TFs and target genes were specific to the cell type. Higher overlap was observed between the TRNs for adipocytes and myocytes compared to their overlaps with the TRNs for hepatocytes (Figures S1B and S1C). We also found that the TRNs of hepatocytes and myocytes had higher similarities with the TRNs of the adjacent cells, including hepatic stellate cells and skeletal myoblasts and myotubes (Figures S1D–S1F).
To evaluate the cell-specific TRNs for hepatocytes, myocytes, and adipocytes, we compared the regulatory interactions of TFs with the interactions in previously presented TRNs generated based on ENCODE ChIP-seq data (Gerstein et al., 2012) and published literature in the Pathway Commons database (Cerami et al., 2011). We only compared the interactions among 64 TFs that were consistently present in all three sources and compared their corresponding regulatory interactions (Figure 1D). We compared the regulatory interactions in our TRNs with the interactions in the TRNs generated based on ENCODE ChIP-seq data and found that 53.9% of the interactions in our TRNs (Jaccard index [JI]: 0.125) were shared between these two networks. We also compared the TRNs generated herein as well as the TRNs generated based on ENCODE chromatin immunoprecipitation sequencing (ChIP-seq) data with the TRNs retrieved from the Pathway Commons database and found minor similarities among these networks (JI: 0.0301 and JI: 0.0290, respectively). The relatively low coverage of cell-specific regulatory interactions in the knowledge-based databases illustrates the importance of generating cell-specific TRNs based on DNase-seq and ChIP-seq data.
Refinement of Cell-Specific Networks Using Proteomic and Transcriptomic Data
DNase-seq data can be used to identify the binding regions of TFs; however, all of the regulatory interactions in the reference TRNs may not be observed in the biological samples. To remove false-positive regulatory interactions from the reference TRNs, we refined our cell-specific TRNs using cell-specific antibody-based proteomic data from hepatocytes, myocytes, and adipocytes as well as tissue-specific RNA-seq data from liver, muscle, and adipose tissues obtained from healthy subjects in the Human Protein Atlas (HPA) (ver.14) (Uhlén et al., 2015) (Figure S2A). The presence or absence of 17,005 unique proteins in each cell type was evaluated using 25,039 antibodies, and the abundance or absence of the proteins was evaluated as “high,” “medium,” “low,” or “not detected.”
We removed the TFs and target genes from the cell-specific TRNs if there was negative evidence (Not detected) for the presence of the protein in the cell type and lower mRNA abundance (fragments per kilobase of exon per million fragments mapped [FPKM] <1) in the corresponding tissues (Table S2). We compared the overlap of the target genes in the TRNs with the metabolism-related (metabolic) genes in the GEMs and found that more than 26% of the metabolic genes were present in the TRNs for each cell type (Figure 2A). Moreover, we analyzed the topological characteristics of the TRNs by examining the out-degrees of TFs and the in-degrees of target genes and comparing their degrees across the cell types (Figures S2C–S2H) and observed that while the out-degrees of the TFs remained relatively stable across cell types, the in-degrees of target genes revealed that TFs were binding to different sets of target genes in each cell type.
Figure 2. Generation of Cell-Specific Integrated Networks.
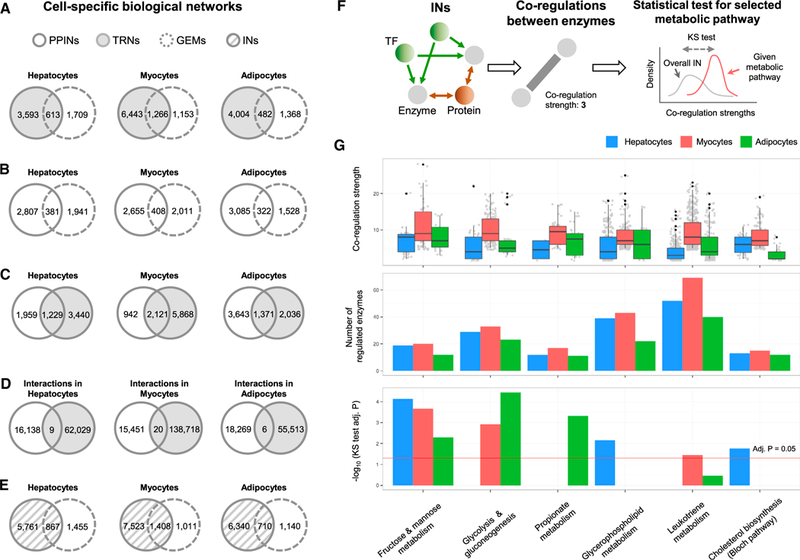
(A) We investigated the overlap between the target genes of cell-specific regulatory networks (RNs) and metabolic genes in their corresponding cell-specific genome-scale metabolic models (GEMs) for hepatocytes (iHepatocytes2322), myocytes (iMyocytes2419), and adipocytes (iAdipocytes1850).
(B) Similarly, we examined the overlap between the protein-coding genes in the cell-specific protein-protein interaction networks (PPINs) with the metabolic genes in the corresponding cell-specific GEMs.
(C) A comparison of the protein-coding genes in the cell-specific PPINs with TFs and target genes in the cell-specific TRNs showed relatively good overlap between the genes.
(D) However, when we examined the overlap of interactions between the cell-specific TRNs and PPINs, we found almost no overlap between these networks.
(E) After integrating the cell-specific TRNs and PPINs, we examined the overlap between the protein-coding genes with the metabolic genes in the GEMs and found relatively higher overlap between them.
(F) We analyzed the co-regulation of all enzymes in the cell-specific integrated networks (INs) by establishing a weighted edge between metabolic genes that had co-regulating TFs and/or proteins.
(G) We identified highly co-regulated metabolic pathways from a co-regulation analysis performed using the metabolic genes involved in each metabolic pathway by a Bonferroni-adjusted Kolmogorov-Smirnov one-sided test. Fructose and mannose metabolism, glycerophospholipid metabolism, and cholesterol biosynthesis were highly co-regulated in the hepatocytes; fructose and mannose metabolism, leukotriene metabolism, and glycolysis were highly co-regulated in the myocytes; and fructose and mannose metabolism, propionate metabolism, and glycolysis were highly co-regulated in the adipocytes.
See also Figures S2–S4, Table S4, and Data S1.
We obtained a recently published generic human PPIN (Rolland et al., 2014) and refined this reference network using cell-specific antibody-based proteomic data and tissue-specific RNA-seq data in HPA (Uhlén et al., 2015). We generated cell-specific PPINs for healthy hepatocytes, myocytes, and adipocytes by removing genes without protein abundance (“Not detected” in the corresponding cell) or low mRNA abundance (FPKM <1) in the corresponding tissue (Table S2).
We compared the protein-coding genes in the resulting cell-specific PPINs and GEMs and found that 381 genes were shared between the PPIN for hepatocytes and iHepatocytes2322, 408 genes were shared between the PPIN for myocytes and iMyocytes2419, and 322 genes were shared between the PPIN for adipocytes and iAdipocytes1850 (Figure 2B). Although there was relatively good overlap between the genes in the cell-specific PPINs and the TFs and target genes in the cell-specific TRNs (Figure 2C), we detected almost no overlap between the interactions in these networks (Figure 2D), which suggests that PPINs and TRNs should be integrated with GEMs to elucidate all of the molecular mechanisms that govern cell/tissue behaviors.
Highly Co-regulated Metabolic Pathways in Healthy Subjects
We first integrated the TRNs and PPINs for hepatocytes, myocytes, and adipocytes (Table S2) and found that 37.3% of the genes in iHepatocytes2322, 58.2% of the genes in iMyocytes2419, and 38.4% of the genes in iAdipocytes1850 were regulated by TFs or other target genes in the TRNs and PPINs (Figure 2E). To gain further insights into the metabolism of cells and tissues, we generated cell-specific INs for hepatocytes, myocytes, and adipocytes by integrating the cell- specific GEMs, TRNs, and PPINs (Table S2). We examined the degree to which each metabolic pathway in a cell-specific GEM was concurrently regulated by TFs and targets in the TRNs and PPINs by analyzing the co-regulated functions (Figure 2F). We established a weighted edge between the metabolic genes that we call the co-regulation strength (counting the number of co-regulating TFs and proteins as weight) and examined the co-regulation strengths of the metabolic pathways of each cell type.
We identified the highly co-regulated metabolic pathways in each cell type based on the co-regulation strengths and then examined the global characteristics of the co-regulation strengths of all gene pairs in the INs (Figure S3) and tested each metabolic pathway to see if its co-regulation strength was greater than or less than the overall co-regulation of the INs (Figure 2G; Data S1A) (see the Experimental Procedures). For each cell type, we found different sets of highly co-regulated pathways (adjusted p value < 0.05). During the co-regulation analysis, we used the metabolic genes involved in a specific pathway and their surrounding TFs or proteins in the cell-specific INs. We found that fructose and mannose metabolism, glycerophospholipid metabolism, and cholesterol biosynthesis were highly co-regulated in hepatocytes; fructose and mannose metabolism, glycolysis, and leukotriene metabolism were highly co-regulated in myocytes; and fructose and mannose metabolism, glycolysis, and propionate metabolism were highly co-regulated in adipocytes.
These top co-regulated metabolic pathways are highly relevant for tissue-specific functions. For example, glycerophospho-lipid metabolism and cholesterol biosynthesis, which were found to be significantly and specifically co-regulated in hepatocytes, are known to be regulated by the liver through lipoprotein secretion (Tijburg et al., 1989). Similarly, leukotriene metabolism and glycolysis, which were found to be significantly and specifically co-regulated in myocytes, are known to play a major role in triggering and fueling muscle contractions (Balsom et al., 1995; Setoguchi etal., 2001). Moreover, propionate metabolism, which was found to be significantly and specifically co-regulated in adipocytes, is known to control the lipid-buffering capacity of adipose tissue (Canfora et al., 2015). These findings strongly suggest that co-regulation strength is a good indicator of tissue-specific metabolism regulation.
Dysregulation of Fructose and Mannose Metabolism in Obese Subjects
After generating the detailed INs for healthy subjects, we next sought to determine how INs differed in obesity. To generate obese-specific INs, we obtained liver samples and three different adipose tissue depots, including subcutaneous, omental, and mesenteric, from 12 morbidly obese (BMI >40) subjects undergoing bariatric surgery. We then performed global gene expression profile analyses using RNA sequencing. The clinical characteristics of all of the subjects involved in our study are presented in Table 1.
Table 1.
Clinical Characteristics of the 12 Obese Subjects Undergoing Bariatric Surgery
| Clinical Variable | Obese Subjects |
|---|---|
| Number of subjects | 12 |
| Age (years) | 39.3 ± 10.9 |
| Weight (kg) | 122.9 ± 12.8 |
| BMI (kg/m2) | 43.6 ±3.6 |
| Fasting plasma glucose (mmol/l) | 5.6 ±0.6 |
| Fasting plasma insulin (FPI) (pmol/l) | 128.7 ±49.9 |
| HOMA-IR | 4.7 ± 1.9 |
| Plasma triglycerides (TG) (mmol/l) | 1.5 ±0.5 |
| Total cholesterol (mmol/l) | 5.1 ± 0.7 |
| LDL cholesterol (mmol/l) | 3.1 ± 0.7 |
| HDL cholesterol (mmol/l) | 1.3 ±0.3 |
| Alanine aminotransferase (ALT) (U/l) | 25.3 ± 16.3 |
| γ-Glutamyl transferase (μGT) (U/l) | 30.7 ±23.2 |
The data are presented as the mean ± SD.
As we did previously for the healthy subjects, we generated cell-specific TRNs and PPINs for the obese subjects by refining the reference networks using the mRNA expression levels of the genes in the liver and adipose tissue samples. Genes with low mRNA expression (FPKM <1) were excluded from the reference networks for the hepatocytes and adipocytes (Table S2). During the generation of adipocyte-specific networks for the obese subjects, we accounted for differences in the three adipose tissue depots, including subcutaneous, mesenteric, and omental fat (Figure S4A) and excluded genes from the reference networks only if the RNA expression level of the genes was low (FPKM <1) in all three adipose tissue samples in all of the obese subjects (Table S2).
We performed a co-regulation analysis for each metabolic pathway in the healthy and obese subjects based on the network topology provided by the cell-specific INs. Next, we compared their co-regulation strengths in the hepatocytes (Figure 3A) and adipocytes (Figure 3B) based on the number of co-regulators and identified the metabolic pathways for which co-regulation decreased or increased. We found that the co-regulation of fructose and mannose metabolism was significantly decreased (Kolmogorov-Smirnov [KS] one-sided test, p < 0.05) in the hepatocytes of the obese subjects (Figure 3A; Data S1B), whereas the co-regulation of propionate metabolism was slightly increased in the adipocytes of the obese subjects (Figure 3B; Data S1C).
Figure 3. Revealing Biological Processes Altered between Healthy and Obese Subjects.
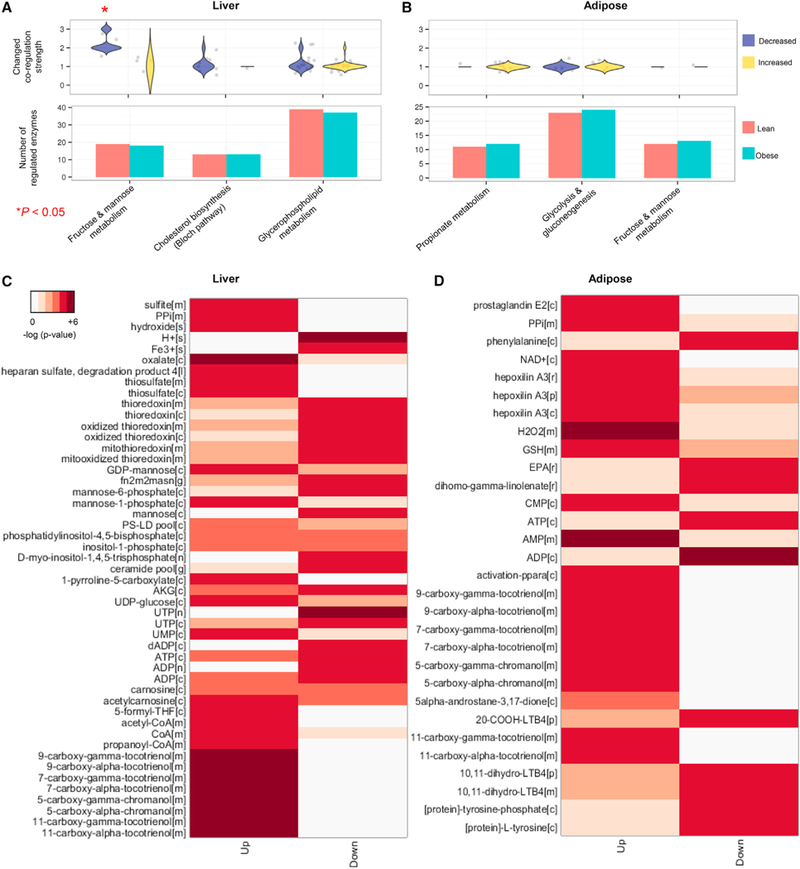
We refined the reference cell-specific networks using transcriptomic data from the obese subjects and compared the co-regulations of the metabolic pathways between the (A) hepatocytes and (B) adipocytes of the healthy and obese subjects. We also compared the number of regulated enzymes in the INs of hepatocytes and adipocytes from the healthy and obese subjects and did not observe significant differences. Among the significantly co-regulated metabolic pathways (Kolmogorov-Smirnov [KS] test, Bonferroni-adjusted p value < 0.05) in Figure 2G, we examined the changes in co-regulation strength between the healthy and obese subjects based on the number of co-regulators. We found that the highly co-regulated metabolism of fructose and mannose presented substantially decreased co-regulation in the hepatocytes (KS one-sided test p < 0.05), whereas propionate metabolism presented weakly increased co-regulation in the adipocytes. Reporter metabolites were identified in the (C) liver and (D) adipose tissues of the lean and obese subjects. Detailed metabolic differences between the lean and obese subjects were investigated through a comparative analysis of the gene expression profiles (RNA-seq) of the liver and adipose tissues and cell-specific genome-scale metabolic models (GEMs). P values for each reporter metabolite were calculated for the upregulated and downregulated genes.See also Figures S5 and S6 and Data S1.
Moreover, we compared the mRNA expression levels of the protein-coding genes from the human liver and subcutaneous adipose tissue samples obtained from 12 obese subjects (Table 1 ) with seven liver and five subcutaneous adipose tissue samples obtained from the healthy subjects (previously described in Uhlen et al., 2015). We first clustered the biological samples using the mRNA expression level of the protein-coding genes and found that the liver samples from the obese and healthy subjects as well as the adipose tissue samples from the obese and healthy subjects clustered together (Figure S4B). We identified differentially expressed genes (adjusted p value < 0.05) to reveal the global biological differences between the liver and subcutaneous adipose tissue of the obese and healthy subjects, and the results indicated that 6,496 genes in the liver tissue (Data S1D) and 1,298 genes in the subcutaneous adipose tissue (Data S1E) were differentially expressed. To gain greater insight into the biological processes altered in the liver and adipose tissue of the obese subjects relative to the healthy individuals, we performed a gene set analysis (GSA) for gene ontology (GO) biological process (BP) terms. We found that the gene sets that included the terms regulation of cellular metabolic process, post translational protein modification, negative regulation of biological process and immune response were associated with downregulated genes, whereas the metabolism-related BP terms, including lipid metabolic process, phospholipid metabolic process, phosphoinositide metabolic process, and fatty acid β-oxidation, were associated with upregulated genes in the liver tissue of the obese subjects (Figure S5). We also found that the gene sets that included the terms regulation of RNA metabolic process, regulation of cell proliferation, and positive regulation of biological process were associated with downregulated genes, whereas metabolism-related BP terms including carboxylic acid metabolic process and lipid metabolic process were associated with upregulated genes in the subcutaneous adipose tissue of the obese subjects (Figure S6). Taken together, our data indicated an increased activity in lipid metabolism in both the liver and fat in obese subjects as well as novel differences in pathways such as the immune response and RNA metabolism.
To evaluate the detailed metabolic differences between the lean and obese subjects, we identified reporter metabolites (Patil and Nielsen, 2005) using the differentially expressed genes in the liver and adipose tissue as well as the network topology provided by the cell-specific GEMs. Reporter metabolite analyses are used as statistical tests to determine whether a significant change has occurred in the expression of the genes surrounding a metabolite. We identified mannose, mannose-1-phosphate, mannose-6-phosphate, GDP-mannose involved in fructose, and mannose metabolism as well as other metabolites involved in inositol phosphate, glycerophospholipid, and vitamin E metabolism as reporter metabolites in the liver tissue (Figure 3C; Data S1F). We also identified the metabolites involved in leukotriene, vitamin E, and lipid metabolism as reporter metabolites in adipose tissue (Figure 3D; Data S1G).
Plasma Mannose Levels Are Increased in Response to Obesity
We observed that the co-regulation of the highly co-regulated metabolism of fructose and mannose was significantly decreased in the liver of the obese subjects compared with that of the lean subjects. Moreover, mannose, an essential hexose that is required for glycoprotein synthesis (Davis and Freeze, 2001), as well as other associated metabolites were identified as top-ranked reporter metabolites in the liver tissue of the obese subjects. We examined the expression of genes associated with the utilization of mannose and found significant (adjusted p value < 0.05) differences in the expression of these genes in the liver tissue of the obese subjects compared with that of the lean subjects (Figure 4A). The expression levels of HK1 and HK2, which convert mannose to mannose-6-phosphate, were significantly downregulated in the liver tissue of the obese subjects (Figure 4B). However, we found that the expression of enzymes involved in the metabolism of glucose into fructose and mannose (e.g., PMM1) was significantly upregulated. Notably, when we analyzed the co-regulations among these enzymes, we found that PMM1, PFKL, GMPPB, and GMPPA were highly co-regulated (Figure 4C) and the expression levels of these regulators were significantly changed (Figure 4D). Hence, we observed that there was a shift in the utilization of carbohydrates in the liver tissue of the obese subjects. Because the liver is one of the main organs responsible for plasma mannose consumption (Davis and Freeze, 2001), we hypothesized that due to the downregulation of genes responsible for mannose processing in the liver, the levels of mannose in the plasma would be increased.
Figure 4. Substrate Shifts in the Liver Tissue of the Obese Subjects.
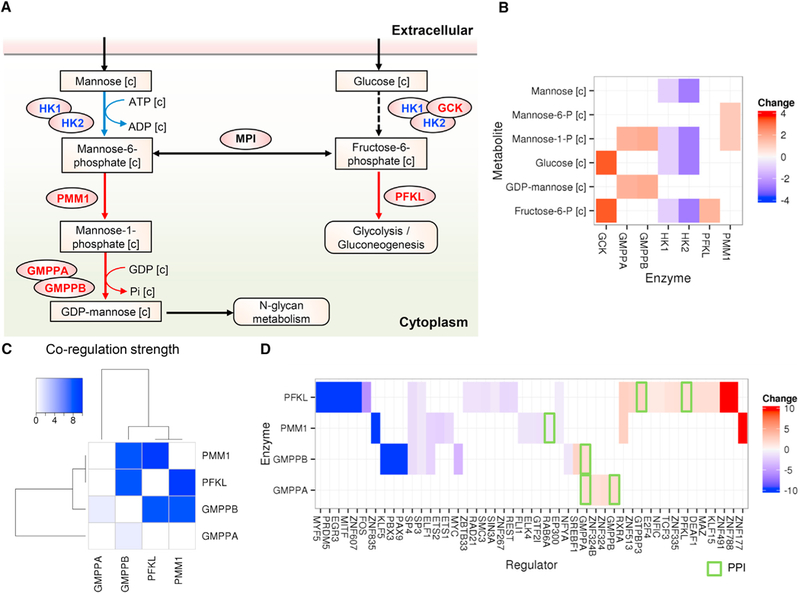
(A) We identified a metabolic pathway for the utilization of mannose and glucose in the liver tissue. Upregulated and downregulated enzymes and reactions in the obese subjects are represented as red and blue, respectively. The changed reactions implied that the consumption of mannose was decreased and the utilization of carbohydrates was altered (A).
(B) Fold changes of the corresponding enzyme expression levels.
(C and D) We found that the enzymes involved in mannose and glucose utilization were highly co-regulated with each other, and their regulators (TFs or proteins physically interacting with them [green rectangle]) were differentially expressed. Non-expressing regulators (FPKM <1) in the obese subjects or healthy subjects were identified as a maximal fold change of 10 or −10, respectively.
To evaluate our hypothesis, we detected the plasma levels of mannose, glucose, and fructose in 399 subjects involved in the Relationship between Insulin Sensitivity and Cardiovascular Disease (RISC) Study (Balkau et al., 2008; Hills et al., 2004). The RISC Study is being conducted in 19 European recruiting centers, and the insulin sensitivity (IS) of each subject is measured with the euglycemic clamp technique. The clinical characteristics of all of the lean, overweight, and obese subjects involved in the study are presented in Table 2. We measured the plasma mannose, fructose, and glucose levels in these subjects by liquid chromatography-mass spectrometry and found that the mannose and glucose plasma levels were significantly (p value < 0.05) higher in the obese subjects compared with the lean subjects, whereas significant changes were not observed in the fructose plasma levels (Figure 5A).
Table 2.
Clinical Characteristics of the Subjects Involved in the Studies
| Clinical variable | RISC Study | Leipzig Study | EUGENE2/Kuopio Study | ||||
|---|---|---|---|---|---|---|---|
| All Subjects | Lean 18.5< BMI< 25 | Obese 30 < BMI | Obese IS | Obese IR | High Dl | Low Dl | |
| Number of subjects (M/F) | 194/205 | 80/112 | 21/27 | 19/20 | 20/20 | 24/16 | 23/17 |
| Age (years) | 44.75 ± 8.22 | 43.8 ± 8.23 | 44.4 ± 8.42 | 51.36 ±9.43 | 51.53 ±9.67 | 35.5 ±6.01 | 35.85 ± 6.68 |
| Body mass index (kg/m2) | 25.51 ±4.09 | 22.4 ± 1.61 | 33.1 ± 2.77 | 45.13 ± 1.77 | 45.44 ± 1.42 | 26.16 ±4.19 | 26.70 ± 4.68 |
| Fasting plasma insulin (FPI) (pmol/l) | 35.96 ± 21.38 | 27.0 ± 14.8 | 66.0 ±27.7 | 31.11 ± 21.1 | 109.7 ±33.0 | 9.75 ± 4.84 | 8.19 ±3.72 |
| Fasting plasma glucose (mmol/l) | 5.16 ±0.58 | 5.01 ±0.50 | 5.32 ± 0.71 | 5.262 ± 0.31 | 5.748 ± 0.35 | 5.09 ±0.31 | 5.42 ± 0.49 |
| HOMA-IR | 1.50 ± 0.50 | 0.87 ± 0.53 | 2.26 ± 1.02 | 1.074 ±0.85 | 4.052 ± 1.28 | 2.22 ±1.14 | 2.01 ±1.01 |
| Fasting plasma mannose (relative) | - | 1.00 ±0.22 | 1.25 ±0.27 | 1.00 ±0.37 | 1.38 ±0.59 | 1.00 ±0.22 | 1.16 ±0.23 |
| Plasma triglycerides (mmol/l) | 1.13 ±0.83 | 0.93 ±0.51 | 1.42 ± 0.68 | 1.368 ± 0.55 | 2.067 ± 1.10 | 1.35 ±0.79 | 1.15 ±0.56 |
| Total cholesterol (mmol/l) | 4.86 ± 0.89 | 4.73 ± 0.85 | 5.17 ±0.95 | 5.153 ±0.93 | 5.284 ± 1.04 | 5.13 ±1.10 | 5.02 ± 0.78 |
| LDL cholesterol (mmol/l) | 2.94 ±0.80 | 2.76 ±0.74 | 3.31 ± 0.83 | 2.986 ± 0.85 | 3.196 ±0.82 | 3.33 ± 0.93 | 3.35 ± 0.68 |
| HDL cholesterol (mmol/l) | 1.41 ±0.38 | 1.53 ± 0.39 | 1.20 ±0.25 | 1.408 ±0.24 | 0.981 ±0.25 | 1.21 ±0.25 | 1.24 ±0.28 |
| Alanine aminotransferase (ALT) (μkat/l) | 0.39 ±0.18 | 0.34 ±0.15 | 0.49 ±0.21 | 0.450 ±0.17 | 0.505 ±0.13 | 32.30 ± 33.08 | 0.505 ±0.13 |
| γ-Glutamyl transferase (μGT) (μkat/l) | 0.35 ±0.13 | 0.31 ±0.19 | 0.46 ± 0.28 | 0.414 ±0.17 | 0.595 ± 0.20 | - | - |
Clinical characteristics of the lean, overweight, and obese subjects involved in the RISC study, obese subjects with and without insulin resistance involved in the Leipzig study, as well as the subjects with high and low disposition indices involved in the EUGENE2/Kuopio study. The data are presented as the mean ± SD.
Figure 5. Detection of Plasma Mannose Levels in Obese Subjects.
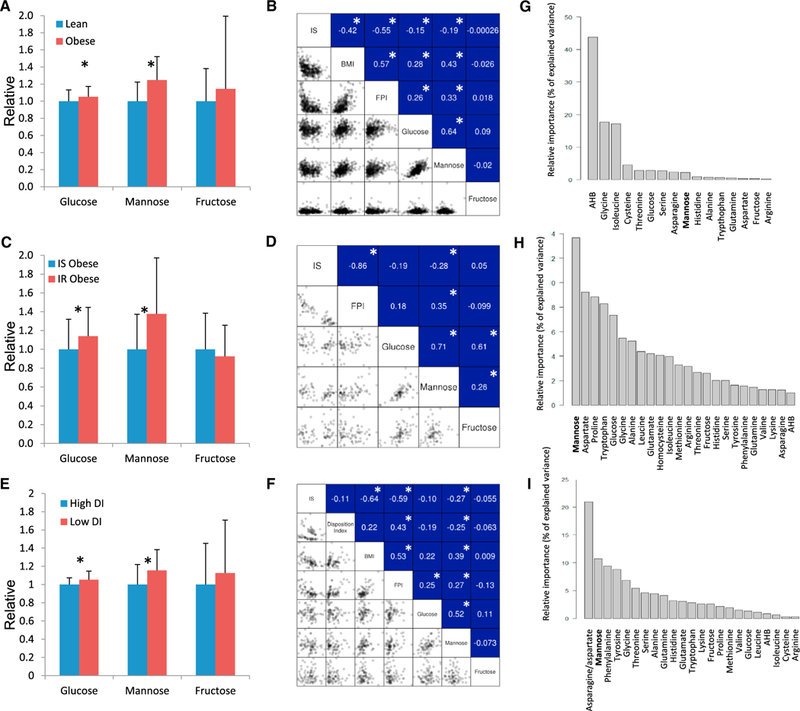
(A) We detected the plasma glucose, mannose, and fructose levels in the lean and obese subjects and found that glucose, mannose levels were significantly upregulated in the obese subjects compared with the lean subjects.
(B) We observed significant correlations between the BMI and the plasma mannose levels as well as the plasma glucose levels. We also calculated the correlations between insulin resistance (IR) and the plasma mannose levels as well as the plasma glucose levels and found significantly higher correlations between IR and the plasma mannose levels.
(C) We detected the plasma glucose, mannose, and fructose levels in the obese subjects with and without insulin resistance (IR). We found that the plasma glucose and mannose levels were significantly upregulated in the obese subjects with IR compared with the obese subjects with insulin sensitivity (IS).
(D) We observed significantly higher correlations between IR and the plasma mannose levels relative to the correlations between IR and the plasma glucose levels.
(E) We detected the plasma glucose, mannose, and fructose levels in the subjects with varying disposition indices (DIs). We found that the plasma glucose and mannose levels were significantly upregulated in the subjects with low DIs compared with the subjects with high DIs.
(F) We observed significantly correlations between IS and DIs and the plasma mannose levels and did not observe correlations between the DI or IS and glucose or fructose in this subject group.
(G–I) The results of the relative importance analysis that has been performed to estimate how much various plasma metabolite levels contributed in explaining the variance in IS. Plasma measurements of mannose; glucose, fructose, and 20aminoacids (available in the datasets) as well as a-hydroxybutyrate (AHB) were used in the analysis and the importance of mannose in (G) lean and obese subjects (H) obese subjects with IR and IS (I) in subjects with varying DIs was presented.
Moreover, we calculated Spearman’s correlation coefficient (r) between the BMI and plasma mannose levels as well as the glucose levels in 399 subjects and found that the BMI was significantly correlated with the plasma mannose levels (r = 0.43, p value < 0.05) and plasma glucose levels (r = 0.28, p value < 0.05), whereas significant correlations were not observed with the plasma fructose levels (Figure 5B). Considering the negative correlation between the BMI and IS (r = −0.42, p value < 0.05) (Figure 5B), we also examined the correlations between IS and the plasma mannose and glucose levels. Our correlation analysis indicated that slightly lower negative correlations occurred between the IS and the mannose levels (r = −0.19, p value <0.05) relative to the IS and the glucose levels (r = −0.15, p value < 0.05) (Figure 5B).
Moreover, we found significantly high correlations between glucose and mannose (r = 0.64, p value < 0.05). To eliminate the effect of glucose, we performed partial correlation analysis, which can control confounding variables’ effect (i.e., glucose) removed within ordinary correlation analysis. Partial correlation analysis showed that mannose was significantly correlated with both BMI (r = 0.34, p value <0.05) and IS (r = −0.13, p value < 0.05) without the effect of glucose in the RISC study (399 lean and obese subjects). In contrast, we found that glucose is not significantly correlated with BMI and IS after eliminating the effect of mannose (p values = 0.83 and 0.44, respectively).
Plasma Mannose Levels Are Associated with Insulin Resistance
We observed a significantly positive correlation between the plasma mannose levels and BMI and a significantly negative correlation between the plasma mannose levels and IS. To reveal the distinct association between the plasma mannose levels and IS without the effect of BMI, we recruited an additional 79 male and female obese patients involved in the Leipzig study (Klöting et al., 2010). The IS of each subject involved in the study was measured with the euglycemic clamp technique, and the clinical characteristics of all of the obese subjects involved in the study are presented in Table 2. We separated the obese subjects into two groups based on their IS using previously described criteria (Klöting et al., 2010) and then measured their plasma glucose, mannose, and fructose levels. We found that plasma mannose and glucose levels were significantly higher in the IR obese subjects compared with the IS obese subjects, whereas we did not detect significant differences in the plasma fructose levels between the two groups of obese subjects (Figure 5C).
We calculated the correlations between the IS and plasma mannose, fructose, and glucose levels. We observed that the IS was negatively correlated with the plasma mannose level (r = −0.28, p value < 0.05), whereas significant correlations were not observed between the IS and plasma glucose and fructose levels in the obese subjects (Figure 5D). After eliminating the effect of glucose, we found that mannose is significantly correlated with IS (r = −0.20, p value < 0.05), whereas glucose is not significantly correlated with IS after eliminating the effect of mannose (p value = 0.95). Hence, we observed that the plasma mannose levels could be used to identify obese subjects with high risk factors.
Plasma Mannose Levels Are Associated with Insulin Secretion
We also detected the plasma levels of mannose, glucose, and fructose in another independent cohort of 80 male and female subjects involved in the population-based EUGENE2/Kuopio Study, where the phenotype of individuals with impaired fasting glucose and/or impaired glucose tolerance with regard to insulin secretion and IR was examined (Laakso et al., 2008). Evaluations of insulin secretion (InsAUC0–30/GlucAUC0–30) and IS have been previously described (Matsuda and DeFronzo, 1999; Stancáková et al., 2009). The subjects presented varying disposition indices (DIs), which were calculated as the product of the indices of IS and insulin secretion (InsAUC0–30/GlucAUC0–30). The characteristics of the study participants are presented in Table 2.
We classified the patients as low or high DI and found that the plasma glucose and mannose levels were significantly higher in the subjects with low DI relative to the subjects with high DI. Significant changes were not observed in the plasma fructose levels (Figure 5E). Moreover, we calculated the correlations (r) between the BMI, IS, DI, and plasma mannose, glucose, and fructose levels and found that the plasma mannose level was significantly correlated with the IS (r = −0.27, p value <0.05) and DI (r = −0.25, p value < 0.05) (Figure 5F). In addition, significant correlations were not observed between the Dl or IS and glucose or fructose in this subject group. We finally observed that mannose is significantly correlated with BMI (r = 0.34, p value < 0.05) and IS (r = −0.25, p value < 0.05) after eliminating the effect of glucose whereas glucose is not significantly correlated with BMI or IS after eliminating the effect of mannose (p values = 0.89 and 0.72, respectively).
The Use of Plasma Mannose Level in Explaining the Variance in IR
We performed relative importance analysis (Lindeman et al., 1980) to estimate how much various plasma metabolite levels contributed in explaining the variance in IR (as measured with the euglycemic clamp technique). During the analysis, we included the plasma measurements of mannose, glucose, fructose, and 20 amino acids (available in the datasets) as well as a-hydroxybutyrate (AHB). The plasma levels of some amino acids, including branched-chain amino acids, leucine, isoleucine, and valine, have already been associated with IR (Newgard et al., 2009). AHB has also been proposed as an early biomarker of IR and glucose intolerance in a nondiabetic subjects (Gall et al., 2010). The contributions of the metabolites were summed up to 100 and divided up per metabolite so that the relative contribution to explain the variance in IR (as percentage of the total explained variance) is displayed in lean and obese subjects (Figure 5G) in obese subjects with IR and IS (Figure 5H) and in subjects with varying DIs and matched BMI (Figure 5i). We found that mannose is the best metabolite whose plasma level can be used in explaining the variance of IR in subject groups with matched BMI (Figures 5H and 5I).
Plasma levels of mannose, glucose, and other metabolites allowed us to compare not only mannose and glucose levels, but also the level other potential markers of IR. Considering that plasma mannose levels outperformed the level of metabolites that have been associated with IR, we suggested to use of plasma mannose levels in stratification of the obese subjects with high risk factors independent of BMI.
DISCUSSION
Recent advances in sequencing technologies and methods for genomic analyses (e.g., digital genomic foot printing) have allowed for the massive profiling of in vivo regulatory events in human cells and tissues. In our study, we examined the regulatory events in hepatocytes, myocytes, and adipocytes and generated cell-specific TRNs. We integrated the TRNs with GEMs and PPINs and eventually generated cell-specific INs to investigate the biological processes that are altered in the patients presenting obesity and IR based on the network topology provided by the networks. We refined the topology of the cell-specific INs using the cell-specific antibody proteomic and tissue-specific transcriptomic data in the HPA (Uhlén et al., 2015). Moreover, we generated RNA-seq data from the liver samples and three different adipose tissue depots obtained from the obese subjects and used these data in the generation of INs for the obese subjects. We performed a comparative analysis between the lean and obese subjects to reveal the biological processes that are altered in response to obesity and validated our predictions by performing a metabolomic analysis in three independent cohorts of subjects.
The power of the IN strategy allowed us to take a more complete look at the role metabolism plays in obesity across multiple tissues. We compared the cell-specific INs between lean and obese subjects and investigated the biological differences in response to obesity. The integration of complex biological networks enables the elucidation of metabolic adaptations as well as other hallmarks of IR. We found that the co-regulation of fructose and mannose metabolism was decreased in the obese subjects, and metabolites involved in fructose and mannose metabolism were identified as a reporter metabolite. This finding implies that mannose may be used as a marker in identification of obese subjects with high risk factors, e.g., insulin resistance and secretion. We also found a significant downregulation of mannose phosphorylation (HK1 and HK2) in the liver tissue, which may lead to decreased plasma mannose utilization in the liver tissue. The downregulation of HK1 and HK2 might also lead to decreased glucose utilization. However, this decrease could be compensated for by the upregulation of glucose-specific phosphorylase GCK in obese patients. GCK is an enzyme with a relatively high half-saturated concentration (KM) (GCK = ~10 mM) compared with other hexokinases (~0.1 mM) (Ahn et al., 2009; Magnani et al., 1988; Xu et al., 1995). Thus, the utilization of glucose in the liver tissue was not decreased in the obese patients as previously reported (Caro et al., 1984). This finding can help explain why mannose had better correlation with IR than glucose.
The liver has been reported as the main organ for mannose consumption (Davis and Freeze, 2001). Therefore, altered mannose utilization in the liver tissue could lead to differences in plasma mannose levels. To validate our predictions, we first measured the plasma mannose levels in the lean and obese subjects and found that the plasma mannose levels were significantly upregulated in the obese subjects. Alterations in mannose metabolism may affect glycoprotein synthesis and secretion since mannose is the main source of glycoprotein synthesis (Panneerselvam et al., 1997; Panneerselvam and Freeze, 1996). Abnormal glycosylation could also affect the insulin receptors in the liver tissue and increase the resistance of liver tissue to insulin (Caro et al., 1984). Therefore, we speculated that the plasma mannose levels could potentially play a supplementary role in the development of IR. In accordance with this hypothesis, we found that the plasma mannose levels were significantly negatively correlated with the IS of lean and obese subjects. In addition, we investigated whether plasma mannose level was significantly different between IR and IS independent of BMI and measured the plasma mannose levels of 79 obese subjects classified into two groups based on their IR and IS. We found that the plasma mannose levels were significantly higher in the IR obese subjects compared with the IS obese subjects, and this level was significantly negatively correlated with the patient’s IS. We also measured the plasma mannose levels of 80 subjects classified into two BMI matched groups based on their DI. We found that the plasma mannose levels were significantly higher in the subjects with low DI compared with the subjects with high DI, and this level was significantly negatively correlated with the patient’s DI and IS. Finally, we compared the plasma mannose levels with the levels of glucose, fructose, AHB and amino acids in three independent cohorts of subjects and found that mannose outperformed most other metabolites across a variety of conditions in explaining the variance in obesity-independent insulin resistance.
It has been previously shown that plasma mannose levels correlate with glucose levels but that the level of mannose is unlike glucose not different between fasting and postprandial state (Sone et al., 2003). This observation coupled with the closer correlation of mannose to IR could indicate that using plasma mannose levels instead of glucose as a marker of IR in the clinic could have the potential of both improving the accuracy of assessing IR and being independent of meal-timing. In conclusion, through the integration of biological networks, we revealed the biological processes altered in response to obesity and observed an association between plasma mannose levels and BMI as well as insulin sensitivity and secretion. Moreover, we demonstrated a strategy for how cell-specific INs can be employed to identify dysregulation of biological functions in response to a disease, to reveal the consequences on relevant metabolites in plasma and eventually to identify new candidate disease biomarkers.
EXPERIMENTAL PROCEDURES
Identification of Genomic Footprints from DNase-Seq Data
We retrieved DNase-seq data from the hepatocytes of liver tissue (two samples), myocytes of skeletal muscle tissue (three samples), adipocytes of subcutaneous adipose tissue (four samples) and stellate cells (two samples), skeletal myoblasts (three samples), and myotubes (two samples) from the ENCODE repository (ENCODE Project Consortium, 2012) (Table S1). We mapped the DNase-seq data with the unaligned reads (i.e., FASTQ files) to the reference human genome (build GRCh37/hg19) by Bowtie (ver. 1.1.1) (Lang-mead et al., 2009) as previously described by the ENCODE Consortium (Neph et al., 2012c) (Figure S1A). We verified the quality of the DNase-seq samples using SPOT scores that were calculated by the HOTSPOT program (John et al., 2011) and found that all of the samples had SPOT scores >0.4 (average 0.608 per sample) (Table S1). After counting the DNase I cleavages per base from the mapped reads, we identified genomic footprints with their footprint occupancy scores (FOS), and a low FOS score indicated a strong footprint site. We found optimized footprints with a 6–40 base pair (bp) central component and 3–10 bp flanking component. Finally, we selected footprints within highly DNase I-sensitive regions (hotspot regions) as identified by the HOTSPOT program (false discovery rate [FDR] <0.01) (John et al., 2011) and selected the overlapping footprints with hotspot regions using the BEDOPS program (Neph etal., 2012a) (see the Supplemental Experimental Procedures).
Transcript Profiling—RNA-Seq—of Liver and Adipose Tissue Samples
Patients had low calorie diet weight reduction run in prior to the day of surgery. The human liver and subcutaneous, omental, and mesenteric tissue samples were obtained from 12 obese subjects undergoing bariatric surgery and then used for the mRNA expression analyses. The tissue samples were collected and handled in accordance with the laws and regulations of the Netherlands as part of the sample collection. All of the human samples used in the present study were anonymized in accordance with the approval and advisory report from the Ethical Review Board.
A total of 48 samples from the four tissue types of the obese subjects were sequenced using Illumina HiSeq 2000 and HiSeq 2500 systems with the standard Illumina RNA-seq protocol. The healthy samples included seven liver samples and five adipose (subcutaneous) samples from the lean subjects (Fagerberg et al., 2014; Uhlén et al., 2015), and they were compared to the 12 liver samples and 36 adipose (12 subcutaneous, 12 mesenteric, and 12 omental) samples from the obese subjects. Consequently, the fragments per kilobase of exon model per million mapped reads (FPKM) values were obtained for 19,709 genes for all samples. Throughout this study, the genes with an expression level of FPKM <1 were considered to have no expression.
A differential expression (DE) analysis was performed following a standard protocol using the differential expression sequencing (DESeq) v2.0 package (Anders and Huber, 2010). In brief, the DE analysis was performed between obese and lean tissue (obese liver versus lean liver and obese adipose [subcutaneous] versus lean adipose [subcutaneous]). In this study, expression differences with a p value < 0.05 (after Bonferroni correction) were regarded as significantly changed.
Detection of Plasma Metabolite Levels
Measurement of plasma levels of glucose, mannose, fructose, amino acids, and AHB was performed. Briefly, the liquid chromatography-tandem mass spectrometry (LC-MS/MS) platform was based on a Waters ACQUITY ultra-performance liquid chromatography (UPLC) system and a Thermo-Finnigan LTQ mass spectrometer operated at nominal mass resolution, which was equipped with an electrospray ionization (ESI) source and a linear ion trap (LIT) mass analyzer.
Supplementary Material
Highlights.
We generated cell-specific integrated networks for lean and obese subjects
We found dysregulations in the mannose metabolism in obese subjects
Plasma mannose level was associated with insulin resistance independent of BMI
Mannose is used in explaining the variance in obesity-independent insulin resistance
ACKNOWLEDGMENTS
This work was financially supported by the Knut and Alice Wallenberg Foundation, Bill and Melinda Gates Foundation, Swedish Research Foundation, and EU Seventh Framework Programme RESOLVE. The research leading to these results has received support from the Innovative Medicines Initiative Joint Undertaking under EMIF grant agreement 115372. The computations of network generations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at C3SE and UPPMAX.
Footnotes
ACCESSION NUMBERS
The accession number for the raw and processed RNA-seq data reported in this paper is available at Gene Expression Omnibus (GEO): GSE83322. The networks generated in this study are also publicly available at http://sysmedicine.com/integratednetworks.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, four tables, and one data file and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2016.05.026.
REFERENCES
- Ahn KJ, Kim J, Yun M, Park JH, and Lee JD (2009). Enzymatic properties of the N-and C-terminal halves of human hexokinase II. BMB Rep. 42, 350–355. [DOI] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkau B, Mhamdi L, Oppert JM, Nolan J, Golay A, Porcellati F, Laakso M, and Ferrannini E; EGIR-RISC Study Group (2008). Physical activity and insulin sensitivity: the RISC study. Diabetes 57, 2613–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsom PD, Söderlund K, Sjödin B, and Ekblom B (1995). Skeletal muscle metabolism during short duration high-intensity exercise: influence of creatine supplementation. Acta Physiol. Scand. 154, 303–310. [DOI] [PubMed] [Google Scholar]
- Björnson E, Mukhopadhyay B, Asplund A, Pristovsek N, Cinar R, Romeo S, Uhlen M, Kunos G, Nielsen J, and Mardinoglu A (2015).Stratification of hepatocellular carcinoma patients based on acetate utilization. Cell Rep. 13, 2014–2026. [DOI] [PubMed] [Google Scholar]
- Bordbar A, Monk JM, King ZA, and Palsson BO (2014). Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 15, 107–120. [DOI] [PubMed] [Google Scholar]
- Canfora EE, Jocken JW, and Blaak EE (2015). Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 11, 577–591. [DOI] [PubMed] [Google Scholar]
- Caro JF, Cecchin F, and Sinha MK (1984). Is glycosylation in the liver needed for insulin binding, processing, and action? Evidence for heterogeneity. J. Biol. Chem. 259, 12810–12816. [PubMed] [Google Scholar]
- Cerami EG, Gross BE, Demir E, Rodchenkov I, Babur O, Anwar N, Schultz N, Bader GD, and Sander C (2011). Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 39, D685–D690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford GE, Holt IE, Whittle J, Webb BD, Tai D, Davis S, Margulies EH, Chen Y, Bernat JA, Ginsburg D, et al. (2006). Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res. 16, 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JA, and Freeze HH (2001). Studies of mannose metabolism and effects of long-term mannose ingestion in the mouse. Biochim. Biophys. Acta 1528,116–126. [DOI] [PubMed] [Google Scholar]
- De Las Rivas J, and Fontanillo C (2010). Protein-protein interactions essentials: key concepts to building and analyzing interactome networks. PLoS Comput. Biol. 6, e1000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall WE, Beebe K, Lawton KA, Adam KP, Mitchell MW, Nakhle PJ, Ryals JA, Milburn MV, Nannipieri M, Camastra S, et al. ; RISC Study Group (2010). alpha-Hydroxybutyrate is an early biomarker of insulin resistance and glucose intolerance in a nondiabetic population. PLoS ONE 5, e10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et al. (2012). Architecture of the human regulatory network derived from ENCODE data. Nature 489, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hills SA, Balkau B, Coppack SW, Dekker JM, Mari A, Natali A, Walker M, and Ferrannini E; EGIR-RISC Study Group (2004). The EGIR-RISC STUDY (The European group forthe study of insulin resistance: relationship between insulin sensitivity and cardiovascular disease risk): I. Methodology and objectives. Diabetologia 47, 566–570. [DOI] [PubMed] [Google Scholar]
- Hyötylainen T, Jerby L, Petäjä EM, Mattila I, Jäntti S, Auvinen P, Gastaldelli A, Yki-Järvinen H, Ruppin E, and Orešič M (2016). Genomescale study reveals reduced metabolic adaptability in patients with non-alcoholic fatty liver disease. Nat. Commun. 7, 8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, Hager GL, and Stamatoyannopoulos JA (2011). Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 43, 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klöting N, Fasshauer M, Dietrich A, Kovacs P, Schön MR, Kern M, Stumvoll M, and Blüher M (2010). Insulin-sensitive obesity. Am. J. Physiol. Endocrinol. Metab. 299, E506–E515. [DOI] [PubMed] [Google Scholar]
- Laakso M, Zilinskaite J, Hansen T, Boesgaard TW, Vänttinen M, Stancáková A, Jansson PA, Pellme F, Holst JJ, Kuulasmaa T, et al. ; EUGENE2 Consortium (2008). Insulin sensitivity, insulin release and glucagon-like peptide-1 levels in persons with impaired fasting glucose and/ or impaired glucose tolerance in the EUGENE2 study. Diabetologia 51, 502–511. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang W, and Chen J (2015). From pathways to networks: connecting dots by establishing protein-protein interaction networks in signaling pathways using affinity purification and mass spectrometry. Proteomics 15, 188–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeman RH, Merenda PF, and Gold RZ (1980). Introduction to Bivariate and Multivariate-Analysis (The University of Michigan; ). [Google Scholar]
- Magnani M, Stocchi V, Serafini G, Chiarantini L, and Fornaini G (1988). Purification, properties, and evidence for two subtypes of human placenta hexokinase type I. Arch. Biochem. Biophys. 260, 388–399. [DOI] [PubMed] [Google Scholar]
- Mardinoglu A, and Nielsen J (2012). Systems medicine and metabolic modelling. J. Intern. Med. 271, 142–154. [DOI] [PubMed] [Google Scholar]
- Mardinoglu A, and Nielsen J (2015). New paradigmsfor metabolic modeling of human cells. Curr. Opin. Biotechnol. 34, 91–97. [DOI] [PubMed] [Google Scholar]
- Mardinoglu A, Agren R, Kampf C, Asplund A, Nookaew I, Jacobson P, Walley AJ, Froguel P, Carlsson LM, Uhlen M, and Nielsen J (2013). Integration of clinical data withagenome-scale metabolic model ofthe human adipocyte. Mol. Syst. Biol. 9, 649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardinoglu A, Agren R, Kampf C, Asplund A, Uhlen M, and Nielsen J (2014a). Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 5, 3083. [DOI] [PubMed] [Google Scholar]
- Mardinoglu A, Kampf C, Asplund A, Fagerberg L, Hallström BM, Edlund K, Blüher M, Pontén F, Uhlen M, and Nielsen J (2014b). Defining the human adipose tissue proteome to reveal metabolic alterations in obesity. J. Proteome Res. 13, 5106–5119. [DOI] [PubMed] [Google Scholar]
- Mardinoglu A, Heiker JT, Gärtner D, Bjornson E, Schon MR, Flehmig G, Klöting N, Krohn K, Fasshauer M, Stumvoll M, et al. (2015a). Extensiveweight loss reveals distinct gene expression changes in human subcutaneous and visceral adipose tissue. Sci. Rep. 5, 14841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardinoglu A, Shoaie S, Bergentall M, Ghaffari P, Zhang C, Larsson E, Bäckhed F, and Nielsen J (2015b). The gut microbiota modulates host amino acid and glutathione metabolism in mice. Mol. Syst. Biol. 11, 834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda M, and DeFronzo RA (1999). Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 22, 1462–1470. [DOI] [PubMed] [Google Scholar]
- Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK, Rynes E, Maurano MT, Vierstra J, Thomas S, et al. (2012a). BEDOPS: high-performance genomic feature operations. Bioinformatics 28, 1919–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, and Stamatoyannopoulos JA (2012b). Circuitry and dynamics of human transcription factor regulatory networks. Cell 150, 1274–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK, et al. (2012c). An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, et al. (2009). A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 9, 311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien EJ, Monk JM, and Palsson BO (2015). Using genome-scale models to predict biological capabilities. Cell 161, 971–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S, Kerrien S, Abbani S, Aranda B, Bhate J, Bidwell S, Bridge A, Briganti L, Brinkman FS, Cesareni G, et al. (2012). Protein interaction data curation: the International Molecular Exchange (IMEx) consortium. Nat. Methods 9, 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panneerselvam K, and Freeze HH (1996). Mannose corrects altered N-glycosylation in carbohydrate-deficient glycoprotein syndrome fibroblasts. J. Clin. Invest. 97, 1478–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panneerselvam K, Etchison JR, and Freeze HH (1997). Human fibroblasts prefer mannose over glucose as a source of mannose for N-glycosylation. Evidence for the functional importance of transported mannose. J. Biol. Chem. 272, 23123–23129. [DOI] [PubMed] [Google Scholar]
- Patil KR, and Nielsen J (2005). Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc. Natl. Acad. Sci. USA 702, 2685–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechenick DA, Payne JL, and Moore JH (2014). Phenotypic robustness and the assortativity signature of human transcription factor networks. PLoS Comput. Biol. 70, e1003780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland T, Tasan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, et al. (2014).Aproteome-scalemap of the human interactome network. Cell 759, 1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setoguchi H, Nishimura J, Hirano K, Takahashi S, and Kanaide H (2001). Leukotriene C(4) enhances the contraction of porcine tracheal smooth muscle through the activation ofY-27632, a rho kinase inhibitor, sensitive pathway. Br. J. Pharmacol. 732, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoaie S, Ghaffari P, Kovatcheva-Datchary P, Mardinoglu A, Sen P, Pujos-Guillot E, de Wouters T, Juste C, Rizkalla S, Chilloux J, et al. ; MICRO-Obes Consortium (2015). Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 22, 320–331. [DOI] [PubMed] [Google Scholar]
- Snider J, Kotlyar M, Saraon P, Yao Z, Jurisica I, and Stagljar I (2015). Fundamentals of protein interaction network mapping. Mol. Syst. Biol. 77,848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sone H, Shimano H, Ebinuma H, Takahashi A, Yano Y, Iida KT, Suzuki H, Toyoshima H, Kawakami Y, Okuda Y, et al. (2003). Physiological changes in circulating mannose levels in normal, glucose-intolerant, and diabetic subjects. Metabolism 52, 1019–1027. [DOI] [PubMed] [Google Scholar]
- Stancáková A, Javorsky M, Kuulasmaa T, Haffner SM, Kuusisto J, and Laakso M (2009). Changes in insulin sensitivity and insulin release in relation to glycemia and glucose tolerance in 6,414 Finnish men. Diabetes 58, 1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijburg LB, Geelen MJ, and van Golde LM (1989). Regulation of the biosynthesis of triacylglycerol, phosphatidylcholine and phosphatidylethanolamine in the liver. Biochim. Biophys. Acta 7004, 1–19. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Hallström BM, Lindskog C, Mardinoglu A, Pontén F, and Nielsen J (2016). Transcriptomics resources of human tissues and organs. Mol. Syst. Biol. 72, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Väremo L, Scheele C, Broholm C, Mardinoglu A, Kampf C, Asplund A, Nookaew I, Uhlén M, Pedersen BK, and Nielsen J (2015). Proteome-and transcriptome-driven reconstruction of the human myocyte metabolic network and its use for identification of markers for diabetes. Cell Rep. 77, 921–933. [DOI] [PubMed] [Google Scholar]
- Xu LZ, Harrison RW, Weber IT, and Pilkis SJ (1995). Human beta-cell glucokinase. Dual role of Ser-151 in catalysis and hexose affinity. J. Biol. Chem. 270, 9939–9946. [DOI] [PubMed] [Google Scholar]
- Yizhak K, Gabay O, Cohen H, and Ruppin E (2013). Model-based identification of drug targets that revert disrupted metabolism and its application to ageing. Nat. Commun. 4, 2632. [DOI] [PubMed] [Google Scholar]
- Yizhak K, Gaude E, Le Dévédec S, Waldman YY, Stein GY, van de Water B, Frezza C, and Ruppin E (2014a). Phenotype-based cell-specific metabolic modeling reveals metabolic liabilities of cancer. eLife 3, e03641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhak K, Le Devedec SE, Rogkoti VM, Baenke F, de Boer VC, Frezza C, Schulze A, van de Water B, and Ruppin E (2014b). A computational study of the Warburg effect identifies metabolic targets inhibiting cancer migration. Mol. Syst. Biol. 70, 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Ji B, Mardinoglu A, Nielsen J, and Hua Q (2015). Logical transformation of genome-scale metabolic models for gene level applications and analysis. Bioinformatics 37, 2324–2331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.