

Abstract
During brain development, young neurons closely associate with radial glial while migrating from the ventricular zone (VZ) to the cortical plate (CP) of the neocortex. It has been shown previously that gap junctions are needed for this migration to occur properly, but the precise mechanism responsible is still in question. Here, we used Cre recombinase, driven by the nestin promoter, to conditionally knock-out a floxed coding DNA of the connexin43 (Cx43) gene in mice. Radial glia in the VZ normally express connexin43. They undergo divisions that produce neurons and astrocytes and serve as migratory guides for the daughter cells that they produce. Based on histological analysis, we suggest that removing Cx43 from radial glia alters the normal lamination of the mouse neocortex. To monitor newborn neurons during development, we introduced a plasmid containing green fluorescent protein driven by a neuronal (Tα1 tubulin) promoter into the embryonic neocortex using in utero electroporation. The transfected migrating neurons remain in the VZ/intermediate zone (IZ) of the Cx43 conditional knock-out (Cx43cKO) animals, whereas in Cx43fl/fl mice, neurons migrate through the IZ into the CP, indicating that deletion of Cx43 from nestin-positive cells disrupts neuronal migration. We were able to rescue migration of Cx43cKO neurons by electroporating a cytomegalovirus–Cx43 expression plasmid into the embryonic cortex. In contrast, a C-terminal truncated form of Cx43 failed to rescue neuronal migration. In addition, Cx43K258stop mice, in which Cx43 lacks the last 125 amino acid residues of the cytoplasmic C-terminal domain, gave results similar to those seen with the Cx43cKO mice. This study illustrates that deletion of the C-terminal domain of Cx43 alters neuronal migration in the neocortex.
Keywords: development, cortex, gap junction, migration, glia, neuron
Introduction
During embryonic development, radial glial cells divide and give rise to astrocytes and neurons in the cerebral cortex (Noctor et al., 2001; Malatesta et al., 2003; Casper and McCarthy, 2006; Noctor et al., 2008). Radial glia are bipolar cells originating in the ventricular zone (VZ) of the dorsal telencephalon, with one process extending to the ventricular surface, whereas the other process projects outward to the pial surface. As the neocortex forms, the radial glia undergo divisions that produce neurons and build a dynamic scaffold that facilitates migration from the VZ to their location in the cortical plate (CP) (Anthony et al., 2004; Götz and Sommer, 2005).
Within the developing brain, neural progenitor cells are extensively coupled by gap junctions (Lo Turco and Kriegstein, 1991; Bittman et al, 1997). Gap junctions are intercellular membrane channels that provide cytoplasmic continuity between adjacent cells and permit the exchange of molecules <2 kDa in size (Simon and Goodenough, 1998). Gap junction channels are formed by docking of two connexons in contacting plasma membranes of adjacent cells. The protein subunits of these hemichannels are called connexins (Cx) (Goodenough and Paul, 2003).
Two gap junction proteins, Cx43 and Cx26, are widely expressed at the points of contact between radial glia and migrating neurons (Nadarajah et al., 1997; Cina et al., 2007), suggesting their role in neuronal migration. Indeed, a recent study showed that Cx43 and Cx26 are involved in neuronal migration by mediating adhesion and not by formation of intercellular gap junction channels (Elias et al., 2007).
The targeted deletion of Cx genes in mice (accomplished through homologous recombination) has provided preliminary insights into the role of gap junctions in neuronal migration. For instance, we have shown previously that embryonic mice lacking Cx43 expression phenotypically present significant accumulation of bromodeoxyuridine-positive (BrdU+) pulse-labeled cells in the intermediate zone (IZ), consistent with a disruption in neuronal migration, whereas these cells establish themselves in the CP of wild-type littermates (Fushiki et al., 2003). Unfortunately, Cx43 is widely expressed both temporally and spatially, making it difficult to attribute the migrational phenotype to radial glia alone when presented in a complete knock-out. To further complicate the situation, complete knock-out of Cx43 is neonatal lethal attributable to abnormalities in heart development, preventing a comprehensive study of the long-term effects of such a deletion. To circumvent the lethality associated with total Cx43 knock-out, we have made conditional knock-out (Cx43cKO) mice produced by crossing Cx43fl/fl mice (Theis et al., 2001), with nestin–Cre mice, driving the expression of Cre recombinase in radial glia (Bérubé et al., 2005).
In addition to Cx43cKO mice, Cx43K258stop mice, in which the last 125 amino acid residues of the cytoplasmic C-terminal domain of Cx43 are lacking (Maass et al., 2004), have been used in this study to examine the significance of Cx43 and its C terminus in neuronal migration.
Our data demonstrate that conditional knock-out of Cx43 in radial glia disrupts neuronal migration, but this defect is rescued by reconstitution of full-length Cx43, but not by C-terminal truncated Cx43, into the neocortex.
Materials and Methods
Animals.
The production of the Cx43 null mutant (Reaume et al., 1995), nestin–Cre (Bérubé et al., 2005), floxed Cx43 (Theis et al., 2001), and Cx43K258stop (Maass et al., 2004) transgenic mice have been reported previously. Nestin–Cre mice were crossed with floxed Cx43 mice, maintained in an animal facility with a 12 h light/dark cycle, and were provided food and water ad libitum. The animals were maintained according to Canadian Council on Animal Care guidelines for the care and use of laboratory mice at the University of British Columbia. After mating of adult mice, the day on which a vaginal plug was identified was considered embryonic day 0 (E0).
Genotype analysis by PCR.
For detection of the Cx43 floxed (Cx43fl) allele and the Cx43 wild type (Cx43+) allele, forward primers (5′-TCA TGC CCG GCA CAA GTG AGA C-3′) and reverse primers (5′-TCA CCC CAA GCT GAC TCA ACC G-3′) were applied to generate a product of 1 kb floxed amplicon and a 900 bp wild-type amplicon (Theis et al., 2001). To detect the deleted Cx43 (Cx43−) allele, forward primers Cx43 (5′-AT TTT GCC GCC GCC TAG CTA TCC C-3′) and reverse primers (5′-GCT TGC CGA ATA TCA TGG TGG A-3′) were used to generate a product of 1 kb for the deleted amplicon and 500 bp for the wild-type amplicon (Perez Velazquez et al., 1996). For the detection of the nestin–Cre transgene, forward primers (5′-TGA CCA GAG TCA TCC TTA GCG-3′) and reverse primers (5′-AAT GCT TCT GTC CGT TTG CC-3′) were applied, generating a 300–500 bp product (Tronche et al., 1999). To detect the C-terminally truncated Cx43 allele, forward primers (5′-GCA TCC TCT TCA AGT CTG TCT TCG-3′) and reverse primers (5′-CAA AAC ACC CCC CAA GGA ACC TAG-3′) were used to generate of 851 bp amplicon for the Cx43 allele and 452 bp amplicon for the Cx43K258stop allele (Maass et al., 2004).
Immunohistochemistry.
Brains obtained from embryonic day 14 and 18 and postnatal day 16 (P16) (the birth date is defined as P0) mice were fixed by immersion in 4% paraformaldehyde (in PBS, pH 7.4) overnight at 4°C. The fixed brains were cryoprotected in 30% sucrose, mounted in OCT (Tissue-Tek), and cut into 10 μm coronal sections using a cryostat. Cut brain sections were mounted on coverslips and blocked for nonspecific antibody binding with IgG blocking solution (M.O.M. kit; Vector Laboratories), and embryonic sections were subsequently incubated with polyclonal antibodies for wild-type Cx43 (Nadarajah et al., 1997) (1:200; epitope spanning amino acids 363–382 located at the C-terminal region of Cx43; Sigma-Aldrich) or C-terminally truncated Cx43 (1:50; epitope spanning amino acids 120–140; Abgent), at 4°C overnight. Selected samples were also double labeled with antibodies to the neuronal marker microtubule associated protein- 2 (MAP-2) (1:400; Sigma-Aldrich) (Huber and Matus, 1984), nestin (a radial glia marker) (1:400; BD Biosciences) (Hockfield and McKay, 1985), green fluorescent protein (GFP) (1:800; Millipore Bioscience Research Reagents) (Paquin et al., 2005), or β-galactosidase (β-gal) (1:200; Biogenesis) (Mizutani and Saito, 2005). Antibodies used on P16 tissue sections were specific for neuronal-specific nuclear protein (NeuN) (a neuronal marker) (1:200; Millipore Bioscience Research Reagents), Ki67 (a proliferation marker) (1:1000; Vector Laboratories), and myelin basic protein (MBP) (1:250; Millipore Bioscience Research Reagents) (Groome et al., 1986). The VZ, the IZ, and the CP of embryonic cerebral cortex as well as the subventricular zone (SVZ), the white matter (WM), and the cortex of postnatal forebrain, were processed for imaging. Specificity of the primary antibodies was assessed by omitting them from the labeling protocol on selected slices. The specificity of antibodies recognizing C-terminally truncated Cx43 was confirmed by conducting immunostaining on brain tissue sections of Cx43 null mice. Slices were then incubated in Alexa Fluor-tagged goat anti-mouse, goat anti-rabbit, and goat anti-rat IgG antibodies (Invitrogen) for 1 h and mounted in ProLong Gold containing 4′,6′-diamidino-2-phenylindole (DAPI) (Invitrogen). Labeled sections were examined and analyzed using a Zeiss Axioskop2 epifluorescent microscope and AxioVision 4.2 software (Carl Zeiss). To generate the final figures, all images were stored as TIFF files and processed in Adobe Photoshop 7.0 (Adobe Systems).
5-Bromo-4-chloro-3-indoly-β-galactoside staining.
For 5-bromo-4-chloro-3-indoly-β-galactoside (X-Gal) staining, sections were processed as described previously (Akagi et al., 1997), and embryos were X-Gal stained using established procedures (Brenner et al., 1994; Hogan et al., 1994).
Protein isolation and Western blot analysis.
Western blot analysis on embryonic brain tissue was conducted as described previously (Cina et al., 2007). Briefly, protein was isolated from cerebral cortices of E18 mice using radioimmune precipitation lysis buffer (50 mm Tris-HCl, pH 8.0, and 1% IGEPAL) supplemented with Mini Complete protease inhibitors (Roche Applied Science) and phosphatase inhibitors (Sigma). Protein concentrations were determined using the BCA protein quantification kit (Pierce). Protein samples (30 μg) were boiled for 2 min in SDS sample buffer, pH 8.0, and separated on a 12% polyacrylamide gel in parallel with molecular weight markers (Bio-Rad). Subsequently, the electrophoresed proteins were transferred onto a nitrocellulose membrane (Bio-Rad) at 100 V for 1 h. The blots were then blocked with 5% dry milk in PBS (pH 7.4, with 1% Tween 20) for 1 h and subsequently incubated with an antibody against Cx43 at 4°C overnight. After rinsing with PBS, the blots were incubated in horseradish peroxidase-tagged secondary antibody (Cedarlane Laboratories) for 1 h at room temperature, followed by incubation in SuperSignal chemiluminescent substrate (Pierce). The labeled blots were then exposed to Kodak X-Omat x-ray film to visualize antibody binding. To ensure equal loading of protein samples, the blots were stripped of their Cx43 antibody and reprobed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Cedarlane Laboratories).
Cortical layer thickness.
Cortical layer thickness was measured on 10 μm coronal sections of at least four of E18 or P16 control and Cx43cKO littermates. Sections were stained with DAPI and NeuN, and the outline of each zone was drawn. Pictures were analyzed in AxioVision 4.2 software. Mean values of three measurements per section were used for calculating statistical significance.
BrdU labeling, proliferation, and migrational analysis.
Male Cx43fl/fl; nestin–Cre mice were crossed with female Cx43fl/fl mice, and pregnant mice at gestational day 14 were used for conducting BrdU experiments. For injections, 200 mg/kg BrdU was dissolved in saline and injected intraperitoneally (Redila et al., 2006). Pregnant female mice were injected with BrdU once, and animals were allowed to survive for either 2 or 96 h. Embryonic brains obtained from either E14 or E18 mice were sectioned and mounted on coverslips, and DNA was denatured by incubating the slides in HCl for 30 min at 37°C. The acid was neutralized by immersing the slides in 0.1 m borate buffer, pH 8.5 (Sigma), for 20 min. Brain sections were then blocked for nonspecific antibody binding with IgG blocking solution (M.O.M. kit; Vector Laboratories) and subsequently processed for immunohistochemical staining using a mouse monoclonal antibody against BrdU (Sigma). Selected samples were also double labeled with polyclonal antibodies for Ki67 (1:1000; Vector Laboratories). From each animal, 10 coronal sections were assessed spanning the rostral to the caudal end of the neocortex. Based on DAPI staining, the sections of the neocortex were divided into three zones: the VZ, the IZ, and the CP. To define each zone, the outline of the VZ, the IZ, and the CP of each section was traced using the AxioVision 4.2 software. Immunopositive cells were manually counted with the investigator blinded as to group identity (control and Cx43cKO). To measure a migration change, the number of BrdU+ labeled cell nuclei was counted in a 16× optical view of each zone per 10 μm section, whereas BrdU+ or Ki67+ cells were counted in the VZ of each section to assess proliferation. BrdU+ cells were counted if they had round nuclei that were evenly stained; thus elongated endothelial nuclei were excluded on the basis of shape (Fushiki et al., 2003). Punctated nuclei were also not counted because these cells may have entered or exited the S-phase at the time of labeling (Fushiki et al., 2003). The mean number of BrdU+ or Ki67+ cells per section was calculated for both proliferation and migration. These numbers were then analyzed for statistical significance using an ANOVA.
In utero electroporation.
A plasmid expressing nuclear-localized GFP driven by a neuronal promoter (Tα1 tubulin) (gift from Dr. Freda D. Miller, University of Toronto, Toronto, Ontario, Canada) (Gloster et al., 1994) was used either alone or with each of the following plasmids: (1) full-length Cx43 driven by the cytomegalovirus (CMV) promoter (CMV–Cx43) (Fu et al., 2004); (2) C-terminally truncated Cx43 tagged with GFP [Cx43Δ244–382GFP (Cx43t–GFP)] (Bates et al., 2007); (3) U6–Cx43siRNA [sequence, CAATTCCTCCTGCCGCAAT (Iacobas et al., 2008); gift from Dr. Eliana Scemes, Albert Einstein College of Medicine, Bronx, NY]; and (4) U6-control–siRNA (sequence, CTCCTTTTTT; gift from Dr. Timothy O'Connor, University of British Columbia, British Columbia, Canada). The introduction of DNA by in utero electroporation was conducted as described previously (Tabata and Nakajima, 2002; Barnabé-Heider et al., 2005). Briefly, pregnant female Cx43cKO mice, Cx43 null mice, or Cx43K258stop mice were anesthetized with isoflurane and nitrous oxide. A midline incision was performed to reach the embryos, and 1 μl of DNA was injected into the E14 lateral ventricle. After injection, electroporation was conducted using a square electroporator CUY21 EDIT (Protech) to deliver five 50 ms pulses of 50 V with 950 ms intervals per embryo. Embryos were reinserted in utero, and the incision was closed. Four days later, at E18, the embryonic brains were harvested and fixed in 4% paraformaldehyde at 4°C overnight. The fixed brains were cryoprotected in 30% sucrose, mounted in OCT, cryosectioned (10 μm), and analyzed.
Intensity measurements of GFP across neocortical layers.
NIH Image J software (http://rsb.info.nih.gov/ij/) was used to quantify GFP fluorescent intensity of neocortical layers and data analyzed in Microsoft Excel. For each image, the outlines of the VZ, the IZ, and the CP layers were drawn, and then a threshold was set to isolate GFP labeling to match the size and distribution of cells perceived by eye in the original grayscale image. The average pixel intensity from each layer was measured and subtracted from the background intensity. Six sections from at least three brains per condition were used, and the results were expressed as the percentage of total GFP intensity, for each condition, within each layer of the neocortex (Mizuno et al., 2007).
Data analysis.
All experiments were performed at least three times. When appropriate, values are presented as mean ± SEM, and the results were analyzed by one-way ANOVA, followed by the Student's t test or Mann–Whitney test. A p value of <0.05 was considered statistically significant.
Results
Cre is active in Cx43fl/fl; nestin–Cre mice and deletes Cx43 from VZ radial glia and immature neurons
During early neurogenesis, nestin-expressing radial glia are the foundation of the developing neocortex and give rise to neurons, astrocytes, and oligodendrocytes as the cortex matures (Anthony et al., 2004; Götz and Sommer, 2005). Nestin is highly expressed in radial glial progenitor cells located in the VZ and is expressed from E10 onward (Malatesta et al., 2003). Taking advantage of this expression profile, we specifically inactivated a floxed Cx43 allele (Cx43fl allele) (Theis et al., 2001) in radial glial progenitors at the beginning of neurogenesis using the Cre/LoxP system in Cx43fl/fl; nestin–Cre (Cx43cKO) mice. Activation of Cre resulted in the complete knock-out of several floxed target genes expressed in the CNS (Tronche et al., 1999; Bérubé et al., 2005). The Cx43fl/fl mice are phenotypically indistinguishable from wild-type littermates and are born at predicted Mendelian frequency (Theis et al., 2001). They are referred to as “control” in the following. By conducting β-gal staining, immunohistochemistry, and Western blotting, we confirmed activity of the Cre recombinase in the VZ; additionally, we showed localization of Cre recombinase in radial glial cells in embryonic brain of Cx43cKO mice. β-Gal staining (β-galactosidase is expressed instead of Cx43 after Cre-mediated recombination) of the neocortex of E14 embryos confirms Cre is being expressed in the brain of Cx43cKO mice but not in control mice (Fig. 1a).
Figure 1.
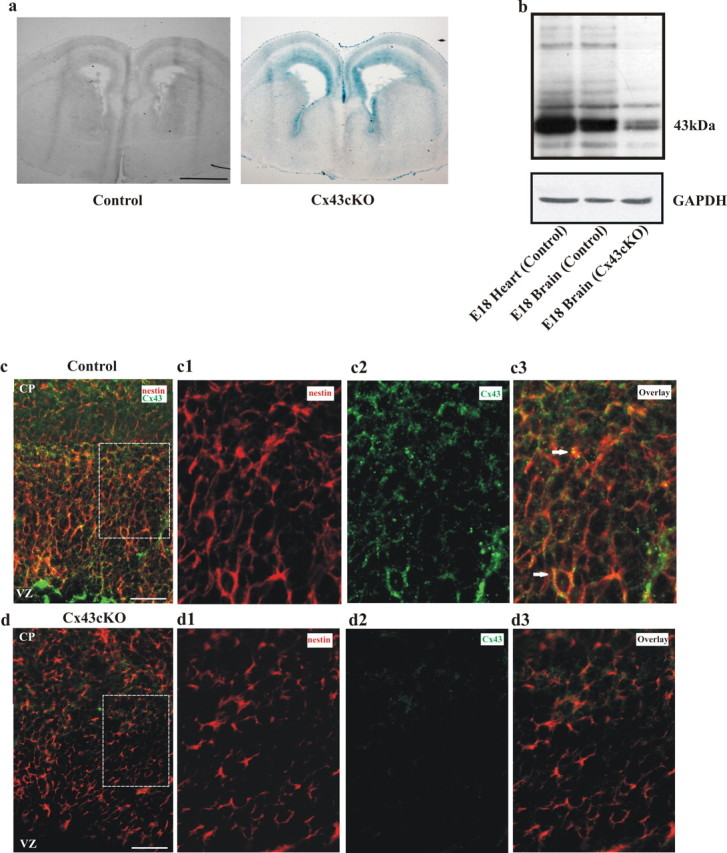
Cre activity in Cx43fl/fl; nestin–Cre (Cx43cKO) mice. a, Coronal sections of E14 embryonic forebrain from control Cx43fl/fl (left) and Cx43cKO (right) mice were stained for lacZ, indicating Cre-mediated loss of Cx43 expression. Cre activity was observed in Cx43cKO mice but not in control mice. n = 4. b, Representative immunoblot showing the reduction of Cx43 protein in the neocortex of Cx43cKO mice. Equal amounts of embryonic heart and cortical tissue protein were immunoblotted and probed with antibodies recognizing Cx43 (43 kDa). GAPDH was used as a loading control. n = 3. c, d, Cx43cKO mice display a strong decrease in the level of Cx43 expression throughout the neocortex compared with control mice. Coronal brain sections of E18 littermates were stained for nestin and Cx43. Cx43 showed association (arrows) with nestin-expressing cells in control animals (c) and was absent in Cx43cKO sections (d). c1–d3 are higher-magnification micrographs of the areas outlined in c and d. n = 4. Scale bars: a, 1 mm; c, d, 100 μm; c1–d3, 40 μm.
To demonstrate the removal of Cx43 from the brain, we used immunoblot analysis. Tissue lysates from E18 cortex and heart were probed with anti-Cx43 and GAPDH antibodies. Cx43 protein was strongly decreased in lysates of E18 Cx43cKO cortex relative to control (Fig. 1b). Residual expression of Cx43 was most likely attributable to Cx43 gene activity in cells not targeted by nestin–Cre. Embryonic heart tissue was used as a positive control for the expression of Cx43 (Fig. 1b).
Double-immunofluorescent labeling with Cx43 and nestin antibodies further confirmed deletion of Cx43 from the radial glia. As can be seen in Figure 1, c and d, there was a dramatic reduction in the classical punctate Cx43 signals in the plasma membrane of nestin+ cells in the Cx43cKO animals.
Having verified efficient Cre activity in Cx43cKO mice, we then addressed the question whether Cre was expressed in both cell types of interest, the radial glia and the early born neurons. Therefore, double-immunofluorescence labeling experiments were performed on tissue sections of control and Cx43cKO cortices at E14, using antibodies against β-gal together with either nestin or MAP-2 antibodies, specific for radial glia and neurons, respectively. Brain sections of control mice were devoid of β-gal labeling (Fig. 2a), whereas brain sections of Cx43fl/fl; nestin–Cre mice showed extensive β-gal labeling, indicating Cre-mediated loss of Cx43 expression, which was associated with nestin+ and MAP-2+ cells (Fig. 2b,c). Thus, in Cx43cKO mice, Cre is expressed in radial glia and early born neurons.
Figure 2.
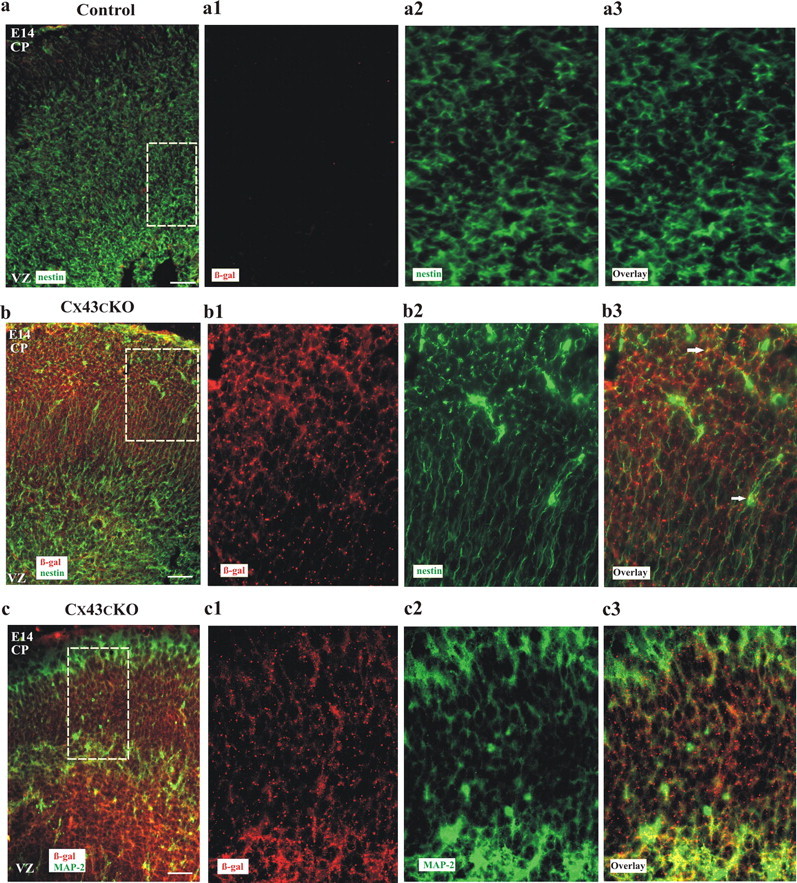
Cre activity is observed in radial glia and MAP-2-positive neurons in Cx43cKO brains. Coronal sections of control (a) and Cx43cKO (b, c) neocortices were stained for β-gal (red), MAP-2, and/or nestin (green). Control brains were devoid of β-gal labeling compared with Cx43cKO littermates (a–c). a1–c3 represent higher-magnification micrographs of the section in a–c. Note that β-gal staining is associated with both nestin and MAP-2 expression in Cx43cKO mice. Arrows represent association of nestin- or MAP-2-expressing cells with β-gal. n = 3. Scale bars: a–c, 100 μm; a1–c3, 40 μm.
Cx43 is required for normal lamination of the neocortex
The majority of neuronal migration in the mouse cerebral cortex occurs between developmental stages E14 and E18. Therefore, any abnormalities in the laminar formation of this structure in Cx43cKO mice should be observed by E18. Cortical sections of Cx43cKO (Cx43fl/fl; nestin–Cre) and control (Cx43fl/fl) E18 mice were treated with the fluorescent nuclear stain DAPI, to measure the entire thickness of the cortical hemispheres, as well as that of the VZ, the IZ, and the CP sublayers of each hemisphere. In addition, the thickness of the cortical layers was measured and compared between control and Cx43cKO mice. As shown in Figure 3, there was a significant change in the thickness of all layers except the VZ. The thickness of the CP was greater than the thickness of the IZ in the control neocortex, whereas this pattern was reversed in the Cx43cKO neocortex, consistent with the accumulation of cells in the IZ (Fig. 3a,b). The total cortical thickness of cerebral hemisphere in Cx43cKO mice appeared to be unchanged when compared with control mice (control, 799 ± 12 μm vs Cx43cKO, 756 ± 11 μm; p > 0.05). Therefore, these findings suggest that absence of Cx43 from radial glial fibers and immature neurons interrupts normal formation of the neocortical layers by accumulation of cells in the IZ.
Figure 3.
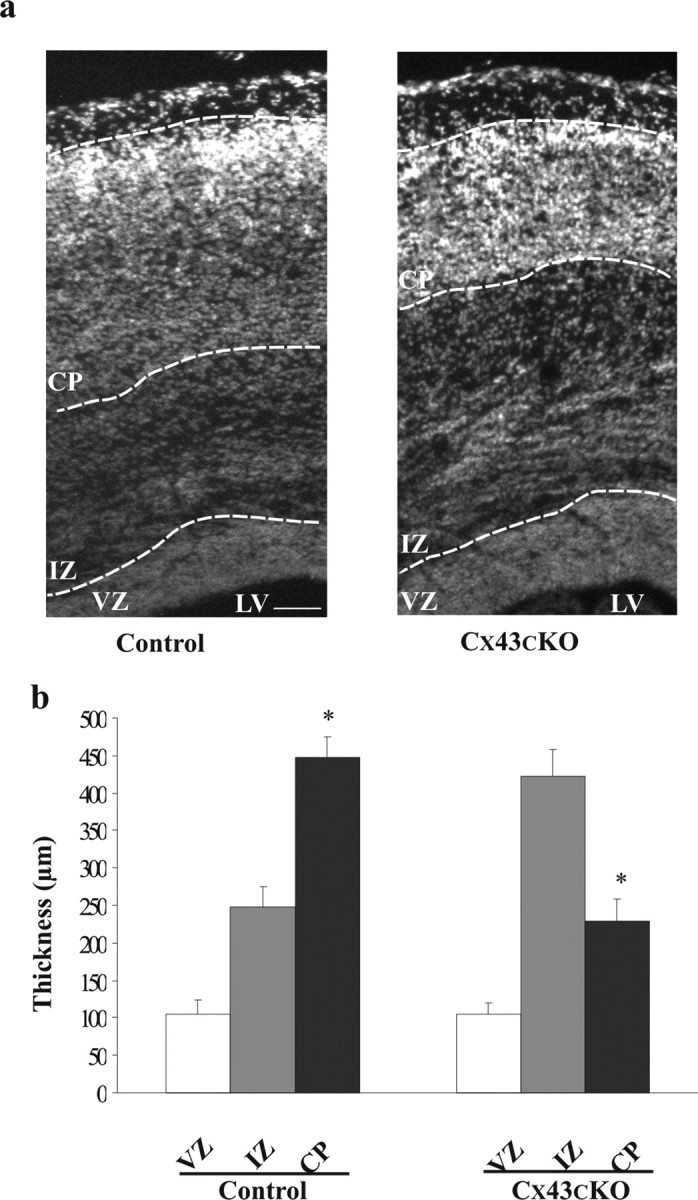
Deletion of Cx43 in the developing neocortex perturbs normal cortical lamination. a, Thickness of the VZ, IZ, and CP was measured in the neocortices of control and Cx43cKO mice. The cortical sections were stained with DAPI, and the cortical layers were identified based on their anatomical cell distribution. Using AxioVision software, the identified border lines between layers were labeled. In the cerebral cortices of E18 Cx43cKO mice, the thickness of the IZ was increased and the CP decreased compared with the IZ of control mice. b, The representative graph shows that the IZ in the Cx43cKO mice is increased in size compared with the control mice, whereas in control mice the CP is larger. *p < 0.05, Student's t test. n = 4. LV, Lateral ventricle. Scale bar, 100 μm.
To address the question whether the phenotype that was seen in the neocortex of E18 Cx43cKO mice persists into the postnatal stage or whether it recovers as the neocortex matures, the entire thickness of the postnatal cerebral hemisphere, as well as that of the sublayers of each hemisphere, was measured and compared between control and Cx43cKO littermates. To define the SVZ, the WM, and the cortex of postnatal forebrain, we performed immunohistochemistry by using a cell proliferation marker (Ki67), MBP, and NeuN, respectively. From each animal, 10 coronal sections spanning the rostral to the caudal end of the cerebral cortex were assessed, and the thickness of each lamina was measured. As seen in Figure 4a, in general, the lamination of the cerebral cortex in Cx43cKO mice seemed to be proper, but compared with the control littermates at P16, the cortex and cerebral hemisphere were significantly thinner (15.5, 16, and 17% respectively; p < 0.05) (Fig. 4b). The thickness of the neocortex (including the cortical layers, white matter, and the subventricular zone) in Cx43cKO mice was smaller than that of the control littermates (Cx43cKO, 1410 ± 61 μm vs control, 1692 ± 60 μm; p < 0.05). Furthermore, the thickness of the upper layers (I–IV) and the lower layers (V–VI) in Cx43cKO mice was defined, measured, and then compared with the control littermates. As shown in Figure 4c, in the control forebrain, the thickness of the layers I–IV was significantly greater than the thickness of the layers V–VI (36%), whereas this pattern was reversed in the Cx43cKO mice, consistent with the greater thickness of the layers V–VI compared with the layers I–IV (30%). Collectively, these findings suggest that the deletion of Cx43 from radial glia causes an accumulation of cells in the IZ during development, which leads to a smaller cortex in postnatal mice.
Figure 4.
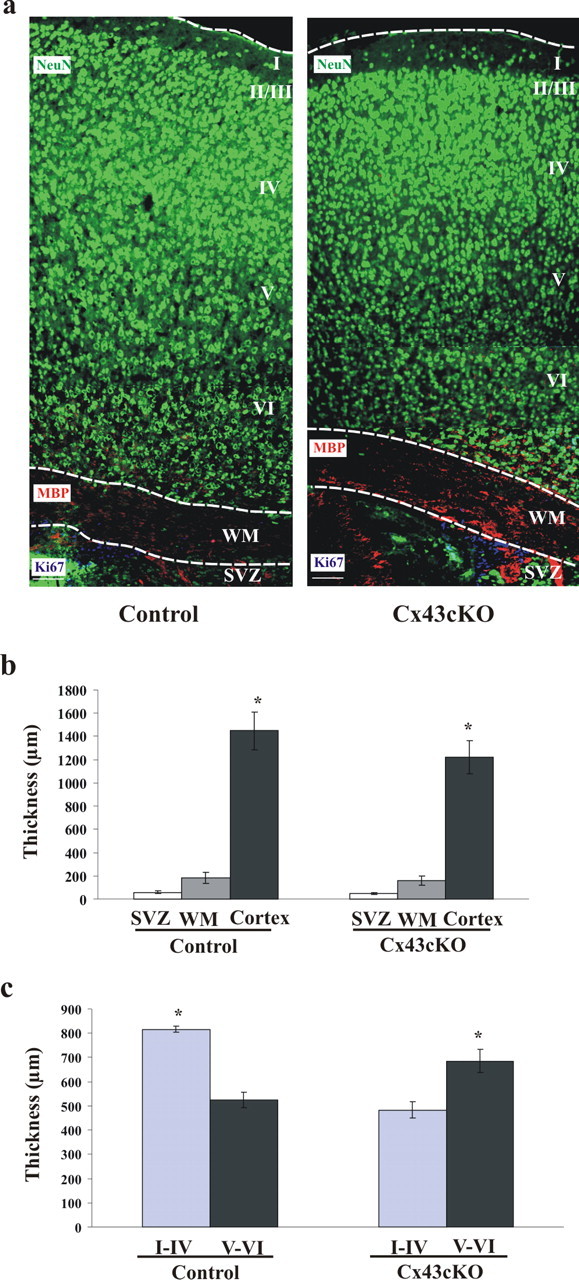
Loss of Cx43 in the developing neocortex leads to a reduction in the thickness of the postnatal neocortex. a, Thickness of the SVZ, WM, and the cortex was measured in the P16 cortices of control and Cx43cKO mice. From each animal, 10 cortical sections spanning rostral end to the caudal end were stained for NeuN (green) to label mature neurons in the cortex (lamina I–VI), MBP (red) to label myelin fibers in the WM, and Ki67 (blue) to label proliferating cells in the SVZ. Using AxioVision software, the outline of each layer was drawn and the thickness was measured. b, The representative graph shows that, compared with the control littermates at P16, the cortex of the Cx43cKO mice was thinner. c, The representative graph shows significantly reduced thickness of upper cortical layers and increased thickness of lower cortical layers in Cx43cKO, but the normal layering is maintained. *p < 0.05, Mann–Whitney test. n = 5. Scale bars, 100 μm.
Differences in cortical formation between Cx43cKO and control mice are a result of neuronal migration but not altered cellular proliferation
BrdU integration into newly synthesized DNA occurs in the VZ and the SVZ of the developing cortex as a result of proliferation of progenitor cells and intermediate progenitor cells, respectively. Here, we performed a 2 h labeling of BrdU to look at the effect of Cx43 in the neural progenitor cell population. Immunohistochemistry for BrdU and Ki67 was conducted to show the proliferative cells during S-phase and all phases of the cell cycle, respectively (Fig. 5a,b). As shown in the representative pictures, a normal distribution of proliferative cells was observed in Cx43cKO mice. Based on DAPI staining, the sections of the neocortex were divided into three zones: the VZ, the IZ, and the CP. To define each zone, the outline of the VZ, the IZ, and the CP of each section was traced using the AxioVsion 4.2 software. The number of BrdU+ or Ki67+ cells was calculated in the VZ of 10 rostral, medial, and caudal sections from four brains per group. There was no significant difference in the number of Brdu+ or Ki67+ cells in the VZ during proliferation between control and Cx43cKO mice (BrdU: control, 1386 ± 75 vs Cx43cKO, 1295 ± 91 cells per section; Ki67: control, 1870 ± 56 vs Cx43KO, 1650 ± 130 cells per section; p > 0.05). These findings indicate that the accumulation of cells in the IZ of the Cx43cKO mice is not a result of disrupted cellular proliferation.
Figure 5.
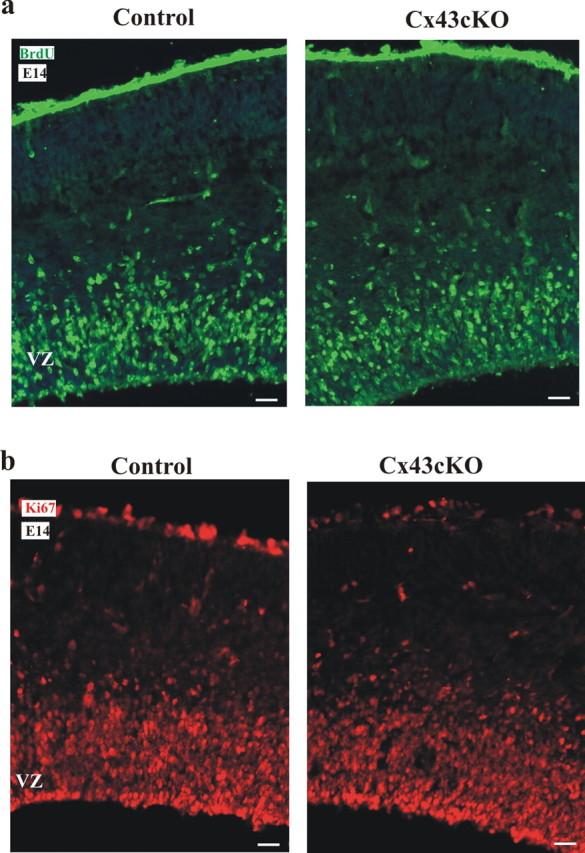
Deletion of Cx43 in radial glia of Cx43cKO mice does not produce a change in the rate of proliferation. Representative sections of BrdU labeling at E14 are shown in a. BrdU was administered at E14, and brains were fixed 2 h after labeling. Immunohistochemistry for BrdU was performed. From at least four brains per group, 10 coronal sections were assessed spanning the rostral to the caudal end of the neocortex. Based on DAPI staining, the sections of the neocortex were divided into three zones: the VZ, the IZ, and the CP. To define each zone, the outline of the VZ, the IZ, and the CP of each section was traced using the AxioVsion 4.2 software. Selected samples were also double labeled with polyclonal antibodies for Ki67 and showed a normal distribution of proliferative cells in the Cx43cKO mice (b). n = 4. Scale bars, 100 μm.
Our previous BrdU studies investigating embryonic neocortical cell migration in Cx43 knock-out mice in vivo showed an accumulation of immature neurons in the outer IZ and fewer in the inner CP, suggesting delayed neuronal migration (Fushiki et al., 2003). Here, in a similar approach, embryos were exposed to BrdU on E14 and analyzed at E18 in control and Cx43cKO mice. In control mice, the majority of labeled cells were detected in the CP, whereas in Cx43cKO mice, more BrdU-labeled cells were observed in the IZ (Fig. 6a).
Figure 6.
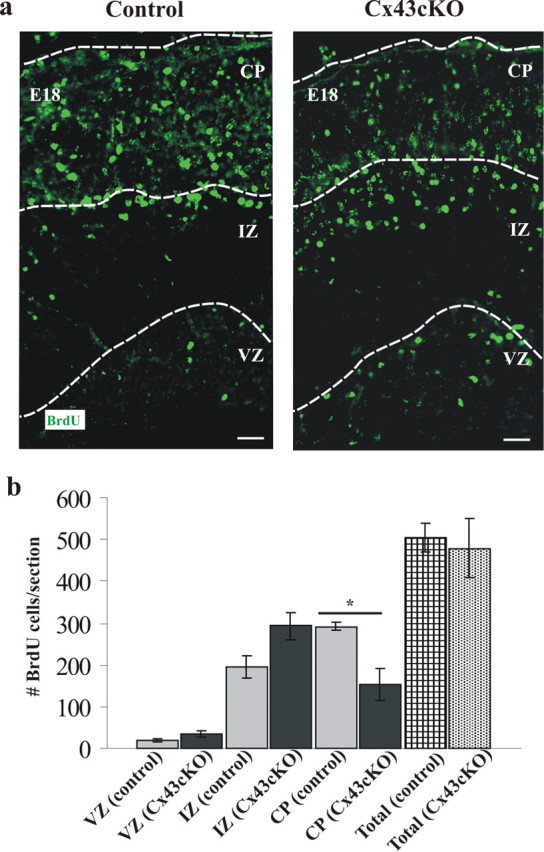
Loss of Cx43 in radial glia causes accumulation of BrdU+ cells in the VZ/IZ of Cx43cKO forebrains. a, Representative sections of BrdU labeling in E18 cortex of mice exposed to BrdU at E14. In Cx43cKO mice, there were still BrdU-labeled cells positioned in the VZ compared with the control mice. b, Distribution of BrdU-labeled cells in the IZ and the CP of the neocortex at E18. In the Cx43cKO mice, the number of BrdU-labeled cells in the CP was decreased and in the IZ increased compared with the control mice. Total number of cells in all zones was counted, and no significant difference was found among all the animals studied. *p < 0.05, Student's t test. n = 6. Scale bars, 100 μm.
In control animals, >60% of the labeled cells resided in the CP and only 38% in the IZ. In contrast, the CP of the Cx43cKO mice contained only 30% of all labeled cells, with 65% residing in the IZ (Fig. 6b).
Exogenous wild-type Cx43 is necessary for neuronal migration in vivo
We predicted that the accumulation of cells in the VZ/IZ is attributable to disrupted neuronal migration. To test this prediction, we monitored the migration of newborn neurons leaving the VZ in Cx43cKO mice. We made use of a transgene expressing GFP under control of a Tα1 tubulin promoter, because the majority of cells expressing Tα1 tubulin are neuronal progenitor cells (Gloster et al., 1999). The transgene was injected into the lateral ventricle of E14 control and Cx43cKO mice, followed by in utero electroporation; the embryos were allowed to develop until E16 or E18. Subsequently, the embryos were removed, and the brains were isolated and sectioned. One day after electroporation, only cells within the VZ or the IZ displayed GFP expression (data not shown), as reported previously (Ohtsuka et al., 1999; Barnabé-Heider et al., 2005). We quantified and compared the distribution of GFP-labeled cells as they migrate from the VZ through the IZ and into the CP between control and Cx43cKO mice. The neocortex was divided into three zones (the VZ, the IZ, and the CP), and the percentage intensity of GFP labeling within each zone was determined (see Materials and Methods). At E16, most cells accumulated in the IZ with no significant difference in the fraction of GFP intensity between control and Cx43cKO, as shown in the representative images (76 and 73%, respectively; p > 0.05) (Fig. 7a,c). At E18, however, GFP+ cells had migrated out of the VZ, passed through the IZ (15.34% GFP intensity), and into the CP (79.9% GFP intensity) of control animals, in which they differentiate into mature neurons. This pattern was not observed in the cortex of Cx43cKO mice, in which GFP+ cells accumulate in the IZ (65.09% GFP intensity), with very few cells reaching the CP (18.17% GFP intensity) by E18 (p < 0.05) (Fig. 7a,c). Furthermore, the majority of cells expressing GFP were positive for NeuN and MAP-2, confirming those as neurons [Fig. 7b and supplemental Fig. 1 (available at www.jneurosci.org as supplemental material), respectively]. These results indicate that Cx43 is necessary for proper neuronal migration.
Figure 7.
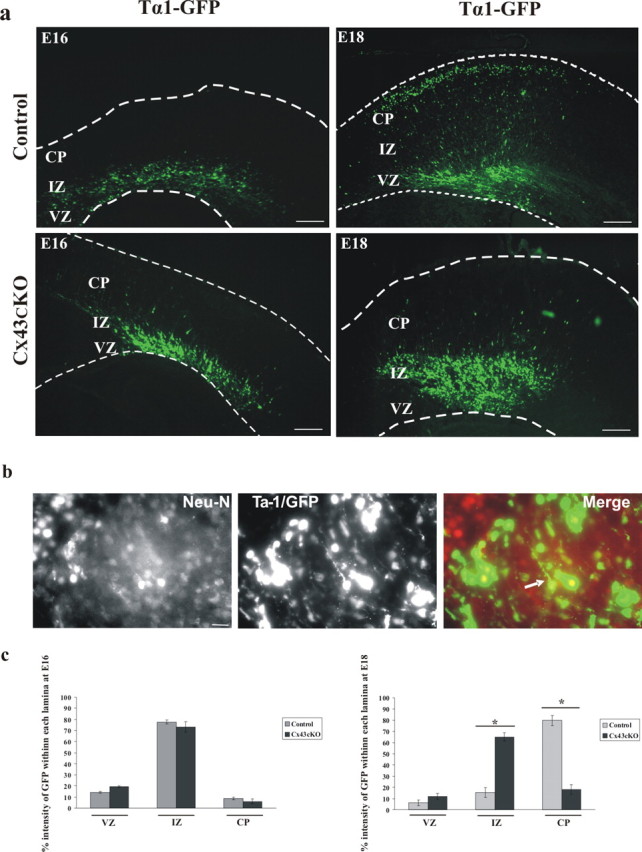
Deletion of Cx43 in radial glial cells and newborn neurons of Cx43cKO mice interrupts neuronal migration. a, At E14, a transgene expressing GFP under control of a neuronal promoter (Tα1 tubulin) was injected into the lateral ventricle of control or Cx43cKO forebrains, followed by in utero electroporation. The brains were removed at E16 or E18, 10 μm sections of cerebral cortices were collected, and subsequently the GFP+ cells were visualized under an epifluorescent microscope. At E16, most GFP+ cells were still accumulated in the IZ with no significant difference between control and Cx43cKO. In control mice, at E18, GFP+ cells were able to migrate to their destination in the CP, whereas in Cx43cKO cerebral cortices, the neurons failed to migrate to the CP. Four days after electroporation, coronal sections of the embryonic cerebral cortex were analyzed for coexpression of GFP and NeuN. Note that the majority of GFP+ cells also express NeuN, confirming them as neurons (b). The percentage intensity of GFP-labeled cells in the VZ, the IZ, and the CP of the E16 and E18 forebrain sections was calculated and plotted as the mean ± SEM as shown in c. *p < 0.05, Student's t test. n = 5. Scale bars: a, 200 μm; b, 50 μm.
The C-terminal domain of Cx43 is required for neuronal migration
We have shown previously that, during embryonic development, several connexins, including Cx43, are expressed not only in the VZ but also throughout the IZ and the CP (Cina et al., 2007). Furthermore, we found that Cx43 expression is closely associated with the radial glia and the migrating neurons (Cina et al., 2007), suggesting that Cx43 might play a significant role in the interactions between migrating neurons and the radial glial scaffold.
Additionally, in a more recent study by Elias et al. (2007), Cx43 was demonstrated to provide necessary adhesion between migrating neurons and radial glial fibers for proper neuronal migration. Thus, to rescue the Cx43-dependent migration/development paradigm, we introduced Cx43 into the cortices of Cx43-deficient mice by in utero electroporation. Cx43 was expressed under the control of the CMV promoter (CMV–Cx43).
As demonstrated in Figure 8a, when the CMV–Cx43 plasmid was coinjected with a plasmid encoding Tα1–GFP and electroporated into the lateral ventricle of E14 Cx43cKO embryos, the migrating neurons were able to reach the CP at E18. In addition, brain sections of Cx43cKO mice subjected to this in utero electroporation were immunostained for Cx43 and showed extensive labeling as seen in Figure 8b. These results strongly suggest that the reconstitution of full-length Cx43 into the lateral ventricle of Cx43cKO embryos at E14 rescued the defected neuronal migration.
Figure 8.
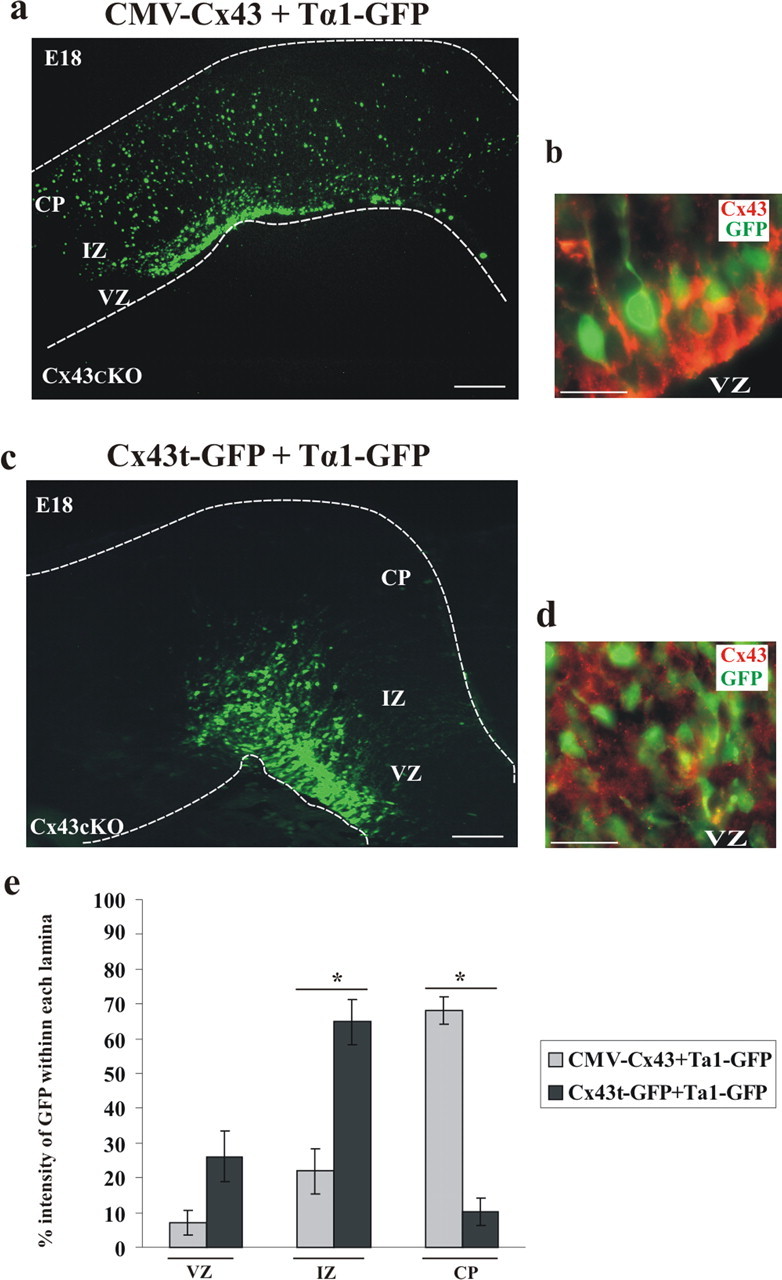
Rescue of radial migration phenotype by wild-type Cx43. a, CMV–Cx43 and Tα1–GFP plasmids were coinjected into the lateral ventricle of control and Cx43cKO mice, followed by in utero electroporation at E14. The brains were removed at E18, and the GFP-positive cells were visualized under an epifluorescent microscope. The insertion of wild-type Cx43 into the lateral ventricle resulted in proper neuronal migration. b, Coronal sections were stained for Cx43 (shown in red). n = 4. c, C-terminal region of Cx43 is necessary for radial migration. Truncated form of Cx43 (Cx43t–GFP) failed to rescue deficient migration. d, Frozen sections were stained for Cx43 (N terminal, shown in red). n = 3. Scale bars: a, c, 200 μm; b, d, 50 μm. e, The percentage intensity of GFP-labeled cells in the VZ, the IZ, and the CP of the E18 forebrain sections of Cx43cKO mice, electroporated with either CMV–Cx43 or Cx43t–GFP, was calculated and plotted as the mean ± SEM. The percentage intensity of GFP in the CP of the mice electroporated with CMV–Cx43 was found to be significantly higher when compared with mice electroporated with Cx43t–GFP. *p < 0.05, Student's t test. n = 5.
Previous studies have investigated the consequence of deleting the C-terminal domain of Cx43 on cell motility. In C6 glioma cells, the C-terminal region of Cx43 has been shown to play a significant role in enhancing motility (Moorby, 2000; Bates et al., 2007), but Elias et al. (2007) found the C-terminal of Cx43 not to be essential for this role, because their experiments showed that truncated Cx43 was able to rescue the neuronal migration defect in developing rat neocortex after small interfering RNA (siRNA) knockdown of endogenous Cx43. To elucidate the contribution of the C terminus in Cx43-mediated neuronal migration, we electroporated E14 Cx43cKO embryos with a construct coding for a C-terminally truncated form of Cx43 tagged with GFP (Cx43t–GFP). Interestingly, the truncated form of Cx43 failed to rescue migration; the cells remained in the VZ/IZ, suggesting that the C-terminal domain of Cx43 is necessary for radial migration in Cx43cKO mouse neocortex (Fig. 8c). We performed immunohistochemistry for Cx43 using an antibody directed against the N terminal. The punctate expression of Cx43t and its association with the Tα1–GFP transgene in the VZ is shown in Figure 8d. Immunostaining for Cx43 N-terminal domain on Cx43KO tissue sections was negative, whereas wild-type Cx43 tissue sections showed punctate staining demonstrating the specificity of the antibody (data not shown).
We next quantified the effect of the CMV–Cx43 transgene in Cx43cKO mice and compared it with the effect of the Cx43t–GFP transgene in the same genotype. By E18, 68% of the intensity of GFP+ cells was observed in the CP when electroporated with CMV–Cx43, whereas in embryos electroporated with Cx43t–GFP, the majority of the cells (65% cell intensity) accumulated in the IZ with significantly less GFP intensity (10.28%) in the CP (p < 0.05) (Fig. 8e), as shown in the representative images (Fig. 8a,c). These data suggest that the C-terminal domain of Cx43 is required for neuronal migration.
To further examine the contribution of the C-terminal domain of Cx43 in neuronal migration, we performed in utero electroporation experiments using Cx43K258stop/− mice that endogenously lack the last 125 amino acid residues of the Cx43 C-terminal domain (Maass et al., 2004). E14 cortices were electroporated with the Tα1–GFP plasmid, and embryos were analyzed 4 d later. The Cx43K258stop/− cortices showed results similar to Cx43cKO mice. Specifically, the vast majority of the Tα1–GFP-expressing neurons remained in the VZ/IZ at E18 (Fig. 9a). In contrast, in Cx43K258stop/+ mice, neurons were able to reach their final location in the CP when electroporated with the Tα1–GFP plasmid at E14 (Fig. 9b). Quantitative analysis demonstrated that, in Cx43K258stop/− mice, 71.16% intensity of GFP+ cells was seen in the IZ with only 3.55% cell intensity in the CP. In contrast, the majority of the GFP+ cells (64.59% cell intensity) reached the CP in Cx43K258stop/+ mice, implying that neuronal migration was not perturbed in these mice as shown in the representative image (p < 0.05) (Fig. 9b). Thus, we propose that the C-terminal tail of Cx43 is essential to promote neuronal migration in the embryonic mouse neocortex.
Figure 9.

The C terminus is necessary for the function of Cx43 during neuronal migration. At E14, Tα1–GFP plasmid was injected alone or coinjected with CMV–Cx43 plasmid into the lateral ventricle of Cx43−/−, Cx43K258stop/+, or Cx43K258stop/− mice, followed by electroporation (a–d). Four days later, the electroporated brains were removed, 10 μm sections of cerebral cortices were collected, and subsequently the GFP+ cells were visualized under an epifluorescent microscope. Although in Cx43K258stop/− mice neurons failed to migrate to the CP (a), Cx43K258stop/+ mice showed normal neuronal migration (b). The bar graph on the right indicates a significant difference in neuronal migration between Cx43K258stop/− and Cx43K258stop/+ mice. In Cx43−/− mice, neurons electroporated with Tα1–GFP failed to migrate to their final location in the CP (c), whereas CMV–Cx43 rescued neuronal migration in these mice (d). The bar graph on the right shows that the percentage intensity of GFP in the CP of Cx43−/− mice, rescued with CMV–Cx43, is significantly higher than of those injected with only Tα1–GFP. Disrupted neuronal migration by Cx43siRNA is shown in e and f. In Cx43+/+ mice, 4 d after electroporation, cells transfected with Cx43–siRNA were positioned in the VZ/IZ, and no cells reached the CP (e), whereas cells transfected with control–siRNA were able to migrate to the CP (f). The bar graph on the right demonstrates that Cx43–siRNA causes a significant loss in the CP when compared with the control–siRNA. *p < 0.05, Student's t test. n = 3. Scale bars, 200 μm.
To further confirm the role of Cx43 in neuronal migration, we conducted in utero electroporation experiments on Cx43KO (Cx43−/−) mice. Tα1–GFP was expressed alone or coexpressed with CMV–Cx43 in E14 neocortices, and embryos were analyzed at E18. The data demonstrated that, although newborn neurons electroporated with Tα1–GFP plasmid failed to migrate to the CP (Fig. 9c), exogenous Cx43 was able to rescue neuronal migration (Fig. 9d). The results were confirmed by quantitative analysis demonstrating that, in Cx43KO mice, 58.99% GFP intensity was present in the IZ and 8.16% in the CP when electroporated with Tα1–GFP, whereas the fraction of GFP intensity in the IZ decreased to 34.64% and in the CP increased to 50.79% when Cx43KO mice were electroporated with CMV–Cx43 transgene (p < 0.05) (Fig. 9d). Additionally, either an siRNA directed against Cx43 (U6–Cx43–siRNA) or a control–U6–siRNA plasmid was cointroduced using electroporation with Tα1–GFP into the developing neocortex of E14 wild-type embryos. By E18, an accumulation of GFP+ cells was observed in the VZ and the IZ of the wild-type mice electroporated with Cx43–siRNA (Fig. 9e), whereas mice electroporated with U6–control–siRNA showed normal neuronal migration (Fig. 9f). As shown in Figure 9, quantitative analysis confirmed that there was a significant change in the fraction of intensity of GFP in the CP when Cx43+/+ embryos were electroporated with Cx43–siRNA plasmid compared with control–siRNA plasmid. Most clearly, there was a loss of GFP intensity in the cortical plate (3.15% intensity), whereas the majority of the intensity was seen in the VZ and the IZ (26.41 and 68.70%, respectively). However, in mice electroporated with control–siRNA, the fraction of GFP intensity was more abundant in the CP (87.41%) than in the VZ or the IZ.
Because exogenous expression of Cx43 in radial glial progenitors of Cx43cKO and Cx43KO mice restores neuronal migration to levels seen in control mice, our study strongly supports the concept that Cx43 plays a vital role in neuronal migration. Furthermore, because a C-terminally truncated Cx43 failed to rescue migration, the C-terminal region appears necessary for this function of Cx43.
Discussion
Over the past several years, a number of research groups have used in vitro models to elucidate the role of Cx43 in cell migration. For example, astrocytes normally form a layer that radiates away from organotypic slice cultures of embryonic mouse brain, but we have shown previously that astrocytes in Cx43KO slices remain confined to the central regions of similar slice cultures (Perez Velazquez et al., 1996). Aggregation assays have shown Cx43 enhancing adhesion between astrocytes and increasing the invasiveness of glial tumor cells (Lin et al., 2002), whereas Cx43KO cardiac neural crest cells were observed to migrate slower than normal with reduced directionality (Xu et al., 2006). Together, these studies suggest that Cx43 may contribute to cellular migration during development.
Despite the many advances in our understanding about cellular motility and the role Cx43 has to play in directing it, there have been relatively few successful attempts to document these effects in vivo. We have striven to address this issue by investigating the role of Cx43 in the neocortical development of three distinct mouse lines. Cx43 null mice represent a complete knock-out model. Although general homozygous Cx43 ablation is lethal, death occurs postnatally, allowing us to trace the effect of Cx43 on neuronal migration during embryogenesis (Reaume et al., 1995; Fushiki et al., 2003). Cx43K258stop mice lack the last 125 amino acid residues of the cytoplasmic C-terminal domain of Cx43. This is also a homozygous lethal phenotype, resulting in postnatal death from a complete loss of the epidermal permeability barrier (Maass et al., 2004). The Cx43cKO mice used in the current study allow selective knock-out of Cx43 only in the radial glial progenitors.
The current work represents significant evidence for Cx43 modulating the motility of the neuronal daughter cells that are produced by the radial glial daughter cells and suggests a possible mechanism by which this occurs.
Deletion of Cx43 from radial glial progenitors causes abnormal cortical development
Our results demonstrate that expression of Cx43 in radial glial progenitors is necessary for normal neocortical development. The spatial restricted ablation of Cx43 in Cx43cKO mice leads to a thickening of the IZ and a concomitant thinning of the CP, both of which appear to be the result of newly born neurons accumulating in the IZ. To show this is in fact the case, we labeled radial glial progenitor cells in the VZ with BrdU. As expected, the number of BrdU-labeled cells in the CP of Cx43cKO mice was significantly lower than was observed in the control mice. This was not a result of reduced cellular proliferation, however, because the average number of BrdU+ cells was the same for both groups when a short survival BrdU experiment was conducted to assess the S-phase. Similar results have been reported in our previous studies using Cx43 null mice, in which the majority of newly born neurons labeled with BrdU in the VZ tended to accumulate in the IZ (Fushiki et al., 2003). This suggests that the migrational defect observed in Cx43 null mice results from an absence of Cx43 in radial glial progenitor cells. Furthermore, expression of exogenous wild-type Cx43 restores normal migration patterns in Cx43cKO mice when introduced in vivo. Therefore, our findings strongly suggest that Cx43 acts as a regulator for neuronal migration.
Loss of Cx43 alters the ability of newborn neurons to migrate along the radial fibers extending into the CP. As a result, accumulation of neurons disrupts the normal lamination of the cerebral cortex. To further explore these cortical abnormalities, we analyzed the cortical thickness during postnatal development and demonstrated that, compared with the control littermates at P16, in Cx43cKO mice, the upper-layer (I–IV) thickness was markedly reduced; the size of the entire cortical hemisphere was also reduced, but the lamination of the forebrain seemed unchanged. Similarly, a previous study demonstrated that, during postnatal development in Cx43cKO mice, removal of Cx43 in GFAP-expressing cells leads to a smaller cerebral cortex by disrupting cellular organization (Wiencken-Barger et al., 2007). Interestingly, this phenotype was dependant on the genetic background of the experimental mice. C57BL/6 mice suffered no ill effects when Cx43 was removed from radial glia, whereas crossing into the 129SEV background resulted in developmental defects even more severe than we observed in our own study. GFAP and nestin are both radial glial markers, but GFAP expression is not observed until E12.5 (Brenner and Messing, 1996; Zhuo et al., 2001; Kriegstein and Gotz, 2003). Nestin, however, is first expressed in neuroepithelial cells surrounding the neural tube at approximately E10, which later mature into radial glia (Hartfuss et al., 2001; Malatesta et al., 2003). By driving Cre expression with the nestin promoter, we block Cx43 expression at the very beginning of neurogenesis. Given that all of our experiments were performed in the C57BL/6 background and we still observed a robust alteration in neuronal migration, we predict that subsequent experiments in which nestin–Cre is crossed into a Cx43fl/fl 129SEV background will further enhance developmental defects beyond those seen in the Wiencken-Barger study.
Although the importance of Cx43 on neuronal migration is fairly established, the mechanisms regulating these effects are still in question. Gap junctional coupling is not likely to be an important factor driving migration, because fluorescent recovery after photo-bleaching and dye-coupling techniques have not detected major differences in gap junctional coupling between wild-type and Cx43cKO mice (Wiencken-Barger et al., 2007). It has been suggested that gap junctional plaques act as dynamic adhesive contact points between radial glial fibers and newborn neurons moving into the CP (Elias et al., 2007); the current study demonstrates that the C-terminal tail is another important factor.
The cytoplasmic C-terminal tail of Cx43 is crucial in cell migration
Motility of adherent cancer cells is already known to be modulated by the C-terminal tail of Cx43 (Bates et al., 2007), and here we have shown that this is the case for neuronal migration during development as well. Electroporetic delivery of full-length Cx43 into the VZ of Cx43cKO embryos is able to restore normal migration of neurons into the CP. Similar delivery of Cx43 with the C-terminal truncated does not rescue migration. Even more compelling is our observation that neurons in Cx43K258stop embryos also fail to move into the CP normally. These results are in conflict with the recent study from Elias et al. (2007), in which short hairpin RNA (shRNA) was used to knockdown Cx43 expression, and they reported that a C-terminal truncated version of Cx43 was able to return migration rates to normal. It is not entirely clear why the results of their experiments differ from ours, but these discrepancies may actually hold clues as to the mechanism regulating motility. Their method of delivering shRNA against Cx43 via electroporation would have knocked down expression in only a subset of radial glial cells. It is possible then that the cells that retained normal Cx43 levels acted as a support network, facilitating adhesion-mediated migration of truncated Cx43-expressing cells. In our system, in which nearly all radial glia are Cx43 deficient, such a support system would not exist; thus, the expression of truncated Cx43 becomes insufficient to support normal migration.
A previous study from van Rijen et al. (2004), in which an inducible system was used to conditionally delete the expression of cardiac Cx43 in adult mice, reported that a decrease of Cx43 protein up to 95% is needed to induce cardiac-related death. Thus, one could speculate that the siRNA technique is limited in its ability to reduce Cx43 expression and cause a major defect. Regardless of the reason behind these differences, we have gathered compelling evidence that the C-terminal tail of Cx43 is in fact responsible for directing movement of newborn neurons into the CP.
In summary, our data supports two major conclusions. First, in Cx43cKO mice, when Cx43 is deleted from radial glia, many newborn neurons remain in the IZ, which causes an increase in the thickness of the IZ and a subsequent decrease in the thickness of the CP compared with control mice. Second, Cx43, and its cytoplasmic C-terminal domain, play a functional role in radial guided neuronal migration during neocortical development. The detailed molecular mechanisms behind this activity are still unclear. Many proteins have been shown to interact with the C-terminal domain of Cx43, linking it to cytoskeletal organization. These proteins include CCN3/NOV (McLeod et al., 2001; Fu et al., 2004), zona occludens-1 (ZO-1) (Giepmans and Moolenaar, 1998), and N-cadherins (Xu et al., 2001), and each represents a key component of a different cellular structure or function, any one of which could assist in regulating migration. Scaffolding proteins such as actin are linked to tight junctions and adherence junctions via members of the membrane-associated guanylate kinase family of proteins such as ZO-1 (Itoh et al., 1997; Perez-Moreno et al., 2003). Furthermore, ZO-1 interacts with the C-terminal domain of Cx43 as removal of the last four amino acids completely abolishes the binding (Giepmans et al., 2001). Therefore, it is tempting to speculate that scaffolding proteins such as ZO-1 stabilize the actin cytoskeleton to the Cx43 C-terminal domain, thus involving Cx43 in cytoskeletal anchorage. A better understanding of these mechanisms will facilitate the study of a number of neurodevelopmental malformations and diseases.
Footnotes
This work was supported by the Canadian Institutes of Health Research. C.C.N. is a recipient of a Canada Research Chair. We thank Dr. F. Miller for assistance in establishing the in utero electroporation technique, A. Webber and B. Chan for their help with the BrdU experiments, and Dr. W. C. Sin and S. Bond for helpful discussions.
References
- Akagi K, Sandig V, Vooijs M, Van der Valk M, Giovannini M, Strauss M, Berns A. Cre-mediated somatic site-specific recombination in mice. Nucleic Acids Res. 1997;25:1766–1773. doi: 10.1093/nar/25.9.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony TE, Klein C, Fishell G, Heintz N. Radial glia serve as neuronal progenitors in all regions of the central nervous system. Neuron. 2004;41:881–890. doi: 10.1016/s0896-6273(04)00140-0. [DOI] [PubMed] [Google Scholar]
- Barnabé-Heider F, Wasylnka JA, Fernandes KJ, Porsche C, Sendtner M, Kaplan DR, Miller FD. Evidence that embryonic neurons regulate the onset of cortical gliogenesis via cardiotrophin-1. Neuron. 2005;48:253–265. doi: 10.1016/j.neuron.2005.08.037. [DOI] [PubMed] [Google Scholar]
- Bates DC, Sin WC, Aftab Q, Naus CC. Connexin43 enhances glioma invasion by a mechanism involving the carboxy terminus. Glia. 2007;55:1554–1564. doi: 10.1002/glia.20569. [DOI] [PubMed] [Google Scholar]
- Bérubé NG, Mangelsdorf M, Jagla M, Vanderluit J, Garrick D, Gibbons RJ, Higgs DR, Slack RS, Picketts DJ. The chromatin-remodelling protein ATRX is critical for neuronal survival during corticogenesis. J Clin Invest. 2005;115:258–267. doi: 10.1172/JCI22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittman K, Owens DF, Kriegstein AR, LoTurco JJ. Cell coupling and uncoupling in the ventricular zone of the developing neocortex. J Neurosci. 1997;17:7037–7044. doi: 10.1523/JNEUROSCI.17-18-07037.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner M, Messing A. GFAP transgenic mice. Methods. 1996;10:351–364. doi: 10.1006/meth.1996.0113. [DOI] [PubMed] [Google Scholar]
- Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A. GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci. 1994;14:1030–1037. doi: 10.1523/JNEUROSCI.14-03-01030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper KB, McCarthy KD. GFAP-positive progenitor cells produce neurons and oligodendrocytes throughout the CNS. Mol Cell Neurosci. 2006;31:676–684. doi: 10.1016/j.mcn.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Cina C, Bechberger JF, Ozog MA, Naus CCG. Expression of connexins in embryonic mouse neocortical development. J Comp Neurol. 2007;504:298–313. doi: 10.1002/cne.21426. [DOI] [PubMed] [Google Scholar]
- Elias LA, Wang DD, Kriegstein AR. Gap junction adhesion is necessary for radial migration in the neocortex. Nature. 2007;448:901–907. doi: 10.1038/nature06063. [DOI] [PubMed] [Google Scholar]
- Fu CT, Bechberger JF, Ozog MA, Perbal B, Naus CCG. CCN3 (NOV) interacts with connexin43 in C6 glioma cells. J Biol Chem. 2004;279:36943–36950. doi: 10.1074/jbc.M403952200. [DOI] [PubMed] [Google Scholar]
- Fushiki S, Perez Velazquez JL, Zhang L, Bechberger JF, Carlen PL, Naus CC. Changes in neuronal migration in neocortex of connexin43 null mutant mice. J Neuropathol Exp Neurol. 2003;62:304–314. doi: 10.1093/jnen/62.3.304. [DOI] [PubMed] [Google Scholar]
- Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol. 1998;8:931–934. doi: 10.1016/s0960-9822(07)00375-2. [DOI] [PubMed] [Google Scholar]
- Giepmans BN, Verlaan I, Hengeveld T, Janssen H, Calafat J, Falk MM, Moolenaar WH. Gap junctions protein connexin-43 interacts directly with microtubules. Curr Biol. 2001;11:1364–1368. doi: 10.1016/s0960-9822(01)00424-9. [DOI] [PubMed] [Google Scholar]
- Gloster A, Wu W, Speelman A, Weiss S, Causing C, Pozniak C, Reynolds B, Chang E, Toma JG, Miller FD. The Tα1 α-tubulin promoter specifies gene expression as a function of neuronal growth and regeneration in transgenic mice. J Neurosci. 1994;14:7319–7330. doi: 10.1523/JNEUROSCI.14-12-07319.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloster A, El-Bizri H, Bamji SX, Rogers D, Miller FD. Early induction of Tα1 α-tubulin transcription in neurons of the developing nervous system. J Comp Neurol. 1999;405:45–60. doi: 10.1002/(sici)1096-9861(19990301)405:1<45::aid-cne4>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Goodenough DA, Paul DL. Beyond the gap: functions of unpaired connexon channels. Nat Rev Mol Cell Biol. 2003;4:285–294. doi: 10.1038/nrm1072. [DOI] [PubMed] [Google Scholar]
- Götz M, Sommer L. Cortical development: the art of generating cell diversity. Development. 2005;132:3327–3332. doi: 10.1242/dev.01931. [DOI] [PubMed] [Google Scholar]
- Groome NP, Dawkes A, Gales M, Hruby S, Alvord EC., Jr Region-specific immunoassays for human myelin basic protein. J Neuroimmunol. 1986;12:253–264. doi: 10.1016/0165-5728(86)90032-9. [DOI] [PubMed] [Google Scholar]
- Hartfuss E, Galli R, Heins N, Götz M. Characterization of CNS precursor subtypes and radial glia. Dev Biol. 2001;229:15–30. doi: 10.1006/dbio.2000.9962. [DOI] [PubMed] [Google Scholar]
- Hockfield S, McKay RD. Identification of major cell classes in the developing mammalian nervous system. J Neurosci. 1985;5:3310–3328. doi: 10.1523/JNEUROSCI.05-12-03310.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Costantini F, Lacy E. Plainview, NY: Cold Spring Harbor Laboratory; 1994. Manipulating the mouse embryo: a laboratory manual. [Google Scholar]
- Huber G, Matus A. Differences in the cellular distribution of two microtubule- associated proteins MAP1 and MAP2 in rat brain. J Neurosci. 1984;4:151–160. doi: 10.1523/JNEUROSCI.04-01-00151.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobas DA, Iacobas S, Urban-Maldonado M, Scemes E, Spray DC. Similar transcriptomic alterations in Cx43 knock-down and knock-out astrocytes. Cell Commun Adhes. 2008;15:195–206. doi: 10.1080/15419060802014222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M, Nagafuchi A, Moroi S, Tsukita S. Involvement of ZO-1 in cadherin-based cell adhesion through its direct binding to α catenin and actin filaments. J Cell Biol. 1997;138:181–192. doi: 10.1083/jcb.138.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein AR, Götz M. Radial glia diversity: a matter of cell fate. Glia. 2003;43:37–43. doi: 10.1002/glia.10250. [DOI] [PubMed] [Google Scholar]
- Lin JH, Takano T, Cotrina ML, Arcuino G, Kang J, Liu S, Gao Q, Jiang L, Li F, Lichtenberg-Frate H, Haubrich S, Willecke K, Goldman SA, Nedergaard M. Connexin43 enhances the adhesivity and mediates the invasion of malignant glioma cells. J Neurosci. 2002;22:4302–4311. doi: 10.1523/JNEUROSCI.22-11-04302.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Turco JJ, Kriegstein AR. Clusters of coupled neuroblasts in embryonic neocortex. Science. 1991;252:563–566. doi: 10.1126/science.1850552. [DOI] [PubMed] [Google Scholar]
- Maass K, Ghanem A, Kim JS, Saathoff M, Urschel S, Kirfel G, Grümmer R, Kretz M, Lewalter T, Tiemann K, Winterhager E, Herzog V, Willecke K. Defective epidermal barrier in neonatal mice lacking the C-terminal region of coneixn43. Mol Biol Cell. 2004;15:4597–4608. doi: 10.1091/mbc.E04-04-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malatesta P, Hack MA, Hartfuss E, Kettenmann H, Klinkert W, Kirchhoff F, Götz M. Neuronal or glial progeny: regional differences in radial glia fate. Neuron. 2003;37:751–764. doi: 10.1016/s0896-6273(03)00116-8. [DOI] [PubMed] [Google Scholar]
- McLeod TL, Bechberger JF, Naus CC. Determination of a potential role of the CCN family of growth regulators in connexin43 transfected C6 glioma cells. Cell Commun Adhes. 2001;8:441–445. doi: 10.3109/15419060109080767. [DOI] [PubMed] [Google Scholar]
- Mizuno H, Hirano T, Tagawa Y. Activity-dependent cortical wiring: Formation of interhemispheric connections in neonatal mouse visual cortex requires projection neuron activity. J Neurosci. 2007;27:6760–6770. doi: 10.1523/JNEUROSCI.1215-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani K, Saito T. Progenitors resume generating neurons after temporary inhibition of neurogenesis by notch activation in the mammalian cerebral cortex. Development. 2005;132:1295–1304. doi: 10.1242/dev.01693. [DOI] [PubMed] [Google Scholar]
- Moorby CD. A connexin43 mutant lacking the carboxyl cytoplasmic domain inhibits both growth and motility of mouse 3T3 fibroblasts. Mol Carcinog. 2000;28:23–30. [PubMed] [Google Scholar]
- Nadarajah B, Jones AM, Evans WH, Parnavelas JG. Differential expression of connexins during neocortical development and neuronal circuit formation. J Neurosci. 1997;17:3096–3111. doi: 10.1523/JNEUROSCI.17-09-03096.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409:714–720. doi: 10.1038/35055553. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Kriegstein AR. Distinct behaviors of neural stem and progenitor cells underlie cortical neurogenesis. J Comp Neurol. 2008;508:28–44. doi: 10.1002/cne.21669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka T, Ishibashi M, Gradwohl G, Nakanishi S, Guillemot F, Kageyama R. Hes1 and Hes5 as notch effectors in mammalian neuronal differentiation. EMBO J. 1999;18:2196–2207. doi: 10.1093/emboj/18.8.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquin A, Barnabé-Heider F, Kageyama R, Miller FD. CCAAT/enhancer-binding protein phosphorylation biases cortical precursors to generate neurons rather than astrocytes in vivo. J Neurosci. 2005;25:10747–10758. doi: 10.1523/JNEUROSCI.2662-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Moreno M, Jamora C, Fuchs E. Sticky business: orchestrating cellular signals at adherence junctions. Cell. 2003;112:535–548. doi: 10.1016/s0092-8674(03)00108-9. [DOI] [PubMed] [Google Scholar]
- Perez Velazquez JL, Frantseva M, Naus CC, Bechberger JF, Juneja SC, Velumian A, Carlen PL, Kidder GM, Mills LR. Development of astrocytes and neurons in cultured brain slices from mice lacking connexin43. Brain Res Dev Brain Res. 1996;97:293–296. doi: 10.1016/s0165-3806(96)00156-3. [DOI] [PubMed] [Google Scholar]
- Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, Juneja SC, Kidder GM, Rossant J. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267:1831–1834. doi: 10.1126/science.7892609. [DOI] [PubMed] [Google Scholar]
- Redila VA, Olson AK, Swann SE, Mohades G, Webber AJ, Weinberg J, Christie BR. Hippocampal cell proliferation is reduced following prenatal ethanol exposure but can be reduced with voluntary exercise. Hippocampus. 2006;16:305–311. doi: 10.1002/hipo.20164. [DOI] [PubMed] [Google Scholar]
- Simon AM, Goodenough DA. Diverse functions of vertebrate gap junctions. Trends Cell Biol. 1998;8:477–483. doi: 10.1016/s0962-8924(98)01372-5. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Neurons tend to stop migration and differentiate along the cortical internal plexiform zones in the reelin signal-deficient mice. J Neurosci Res. 2002;69:723–730. doi: 10.1002/jnr.10345. [DOI] [PubMed] [Google Scholar]
- Theis M, de Wit C, Schlaeger TM, Eckardt D, Krüger O, Döring B, Risau W, Deutsch U, Pohl U, Willecke K. Endothelium-specific replacement of the connexin43 coding region by a lacZ reporter gene. Genesis. 2001;29:1–13. doi: 10.1002/1526-968x(200101)29:1<1::aid-gene1000>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- van Rijen HV, Eckardt D, Degen J, Theis M, Ott T, Willecke K, Jongsma HJ, Opthof T, de Bakker JM. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation. 2004;109:1048–1055. doi: 10.1161/01.CIR.0000117402.70689.75. [DOI] [PubMed] [Google Scholar]
- Wiencken-Barger AE, Djukic B, Casper KB, McCarthy KD. A role for connexin43 during neurodevelopment. Glia. 2007;55:675–686. doi: 10.1002/glia.20484. [DOI] [PubMed] [Google Scholar]
- Xu X, Li WE, Huang GY, Meyer R, Chen T, Luo Y, Thomas MP, Radice GL, Lo CW. Modulation of mouse neural crest cell motility by N-cadherin and connexin43 gap junctions. J Cell Biol. 2001;154:217–230. doi: 10.1083/jcb.200105047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Francis R, Wei CJ, Linask KL, Lo CW. Connexin43-mediated modulation of polarized cell movement and the directional migration of cardiac neural crest cells. Development. 2006;133:3629–3639. doi: 10.1242/dev.02543. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-Cre transgene mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]