

Abstract
Nervous system functions in all animals rely upon synaptic connectivity that is established during early development. Whereas cell–cell signaling plays a critical role in establishing synapse specificity, the involvement of extrinsic growth factors cannot, however, be undermined. We have previously demonstrated that trophic factors are required for excitatory but not inhibitory synapse formation between Lymnaea neurons. Moreover, in the absence of trophic factors, neurons from a number of species establish inappropriate inhibitory synapses, which can, however, be corrected by the addition of trophic factors. The precise site of trophic factor actions (presynaptic versus postsynaptic) and the underlying mechanisms remain, however, undefined. Here, we provide the first direct evidence that the trophic factor-mediated excitatory synapse formation involves activity-induced calcium (Ca2+) oscillations in the postsynaptic left pedal dorsal 1 (LPeD1) but not the presynaptic visceral dorsal 4 (VD4, cholinergic) neuron. These oscillations involved Ca2+ influx through voltage-gated Ca2+ channels and required receptor tyrosine kinase activity which was essential for the expression of excitatory, nicotinic acetylcholine receptors in the postsynaptic cell during synapse formation. We also demonstrate that selectively blocking the electrical activity presynaptically did not perturb trophic factor-induced synapse formation between the paired cells, whereas hyperpolarizing the postsynaptic cell prevented appropriate synaptogenesis between VD4 and LPeD1 cells. Together, our data underscore the importance of extrinsic trophic factors in regulating the electrical activity of the postsynaptic but not the presynaptic cell and that the resulting Ca2+ oscillations are essential for the expression of postsynaptic receptors during specific synapse formation.
Keywords: trophic factors, synapse formation, electrical activity, intracellular Ca2+, nAChRs, Lymnaea
Introduction
Synapse formation, an essential step in the development of a functional nervous system, involves interactions between specialized machineries of the presynaptic and postsynaptic partners. Although some components of the synaptic machinery are preassembled before contact between synaptic partners (Haydon and Drapeau, 1995; Friedman et al., 2000; Munno and Syed, 2003; Meems and Syed, 2006), both the cell–cell signaling (Scheiffele, 2003) and extrinsic molecules (Vicario-Abejón et al., 1998; Craig et al., 2006) present in the extracellular milieu are deemed necessary for the establishment, maturation and remodeling of synapses. Studies have found that intrinsic cell–cell interactions that involve activity-dependent mechanisms are required for specific synapse formation (Shatz, 1990; Katz and Shatz, 1996; Dan and Poo, 2004). Similarly, extrinsic trophic factors have also been reported to regulate synapse formation and synaptic plasticity (Martínez et al., 1998; Vicario-Abejón et al., 1998; Hamakawa et al., 1999). In contrast to cell–cell signaling, however, the precise site of trophic factor actions (presynaptic versus postsynaptic) and the underlying mechanisms (activity dependent versus independent) remain poorly defined.
We have adopted the soma–soma synapse model where synapses between two cholinergic neurons can be reconstructed between the somata in the absence of neurite outgrowth. The soma–soma synapses between Lymnaea neurons are morphologically and electrophysiologically similar to those seen in vivo (Feng et al., 1997). Moreover, the soma–soma synapses are target cell contact specific and require new gene transcription and de novo protein synthesis. Interestingly, we discovered that synapse formation between excitatory (Hamakawa et al., 1999; Woodin et al., 1999) but not the inhibitory (Feng et al., 1997) partners required extrinsic trophic factors present in the brain-conditioned medium (CM). These effects were mimicked by purified, Lymnaea epidermal growth factors (LEGFs) and were mediated through receptor tyrosine kinases (RTKs) (Hamakawa et al., 1999; Meems et al., 2003). Moreover, in the absence of appropriate trophic factors, Lymnaea neurons establish inappropriate (not seen in vivo) inhibitory synapses, which can, however, be restored to appropriate excitatory synapses after the addition of trophic factors. However, neither the precise site of trophic factor action nor the underlying mechanisms have yet been defined.
Here, we demonstrate that the trophic factor-induced excitatory synapse formation between Lymnaea presynaptic neuron visceral dorsal 4 (VD4) and its postsynaptic partner left pedal dorsal 1 (LPeD1) involves activity-mediated Ca2+ influx in LPeD1 only. Specifically, we show that within minutes of CM perfusion, the spontaneous activity in the postsynaptic cell was significantly enhanced, which in turn gave rise to spontaneous Ca2+ oscillations mediated through voltage-gated Ca2+ channels (VGCCs) and required RTK activity. These postsynaptic Ca2+ oscillations were essential for the expression of excitatory, nicotinic acetylcholine receptors (nAChRs), which required de novo protein synthesis but not new gene transcription. Together, our data underscore the importance of trophic factor-induced changes during synapse formation in the postsynaptic cell, independent of presynaptic neuron. These changes were initiated by Ca2+ oscillations, which subsequently triggered a cascade of protein synthesis-dependent events leading to the expression of appropriate excitatory postsynaptic receptors.
Materials and Methods
Animals and cell culture.
Laboratory-raised stock of the freshwater snail Lymnaea stagnalis were maintained in well aerated aquarium at room temperature (20–22°C) and fed lettuce. Snails 10–15 mm in length (1–2 months old) were used for experiments involving cell isolation, whereas older animals, 20–30 mm in length (2–4 months old), were used to prepare the CM. Detailed cell isolation and cell culture procedures have previously been described previously (Syed et al., 1990; Ridgway et al., 1991). In brief, the animals were first deshelled and anesthetized with 10% Listerine solution (ethanol, 21.9%; and methanol, 0.042%) in normal Lymnaea saline (in mm: 51.3 NaCl, 1.7 KCl, 4.0 CaCl2, and 1.5 MgCl2) buffered to pH 7.9 with HEPES. The central ring ganglia were then removed and washed with normal saline containing antibiotic (gentamicin, 50 μg/ml) three times (10 min each). The central ring ganglia were then treated with trypsin (2 mg/ml) dissolved in defined medium (DM; L-15; Life Technologies; special order), for 23 min at room temperature. The action of enzyme was stopped by replacing trypsin with trypsin inhibitor (type I-S; 2 mg/ml). After enzymatic treatment, the central ring ganglia were pinned down into a dissection dish containing high osmolarity DM (by adding 750 μl of 1 m glucose to 20 ml DM; raise normal osmolarity of the DM from 130–145 to 180–195 mOsm).
Identified, respiratory neurons VD4 and LPeD1 were isolated by applying gentle suction through a fire-polished and Sigmacote (Sigma)-treated pipette. Isolated cells were then plated onto poly-l-lysine-coated dishes in the presence of either DM or CM. Soma–soma synapses were prepared by placing VD4 (presynaptic, cholinergic) and LPeD1 (postsynaptic) cell bodies or their axons juxtaposed with each other. The synapses developed overnight as described previously (Feng et al., 1997).
CM was prepared by incubating gentamicin-treated ganglia in Sigmacote-treated glass Petri dishes containing DM (12 ganglia/6.5 ml DM). DM consisted of serum-free 50% L-15 medium with added inorganic salts (in mm: 40 NaCl, 1.7 KCl, 4.1 CaCl2, 1.5 MgCl2, and 10 HEPES, pH 7.9) and 20 μm gentamicin. The ganglia were first maintained in a humidified incubator for 3 d (first time CM), and subsequently removed and the resulting CM was collected and frozen (−20°C) until used (Syed et al., 1999). After several washes with antibiotic saline, the ganglia underwent two incubations in DM to prepare two additional batches of CM (second and third time CM, respectively). Previous studies have shown that although CM-induced effects on outgrowth and excitatory synapse formation are mimicked by LEGF (Hamakawa et al., 1999; Meems et al., 2003), in this study, however, to ensure that a full complement of the trophic factors are presented to the paired cells, we opted to use the CM in all experiments.
Electrophysiology.
Intracellular recording techniques were used to investigate synaptic physiology between the paired cells. Glass microelectrodes (1.5 mm internal diameter; World Precision Instruments) were pulled using vertical pipette puller (Model 700C, David Kopf Instruments). The electrodes were back filled with a saturated solution of K2SO4 to yield a tip resistance ranging from 30 to 60 mega ohms. Neurons were viewed under an inverted microscope (Axiovert 200M; Zeiss) and impaled by Narishige micromanipulators (MO-202, Narishige). Electrical signals were amplified with a Neuro data amplifier (Neuron Data Instrument) and recorded with Axoscope program (Axon Instruments). Acetylcholine (ACh, 1 μm) was pressure applied (10–20 psi, 0.5–1 s duration) directly to the somata through a glass pipette (2–4 μm tip in diameter) connected to a PV800 pneumatic picopump (World Precision Instruments).
ACh was obtained from Research Biochemicals (product A-112). Ryanodine, Lavendustin A and Lavendustin B were obtained from Calbiochem. All other chemicals were obtained from Sigma unless stated otherwise.
Ca2+ imaging.
The Ca2+-sensitive, membrane-permeable Fura-2 AM (Invitrogen) was used to determine changes in intracellular Ca2+ levels. Cells were loaded with Fura-2 AM (10 μm) in DM at room temperature for 45 min. Fura-2 AM was washed off with DM and cells were then observed under an inverted microscope. Fura-2 was excited sequentially at 340 and 380 nm wavelengths delivered from a mercury-arc lamp via a 40×, 1.3 NA Zeiss Fluor oil objective. Emitted fluorescence light was collected at 510 nm by a Retiga EXi camera. The ratio of fluorescence, R (340/380), from individual cells was stored on a computer running Northern Eclipse (Empix Imaging) and was used to calculate free intracellular calcium concentration ([Ca2+]i). Ca2+ calibration was performed with a fura-2 Ca2+ imaging calibration kit (F-6774, Invitrogen) and free [Ca2+]i was calculated from the equation [Ca2+]i = Kd × ((R − Rmin)/Rmax − R)) (F380max/F380 min) (Kao, 1994), where Rmin is the ratio at zero free Ca2+, Rmax is the ratio at saturating Ca2+ (e.g., 39 μm), F380max is the fluorescence intensity exciting at 380 nm, for zero Ca2+, F380 min is the fluorescence intensity at saturating free Ca2+, and Kd is the dissociation constant which was obtained by using the available dissociation constant calculator provided on the website of Invitrogen.
All experiments were performed at room temperature (20–23°C). Values given in the text and figures are mean ± SE. Statistical difference was determined by an independent Student's t test. A difference with p < 0.05 was considered statistically significant and was marked with an asterisk on the figures.
Results
CM-derived trophic factors are required for excitatory synapse formation
We (Syed et al., 1996; Hamakawa et al., 1999; Meems et al., 2003), and others (Barker et al., 1982; Miller et al., 1994; Fiumara et al., 2005), have demonstrated the requirements of brain CM for neurite outgrowth and specific synapse formation between cultured neurons. Although the precise identity of various trophic factors present in CM has not yet been fully deduced, their effects on growth and synapse formation can, however, be mimicked, in part by a number of mammalian counterparts and their Lymnaea homolog such as the Lymnaea LEGF (Hamakawa et al., 1999; Meems et al., 2003). Interestingly, in the absence of CM-derived trophic factors, Lymnaea neurons can only form inhibitory synapses (not seen in vivo), which we have defined previously as the “default synapses” (Woodin et al., 1999). These default synapses can, however, be corrected within hours of adding trophic factors to the culture dish (Hamakawa et al., 1999; Woodin et al., 1999). To examine the time required by CM to correct inappropriate inhibitory synapses, we first paired VD4 (presynaptic) and its postsynaptic partner LPeD1 in DM and the pairs were maintained overnight. After 12–18 h of pairing, direct intracellular recordings were made. In all instances (100%), intracellular stimulation of VD4 elicited an inhibitory response in its postsynaptic partner LPeD1 cell (n = 7) (Fig. 1). The DM was then replaced with CM, and the cells were reimpaled either after 1, 4, or 12–18 h of medium exchange. We discovered that over time (1 h, 14%; 4 h, 86%; and 12–18 h, 100%), a complete switch from inappropriate inhibitory to appropriate excitatory synapses had occurred, whereas cells maintained in DM remained inhibitory (data not shown). These data show that CM does correct default synapses formed in DM as was shown previously (Hamakawa et al., 1999; Woodin et al., 1999). However, the precise site of CM actions and the underlying mechanisms by which trophic factors switched default inappropriate inhibitory synapses to proper excitatory synapse formation remained largely unknown.
Figure 1.

Excitatory synapse formation between Lymnaea neurons requires brain CM. When paired overnight in DM (does not contain trophic factors), the identified neuron VD4 (presynaptic) and LPeD1 (postsynaptic) developed inappropriate inhibitory synapses (not seen in vivo), which switched to appropriate excitatory synapses after the addition of CM. Cells were paired in DM overnight and synapses tested by direct intracellular recordings. All pairs cultured in DM exhibited inhibitory synapses (n = 7). DM was then replaced with CM and synapses were sampled electrophysiologically at hour 1, 4 and 12–18. We found that a 1 h exposure to CM resulted in 14% (1 out 7) of cell pairs exhibiting appropriate excitatory synapses. The incidence of excitatory synapse formation increased with increasing time points and 86% (6 of 7) pairs exhibited excitatory synapses after 4 h of CM addition, whereas by 12–18 h, all (7 of 7) pairs examined had established appropriate excitatory synapses.
Acute CM exposure increased spontaneous activity of LPeD1 but not VD4
Because trophic factors have previously been shown to alter neuronal excitability (Kim et al., 1994; Blum et al., 2002), we hypothesize that CM-induced excitatory synapse formation may involve activity-dependent mechanisms. To test this hypothesis, cells paired overnight in DM were exposed to CM during intracellular recordings. We found that within 10 min of CM perfusion, the spontaneous activity in LPeD1 but not VD4 was significantly enhanced (Fig. 2A,B). Specifically, neuronal activity was first monitored for 20 min in DM and then CM was superfused and the cells were allowed to settle for 10 min during continuous intracellular recordings. Neurons were then recorded for an additional 20 min in CM. The total number of action potentials recorded in DM (for 20 min) were compared with those recorded in CM (for 20 min) (Fig. 2B). In instances where LPeD1 was spontaneously active, CM exposure significantly enhanced its bursting pattern (Fig. 2Aa), whereas tonically active cells increased their firing rate (Fig. 2Ab). In contrast, CM did not affect spontaneous activity of VD4, which remained quiescent throughout the experiment (Fig. 2A). In almost all instances, the total number of spontaneous action potentials in LPeD1 increased significantly in response to CM (n = 12/13) (Fig. 2B), whereas superfusion of defined medium (DM to DM) did not affect the total number of action potentials in LPeD1 (n = 4) (Fig. 2C).
Figure 2.
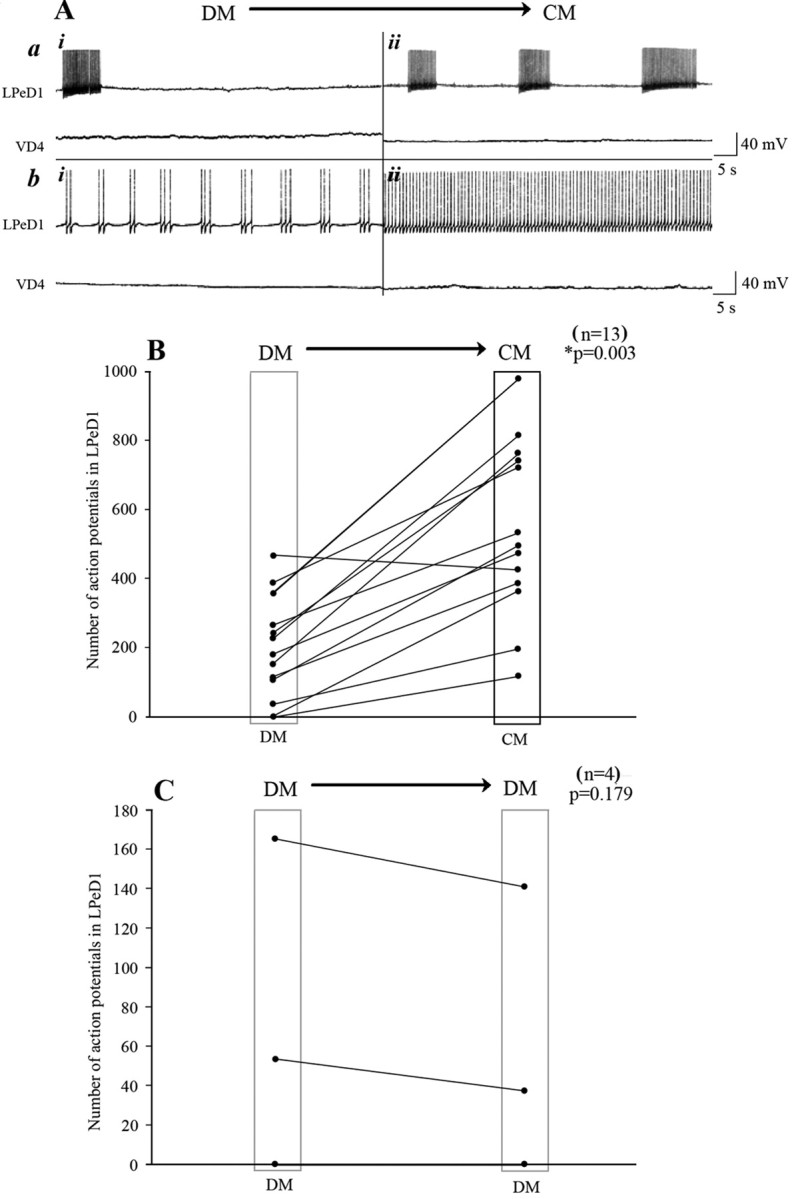
CM enhanced spontaneous electrical activity of the postsynaptic but not the presynaptic cell. To test whether CM affects the activity patterns of LPeD1 and VD4, cells were first paired overnight in DM, and on day 2, both neurons were impaled with sharp intracellular electrodes. A baseline for spontaneous activity was established for 20–30 min in DM (Ai), and DM was replaced with CM (Aii). Within minutes of CM perfusion, the spontaneous activity of LPeD1 was significantly enhanced (A). Specifically, regardless of LPeD1 's activity status, i.e., bursting (Aa) or tonic activity (Ab), the total number of action potentials increased significantly in CM. In contrast, the CM perfusion did not affect the activity of the presynaptic cell VD4 (A). B, C, Summary data showing the total number of action potentials recorded from LPeD1 [DM to CM (B) and DM to DM (C)]. The spontaneous activity in 93% of LPeD1 cells was significantly enhanced over a recording period of 20 min, whereas DM to DM perfusion did not affect the total number of action potentials in LPeD1 (n = 4). The asterisk indicates significant difference (t test; p < 0.05).
The CM-induced electrical activity in LPeD1 triggered Ca2+ oscillations
Previous studies on Lymnaea neurons have demonstrated that nerve growth factor acutely modulates voltage-gated Ca2+ currents (Wildering et al., 1995). As membrane depolarization is usually associated with the opening of VGCCs, we therefore hypothesized that CM-induced activity may trigger intracellular Ca2+ oscillations in LPeD1. To test this hypothesis, both single and paired (with VD4) LPeD1 neurons were loaded with the Ca2+ indicator dye Fura-2 AM, and imaged either in the absence or presence of CM. Specifically, LPeD1 cells were cultured in DM overnight either as single cells or paired with VD4. On day 2, all neurons were first loaded with Fura-2 AM (10 μm) for 45 min at room temperature and ratiometric, fluorescent Ca2+ images were acquired in DM for ∼3 min to establish a baseline (Fig. 3B). We found that regardless of their pairing status (single or paired), within minutes of exposure to CM, [Ca2+]i in LPeD1 but not VD4 increased significantly (n = 30) (Fig. 3A,B). These Ca2+ levels remained elevated throughout CM exposure and returned to their baseline once the CM was replaced with DM (Fig. 3A,B). It is important to note that although the baseline Ca2+ levels also increased in LPeD1 in most instances, the major Ca2+ signal could only be attributed to large Ca2+“spikes” that gave rise to its oscillations (Fig. 3B). These Ca2+ oscillations were primarily observed in LPeD1, whereas the presynaptic cell VD4 exhibited only modest rise in [Ca2+]i. For instance, all (100%, n = 30) LPeD1 cells monitored in these experiments exhibited a rise in [Ca2+]i upon CM exposure, compared with only 18% (2 of 11) of VD4 cells. The mean [Ca2+]i in LPeD1 cells in DM was 151 ± 31 nm (n = 30) which was significantly increased to 337 ± 49 nm (mean peak amplitude) in CM. However, CM only increased [Ca2+]i from 131 ± 22 nm to 162 ± 20 nm in VD4 cells (n = 11) (Fig. 3C). Although we did observe slight increases in [Ca2+]i, large Ca2+ oscillations were, however, never observed in VD4 cells. The Ca2+ oscillations in LPeD1 returned to their baseline level once the CM was replaced again with DM suggesting that they were indeed trophic factor-dependent.
Figure 3.

CM triggered intracellular Ca2+ oscillations in the postsynaptic (LPeD1) but not presynaptic (VD4) neuron. A. Photomicrographs depicting intracellular Ca2+ concentration ([Ca2+]i) in single and paired cells maintained under various different experimental conditions. Either single or paired cells were maintained in DM overnight. After 12–18 h of culture, cells were loaded with Ca2+ indicator dye Fura-2 AM, and ratiometric images (340/380 nm) were acquired first in DM and during CM perfusion. In DM, we found that the Ca2+ levels were fairly stable with the intracellular concentration in the range of 151 ± 31 nm (n = 30) and 131 ± 22 nm (n = 11) in both LPeD1 and VD4, respectively. It is interesting to note that Ca2+ concentration, as determined by Fura-2, did not vary between single or paired cells (A, B). However, within minutes of CM perfusion, [Ca2+]i in LPeD1 increased significantly, whereas no significant change was observed in VD4. Ca2+ increases in LPeD1 followed an oscillatory pattern which continued as long as the cells were maintained in CM. Upon replacement of CM with DM, the Ca2+ levels returned to their respective baselines in both single and paired LPeD1 cells. These Ca2+ changes in LPeD1 and VD4 as measured through the ratiometric data are depicted in C. The asterisk indicates significant difference (t test; p < 0.05) between the [Ca]i in DM and the [Ca]i in the presence of CM in LPeD1 cells. Error bars indicate SE.
Together, these data demonstrate that CM exposure acutely raises intracellular Ca2+ in the postsynaptic but not the presynaptic cell and that these changes are independent of the neuronal pairing status (single versus paired). We also demonstrated that the CM-induced Ca2+ increases result from Ca2+ spikes, which were also exclusive to the postsynaptic cell.
CM-induced Ca2+ oscillations were activity dependent and involved RTK activity
To deduce whether CM-induced Ca2+ oscillation/electrical activity observed above, involved trophic factor-triggered action potentials reported earlier (Fig. 2), intracellular recordings were made simultaneously with Fura-2 AM Ca2+ imaging. Specifically, single LPeD1 cells cultured in DM overnight were loaded with Fura-2 AM and simultaneously impaled with sharp intracellular electrodes. The baseline electrical activity and the Ca2+ signal were first established in DM as above and the neurons were exposed to CM. At resting conditions (resting potential = −59 ± 2 mV, n = 10), the electrical activity of the neurons was relatively stable and the basel intracellular Ca2+ levels were steady (Fig. 4A). However, within minutes of CM exposures, the neurons began to exhibit spontaneous Ca2+ oscillations, which were almost always accompanied by spontaneous bursts of APs in LPeD1 (n = 10) (Fig. 4A, inset). To determine the causal relationship between the CM-induced electrical activity and the observed Ca2+ oscillations, the LPeD1 cells were first allowed to sit at resting membrane potentials and were then hyperpolarized by injecting negative current before CM exposure (images acquired throughout) (Fig. 4B). We found that injecting 0.05–0.2 nA of hyperpolarizing current (−60 pA in this case) before CM exposure prevented CM-induced activity in LPeD1 concomitant with the disappearance of Ca2+ oscillations (n = 8) (Fig. 4B). These data clearly demonstrate that the observed Ca2+ oscillations indeed resulted from CM-induced electrical activity in LPeD1.
Figure 4.

The CM-induced Ca2+ oscillations in LPeD1 neurons are activity-dependent and involve RTK activity. To determine whether CM-induced Ca2+ oscillations resulted from trophic factor-triggered action potentials, LPeD1 cells were cultured overnight in DM. On day 2, cells were loaded with Fura-2 AM and somata impaled with intracellular electrodes to monitor neuronal activity. Spontaneous action potentials were monitored in concomitant with ratiometric fluorescent images of LPeD1 first in DM and then in CM. A, Within minutes of CM exposure, LPeD1 neurons began to exhibit a bursting pattern and these bursts were almost always accompanied with Ca2+ oscillations, whereby [Ca2+]i raised from 138 ± 20 nm in DM to 286 ± 28 nm in CM (n = 10). The inset on right shows action potential bursts and Ca2+ activity on an extended time scale. B, To demonstrate that the observed Ca2+ oscillations indeed resulted from CM-induced burst of action potentials, LPeD1 neurons loaded with Fura-2 were first hyperpolarized by injecting −60 pA negative current (indicated by arrow) before neuronal exposure to CM. We found that neuronal hyperpolarization before CM exposure not only prevented action potentials but also the Ca2+ oscillations. C, To demonstrate that CM-induced increase in the intracellular Ca2+ involved RTK activity, LPeD1 cells cultured overnight in DM were first treated with RTK inhibitor Lav A (30 μm) and its inactive analog Lav B (30 μm). We found that Lav A, but not Lav B, significantly inhibited CM-induced rise in [Ca2+]i compared with the [Ca2+]i rise induced by CM alone (data from Fig. 3C). The asterisk indicates significant difference (t test; p < 0.05). Error bars indicate SE.
We have previously demonstrated that CM-induced effects on excitatory synapse formation are mediated through RTKs (Hamakawa et al., 1999) and thus strongly support the trophic nature of the molecules that mediate synaptogenesis. To determine whether CM-induced effects on electrical activity and intracellular Ca2+ oscillations involved RTK, its activity was blocked by the antagonist Lavendustin A (Lav A; 30 μm), whereas its inactive analog Lavendustin B (Lav B; 30 μm) was used as a control. Specifically, LPeD1 neurons cultured in DM overnight were incubated with Lav A or B for ∼1 h before CM exposure. We found that Lav A, but not Lav B, could block CM-induced rise in [Ca2+]i in LPeD1 cells (Fig. 4C). Specifically, CM in the presence of Lav B could still increase [Ca2+]i by 147 ± 14 nm (n = 8), which was not significantly different from that induced by CM alone (186 ± 22 nm, n = 30) (data from Fig. 3C). Prior treatment of LPeD1 cells by Lav A, however, completely abolished CM-induced Ca2+ oscillations (n = 8) (Fig. 4C). Consistently, Lav A, but not Lav B, inhibited CM-induced modulation of spontaneous electrical activity in LPeD1 neurons (n = 7, data not shown). These data thus support the notion that the CM-induced effects on Ca2+ oscillations are mediated through RTK.
CM-induced Ca2+ oscillations required Ca2+ influx through VGCCs
To determine whether CM-induced Ca2+ oscillations involved Ca2+ influx through VGCCs, the LPeD1 neurons were exposed to CM either in the presence of Ca2+ channel blocker cadmium (Cd2+, 100 μm) or nifedipine (10 μm) shown previously to essentially block l-type Ca2+ channels in Lymnaea neurons (Spafford et al., 2006). We found that CM-induced Ca2+ increase in LPeD1 was gradually and completely blocked by Cd2+ (n = 8) (Fig. 5A). Similarly, nifedipine significantly reduced both the intensity and the frequency of CM-induced Ca2+ oscillations in LPeD1 (n = 6) (Fig. 5B). The time integral (for 200 s) of the Ca2+ signal (Fig. 5B) induced by CM alone (15 ± 6 μm s; n = 6) was greatly reduced by nifedipine (6 ± 2 μm s; n = 6). Because nifedipine did not completely abolish the CM-induced Ca2+ increase, we next sought to determine the involvement of Ca2+-induced Ca2+ release from the intracellular Ca2+ stores. To test this possibility, LPeD1 cells were pretreated with ryanodine (50 μm), which we have shown previously to perturb release from the intracellular Ca2+ stores (Dunn and Syed, 2006). A 1 h pretreatment of LPeD1 with ryanodine (50 μm) failed to block CM-induced Ca2+ rise in all cells tested (n = 5) (Fig. 5C). The mean peak value of CM-induced increase in intracellular Ca2+ level in ryanodine treated LPeD1 cells was 144 ± 10 nm (n = 5), which was comparable with that of ryanodine nontreated cells (149 ± 20 nm) (n = 5). Together, these data demonstrate that CM-induced Ca2+ oscillations in LPeD1 involve Ca2+ influx through VGCCs and that the intracellular stores may not play a significant role in regulating CM-induced increase in [Ca2+]i.
Figure 5.

CM-induced Ca2+ oscillations in LPeD1 cells involve Ca2+ influx via VGCCs. A, LPeD1 cells were cultured overnight in DM and were loaded with Ca2+ indicator dye Fura-2 AM after 12–18 h. Images were acquired first in CM and then CM containing blockers of VGCCs. As reported earlier, CM perfusion significantly increased [Ca2+]i from 181 ± 42 nm to 334 ± 36 nm (n = 8). Cd2+ (100 μm) was then added to the dish containing CM while continuously monitoring [Ca2+]i. Within minutes of adding Cd2+ to the culture, the CM-induced Ca2+ oscillations were almost completely blocked. Similarly, nifedipine (10 μm) significantly reduced both the amplitude and the frequency of CM-induced Ca2+ oscillations in LPeD1 neurons (B). The shaded areas represent the time integral of Ca2+ signal for 200 s induced by CM alone (15 ± 6 μm s) and CM in combination with nifedipine (6 ± 2 μm s) (n = 6). C, To rule out the involvement of Ca2+-induced Ca2+ release from the intracellular stores, cells were pretreated with ryanodine (50 μm, 1 h), and Ca2+ images were acquired first in DM and then in the presence of CM. We found that ryanodine failed to block CM-induced Ca2+ increase in LPeD1 neurons. Thus, CM-induced Ca2+ elevation in LPeD1 cells mainly involves Ca2+ entry via VGCCs, but not Ca2+-induced Ca2+ release from ryanodine-sensitive intracellular Ca2+ store.
CM-induced de novo protein synthesis was required for the expression of excitatory nAChRs
As mentioned earlier, CM is required for excitatory synapse formation, and in its absence, neurons establish inappropriate inhibitory synapses (Woodin et al., 1999). These inappropriate inhibitory synapses can, however, be corrected by the addition of CM (Hamakawa et al., 1999; Woodin et al., 1999) (Fig. 1), although the precise site (presynaptic versus postsynaptic) for its actions remains unknown. Based on our finding presented above, we next tested the following hypothesis: CM-induced electrical activity and the corresponding Ca2+ influx through VGCCs may activate a protein synthesis-dependent step underlying the synthesis of excitatory nAChR in LPeD1 cells. To test this hypothesis, we first tested the responses of single LPeD1 neurons to exogenously applied ACh. Specifically, cells were cultured overnight in DM and ACh (1 μm) was pressure applied during intracellular recordings. We found that all cells plated in DM, exhibited an inhibitory response as was reported previously (Fig. 6A). CM was then superfused through the dish and the electrophysiological responses to exogenously applied ACh were tested at various different time points. We found that between a 30–120 min time window, the LPeD1 neurons (7 of 15) exposed to CM continued to exhibit an inhibitory response to exogenously applied ACh (Fig. 6A). The other 8 cells exhibited an excitatory response indicating the expression of excitatory receptors. Among these 8 cells, 2 LPeD1 neurons exhibited purely excitatory response to ACh, whereas the other 6 cells exhibited biphasic response whereby excitation was followed by an inhibitory response (Fig. 6ii). After 2–4 h of CM exposure, however, only 3 of 15 cells remained inhibitory, whereas all the other cells either exhibited purely excitatory (7 of 15) or a biphasic (5 of 15) response. More than 80% of LPeD1 neurons exhibited excitatory responses after 4–6 h of CM exposure (Fig. 6A), and within 12–18 h of DM substitution with CM, all neurons exhibited an excitatory response to exogenously applied ACh (data not shown). Moreover, the amplitude of postsynaptic potentials (excitatory versus inhibitory) also changed after CM exposure (Fig. 6B). Specifically, the amplitude of EPSPs was progressively enhanced at the expense of the IPSPs (Fig. 6ii,iii). For instance, the mean amplitude of excitatory potential evoked by ACh was 2.1 ± 0.2 mV (n = 8) after 30–120 min of CM exposure, which increased to 4.2 ± 0.3 mV (n = 7) after 2–4 h, and to 6.2 ± 0.3 mV (n = 5) after 4–6 h of CM exposure. While the mean amplitude of ACh-evoked inhibitory potential in cells cultured in DM was 6.5 ± 0.6 mV (n = 15), and was reduced to 5.5 ± 0.6 mV (n = 6) after 30–120 min, 2.3 ± 0.5 mV (n = 5) after 2–4 h and 1.1 ± 0.4 (n = 3) after 4–6 h of CM exposure (Fig. 6B). These data clearly demonstrate that CM is responsible for switching the inhibitory response to excitatory and may thus be the mechanisms underlying excitatory synapse formation.
Figure 6.

CM-induced new protein synthesis, but not gene transcription, is required for the expression of excitatory nAChRs. A, To determine the time required by CM to switch inhibitory response to appropriate excitatory response, LPeD1 cells were cultured overnight in DM. Intracellular recordings were made after 12–18 h of culture and ACh (1 μm) was applied exogenously to the somata of LPeD1 while neurons were impaled with intracellular sharp electrodes. It is important to note that ACh mimics both inhibitory and excitatory effects observed in LPeD1 neurons cultured in DM and CM respectively. Specifically, responses of LPeD1 cells to exogenously puffed ACh (1 μm) were tested both in DM and then after exposure to CM for 30 min, 2 h, 2–4 h and 4–6 h. We found that within 30–120 min of CM exposure, LPeD1 cells began to switch their inhibitory response to excitatory with there being the presence of an overlapping biphasic response. After 4–6 h of CM exposure, a large majority of the cells exhibited excitatory response (11 of 15 cells were purely excitatory, 2 of 15 cells were biphasic) indicating that the receptor responses had switched from inhibitory to excitatory. B, The amplitude of the inhibitory response gradually decreased with increasing excitation after CM exposure. Error bars indicate SE. Insets (top right, i, ii, iii) depicting A and B are sample traces obtained from LPeD1 cells exhibiting either inhibitory, excitatory or a biphasic response to ACh under various experimental conditions. The LPeD1 cell was held at −56 mV. Arrows indicate the application of ACh (1 μm). The dash line indicates the baseline of membrane potential. Note that cells that fired action potentials in response to ACh were only included in the percentage data but not in the amplitude measurements as these parameters are often difficult to discern from spiking neurons. Furthermore, the amplitude of both excitatory and inhibitory potential that was <0.5 mV was not included in the amplitude statistics. C, To determine whether a switch from inhibitory to excitatory receptor involved the synthesis of new receptors, cells cultured in DM were exposed to CM containing either transcription (actinomycin D, 1 μg/ml) or translation (anisomycin, 12.5 μg/ml) inhibitors for 4–6 h and cholinergic responses were tested electrophysiologically. We found that blocking protein synthesis but not the gene transcription blocked the CM-induced expression of excitatory receptors in LPeD1 cells.
To test the possibility that CM-induced switch from inhibitory to excitatory response involved either gene transcription or de novo synthesis of new nAChR, the cells were cultured in DM overnight and were exposed to trophic factors either in the presence or absence of transcription or nonspecific protein synthesis inhibitors, actinomycin-D (1 μg/ml) and anisomycin (12.5 μg/ml), respectively, for 4–6 h (Fig. 6C). As shown in Figure 6A, 13 of 15 LPeD1 cells exhibited excitatory responses after 4–6 h of CM exposure. To determine whether the expression of these excitatory receptors involves de novo synthesis of new receptors or the targeting/activation of existing receptors, cells were cultured in CM containing protein synthesis inhibitor anisomycin (12.5 μg/ml). The CM-induced expression of the excitatory component was blocked in all LPeD1 cells treated with anisomycin (Fig. 6C) (n = 10), and these cells exhibited only purely inhibitory response to ACh. The mean amplitude of inhibitory potentials was 5.8 ± 0.5 mV (n = 10), which was not significantly different from cells cultured in DM (6.5 ± 0.6 mV, n = 15) (Fig. 6B). These data demonstrate that the expression of excitatory but not the inhibitory receptors requires de novo synthesis of receptor proteins. One cannot, however, rule out the possibility that synthesis of various proteins required to target AChR may have also been affected by anisomycin. To test whether the expression of excitatory receptors is also contingent upon gene transcription, the cells were cultured in CM containing actinomycin-D (1 μg/ml). Actinomycin-D pretreatment, however, failed to block the expression of excitatory receptors (8 of 10) (Fig. 6C). These data thus show that CM-induced effects on excitatory receptor expression involve de novo synthesis of nAChR and that this step does not involve gene transcription.
Inhibiting postsynaptic but not presynaptic activity perturbed CM-induced excitatory synapse formation between VD4 and LPeD1 neurons
The above data demonstrate that CM-induced electrical activity and the corresponding Ca2+ oscillations are likely involved in the expression of postsynaptic, excitatory nAChR and hence the excitatory synapse formation between the paired cells. To test this possibility directly, we paired a single VD4 neuron simultaneously with two LPeD1 neurons (triplet configurations: soma–soma–soma), and the cultures were maintained in DM overnight (Fig. 7A). It is important to note that under such conditions, VD4 successfully synapses with both partners, albeit the efficacy of synaptic strength is reduced (Munno and Syed, 2003). To confirm whether appropriate and dual synapses reform between a single VD4 and its LPeD1 partners, cells were paired in CM in a triplet configuration (Fig. 7A). Intracellular recordings after 12–18 h of pairing revealed that in 9 of 11 instances, VD4 had established excitatory synapses with both LPeD1 cells (Fig. 7C). In one instance, we observed electrical coupling between VD4 and LPeD1 (one cell only; the other cell had an excitatory synapse), and yet in another instance, there was no synapse between VD4 and one of its partners. To test for the role of CM-induced electrical activity in excitatory synapse formation, cells were paired overnight as above, albeit in DM. On day 2, simultaneous triple cell intracellular recordings were made to demonstrate that no excitatory synapses were present between VD4 and any of its partners (n = 10) (Fig. 7C). CM was then superfused, and one postsynaptic LPeD1A was injected with 0.2 nA hyperpolarizing current throughout the course of the experiment (4–6 h) to prevent spiking while the second LPeD1B was allowed to exhibit spontaneous action potentials in response to CM for up to 4–6 h (Fig. 7A,C). At the end of this period, we sought to determine the incidence of synapse formation between VD4 and its partners. To test this, we induced action potentials in VD4 and synapses were tested electrophysiologically (Fig. 7B). We found that action potentials in VD4 generated 1:1 EPSPs in a vast majority of LPeD1B (7 of 10) where no current was injected during CM perfusion (Fig. 7B,C). In contrast, only 2 hyperpolarized LPeD1A exhibited a weak excitatory response to VD4 stimulation, and all the other 8 hyperpolarized LPeD1A exhibited IPSPs (Fig. 7B,C).
Figure 7.

Blocking CM-induced electrical activity in the postsynaptic neurons LPeD1 prevented trophic factor-mediated excitatory synapse formation between the paired cells. To test whether preventing CM-induced activity and the underlying Ca2+ oscillations blocks excitatory synapse formation, cells were paired in a triplet configuration. Specifically, a single VD4 neuron was simultaneously paired with two LPeD1 cells (LPeD1A and LPeD1B) and cells were maintained overnight in DM. A, A phase contrast image depicting the triplet configuration where LPeD1A was injected with hyperpolarizing current during the CM exposure (4–6 h), whereas cell labeled LPeD1B was allowed to fire action potential in response to the trophic factors. While under normal conditions (no current injections), VD4 establishes excitatory synapses with both LPeD1 neurons, hyperpolarizing one cell to prevent CM-induced activity resulted in the formation of inhibitory synapse, whereas cell that was allowed to exhibit normal activity established excitatory synapses after the CM exposure. B, Induced action potentials in VD4 generated 1:1 inhibitory (LPeD1A)and excitatory (LPeD1B) postsynaptic potentials in hyperpolarized versus the control cells. C, Presents summary data showing the percentage of cells exhibiting excitatory and inhibitory synapses under different experimental conditions. Specifically, when cultured in CM overnight, >80% of both LPeD1 neurons could form excitatory synapses with presynaptic VD4 partner, while no excitatory synapses were observed between cells cultured in DM. After replacement of DM with CM, a majority (7 of 10) of control (Ctrl, LPeD1B) neurons established excitatory synapses, whereas cells hyperpolarized (Hyp, LPeD1A) throughout the experiments remained inhibitory (C).
Together, these data demonstrate that CM-induced activity in the postsynaptic but not the presynaptic cell is required for the expression of excitatory nAChRs, which are in turn required for trophic factor-mediated excitatory synapse formation.
Discussion
In this study, we have demonstrated that trophic factors present in the Lymnaea brain CM influence synaptogenic programs by invoking postsynaptic activity-dependent mechanisms. CM-induced activity generated Ca2+ oscillations in the postsynaptic cell by opening VGCCs and promoted de novo synthesis of nAChRs required for excitatory synapse formation between the paired cells VD4 and LPeD1. The CM-induced effects were independent of LPeD1 pairing with its synaptic partner, suggesting strongly that the trophic factor actions were specific to the postsynaptic cell. Blocking Ca2+ influx or postsynaptic activity alone resulted in the perturbation of trophic factor-mediated excitatory synapse formation between the paired cells. These data provide the first direct evidence that trophic factors affecting postsynaptic activity exclusively promote excitatory synapse formation between central neurons. The CM-mediated effects appear to have “primed” the postsynaptic cell for appropriate excitatory synapse formation with its partner by triggering a protein synthesis dependent step needed for the expression of nAChRs.
Trophic factors in CM have previously been shown to regulate the expression of molluscan homolog of the multiple endocrine neoplasia type 1 (MEN 1) tumor suppressor gene, which encodes the transcription factor menin (van Kesteren et al., 2001). Knock-down of MEN 1 specifically in the postsynaptic cell was sufficient to block synapse formation between VD4 and its synaptic partners. Similarly, we have recently demonstrated that an in vitro knock-down of LEGF receptor in LPeD1 renders this cell incapable of responding to either purified LEFG or an EGF-like molecule present in CM thus resulting in the failure of excitatory synapse formation (van Kesteren et al., 2008). In the present study, it would have been ideal for us to use LEGF alone (rather than CM) to explore its effects on electrical activity and hence the expression of excitatory nAChR in LPeD1. However, since LEGF does not completely mimic CM's trophic actions (outgrowth and synapse formation) (Hamakawa et al., 1999) and is scarcely available, we therefore opted to use CM. We have previously deciphered the proteinacious nature of the trophic molecules present in CM (Hamakawa et al., 1999; Meems and Syed, 2006), which are required not only for the formation but also for the maintenance (Woodin et al., 2002) of excitatory synapse formation, and have shown here that these effects are mediated through RTK and blocked by its specific antagonists (Fig. 4C). We are therefore confident that it is indeed the trophic factors and not some other unknown compounds (neurotransmitters, K+, etc.) that affected the excitability of LPeD1 after CM perfusion. Specifically, the CM-induced, acute modulation of LPeD1 electrical activity was neither due to higher concentration of potassium (K+) ions in CM compared with in DM, nor did it involve perfusion artifacts. First, if CM contained higher K+ ions then it should have also depolarized VD4, which was not observed in the present study. Second, DM perfusion into the culture dish did not change neuronal excitability of either LPeD1 or VD4 (Fig. 2C) thus ruling out the notion of perfusion artifacts. We therefore conclude that the CM-induced changes observed in LPeD1 are indeed mediated by trophic factors present in CM. However, the precise mechanisms by which trophic factors present in CM primed LPeD1 for excitatory synapse formation remained largely unknown.
A number of studies have shown that the trophic factors regulate the expression of neuronal transmitter receptors (Kovalchuk et al., 2002). For instance, brain derived neurotrophic factor (BDNF) increases the expression of AMPA-type glutamate receptor proteins (Narisawa-Saito et al., 1999), and perturbation of its tyrosine kinase B-mediated intracellular signaling leads to a loss of postsynaptic receptor clusters (Gonzalez et al., 1999). BDNF has also been shown to increase both the number and the expression of nAChR clustering in hippocampal interneurons over a time course of several hours to days (Kawai et al., 2002; Massey et al., 2006). The above data from a number of vertebrate species are consistent with our observations whereby CM appears to be required for the expression of excitatory nAChRs in LPeD1. In contrast, a recent study has demonstrated that BDNF, acting on the tyrosine kinase TrkB receptor, induces a rapid decrease of α7 nAChR-mediated response in hippocampal interneurons of the CA1 region (Fernandes et al., 2008). These inhibitory effects of BDNF involve actin cytoskeleton and are mediated through phospholipase C/protein kinase C (PLC/PKC) pathway and require Ca2+ ions as a cofactor. Consistent with the observations of Fernandes et al. (2008), we also found that Ca2+ was a common denominator modulating receptor expression function in both vertebrate and invertebrate neurons. Whether the functional expression of nAChR in LPeD1 also involves PLC/PKC or other second message cascade downstream from Ca2+ remains, however, to be investigated. Our data do, nevertheless, allude toward the possibility that either Ca2+ or Ca2+-activated protein kinase (data not shown) may affect synthesis of nAChR proteins in LPeD1, as protein synthesis but not gene transcription of mRNA was required for the expression of nAChR in our model. Because in our model, CM-induced effects involved a protein synthesis-dependent step, our data support the notion that trophic factors are most likely involved in the synthesis of new nAChR and not the membrane trafficking of existing receptors to the LPeD1 membrane. The induction of synapse specific gene transcription and de novo protein synthesis by Ca2+ or its cytoplasmic effectors has also been reported in previous studies (Finkbeiner and Greenberg, 1998; West et al., 2001).
Nerve growth factor has been reported to increase global intracellular Ca2+ level in PC12 cells (Pandiella-Alonso et al., 1986). The [Ca2+]i in rat hippocampal neurons was also rapidly increased by BDNF and neurotrophin-3 (Berninger et al., 1993). Furthermore, BDNF was also found to increase [Ca2+]i in the glial cell (Rose et al., 2003). In these studies, neurotrophins were found to activate PLC/inosital 1,4,5-triphosphate (IP3) pathway to mobilize Ca2+ from IP3-sensitive intracellular stores (Pandiella-Alonso et al., 1986; Berninger et al., 1993). The depletion of intracellular stores resulting in Ca2+ entry through store-operated Ca2+ channels has also been reported in some studies (Rose et al., 2003; Blum and Konnerth, 2005). Although another type of intracellular stores, the caffeine/ryanodine-sensitive Ca2+ stores, is present in a variety of tissues and may contribute to activity-dependent secretion of trophic factors (Balkowiec and Katz, 2002; Blum and Konnerth, 2005), our data rule out the possibility of its involvement in the trophic factors-induced increase in [Ca2+]i. Specifically, we have demonstrated previously that although ryanodine receptors are functionally present in Lymnaea neurons (Dunn and Syed, 2006; Dunn et al., 2009), they do not, however, contribute significantly to CM-induced intracellular Ca2+ oscillations in LPeD1 neurons. These observations are consistent with Kovalchuk et al. (2002) who demonstrated that the BDNF-induced Ca2+ transients in hippocampal neurons are also specific to postsynaptic dendrites and spines (not at presynaptic sites) and that this rise in intracellular Ca2+ relies primarily upon Ca2+ influx via VGCCs.
Our data show that the trophic factor-induced Ca2+ influx was associated with increased electrical activity in LPeD1 thus supporting the important role of trophic factor-induced activity-dependent mechanisms in neuronal connectivity and function (Cohen-Cory, 1999; Thoenen, 2000; Carrasco et al., 2007). For instance, BDNF coincidentally activates both presynaptic and postsynaptic neurons to increase the incidence of synaptogenesis in the developing mouse hippocampus (Aguado et al., 2003). This is consistent with Hebb's postulation that synapses will preferentially form and strengthened between neurons that exhibit coincident activity, that is cells that fire together, wire together (Hebb, 1949; Goodman and Shatz, 1993; Katz and Shatz, 1996). In the present study, we have shown that trophic factors selectively increase the spontaneous spiking activity of the postsynaptic neurons only and this activity is in turn required for the expression of nAChRs in LPeD1 cells. Although our data show that trophic factor-mediated postsynaptic differentiation that involves receptor expression can occur in the absence of its partner, these data do not, however, rule out the possibility that the targeting of nAChR to specific synaptic site may still require presynaptic signaling as has been observed in other models (Sanes and Lichtman, 1999; Dean and Dresbach, 2006). In our model, however, these inductive changes appear to be independent of membrane contacts between VD4 and LPeD1 since the cells paired in DM failed to exhibit excitatory synapse formation. While our data underscore the importance of trophic factors in modulating postsynaptic machinery, they do not, however, undermine the effects of CM on the presynaptic cell vis-à-vis synapse formation. Specifically, we have previously shown that exposure of the presynaptic cell VD4 to CM is essential for synapse formation between this neuron and its postsynaptic partners (Meems et al., 2003). Together, our studies demonstrate that trophic factors are essential for synapse formation between Lymnaea neurons and that these effects involve both presynaptic and postsynaptic cells. It is interesting to note, however, that in contrast to other models where presynaptic activity-dependent changes precede postsynaptic changes (Gerrow et al., 2006), our data show that the trophic factor-mediated actions on presynaptic and postsynaptic cell are activity independent and activity dependent, respectively.
Footnotes
This work was supported by a Canadian Institute of Health Research (CIHR) grant. F.X. is supported by the Alberta Heritage Foundation for Medical Research (AHFMR) Fellowship, and T.K.M.L. was supported by an AHFMR studentship. N.I.S. was a CIHR Investigator. We acknowledge excellent technical support by Wali Zaidi.
References
- Aguado F, Carmona MA, Pozas E, Aguiló A, Martínez-Guijarro FJ, Alcantara S, Borrell V, Yuste R, Ibañez CF, Soriano E. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl- co-transporter KCC2. Development. 2003;130:1267–1280. doi: 10.1242/dev.00351. [DOI] [PubMed] [Google Scholar]
- Balkowiec A, Katz DM. Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci. 2002;22:10399–10407. doi: 10.1523/JNEUROSCI.22-23-10399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DL, Wong RG, Kater SB. Separate factors produced by the CNS of the snail Helisoma stimulate neurite outgrowth and choline metabolism in cultured neurons. J Neurosci Res. 1982;8:419–432. doi: 10.1002/jnr.490080229. [DOI] [PubMed] [Google Scholar]
- Berninger B, García DE, Inagaki N, Hahnel C, Lindholm D. BDNF and NT-3 induce intracellular Ca2+ elevation in hippocampal neurones. Neuroreport. 1993;4:1303–1306. doi: 10.1097/00001756-199309150-00004. [DOI] [PubMed] [Google Scholar]
- Blum R, Konnerth A. Neurotrophin-mediated rapid signaling in the central nervous system: mechanisms and functions. Physiology (Bethesda) 2005;20:70–78. doi: 10.1152/physiol.00042.2004. [DOI] [PubMed] [Google Scholar]
- Blum R, Kafitz KW, Konnerth A. Neurotrophin-evoked depolarization requires the sodium channel Na(V)1.9. Nature. 2002;419:687–693. doi: 10.1038/nature01085. [DOI] [PubMed] [Google Scholar]
- Carrasco MA, Castro P, Sepulveda FJ, Tapia JC, Gatica K, Davis MI, Aguayo LG. Regulation of glycinergic and GABAergic synaptogenesis by brain-derived neurotrophic factor in developing spinal neurons. Neuroscience. 2007;145:484–494. doi: 10.1016/j.neuroscience.2006.12.019. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S. BDNF modulates, but does not mediate, activity-dependent branching and remodeling of optic axon arbors in vivo. J Neurosci. 1999;19:9996–10003. doi: 10.1523/JNEUROSCI.19-22-09996.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AM, Graf ER, Linhoff MW. How to build a central synapse: clues from cell culture. Trends Neurosci. 2006;29:8–20. doi: 10.1016/j.tins.2005.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity of neural circuits. Neuron. 2004;44:23–30. doi: 10.1016/j.neuron.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Dean C, Dresbach T. Neuroligins and neurexins: linking cell adhesion, synapse formation and cognitive function. Trends Neurosci. 2006;29:21–29. doi: 10.1016/j.tins.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Dunn TW, Syed NI. Ryanodine receptor-transmitter release site coupling increases quantal size in a synapse-specific manner. Eur J Neurosci. 2006;24:1591–1605. doi: 10.1111/j.1460-9568.2006.05028.x. [DOI] [PubMed] [Google Scholar]
- Dunn TW, MaCamphill PK, Syed NI. Activity-induced large amplitude postsynaptic mPSPs at soma-soma synapses between Lymnaea neurons. Synapse. 2009;63:117–125. doi: 10.1002/syn.20589. [DOI] [PubMed] [Google Scholar]
- Feng ZP, Klumperman J, Lukowiak K, Syed NI. In vitro synaptogenesis between the somata of identified Lymnaea neurons requires protein synthesis but not extrinsic growth factors or substrate adhesion molecules. J Neurosci. 1997;17:7839–7849. doi: 10.1523/JNEUROSCI.17-20-07839.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes CC, Pinto-Duarte A, Ribeiro JA, Sebastião AM. Postsynaptic action of brain-derived neurotrophic factor attenuates α7 nicotinic acetylcholine receptor-mediated responses in hippocampal interneurons. J Neurosci. 2008;28:5611–5618. doi: 10.1523/JNEUROSCI.5378-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S, Greenberg ME. Ca2+ channel-regulated neuronal gene expression. J Neurobiol. 1998;37:171–189. [PubMed] [Google Scholar]
- Fiumara F, Leitinger G, Milanese C, Montarolo PG, Ghirardi M. In vitro formation and activity-dependent plasticity of synapses between Helix neurons involved in the neural control of feeding and withdrawal behaviors. Neuroscience. 2005;134:1133–1151. doi: 10.1016/j.neuroscience.2005.05.021. [DOI] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Gerrow K, Romorini S, Nabi SM, Colicos MA, Sala C, El-Husseini A. A preformed complex of postsynaptic proteins is involved in excitatory synapse development. Neuron. 2006;49:547–562. doi: 10.1016/j.neuron.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Gonzalez M, Ruggiero FP, Chang Q, Shi YJ, Rich MM, Kraner S, Balice-Gordon RJ. Disruption of Trkb-mediated signaling induces disassembly of postsynaptic receptor clusters at neuromuscular junctions. Neuron. 1999;24:567–583. doi: 10.1016/s0896-6273(00)81113-7. [DOI] [PubMed] [Google Scholar]
- Goodman CS, Shatz CJ. Developmental mechanisms that generate precise patterns of neuronal connectivity. Cell. 1993;72(Suppl):77–98. doi: 10.1016/s0092-8674(05)80030-3. [DOI] [PubMed] [Google Scholar]
- Hamakawa T, Woodin MA, Bjorgum MC, Painter SD, Takasaki M, Lukowiak K, Nagle GT, Syed NI. Excitatory synaptogenesis between identified Lymnaea neurons requires extrinsic trophic factors and is mediated by receptor tyrosine kinases. J Neurosci. 1999;19:9306–9312. doi: 10.1523/JNEUROSCI.19-21-09306.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG, Drapeau P. From contact to connection: early events during synaptogenesis. Trends Neurosci. 1995;18:196–201. doi: 10.1016/0166-2236(95)93901-9. [DOI] [PubMed] [Google Scholar]
- Hebb D. The Organization of Behavior. New York: Wiley; 1949. [Google Scholar]
- Kao JP. Practical aspects of measuring [Ca2+]i with fluorescent indicators. Methods Cell Biol. 1994;40:155–181. doi: 10.1016/s0091-679x(08)61114-0. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kawai H, Zago W, Berg DK. Nicotinic alpha 7 receptor clusters on hippocampal GABAergic neurons: regulation by synaptic activity and neurotrophins. J Neurosci. 2002;22:7903–7912. doi: 10.1523/JNEUROSCI.22-18-07903.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HG, Wang T, Olafsson P, Lu B. Neurotrophin 3 potentiates neuronal activity and inhibits gamma-aminobutyratergic synaptic transmission in cortical neurons. Proc Natl Acad Sci U S A. 1994;91:12341–12345. doi: 10.1073/pnas.91.25.12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalchuk Y, Hanse E, Kafitz KW, Konnerth A. Postsynaptic induction of BDNF-mediated long-term potentiation. Science. 2002;295:1729–1734. doi: 10.1126/science.1067766. [DOI] [PubMed] [Google Scholar]
- Martínez A, Alcántara S, Borrell V, Del Río JA, Blasi J, Otal R, Campos N, Boronat A, Barbacid M, Silos-Santiago I, Soriano E. TrkB and TrkC signaling are required for maturation and synaptogenesis of hippocampal connections. J Neurosci. 1998;18:7336–7350. doi: 10.1523/JNEUROSCI.18-18-07336.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey KA, Zago WM, Berg DK. BDNF up-regulates alpha7 nicotinic acetylcholine receptor levels on subpopulations of hippocampal interneurons. Mol Cell Neurosci. 2006;33:381–388. doi: 10.1016/j.mcn.2006.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meems R, Syed NI. Formation of synaptic contacts between identified molluscan neurons. In: Dityater A, EI-Husseini A, editors. Molecular mechanisms of synaptogenesis. New York: Springer; 2006. pp. 29–42. [Google Scholar]
- Meems R, Munno D, van Minnen J, Syed NI. Synapse formation between isolated axons requires presynaptic soma and redistribution of postsynaptic AChRs. J Neurophysiol. 2003;89:2611–2619. doi: 10.1152/jn.00898.2002. [DOI] [PubMed] [Google Scholar]
- Miller JD, Hadley RD, Hammond CE. Growth cone collapse and neurite retraction from cultured Helisoma neurons in response to antibody Fab fragments against an extracellular matrix protein. Brain Res Dev Brain Res. 1994;79:203–218. doi: 10.1016/0165-3806(94)90125-2. [DOI] [PubMed] [Google Scholar]
- Munno DW, Syed NI. Synaptogenesis in the CNS: an odyssey from wiring together to firing together. J Physiol. 2003;552:1–11. doi: 10.1113/jphysiol.2003.045062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narisawa-Saito M, Carnahan J, Araki K, Yamaguchi T, Nawa H. Brain-derived neurotrophic factor regulates the expression of AMPA receptor proteins in neocortical neurons. Neuroscience. 1999;88:1009–1014. doi: 10.1016/s0306-4522(98)00496-5. [DOI] [PubMed] [Google Scholar]
- Pandiella-Alonso A, Malgaroli A, Vicentini LM, Meldolesi J. Early rise of cytosolic Ca2+ induced by NGF in PC12 and chromaffin cells. FEBS Lett. 1986;208:48–51. doi: 10.1016/0014-5793(86)81529-0. [DOI] [PubMed] [Google Scholar]
- Ridgway RL, Syed NI, Lukowiak K, Bulloch AG. Nerve growth factor (NGF) induces sprouting of specific neurons of the snail, Lymnaea stagnalis. J Neurobiol. 1991;22:377–390. doi: 10.1002/neu.480220406. [DOI] [PubMed] [Google Scholar]
- Rose CR, Blum R, Pichler B, Lepier A, Kafitz KW, Konnerth A. Truncated TrkB-T1 mediates neurotrophin-evoked calcium signalling in glia cells. Nature. 2003;426:74–78. doi: 10.1038/nature01983. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Scheiffele P. Cell-cell signaling during synapse formation in the CNS. Annu Rev Neurosci. 2003;26:485–508. doi: 10.1146/annurev.neuro.26.043002.094940. [DOI] [PubMed] [Google Scholar]
- Shatz CJ. Impulse activity and the patterning of connections during CNS development. Neuron. 1990;5:745–756. doi: 10.1016/0896-6273(90)90333-b. [DOI] [PubMed] [Google Scholar]
- Spafford JD, Dunn T, Smit AB, Syed NI, Zamponi GW. In vitro characterization of L-type calcium channels and their contribution to firing behaviour in invertebrate respiratory neurons. J Neurophysiol. 2006;95:42–52. doi: 10.1152/jn.00658.2005. [DOI] [PubMed] [Google Scholar]
- Syed N, Richardson P, Bulloch A. Ciliary neurotrophic factor, unlike nerve growth factor, supports neurite outgrowth but not synapse formation by adult Lymnaea neurons. J Neurobiol. 1996;29:293–303. doi: 10.1002/(SICI)1097-4695(199603)29:3<293::AID-NEU2>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Syed NI, Bulloch AG, Lukowiak K. In vitro reconstruction of the respiratory central pattern generator of the mollusk Lymnaea. Science. 1990;250:282–285. doi: 10.1126/science.2218532. [DOI] [PubMed] [Google Scholar]
- Syed NI, Zaidi HY, Lovell P. In vitro reconstruction of neuronal networks: a simple model system approach. In: Windhurst U, Johansson H, editors. Modern techniques in neuroscience research. Berlin: Springer; 1999. pp. 361–377. [Google Scholar]
- Thoenen H. Neurotrophins and activity-dependent plasticity. Prog Brain Res. 2000;128:183–191. doi: 10.1016/S0079-6123(00)28016-3. [DOI] [PubMed] [Google Scholar]
- van Kesteren RE, Syed NI, Munno DW, Bouwman J, Feng ZP, Geraerts WP, Smit AB. Synapse formation between central neurons requires postsynaptic expression of the MEN1 tumor suppressor gene. J Neurosci. 2001;21:RC161. doi: 10.1523/JNEUROSCI.21-16-j0004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kesteren RE, Gagatek JS, Hagendorf A, Gouwenberg Y, Smit AB, Syed NI. Postsynaptic expression of an epidermal growth factor receptor regulates cholinergic synapse formation between identified molluscan neurons. Eur J Neurosci. 2008;27:2043–2056. doi: 10.1111/j.1460-9568.2008.06189.x. [DOI] [PubMed] [Google Scholar]
- Vicario-Abejón C, Collin C, McKay RD, Segal M. Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. J Neurosci. 1998;18:7256–7271. doi: 10.1523/JNEUROSCI.18-18-07256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci U S A. 2001;98:11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildering WC, Lodder JC, Kits KS, Bulloch AG. Nerve growth factor (NGF) acutely enhances high-voltage-activated calcium currents in molluscan neurons. J Neurophysiol. 1995;74:2778–2781. doi: 10.1152/jn.1995.74.6.2778. [DOI] [PubMed] [Google Scholar]
- Woodin MA, Hamakawa T, Takasaki M, Lukowiak K, Syed NI. Trophic factor-induced plasticity of synaptic connections between identified Lymnaea neurons. Learn Mem. 1999;6:307–316. [PMC free article] [PubMed] [Google Scholar]
- Woodin MA, Munno DW, Syed NI. Trophic factor-induced excitatory synaptogenesis involves postsynaptic modulation of nicotinic acetylcholine receptors. J Neurosci. 2002;22:505–514. doi: 10.1523/JNEUROSCI.22-02-00505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]