

Abstract
Prominin-1/CD133 (Prom-1) is a commonly used marker of neuronal, vascular, hematopoietic and other stem cells, yet little is known about its biological role and importance in vivo. Here, we show that loss of Prom-1 results in progressive degeneration of mature photoreceptors with complete loss of vision. Despite the expression of Prom-1 on endothelial progenitors, photoreceptor degeneration was not attributable to retinal vessel defects, but caused by intrinsic photoreceptor defects in disk formation, outer segment morphogenesis, and associated with visual pigment sorting and phototransduction abnormalities. These findings shed novel insight on how Prom-1 regulates neural retinal development and phototransduction in vertebrates.
Keywords: degeneration, retina, blindness, knockout, apoptosis, eye
Introduction
The Prominin gene family in mammals contains two members of multispan transmembrane glycoproteins, which are specifically sorted to microvilli and other plasma membrane protrusions (Weigmann et al., 1997; Corbeil et al., 2001; Fargeas et al., 2003a; Dubreuil et al., 2007; Florek et al., 2007). Both prominin molecules interact with plasma membrane cholesterol and are enriched in cholesterol-based membrane microdomains (Röper et al., 2000; Huttner and Zimmerberg, 2001; Janich and Corbeil, 2007). Prominin-1 (Prom-1, alias CD133) was originally identified and cloned in the mouse as a protein selectively concentrated at the apical domain of neuroepithelial progenitor cells and adult kidney proximal tubules (Weigmann et al., 1997), whereas human PROM-1 was first identified as a surface antigen (initially referred to as AC133) in hematopoietic stem and progenitor cells and cloned from WERI-Rb-1 retinoblastoma cell line (Miraglia et al., 1997; Yin et al., 1997).
Mounting evidence indicates that mouse/human Prom-1 (as defined in humans by its AC133 epitope) is expressed by other stem cells originating from various sources (for review, see Fargeas et al., 2007; Mizrak et al., 2008). Clinically, the transplantation of Prom-1+ cells results in efficient engraftment in different tissues, indicating the usefulness of Prom-1 as a somatic stem cell marker (de Wynter et al., 1998; Uchida et al., 2000; Stamm et al., 2003; Kelly et al., 2004; Torrente et al., 2004). Nevertheless, as highlighted from its original cloning, the general expression of Prom-1 is beyond stem cells given that, in both species, the protein has been detected in various differentiated epithelia (Weigmann et al., 1997; Fargeas et al., 2004; Florek et al., 2005; Jászai et al., 2007b; Immervoll et al., 2008; Karbanová et al., 2008; Lardon et al., 2008; Shmelkov et al., 2008) and nonepithelial cells, notably photoreceptor cells (Maw et al., 2000).
We previously analyzed a pedigree, produced by uncle–niece marriage, of eight children, four of whom were affected by retinal degeneration (Maw et al., 2000). Linkage analysis revealed that a 627 kb region on chromosome 4p segregated with visual impairment. Further analysis revealed that this region contained the PROM-1 gene, which exhibited a frameshift mutation in affected patients (Maw et al., 2000). Others have reported additional missense mutations, creating a premature stop codon (1726C>T) (Zhang et al., 2007) or a single amino acid substitution (R373C) (Yang et al., 2008), which cause retinal degeneration.
The importance of Prom-1 in the morphogenesis of photoreceptor cells is substantiated by its specific subcellular localization in the plasma membrane evaginations at the base of the outer segment (OS) (Maw et al., 2000). These membranes emerging from the connecting cilium are the precursor structures of the photoreceptive disks, which exhibit a high turnover, as they are shed from photoreceptors and phagocytosed by retinal pigment epithelium (RPE) cells. The molecular mechanisms underlying disk biogenesis and turnover still remain poorly understood (Steinberg et al., 1980). The genetic findings and particular localization of Prom-1 in photoreceptor cells suggest that it might play a role in the formation and/or stabilization of newly synthesized disks. However, the precise nature and underlying mechanisms of the photoreceptor degeneration in these individuals were not explored. For instance, it remains unknown whether photoreceptor degeneration results from a developmental or an adult defect, and whether it is an intrinsic photoreceptor defect or secondary to defects in other retinal cells. The latter question is relevant, as Prom-1/AC133 is expressed on vascular progenitors, and thus its deficiency could cause photoreceptor degeneration indirectly via vascular defects. This aspect is particularly pertinent given that individuals carrying a PROM-1 mutation do not exhibit any additional significant symptom despite the widespread distribution of the protein.
To address the questions listed above, we generated Prom-1 deficient mice and characterized retinal development and adult photoreceptor survival. Our findings uncover, for the first time, that Prom-1 is expressed very early during retinal development and acts as a key regulator of disk morphogenesis, to the extent that loss of Prom-1 causes photoreceptor degeneration and blindness.
Materials and Methods
Generation of Prom-1−/− mice.
To inactivate the Prom-1 gene in embryonic stem (ES) cells, a targeting vector pPNT. Prom-1 was constructed containing a 7.7 kb BamHI-Ssp fragment in which the region encompassing exon 2, which contains the start codon, was replaced by a neomycin phosphotransferase (neo) cassette (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). A thymidine kinase cassette was cloned outside of the homology region to enable selection against random integration events. The linearized targeting vector was used for electroporation into R1 ES cells as described previously (Stalmans et al., 2002). Homologous recombinant clones were identified by appropriate Southern blot analysis. This included Southern blotting of BamHI/NciI digested genomic DNA with a probe corresponding to the NheI-BamHI fragment located ∼1 kb downstream of the 3′ homology, which identifies a 10.8 kb and a 9.2 kb band for the wild type (WT) and the targeted allele, respectively (Fig. 1a). Prom-1−/− ES clones were generated by culturing Prom-1+/− ES clones in 1000 μg/ml G418.
Figure 1.

Absence of Prom-1 leads to retina degeneration. a, Southern blot of BamHI-NciI-digested genomic DNA from ES cell clones generated a unique 10.8 kb band in WT (left lane) and an additional 9.2 kb allele in Prom-1+/− clones (right lane). b, Western blot analysis with an antibody (13A4) for Prom-1 on extracts from WT (left lane) and Prom-1−/− (right lane) kidney confirmed the deletion of Prom-1 protein (115 kDa) in the knock-out mouse. c–k, A time course morphometric analysis of ONL areas clearly indicates that the ONL (NBL for P0) area progressively decreases in Prom-1−/− mice. Shown are hematoxylin and eosin staining of WT (c–f) and Prom-1−/− (g–j) retinal sections at P0 (c, g), P15 (d, h), P20 (e, i), and 6 months (f, j). The quantification of the ONL area over time confirmed that degeneration becomes morphologically evident in Prom-1−/− mice starting from P15 (k). *p < 0.05 versus WT. l, Semithin sections (1 μm) of 10-week-, 6-month- and 12-month-old mice stained with toluidine blue showing that the thickness of the ONL is severely reduced in KO mice at 10 and 24 weeks of age and completely missing in older Prom-1−/− animals. No significant differences are evident in Prom-1+/− mice. OPL, Outer plexiform layer. Dashed line, external limiting membrane. The asterisk in j and l indicates the degenerated photoreceptor layer. Scale bar, 20 μm.
Correctly targeted Prom-1+/− ES clones were used for embryo aggregation with Swiss morula stage embryos as described (Stalmans et al., 2002) to generate chimeric and transgenic animals. Genotyping of the mice was done by PCR on genomic DNA isolated from tail biopsies. PCR with forward primer A (5′-gaa agt atc cat cat ggc tga tgc aat gcg-3′) and reverse primer B (5′-cgc cgc caa gct ctt cag caa tat c-3′), annealing in the neo cassette, amplify a 340-bp knockout-specific band. PCR with forward primer X (5′-ggc caa caa cta tgg ctc tcg tct tca gtg-3′) and reverse primer Z (5′-gta gag agg gcc aac aat ccc agc att g-3′), annealing in exon 2 of the Prom-1 gene, amplify a 178-bp wild type-specific band.
The original mouse line, having a 50% 129/Sv × 50% Swiss genetic background, was back-crossed into a C57BL/6 background for >10 generations, yielding congenic C57BL/6 WT and Prom-1−/− mice. All animal experiments were approved by the local experimental animal committees at the respective research institutes and adhered to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic Research.
In situ hybridization, immunohistochemistry and morphometric analysis.
Eyes were harvested for morphological analysis as described previously (Stalmans et al., 2002). Briefly, mice were anesthetized, eyes were dissected and fixed overnight in 4% phosphate buffered paraformaldehyde. Eyes were then dehydrated or cryo-protected in 20% sucrose and embedded in paraffin or Tissue-tek, respectively. To analyze the various retinal layers in a precise orientation, the superior pole of each eye was marked with a suture at the time of enucleation (Okoye et al., 2003) and embedded in a fixed position. Serial sections were cut at 10 μm thickness, and one series was stained with hematoxylin and eosin. Immunostainings were performed on paraffin or cryo-sections after incubation of sections in antigen retrieval solution (Dako) for 20 min at 95°C. The following primary antibodies were used: rat anti-prominin-1 (clone 13A4, eBioscience) (Weigmann et al., 1997), mouse anti-rhodopsin (Sigma), rabbit anti-opsin and rabbit anti-recoverin (both Millipore Bioscience Research Reagents), mouse anti-calbindin (Sigma), rabbit anti-calretinin (Swant), mouse anti-glial fibrillary acidic protein (GFAP) (Dako Cytomatation), mouse anti-glutamine synthetase (Millipore), mouse anti-protein kinase C (PKC)-α (Santa Cruz), mouse anti-neuronal nuclei antigen (NeuN) (Millipore), FITC-conjugated mouse anti-peanut agglutinin (PNA) (Vector Technologies). Sections were subsequently incubated with fluorescently conjugated secondary antibodies (Alexa 488 or 546, Invitrogen) or with peroxidase-labeled IgGs (Dako), followed by amplification with the proper tyramide signal amplification systems (Perkin-Elmer Life Sciences). Detection of apoptotic cells in the retinas was performed using the In Situ Cell Death Detection kit (Roche) according to the manufacturer's protocol.
Immunostainings were examined using a Zeiss microscope equipped with epifluorescence illumination (Axioplan 2) and morphometric analyses were performed using KS300 image analysis software. Analysis procedures were adjusted for the growth of eyeballs; every fifth section for ages up to postnatal day 5 (P5), every 10th section for ages from P7 to P15 and every 15th section for ages P20 and older. At least 6 eyes from 6 different mice were analyzed for each time point and 4 measurements each were performed for upper and lower hemisphere starting immediately adjacent to the optic nerve, resulting in 8 measurements per eye. Morphometric analysis to quantify photoreceptor loss was performed by measuring the whole retina thickness from the internal limiting membrane up to the pigmented epithelium and the thickness of neuroblast layer (NBL) (at P0), inner nuclear layer (INL) and outer nuclear layer (ONL) (beyond P5).
Whole-mount immunostainings on isolated retinas were performed as described previously (Stalmans et al., 2002). Retinas were incubated with Alexa-647 conjugated isolectin IB4 (from Griffonia simplicifolia, Invitrogen) for 6 h, or overnight at 4°C with the primary anti-GFAP antibody, followed by incubation with an Alexa-488 labeled secondary antibody (Invitrogen) for 2 h.
In situ hybridization was performed according to standard protocols (Tiveron et al., 1996). Briefly, serial sections were hybridized for 16 h at 70°C with digoxigenin (DIG) labeled cRNA probes (see below) at a concentration of 0.5 ng/μl. Stringency washes were performed at 70°C. The sections were then incubated with anti-DIG antibody (1:4000; Roche Molecular Biochemicals) for 16 h at 4°C. After several washing steps the reaction was visualized using NBT-BCIP (nitro-blue tetrazolium chloride and 5-bromo-4-chloro-3′-indolyphosphate p-toluidine salt) substrate (Roche) resulting in a blue reaction product. After stopping the color reaction by several washes in PBS, the sections were rinsed quickly in dH2O and then mounted with Kaiser's Glycerol-Gelatin (Merck). Images were captured using an Olympus BX61 microscope with the IPLAB software.
For electron microscopy, eyes were prefixed in 4% paraformaldehyde in 0.1 m phosphate buffer on ice, fixed overnight in 1% glutaraldehyde in 0.1 m phosphate buffer at 4°C, contrasted with 2% osmium tetroxide (at room temperature) and 0.5% uranyl acetate (on ice) in water, dehydrated with ethanol and embedded in EmBed resin (Science Services). Sectioning was performed on an Ultracut Microtome (Leica Microsystems). Semithin sections were stained with toluidine blue. Ultrathin sections were poststained with uranyl acetate and lead citrate and viewed under an electron microscope (Morgagni, FEI). Micrographs were taken with a digital camera (MegaviewII, Soft Imaging System). For cone detection, vibratome sections were incubated with PNA (0.2 μg/ml; Invitrogen), followed by an ultrasmall gold-coupled anti-rabbit antibody (Aurion, NL) and silver enhancement (R-GENT SE-EM, Aurion, NL); samples were osmicated, dehydrated in ethanol and embedded as for standard electron microscopy.
Angiography.
SLO imaging was performed with a Heidelberg Retina Angiograph (HRA I; Heidelberg Engineering), a confocal-scanning laser ophthalmoscope (SLO), as previously described (Seeliger et al., 2005). The confocal diaphragm of the SLO allows visualization of different planes of the posterior pole, ranging from the surface of the retina down to the RPE and the choroid. The HRA features two argon wavelengths (488 and 514 nm) in the short wavelength range and two infrared diode lasers (795 and 830 nm) in the long wavelength range. The laser wavelength of 488 nm (795 nm) was used for fluorescein (indocyanin-green, ICG) angiography, with a barrier filter at 500 nm (800 nm).
Electroretinography.
Ganzfeld electroretinograms (ERGs) were obtained from anesthetized mice (ketamine (66.7 mg/kg) and xylazine (11.7 mg/kg)) with dilated pupils as described previously (Seeliger et al., 2001). The ERG equipment consisted of a Ganzfeld bowl, a direct coupling amplifier, and a personal computer-based control and recording unit (Toennies Multiliner Vision; Viasys Healthcare). Bandpass filter cutoff frequencies were 0.1 and 3000 Hz. Single white-flash recordings were obtained under both dark-adapted (scotopic) and light-adapted (photopic) conditions. Light adaptation before the photopic session was performed with a background illumination of 30 cd/m2 for 10 min. Single white-flash stimulus intensities were increased from 1 × 10−4 to 25 cd/m2, divided in 10 steps of 0.5 and 1 log cd/m2. Ten responses were averaged with interstimulus interval of 5 or 17 s (for 1, 3, 10, 25 cd/m2).
Statistics.
Data are expressed as mean ± SEM and analyzed using a two-tailed Student t test or a nonparametric Mann–Whitney U test, with p < 0.05 considered significant.
Results
Generation of Prom-1−/− mice
To generate mice lacking Prom-1, we targetedly deleted exon 2 using standard techniques (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Homozygous Prom-1 deficient (Prom-1−/−) mice, sired by Prom-1+/− or Prom-1−/− breeding pairs, were born at a normal Mendelian inheritance frequency, viable and fertile, and had a normal lifespan (data not shown). No other obvious abnormalities were observed upon macroscopic inspection and histological analysis of various organs (data not shown). Southern and Western blotting confirmed correct targeting of the Prom-1 gene (Fig. 1a,b). Moreover, in Prom-1−/− retinas, Prom-1 mRNA was undetectable by in situ hybridization in the photoreceptor layer, identified by the presence of Crx mRNA (supplemental Fig. S3a–d, available at www.jneurosci.org as supplemental material). Consistently, Prom-1 immunostaining decorated the OS of WT and Prom-1+/−, but not Prom-1−/− mice (supplemental Fig. S2a–c, available at www.jneurosci.org as supplemental material). Of notice, loss of Prom-1 did not induce a compensatory upregulation of Prom-2 expression in Prom-1−/− retinas (supplemental Fig. S3e,f, available at www.jneurosci.org as supplemental material).
Loss of Prom-1 causes photoreceptor cell degeneration
Since retinogenesis in the mouse initiates from midgestation onwards and proceeds until 3 weeks after birth, we first screened retinal sections by hematoxylin and eosin staining at various embryonic and postnatal ages. No major changes in the thickness of the various retinal layers were detected in Prom-1−/− mice, including of the NBL during development (data not shown), or of the ONL, where photoreceptor cells differentiate from retinal progenitors, at birth (Fig. 1c,g) and at P7 (data not shown).
However, beyond P15, the ONL underwent progressive degeneration (Fig. 1d–f,h–j). Morphometric analysis revealed that, compared with WT mice, the thickness of the ONL in Prom-1−/− retinas was reduced by 16 ± 3%, 55 ± 3% and 64 ± 2% at P15, P20 and P90, respectively (n = 8–11, p < 0.0005) (Fig. 1k), indicating that photoreceptors progressively disappeared from the ONL. By 6 months of age, only two or, at most, three rows of photoreceptor nuclei persisted in the ONL of Prom-1−/− mice, while up to 12 rows were present in WT mice (Fig. 1f,j; supplemental Fig. S2, available at www.jneurosci.org as supplemental material). The progressive loss of the ONL was specific, as the INL area was comparable in both genotypes at 3 months of age (0.140 ± 0.003 μm2 in WT vs 0.138 ± 0.005 μm2 in Prom-1−/−; n = 5, p = NS), and no significant differences were observed in any other retinal layer (data not shown). Semithin sections confirmed that the ONL was abnormally thin in Prom1−/− mice at 10 and 24 weeks of age, and was completely missing in older Prom-1−/− animals (Fig. 1l). Only homozygous but not heterozygous loss of Prom-1 caused photoreceptor degeneration (Fig. 1l; supplemental Fig. S2, available at www.jneurosci.org as supplemental material; Table 1), indicating that the phenotype is recessive.
Table 1.
Cell-type composition of retinas from WT, Prom-1+/−, and Prom-1−/− at two different time points during postnatal development as determined by immunostaining for cell-type-specific markers
| Marker | Cell type | P10 |
P20 |
||||
|---|---|---|---|---|---|---|---|
| WT | +/− | −/− | WT | +/− | −/− | ||
| Recoverin | Rods and cones | 62.2 ± 5.3 | 68.7 ± 8.1 | 66.1 ± 7.1 | 74.1 ± 7.4 | 70.2 ± 7.3 | 43.2 ± 6.4 |
| Rhodopsin | Rods | 43.4 ± 5.1 | 40.3 ± 3.9 | 41.4 ± 2.8 | 58.9 ± 5.1 | 54.4 ± 7.7 | 24.4 ± 4.5 |
| PNA | Cones | 1.16 ± 0.2 | 1.21 ± 0.2 | 1.01 ± 0.2 | 1.47 ± 0.3 | 1.42 ± 0.3 | 0.98 ± 0.2 |
| Calbindin | Horizontal cells | 2.28 ± 0.3 | 2.48 ± 0.3 | 2.08 ± 0.5 | 3.85 ± 0.7 | 3.48 ± 0.5 | 3.22 ± 0.6 |
| Calretinin | Amacrine cells | 3.43 ± 0.7 | 2.95 ± 0.8 | 3.28 ± 0.8 | 5.27 ± 0.7 | 4.99 ± 0.9 | 5.96 ± 1.0 |
| PKC-α | Bipolar cells | 24.3 ± 5.6 | 26.4 ± 1.1 | 21.9 ± 1.7 | 42.0 ± 6.9 | 39.4 ± 9.7 | 35.5 ± 3.3 |
| NeuN | RGCs | 3.95 ± 0.2 | 4.14 ± 0.2 | 3.89 ± 0.2 | 4.85 ± 0.9 | 4.40 ± 0.4 | 4.80 ± 0.5 |
| GFAP | Müller glia | 0.90 ± 0.2 | 1.11 ± 0.3 | 1.09 ± 0.3 | 1.46 ± 0.2 | 1.69 ± 0.9 | 14.2 ± 2.5 |
RGCs, Retinal ganglion cells. Bold indicates statistical significance versus WT (p < 0.02).
To better characterize the photoreceptor degeneration in Prom-1−/− mice, we analyzed their retinal ultrastructure by electron microscopy (Fig. 2). At P2, when only the NBL (but not yet the OS) has formed, no genotypic differences could be detected (Fig. 2a,b). At P12, connecting cilia appeared normal in Prom-1−/− retinas (arrows), but disk membranes of both rod and cone OS were underdeveloped and morphologically abnormal (Fig. 2c,d, black asterisk). Beyond P20, disorganization of disk membranes and disappearance of photoreceptors became progressively more evident (shown at 10 weeks in Fig. 2e,f), resulting in the almost complete absence of a normal OS at 7 months (Fig. 2g,h). The OS defect was specific, as the morphology of two other structures, rich in membrane protrusions, such as microvilli (Fig. 2i,j, white arrows) and basal infoldings of RPE cells (Fig. 2k,l, black arrowheads), appeared normal in Prom-1−/− eyes (the integrity of RPE cells at P2, P12 and 12 weeks is also shown in supplemental Fig. S2, available at www.jneurosci.org as supplemental material). We then determined whether OS defects only occurred in rod photoreceptors or simultaneously affected cones. At P12, the disk stacked in the OS of cones appeared already profoundly disorganized in Prom-1−/− (Fig. 2m,n), thus indicating that both photoreceptor types undergo degeneration at the same time. Electron microscopy after incubation of retinal sections with peanut agglutinin [PNA, which labels cone extracellular matrix (Sameshima et al., 1987)], followed by ultrasmall gold detection and silver enhancement, revealed major abnormalities in PNA-labeled cones at 12 weeks (Fig. 2o).
Figure 2.
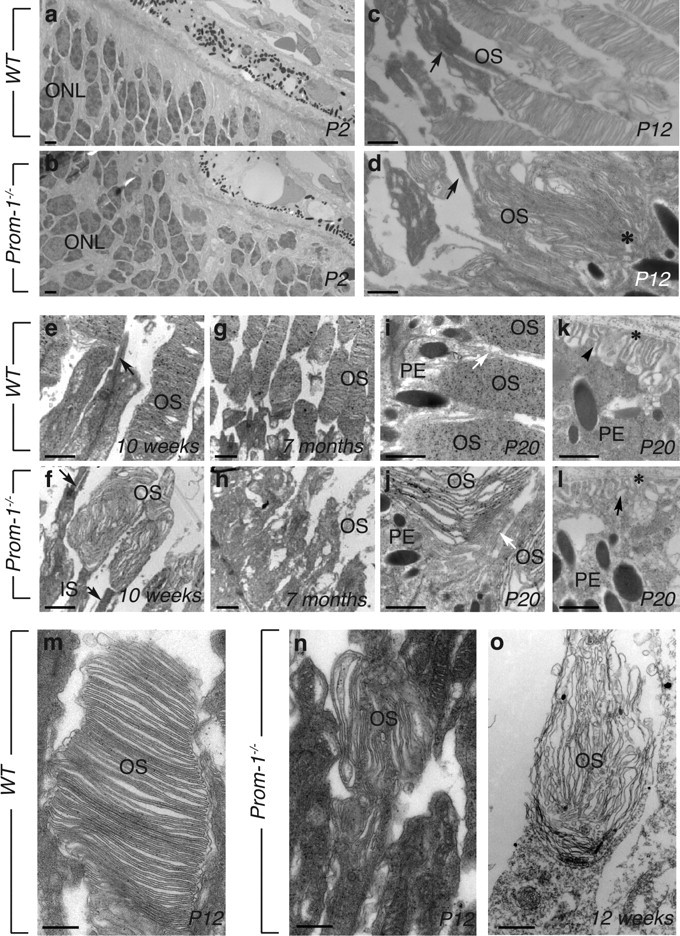
Abnormal OS morphogenesis in Prom-1−/− mice. Time course study of OS morphology by TEM. a, b, Ultrastructure analyses of WT (a) and Prom-1−/− (b) retinas at P2 shows that in both cases the OS is not formed yet. c, d, At P12, the OS of Prom-1−/− mice appears already abnormal (black asterisk in d), indicating that the OS membranes are misfolded early during their development. At later stages, a progressive disorganization of the rod OS is evident in Prom-1−/− mice at 10 weeks (compare f with e) and 7 months (compare h with g), when the photoreceptors have almost disappeared. Black arrows in c–f indicate connecting cilia. In contrast, the RPE layer is well preserved in Prom-1−/− retina, with intact microvilli (white arrows in i and j) and basal infoldings (black arrowheads in k and l; asterisk, Bruch' membrane). m–o, Ultrastracture analysis of cones, as recognized by their position and PNA labeling, followed by ultrasmall gold detection and silver enhancement. Whereas the disks appear regularly stacked in the OS of cones in WT mice at P12 (m), they are completely disarranged in Prom-1−/− retinas (n), indicating that both rods and cones already display abnormal OS structures at this age. Even more profound alterations are evident in PNA-labeled cones at 12 weeks (o). Black dots are silver particles representing the PNA staining of the OS and IS of the cone cells. IS, Inner segment; PE, pigmented epithelial cells. Scale bars: a–l, 1 μm, m–o, 200 nm.
Since retinal gliosis often accompanies photoreceptor degeneration (Semple-Rowland, 1991), we also characterized the expression of GFAP, a marker of reactive glial cells. At P20, the loss of recoverin+ photoreceptors in Prom-1−/− retinas (Table 1; supplemental Fig. S5k,l, available at www.jneurosci.org as supplemental material) was paralleled by a marked upregulation of GFAP+ cells in the Müller glial layer (supplemental Fig. S5m,n, available at www.jneurosci.org as supplemental material). Such a gliosis was not detected earlier in development in Prom-1−/− retinas (Table 1). Overall, loss of Prom-1 caused a specific degeneration of photoreceptors, without affecting other retinal cell types (see also below).
Loss of Prom-1 impairs vision in adult mice
To investigate the functional consequences of these morphological photoreceptor defects, Ganzfeld ERGs were recorded in adult mice under dark and light adapted conditions (Fig. 3). At 6 months of age, rod and cone system responses were almost completely absent. At 1 month, the amplitude of the rod responses, selectively accessible at low stimulus intensities in the scotopic ERG, was reduced by ∼50% in Prom-1−/− mice, without major changes in the overall ERG waveform (Fig. 3a,b). The 6 Hz flicker ERG also revealed a lower sensitivity of rod photoreceptors, leading to a right shift of the amplitude peak (Fig. 3f). The scotopic mixed responses, recorded at higher stimulus intensities in the scotopic ERG, lacked a regular a-wave and had reduced oscillatory potentials, i.e., higher frequent transient wavelets riding on the b-wave (Fig. 3a). The cone photoreceptor system was similarly affected, with a decrease in the amplitude (by ∼50%) (Fig. 3d) and sensitivity of the photopic ERG (Fig. 3c). These visual defects are consistent with and confirm the morphological defects, i.e., loss of photoreceptors and abnormal (irregular, shortened) morphology of the OS, in Prom-1−/− mice.
Figure 3.
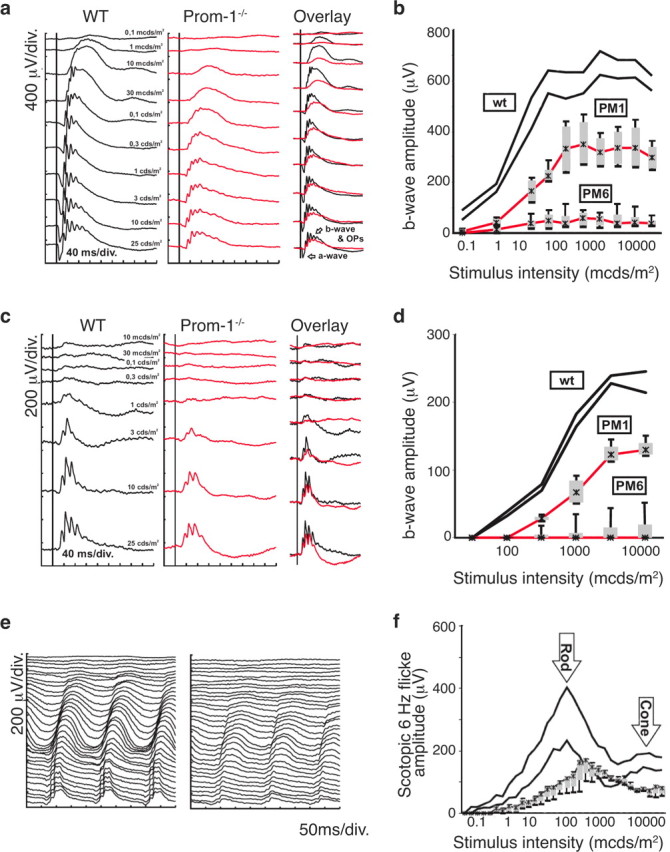
Impaired rod and cone function in Prom-1−/− mice. Functional evaluation of Prom-1−/− mice at 1 month (PM1) and 6 months (PM6) by Ganzfeld ERG. a, b, Scotopic (dark-adapted) single-flash recordings. a, Comparison of representative records between Prom-1−/− mice and WT controls at PM1. b, Statistical evaluation (box-and-whisker plot) of Prom-1−/− mice at PM1 and PM6 compared with WT mice (PM1). The two black lines indicate the normal range (5 to 95% quantile). The red lines give the median, the gray boxes the 25 and 75% quantiles, and the black whiskers the 5 and 95% quantiles of Prom-1−/− mice at PM1 and PM6, respectively. c, d, Photopic (light-adapted) single-flash recordings. c, Comparison of representative records between Prom-1−/− mice and WT controls at PM1. d, Statistical evaluation (box-and-whisker plot as above) of Prom-1−/− mice at PM1 and PM6 compared with WT mice (PM1). e, f, Scotopic flicker recordings at 6Hz flash frequency. e, Comparison of representative records between Prom-1−/− mice (right) and WT controls (left) at PM1. f, Statistical evaluation (box-and-whisker plot) of Prom-1−/− mice compared with WT mice at PM1. The right shift of both rod and cone peaks indicates a reduction in sensitivity to light, and the drop in amplitude an additional reduction of functional cell numbers.
Normal vascular development in Prom-1−/− retinas
Since Prom-1 is expressed by endothelial progenitors (Gehling et al., 2000; Peichev et al., 2000; Uchida et al., 2000) and vascularization occurs coincidentally with neural development in the postnatal murine retina (Mukouyama et al., 2002; Carmeliet and Tessier-Lavigne, 2005), we analyzed retinal vascular development in Prom-1−/− mice. To label blood vessels, we stained whole mount retinas with isolectin IB4 (Stalmans et al., 2002). At P5, the density of the superficial retinal vascular plexus was comparable in both genotypes (IB4+ area, percentage of total retinal area: 48 ± 2% in WT vs 43 ± 3% in Prom-1−/−; n = 3, p = NS) (Fig. 4a,b). Retinal vessels also grew out over a comparable distance (percentage of the distance from the center to the periphery: 67 ± 2% in WT vs 61 ± 5% in Prom-1−/−; n = 3, p = NS). At P10, the superficial vascular plexus reached the periphery in both genotypes, and the vascular density was comparable (Fig. 4c,d). Similar results were obtained for the deep vascular plexus at P10 (IB4+ area, percentage of total retinal area: 69 ± 2% in WT vs 68 ± 3% in Prom-1−/− retinas; n = 3, p = NS). Since vessels in the retina navigate onto astrocytes, we also studied the astrocyte network by GFAP staining. At P5, GFAP+ glial cells developed normally to the retinal periphery in both genotypes, consistent with the normal blood vessel formation (Fig. 4e–h).
Figure 4.
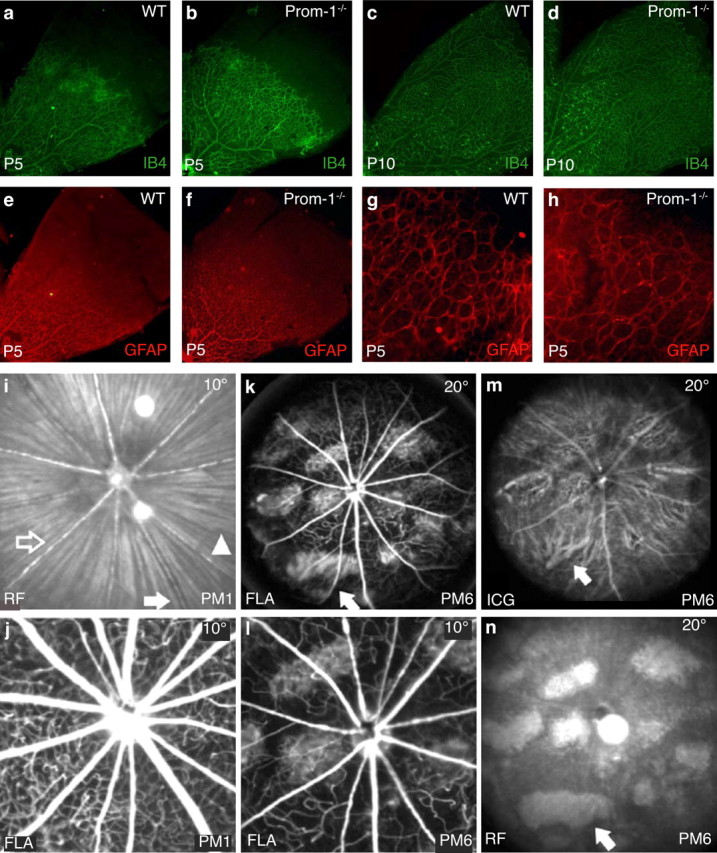
Development and involution of the vascular system in Prom-1−/− retinas. a–h, Normal development of retinal vessels and astrocytic templates in Prom-1−/− retinas. Whole mount staining for isolectin IB4 showing a comparable development of retinal vasculature at P5 in Prom-1−/− retinas (b) and WT mice (a). An equivalent pattern of Isolectin IB4 staining was also observed in WT (c) and Prom-1−/− (d) retinas at P10. GFAP staining at P5 reveals that astrocytic networks also develop similarly in both WT (e) and Prom-1−/− retinas (f). A higher magnification view of peripheral retinas shows that GFAP-positive networks reach fully to the periphery in both WT (g) and Prom-1−/− retinas (h). i–n, Involution of retinal vessels during the course of degeneration in Prom-1−/− mice. Retinal arteries (open arrow) and veins (filled arrow) in Prom-1−/− retinas appeared normal at 1 month postnatally (PM1), as shown by red-free (RF) surface imaging (514 nm, 10°; i), and fluorescence angiography (FLA, 488 nm, barrier at 500 nm, 10°; j). k–n, At 6 months postnatally (PM6), the vasculature appears strongly attenuated. FLA (20°) shows reduction of retinal capillaries, reduced diameter and caliber fluctuations of large retinal vessels, and extended areas of choriocapillaris/RPE loss (arrow) (k). l, Magnification of (k), i.e., FLA (10°), same area as in j. ICG angiography (795 nm, barrier at 800 nm, 20°) enhances the view of choroidal structures because of less absorption by melanin (m). The arrow points to the same area as in (k). An RF surface image (20°) most clearly depicts the lesions (n). The arrow points to the same area as in k and m.
The vasculature of the retina and choroid was also studied in vivo by using scanning laser ophthalmoscopy (native imaging and angiography) (Seeliger et al., 2005). At 1 month, both large and small retinal vessels appeared normal in Prom-1−/− retinas (Fig. 4i,j). However, by 6 months of age, the lumen of the large vessels and the capillary density were decreased in Prom-1−/− retinas (Fig. 4l). These findings are similar to what is observed in humans with retinal degeneration of the Retinitis Pigmentosa type, where vessel regression occurs secondarily to photoreceptor degeneration (Hartong et al., 2006). Native imaging with a green laser (514 nm) further revealed a loss of RPE pigmentation in several areas at 6 months of age (Fig. 4n). As this reduces the absorbance of light, the underlying choroid vessels become better visible at these sites (Fig. 4k–m, “window effect”). In addition, the normal fine haze in ICG angiography associated with the dye in the choriocapillary layer is missing in these regions (Fig. 4m, arrow). Semithin and transmission electron microscopy (TEM) analysis of RPE at this age revealed that RPE cells are still present, albeit with slight morphological alterations, in Prom-1−/− retinas (data not shown). These findings suggest a late alteration of the RPE, presumably secondary to photoreceptor degeneration and reduced choriocapillary perfusion. In summary, retinal and choroidal vessels are abnormal in Prom-1−/− mice at 6 months of age, reminiscent of those found in humans with loss-of-function Prom-1 mutations (Zhang et al., 2007). However, since the vessels initially developed and functioned normally at 1 month, when photoreceptor degeneration had already commenced, the vessel defects are likely secondary to the photoreceptor degeneration.
Expression of Prom-1 in the developing retina
As vascular defects appeared only late in Prom-1−/− mice, it is unlikely that the retinodegeneration is caused by the absence of Prom-1 in endothelial progenitors. Therefore, we characterized Prom-1 expression in the retina in more detail. At birth, Prom-1 expression was detected at the interface between the developing neuroretina and RPE layer (Fig. 5a), where cells of the NBL differentiate to photoreceptors (Fig. 5b). In the postnatal retina (P20), Prom-1 immunoreactivity was detected in photoreceptors, with particular concentration in the region of plasma membrane evaginations at the basal part of the OS (Fig. 5c,d). Double-labeling of retinal sections for Prom-1 and either rhodopsin or PNA confirmed that rod and cone photoreceptors expressed Prom-1, most abundantly between the OS and IS, in close proximity to the connecting cilium (shown at two different magnifications in Fig. 5c,d). Similar expression of Prom-1 in rods and cones was obtained after double-staining of dissociated adult retinal cells, followed by confocal microscopy (supplemental Fig. S4, available at www.jneurosci.org as supplemental material) and fluorescence-activated cell sorting analysis (data not shown). The expression of Prom-1 in the retina was specific, as Prom-2 was not detectable in the embryonic or postnatal retina (supplemental Fig. S3e,f, available at www.jneurosci.org as supplemental material). Thus, Prom-1 is strongly expressed by rod and cone photoreceptors throughout adult life, consistent with the simultaneous occurrence of structural abnormalities in both photoreceptor types observed in Prom-1−/− mice (Fig. 2).
Figure 5.
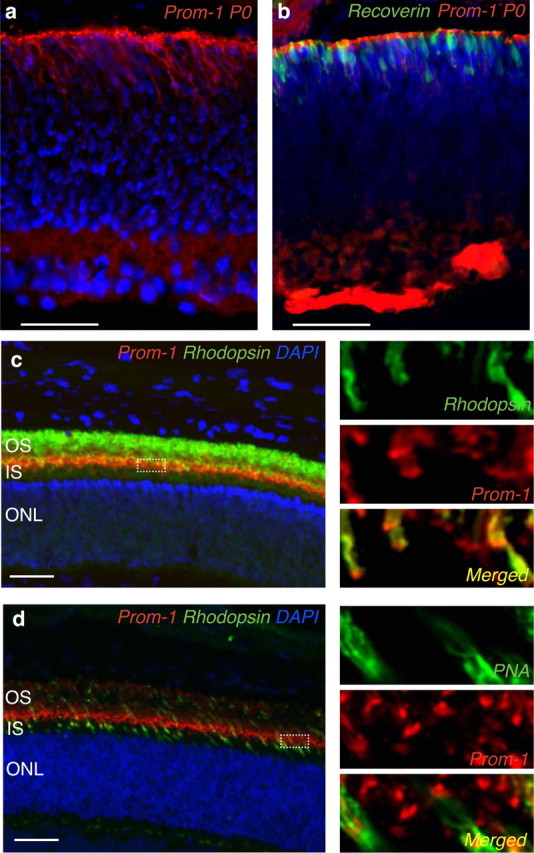
Localization of Prom-1 protein in WT retina. a, b, Immunofluorescent staining (red) shows that at P0 Prom-1 protein is expressed at the interface between the neural and epithelial retina (a), when developing photoreceptors can be recognized by recoverin staining (b). c, d, Confocal microscopy showing Prom-1 expression at P20 at the basal part of the OS of adult rods and cones, as assessed by double immunostaining for Prom-1 in red and either rhodopsin (c) or PNA (d) in green. The higher magnification insets on the right part of each figure better highlight the subcellular localization of Prom-1 in rods and cones, as shown by the yellow color resulting in the merged pictures (yellow, lower insets). IS, Inner segment. Scale bar, 50 μm.
Abnormal distribution of visual pigment in Prom-1−/− photoreceptors
As Prom-1 was concentrated in the region of the connecting cilium, a structure that mediates sorting of OS proteins (Sung and Tai, 2000), we sought to characterize the subcellular distribution of visual pigments in photoreceptors. Correct OS morphogenesis and proper localization of opsins in disk membranes is a prerequisite for normal phototransduction and photoreceptor cell viability. Even more, any disturbance of the polarized sorting of these pigments is toxic for photoreceptor cells, probably because mis-sorted proteins ectopically localize to other membranes, resulting in a deleterious “membrane crowding” at the level of the synapse (Deretic et al., 1998). Immunostaining revealed that all the rhodopsin (i.e., rod opsin) pigment was localized in the rod OS in WT mice at P20. In contrast, and consistent with the abnormal OS morphogenesis, this polarized sorting of the pigment was disturbed in several Prom-1−/− photoreceptors, resulting in ectopic localization of rhodopsin pigment in the ONL, despite normal total amounts of the pigment (Fig. 6a–c) (data not shown). Immunostaining for cone opsin also revealed a shortened length of the OS and disorganized distribution of pigment, with abnormal pigment accumulation in the ONL (Fig. 6d–f). Thus, the presence of Prom-1 in the cilium is required for proper OS morphogenesis and subsequent pigment sorting in photoreceptors.
Figure 6.
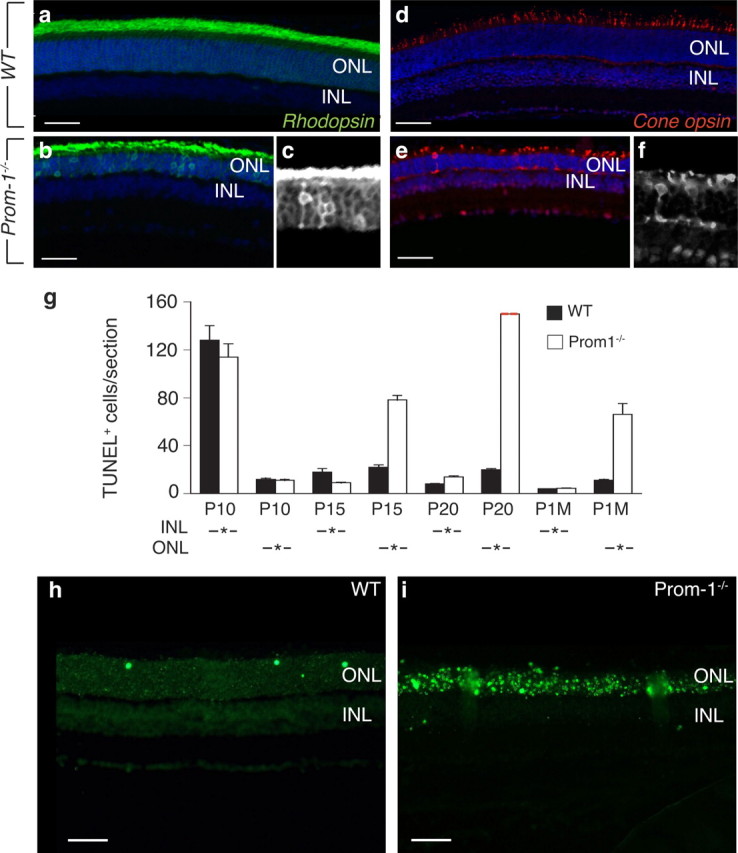
Abnormal pigment sorting and photoreceptor apoptosis in Prom-1−/− mice. a–f, Pigment mislocalization in Prom-1−/− retinas. Rhodopsin immunostaining reveals pigment mislocalization at P20 in Prom-1−/− retinas (b), where the ONL still retains rhodopsin signal, while almost no staining is evident in ONL of WT retinas (a). Similarly, cone opsin staining at P20 reveals shortened OS in cone photoreceptors of Prom-1−/− retinas and pigment mis-sorting to the ONL and the synaptic space (e) compared with WT retinas (d). Black and white magnification insets (c, f) better highlight the abnormal localization of rod and cone opsin, respectively. g, A time course study of TUNEL assay shows an increased number of apoptotic cells in the ONL starting at P15 in Prom-1−/− retinas, with a dramatic peak at P20, when the number of apoptotic cells was not countable (red dashed line). Data are presented as the mean number of TUNEL+ cells per section in the INL or ONL area. h, i, Representative pictures of TUNEL staining, showing the presence of massive apoptosis in Prom-1−/− (i) ONL, compared with WT (h) at P20. Scale bars: a–f, 100 μm; h, i, 50 μm.
Increased apoptosis of photoreceptors in Prom-1−/− mice
Since impaired OS morphogenesis, in combination with ectopic distribution of visual pigment, may lead to photoreceptor degeneration (Jansen and Sanyal, 1984; Bascom et al., 1992), we studied, by TUNEL staining, whether loss of Prom-1 induced excessive retinal cell apoptosis. No genotypic differences were observed in retinal cell apoptosis in the INL and ONL at P10 (n = 6, p = NS) (Fig. 6g). In contrast, by P15, numerous cells in the ONL became apoptotic in Prom-1−/− but not WT retinas, in particular in the region, which contained strongly Prom-1 stained cells in WT retinas. Apoptosis in the INL was comparable in both genotypes (Fig. 6g). By P20, most photoreceptors in the ONL of Prom-1−/− retinas were apoptotic (Fig. 6g–i) (n = 6, p < 0.002). The increased apoptosis was not attributable to opening of eyelids and light exposure, which normally occurs at P13, as increased retinal apoptosis in the ONL was also observed when Prom-1−/− offspring were maintained in the dark until P20 (data not shown).
Finally, we checked whether the degeneration specifically affected the photoreceptors, or whether other retinal cell types were similarly affected, by staining retinal sections for markers of the different retinal cell types. We did not observe genotypic differences in the number of the other retinal cell populations at P10 (Table 1; supplemental Fig. S5a,j, available at www.jneurosci.org as supplemental material), suggesting that Prom-1 deficiency did not affect the overall retinal development. Beyond P15, as expected, both kinds of photoreceptors started to degenerate, as discussed above (Table 1), but no differences in the number of the other retinal cell populations could be observed at any time point, not even at P20, when all cell types were born (Table 1; supplemental Fig. S5, available at www.jneurosci.org as supplemental material). Thus, loss of Prom-1 specifically impaired photoreceptor OS morphogenesis, without affecting the development of any other retinal cell population.
Discussion
Prom-1 is a well known and widely used marker of several types of progenitor cells (Shmelkov et al., 2005), yet its precise function and importance in vivo remain largely unknown. By characterizing retinal development and maintenance in Prom-1−/− mice, we discovered novel roles of Prom-1 in photoreceptor differentiation and survival. The cardinal findings of this study are the following: (1) Prom-1 is expressed by rod and cone photoreceptors; and (2) in the absence of Prom-1, progressive degeneration and functional deterioration of both cone and rod photoreceptors are associated with impaired morphogenesis of the disks and OS.
By scoring retinal cell composition at different time points in vivo, we could not detect major genotypic differences until P10, it is thus unlikely that Prom-1 regulates retinogenesis by controlling the differentiation of the various retinal cell types. A late abnormality in Prom-1−/− retinas was upregulation of GFAP at P20, but this is likely reflecting a secondary response to the retinal degeneration, as is often observed in other models of retinal degeneration (Semple-Rowland, 1991). Of notice, Prom-1 seems to be necessary for proper morphogenesis of disks and OS of photoreceptors and for normal sorting of visual pigment to the OS, associated with disk morphogenesis, although our studies do not exclude the possibility that these defects develop secondary to the photoreceptor degeneration. In mature differentiated photoreceptors, Prom-1 concentrates at the basal part of the OS, a specialized organelle densely packed with disk membranes containing abundant opsin pigment (Sung and Tai, 2000). The OS is linked to the cell body via the connecting cilium, which is critical for the polarized transport of opsins to the OS membrane and acts as a barrier against redistribution (Sung and Tai, 2000). In the absence of Prom-1, disk membranes are disorganized and part of the visual pigment is mislocalized (clearly detectable in the ONL in several photoreceptors), this is a common finding in other models of retinal degeneration (Nir and Papermaster, 1986; Roof et al., 1994; Li et al., 1996, 1998; Hagstrom et al., 1999; Hong et al., 2000). Interestingly, in transgenic rats expressing a mutant rhodopsin, unable of proper sorting to the OS, the rate of rod cell death directly correlates with the severity of rhodopsin mistargeting (Green et al., 2000). Ectopic accumulation of rhodopsin in membranes, that are normally devoid of pigment, has been proposed to interfere with their normal structure and function (Deretic, 2006). Such crowding appears to be particularly relevant at the synapse between the photoreceptor and the ganglion cell, where rhodopsin accumulation disturbs normal synaptic membrane turnover and continuous vesicle recycling (Roof et al., 1994; Li et al., 1995, 1996, 1998; Deretic, 2006; Tam et al., 2006). Indeed, mistargeting of other OS proteins similarly leads to rod degeneration, as shown in a mouse model lacking the regulatory subunit Cngb1 of the rod cyclic nucleotide gated channel (Hüttl et al., 2005). We speculate that the disk dysmorphogenesis, in association with the defects in visual pigment sorting, not only impaired phototransduction, but also may have predisposed photoreceptors to degeneration in Prom-1−/− mice (although, as discussed above, we cannot formally conclude that one is causally linked to the other).
Prom-1−/− retinas also showed cone opsin mis-sorting. Among the few models exhibiting a similar cone opsin misrouting (Hagstrom et al., 1999; Hong et al., 2000; Rohrer et al., 2005), the Retinitis Pigmentosa GTPase Regulator (Rpgr) deficient mouse shares a number of features with Prom-1−/− mice, including disk membrane disorganization and photoreceptor degeneration (Hong et al., 2000). Similar to Prom-1, Rpgr is also localized at the basal part of the OS, and has been proposed to regulate polarized protein distribution across the connecting cilium (Hong et al., 2000). Perhaps, Prom-1 might have a similar function, as it is also preferentially localized in cilia (Dubreuil et al., 2007). A recent study documented that a loss-of-function mutation of Rpgr in humans also results in photoreceptor degeneration, further emphasizing a possible link between abnormal OS formation, pigment redistribution and photoreceptor disease (Adamian et al., 2006). Overall, loss of Prom-1 seems to cause photoreceptor degeneration, at least in part by defective disk morphogenesis and, possibly, also by abnormal distribution of visual pigment.
How Prom-1 regulates disk morphogenesis at the molecular level, remains enigmatic. Prom-1 is specifically sorted to microvilli, cilia and other plasma membrane protrusions (Weigmann et al., 1997; Corbeil et al., 2001; Fargeas et al., 2003a; Dubreuil et al., 2007; Florek et al., 2007), and has been involved in the stabilization of curved membrane protrusions (Huttner and Zimmerberg, 2001; Iglic et al., 2006). In Drosophila melanogaster, rhabdomers are specialized microvillar structures of photoreceptor neurons, that accommodate light receptor molecules (equivalent to disk OS in vertebrates). During evolution of insect compound eyes, rhabdomers partitioned from each other to improve angular sensitivity. Interestingly, a prominin-like molecule (Prom) in the fly decorates the photoreceptor apical surface when rhabdomeres form, and becomes thereafter selectively localized to the stalk membrane and the tips of these microvilli (Zelhof et al., 2006), thus resembling the subcellular localization of Prom-1 in vertebrate photoreceptors. Moreover, Prom may act as a receptor for a secreted protein, called spacemaker, involved in partitioning of rhabdomers (Zelhof et al., 2006). Thus, Prom-1 seems to have retained important evolutionary conserved functions in the animal kingdom to establish highly specialized structures in photoreceptor neurons, required for optimal phototransduction.
Prom-1 is expressed in various stem cells and other cell types (Corbeil et al., 2001; Florek et al., 2005; Jászai et al., 2007a). Why then does Prom-1 deficiency selectively affects retinal development? A possible explanation is that the expression of both Prom-1 and Prom-2 genes, which have redundant functions (Fargeas et al., 2003a), overlaps in many tissues, while, in the retina, only Prom-1 is detectable (this study; Fargeas et al., 2003b). We speculate, however, that future phenotyping of Prom-1−/− mice in stressed conditions is likely to reveal additional roles of Prom-1 in tissue homeostasis. Nonetheless, when considering that Prom-1 is expressed by endothelial progenitor cells (Shmelkov et al., 2005), it was somehow surprising that we did not detect more striking primary vascular defects in Prom-1−/− mice [the vessel abnormalities in mutant mice by 6 months of age, i.e., only after complete photoreceptor loss, are likely secondary to the retinal degeneration, as occurs in human diseases such as Retinitis Pigmentosa (Milam et al., 1998)].
Overall, our data reveal a novel role of Prom-1, in controlling proper OS morphogenesis and maintenance, and provide unprecedented evidence about the endogenous role and importance of the stem cell marker Prom-1 in vertebrates.
Footnotes
This work was supported in part by an unrestricted grant from Bristol-Myers-Squibb, Methusalem Funding, and GOA to P.C.; by grants of the German Research Council [Deutsche Forschungsgemeinschaft (DFG) Se837/4-1, 5-1, 6-1] and the European Union (IP “EVI-GenoRet” LSHG-CT-512036) to M.S. W.B.H. and D.C. were supported by the DFG (SPP 1109, Hu275/7-3; CO 298/2-2; SPP 1111, Hu275/8-3; SFB/TR13-04, B1; SFB 655, A2, A13; Center for Regenerative Therapies Dresden). S.Z. is supported by the European Union Seventh Framework Program via a Marie Curie Intra European fellowship. H.O. was sponsored by the Japan Society for the Promotion of Science. We thank A. Bouché, A. Carton, N. Dai, M. De Mol, M. Deprez, E. Gils, B. Hermans, S. Jansen, L. Kieckens, W. Y. Man, A. Manderveld, M. Nijs, S. Terclavers, C. Vanhuylebroeck, B. Vanwetswinkel, E. Weltens, and S. Wyns (Leuven).
The authors declare no competing financial interests.
References
- Adamian M, Pawlyk BS, Hong DH, Berson EL. Rod and cone opsin mislocalization in an autopsy eye from a carrier of X-linked retinitis pigmentosa with a Gly436Asp mutation in the RPGR gene. Am J Ophthalmol. 2006;142:515–518. doi: 10.1016/j.ajo.2006.03.061. [DOI] [PubMed] [Google Scholar]
- Bascom RA, Manara S, Collins L, Molday RS, Kalnins VI, McInnes RR. Cloning of the cDNA for a novel photoreceptor membrane protein (rom-1) identifies a disk rim protein family implicated in human retinopathies. Neuron. 1992;8:1171–1184. doi: 10.1016/0896-6273(92)90137-3. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Tessier-Lavigne M. Common mechanisms of nerve and blood vessel wiring. Nature. 2005;436:193–200. doi: 10.1038/nature03875. [DOI] [PubMed] [Google Scholar]
- Corbeil D, Röper K, Fargeas CA, Joester A, Huttner WB. Prominin: a story of cholesterol, plasma membrane protrusions and human pathology. Traffic. 2001;2:82–91. doi: 10.1034/j.1600-0854.2001.020202.x. [DOI] [PubMed] [Google Scholar]
- Deretic D. A role for rhodopsin in a signal transduction cascade that regulates membrane trafficking and photoreceptor polarity. Vision Res. 2006;46:4427–4433. doi: 10.1016/j.visres.2006.07.028. [DOI] [PubMed] [Google Scholar]
- Deretic D, Schmerl S, Hargrave PA, Arendt A, McDowell JH. Regulation of sorting and post-Golgi trafficking of rhodopsin by its C-terminal sequence QVS(A)PA. Proc Natl Acad Sci U S A. 1998;95:10620–10625. doi: 10.1073/pnas.95.18.10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wynter EA, Durig J, Cross MA, Heyworth CM, Testa NG. Differential response of CD34+ cells isolated from cord blood and bone marrow to MIP-1 alpha and the expression of MIP-1 alpha receptors on these immature cells. Stem Cells. 1998;16:349–356. doi: 10.1002/stem.160349. [DOI] [PubMed] [Google Scholar]
- Dubreuil V, Marzesco AM, Corbeil D, Huttner WB, Wilsch-Bräuninger M. Midbody and primary cilium of neural progenitors release extracellular membrane particles enriched in the stem cell marker prominin-1. J Cell Biol. 2007;176:483–495. doi: 10.1083/jcb.200608137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fargeas CA, Corbeil D, Huttner WB. AC133 antigen, CD133, prominin-1, prominin-2, etc.: prominin family gene products in need of a rational nomenclature. Stem Cells. 2003a;21:506–508. doi: 10.1634/stemcells.21-4-506. [DOI] [PubMed] [Google Scholar]
- Fargeas CA, Florek M, Huttner WB, Corbeil D. Characterization of prominin-2, a new member of the prominin family of pentaspan membrane glycoproteins. J Biol Chem. 2003b;278:8586–8596. doi: 10.1074/jbc.M210640200. [DOI] [PubMed] [Google Scholar]
- Fargeas CA, Joester A, Missol-Kolka E, Hellwig A, Huttner WB, Corbeil D. Identification of novel Prominin-1/CD133 splice variants with alternative C-termini and their expression in epididymis and testis. J Cell Sci. 2004;117:4301–4311. doi: 10.1242/jcs.01315. [DOI] [PubMed] [Google Scholar]
- Fargeas CA, Huttner WB, Corbeil D. Nomenclature of prominin-1 (CD133) splice variants - an update. Tissue Antigens. 2007;69:602–606. doi: 10.1111/j.1399-0039.2007.00825.x. [DOI] [PubMed] [Google Scholar]
- Florek M, Haase M, Marzesco AM, Freund D, Ehninger G, Huttner WB, Corbeil D. Prominin-1/CD133, a neural and hematopoietic stem cell marker, is expressed in adult human differentiated cells and certain types of kidney cancer. Cell Tissue Res. 2005;319:15–26. doi: 10.1007/s00441-004-1018-z. [DOI] [PubMed] [Google Scholar]
- Florek M, Bauer N, Janich P, Wilsch-Braeuninger M, Fargeas CA, Marzesco AM, Ehninger G, Thiele C, Huttner WB, Corbeil D. Prominin-2 is a cholesterol-binding protein associated with apical and basolateral plasmalemmal protrusions in polarized epithelial cells and released into urine. Cell Tissue Res. 2007;328:31–47. doi: 10.1007/s00441-006-0324-z. [DOI] [PubMed] [Google Scholar]
- Gehling UM, Ergün S, Schumacher U, Wagener C, Pantel K, Otte M, Schuch G, Schafhausen P, Mende T, Kilic N, Kluge K, Schäfer B, Hossfeld DK, Fiedler W. In vitro differentiation of endothelial cells from AC133-positive progenitor cells. Blood. 2000;95:3106–3112. [PubMed] [Google Scholar]
- Green ES, Menz MD, LaVail MM, Flannery JG. Characterization of rhodopsin mis-sorting and constitutive activation in a transgenic rat model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2000;41:1546–1553. [PubMed] [Google Scholar]
- Hagstrom SA, Duyao M, North MA, Li T. Retinal degeneration in tulp1-/- mice: vesicular accumulation in the interphotoreceptor matrix. Invest Ophthalmol Vis Sci. 1999;40:2795–2802. [PubMed] [Google Scholar]
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- Hong DH, Pawlyk BS, Shang J, Sandberg MA, Berson EL, Li T. A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3) Proc Natl Acad Sci U S A. 2000;97:3649–3654. doi: 10.1073/pnas.060037497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüttl S, Michalakis S, Seeliger M, Luo DG, Acar N, Geiger H, Hudl K, Mader R, Haverkamp S, Moser M, Pfeifer A, Gerstner A, Yau KW, Biel M. Impaired channel targeting and retinal degeneration in mice lacking the cyclic nucleotide-gated channel subunit CNGB1. J Neurosci. 2005;25:130–138. doi: 10.1523/JNEUROSCI.3764-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttner WB, Zimmerberg J. Implications of lipid microdomains for membrane curvature, budding and fission. Curr Opin Cell Biol. 2001;13:478–484. doi: 10.1016/s0955-0674(00)00239-8. [DOI] [PubMed] [Google Scholar]
- Iglic A, Hägerstrand H, Veranic P, Plemenitas A, Kralj-Iglic V. Curvature-induced accumulation of anisotropic membrane components and raft formation in cylindrical membrane protrusions. J Theor Biol. 2006;240:368–373. doi: 10.1016/j.jtbi.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Immervoll H, Hoem D, Sakariassen PØ, Steffensen OJ, Molven A. A Expression of the “stem cell marker” CD133 in pancreas and pancreatic ductal adenocarcinomas. BMC Cancer. 2008;8:48. doi: 10.1186/1471-2407-8-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janich P, Corbeil D. GM1 and GM3 gangliosides highlight distinct lipid microdomains within the apical domain of epithelial cells. FEBS Lett. 2007;581:1783–1787. doi: 10.1016/j.febslet.2007.03.065. [DOI] [PubMed] [Google Scholar]
- Jansen HG, Sanyal S. Development and degeneration of retina in rds mutant mice: electron microscopy. J Comp Neurol. 1984;224:71–84. doi: 10.1002/cne.902240107. [DOI] [PubMed] [Google Scholar]
- Jászai J, Fargeas CA, Florek M, Huttner WB, Corbeil D. Focus on molecules: Prominin-1 (CD133) Exp Eye Res. 2007a;85:585–586. doi: 10.1016/j.exer.2006.03.022. [DOI] [PubMed] [Google Scholar]
- Jászai J, Janich P, Farkas LM, Fargeas CA, Huttner WB, Corbeil D. Differential expression of Prominin-1 (CD133) and Prominin-2 in major cephalic exocrine glands of adult mice. Histochem Cell Biol. 2007b;128:409–419. doi: 10.1007/s00418-007-0334-2. [DOI] [PubMed] [Google Scholar]
- Karbanová J, Missol-Kolka E, Fonseca AV, Lorra C, Janich P, Hollerová H, Jászai J, Ehrmann J, Kolár Z, Liebers C, Arl S, Subrtová D, Freund D, Mokry J, Huttner WB, Corbeil D. The stem cell marker CD133 (Prominin-1) is expressed in various human glandular epithelia. J Histochem Cytochem. 2008;56:977–993. doi: 10.1369/jhc.2008.951897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly S, Bliss TM, Shah AK, Sun GH, Ma M, Foo WC, Masel J, Yenari MA, Weissman IL, Uchida N, Palmer T, Steinberg GK. Transplanted human fetal neural stem cells survive, migrate, and differentiate in ischemic rat cerebral cortex. Proc Natl Acad Sci U S A. 2004;101:11839–11844. doi: 10.1073/pnas.0404474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardon J, Corbeil D, Huttner WB, Ling Z, Bouwens L. Stem cell marker prominin-1/AC133 is expressed in duct cells of the adult human pancreas. Pancreas. 2008;36:e1–6. doi: 10.1097/mpa.0b013e318149f2dc. [DOI] [PubMed] [Google Scholar]
- Li T, Snyder WK, Olsson JE, Dryja TP. Transgenic mice carrying the dominant rhodopsin mutation P347S: evidence for defective vectorial transport of rhodopsin to the outer segments. Proc Natl Acad Sci U S A. 1996;93:14176–14181. doi: 10.1073/pnas.93.24.14176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZY, Kljavin IJ, Milam AH. Rod photoreceptor neurite sprouting in retinitis pigmentosa. J Neurosci. 1995;15:5429–5438. doi: 10.1523/JNEUROSCI.15-08-05429.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZY, Wong F, Chang JH, Possin DE, Hao Y, Petters RM, Milam AH. Rhodopsin transgenic pigs as a model for human retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1998;39:808–819. [PubMed] [Google Scholar]
- Maw MA, Corbeil D, Koch J, Hellwig A, Wilson-Wheeler JC, Bridges RJ, Kumaramanickavel G, John S, Nancarrow D, Röper K, Weigmann A, Huttner WB, Denton MJ. A frameshift mutation in prominin (mouse)-like 1 causes human retinal degeneration. Hum Mol Genet. 2000;9:27–34. doi: 10.1093/hmg/9.1.27. [DOI] [PubMed] [Google Scholar]
- Milam AH, Li ZY, Fariss RN. Histopathology of the human retina in retinitis pigmentosa. Prog Retin Eye Res. 1998;17:175–205. doi: 10.1016/s1350-9462(97)00012-8. [DOI] [PubMed] [Google Scholar]
- Miraglia S, Godfrey W, Yin AH, Atkins K, Warnke R, Holden JT, Bray RA, Waller EK, Buck DW. A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood. 1997;90:5013–5021. [PubMed] [Google Scholar]
- Mizrak D, Brittan M, Alison MR. CD133: molecule of the moment. J Pathol. 2008;214:3–9. doi: 10.1002/path.2283. [DOI] [PubMed] [Google Scholar]
- Mukouyama YS, Shin D, Britsch S, Taniguchi M, Anderson DJ. Sensory nerves determine the pattern of arterial differentiation and blood vessel branching in the skin. Cell. 2002;109:693–705. doi: 10.1016/s0092-8674(02)00757-2. [DOI] [PubMed] [Google Scholar]
- Nir I, Papermaster DS. Immunocytochemical localization of opsin in the inner segment and ciliary plasma membrane of photoreceptors in retinas of rds mutant mice. Invest Ophthalmol Vis Sci. 1986;27:836–840. [PubMed] [Google Scholar]
- Okoye G, Zimmer J, Sung J, Gehlbach P, Deering T, Nambu H, Hackett S, Melia M, Esumi N, Zack DJ, Campochiaro PA. Increased expression of brain-derived neurotrophic factor preserves retinal function and slows cell death from rhodopsin mutation or oxidative damage. J Neurosci. 2003;23:4164–4172. doi: 10.1523/JNEUROSCI.23-10-04164.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peichev M, Naiyer AJ, Pereira D, Zhu Z, Lane WJ, Williams M, Oz MC, Hicklin DJ, Witte L, Moore MA, Rafii S. Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells identifies a population of functional endothelial precursors. Blood. 2000;95:952–958. [PubMed] [Google Scholar]
- Rohrer B, Lohr HR, Humphries P, Redmond TM, Seeliger MW, Crouch RK. Cone opsin mislocalization in Rpe65-/- mice: a defect that can be corrected by 11-cis retinal. Invest Ophthalmol Vis Sci. 2005;46:3876–3882. doi: 10.1167/iovs.05-0533. [DOI] [PubMed] [Google Scholar]
- Roof DJ, Adamian M, Hayes A. Rhodopsin accumulation at abnormal sites in retinas of mice with a human P23H rhodopsin transgene. Invest Ophthalmol Vis Sci. 1994;35:4049–4062. [PubMed] [Google Scholar]
- Röper K, Corbeil D, Huttner WB. Retention of prominin in microvilli reveals distinct cholesterol-based lipid micro-domains in the apical plasma membrane. Nat Cell Biol. 2000;2:582–592. doi: 10.1038/35023524. [DOI] [PubMed] [Google Scholar]
- Sameshima M, Uehara F, Ohba N. Specialization of the interphotoreceptor matrices around cone and rod photoreceptor cells in the monkey retina, as revealed by lectin cytochemistry. Exp Eye Res. 1987;45:845–863. doi: 10.1016/s0014-4835(87)80101-x. [DOI] [PubMed] [Google Scholar]
- Seeliger MW, Grimm C, Ståhlberg F, Friedburg C, Jaissle G, Zrenner E, Guo H, Remé CE, Humphries P, Hofmann F, Biel M, Fariss RN, Redmond TM, Wenzel A. New views on RPE65 deficiency: the rod system is the source of vision in a mouse model of Leber congenital amaurosis. Nat Genet. 2001;29:70–74. doi: 10.1038/ng712. [DOI] [PubMed] [Google Scholar]
- Seeliger MW, Beck SC, Pereyra-Muñoz N, Dangel S, Tsai JY, Luhmann UF, van de Pavert SA, Wijnholds J, Samardzija M, Wenzel A, Zrenner E, Narfström K, Fahl E, Tanimoto N, Acar N, Tonagel F. In vivo confocal imaging of the retina in animal models using scanning laser ophthalmoscopy. Vision Res. 2005;45:3512–3519. doi: 10.1016/j.visres.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Semple-Rowland SL. Expression of glial fibrillary acidic protein by Muller cells in rd chick retina. J Comp Neurol. 1991;305:582–590. doi: 10.1002/cne.903050405. [DOI] [PubMed] [Google Scholar]
- Shmelkov SV, St Clair R, Lyden D, Rafii S. AC133/CD133/Prominin-1. Int J Biochem Cell Biol. 2005;37:715–719. doi: 10.1016/j.biocel.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Shmelkov SV, Butler JM, Hooper AT, Hormigo A, Kushner J, Milde T, St Clair R, Baljevic M, White I, Jin DK, Chadburn A, Murphy AJ, Valenzuela DM, Gale NW, Thurston G, Yancopoulos GD, D'Angelica M, Kemeny N, Lyden D, Rafii S. CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors. J Clin Invest. 2008;118:2111–2120. doi: 10.1172/JCI34401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalmans I, Ng YS, Rohan R, Fruttiger M, Bouché A, Yuce A, Fujisawa H, Hermans B, Shani M, Jansen S, Hicklin D, Anderson DJ, Gardiner T, Hammes HP, Moons L, Dewerchin M, Collen D, Carmeliet P, D'Amore PA. Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J Clin Invest. 2002;109:327–336. doi: 10.1172/JCI14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm C, Westphal B, Kleine HD, Petzsch M, Kittner C, Klinge H, Schümichen C, Nienaber CA, Freund M, Steinhoff G. Autologous bone-marrow stem-cell transplantation for myocardial regeneration. Lancet. 2003;361:45–46. doi: 10.1016/S0140-6736(03)12110-1. [DOI] [PubMed] [Google Scholar]
- Steinberg RH, Fisher SK, Anderson DH. Disc morphogenesis in vertebrate photoreceptors. J Comp Neurol. 1980;190:501–508. doi: 10.1002/cne.901900307. [DOI] [PubMed] [Google Scholar]
- Sung CH, Tai AW. Rhodopsin trafficking and its role in retinal dystrophies. Int Rev Cytol. 2000;195:215–267. doi: 10.1016/s0074-7696(08)62706-0. [DOI] [PubMed] [Google Scholar]
- Tam BM, Xie G, Oprian DD, Moritz OL. Mislocalized rhodopsin does not require activation to cause retinal degeneration and neurite outgrowth in Xenopus laevis. J Neurosci. 2006;26:203–209. doi: 10.1523/JNEUROSCI.3849-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiveron MC, Hirsch MR, Brunet JF. The expression pattern of the transcription factor Phox2 delineates synaptic pathways of the autonomic nervous system. J Neurosci. 1996;16:7649–7660. doi: 10.1523/JNEUROSCI.16-23-07649.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrente Y, Belicchi M, Sampaolesi M, Pisati F, Meregalli M, D'Antona G, Tonlorenzi R, Porretti L, Gavina M, Mamchaoui K, Pellegrino MA, Furling D, Mouly V, Butler-Browne GS, Bottinelli R, Cossu G, Bresolin N. Human circulating AC133(+) stem cells restore dystrophin expression and ameliorate function in dystrophic skeletal muscle. J Clin Invest. 2004;114:182–195. doi: 10.1172/JCI20325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, Tsukamoto AS, Gage FH, Weissman IL. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci U S A. 2000;97:14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigmann A, Corbeil D, Hellwig A, Huttner WB. Prominin, a novel microvilli-specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non-epithelial cells. Proc Natl Acad Sci U S A. 1997;94:12425–12430. doi: 10.1073/pnas.94.23.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Chen Y, Lillo C, Chien J, Yu Z, Michaelides M, Klein M, Howes KA, Li Y, Kaminoh Y, Chen H, Zhao C, Chen Y, Al-Sheikh YT, Karan G, Corbeil D, Escher P, Kamaya S, Li C, Johnson S, Frederick JM, Zhao Y, Wang C, Cameron DJ, Huttner WB, Schorderet DF, Munier FL, Moore AT, Birch DG, Baehr W, Hunt DM, Williams DS, Zhang K. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J Clin Invest. 2008;118:2908–2916. doi: 10.1172/JCI35891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, Olweus J, Kearney J, Buck DW. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90:5002–5012. [PubMed] [Google Scholar]
- Zelhof AC, Hardy RW, Becker A, Zuker CS. Transforming the architecture of compound eyes. Nature. 2006;443:696–699. doi: 10.1038/nature05128. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Zulfiqar F, Xiao X, Riazuddin SA, Ahmad Z, Caruso R, MacDonald I, Sieving P, Riazuddin S, Hejtmancik JF. Severe retinitis pigmentosa mapped to 4p15 and associated with a novel mutation in the PROM1 gene. Hum Genet. 2007;122:293–299. doi: 10.1007/s00439-007-0395-2. [DOI] [PubMed] [Google Scholar]