

Abstract
The GABAA receptor has been identified as the single most important target for the intravenous anesthetic propofol. How effects at this receptor are then translated into a loss of consciousness, however, remains a mystery. One possibility is that anesthetics act on natural sleep pathways. Here, we test this hypothesis by exploring the anesthetic sensitivities of GABAergic synaptic currents in three specific brain nuclei that are known to be involved in sleep. Using whole-cell electrophysiology, we have recorded GABAergic IPSCs from the tuberomammillary nucleus (TMN), the perifornical area (Pef), and the locus ceruleus (LC) in brain slices from both wild-type mice and mice that carry a specific mutation in the GABAA receptor β3 subunit (N265M), which greatly reduces their sensitivity to propofol, but not to the neurosteroid alphaxalone. We find that this in vivo pattern of anesthetic sensitivity is mirrored in the hypothalamic TMN and Pef nuclei, consistent with their role as direct anesthetic targets. In contrast, anesthetic sensitivity in the LC was unaffected by the β3N265M mutation, ruling out this nucleus as a major target for propofol. In support of the hypothesis that orexinergic neurons in the Pef are involved in propofol anesthesia, we further show that these neurons are selectively inhibited by GABAergic drugs in vivo during anesthesia, and that a modulation in the activity of Pef neurons alone can affect loss of righting reflex. Overall, our results support the idea that GABAergic anesthetics such as propofol exert their effects, at least in part, by modulating hypothalamic sleep pathways.
Keywords: hypothalamus, sleep, GABAA receptor, anesthesia, histamine, synapse
Introduction
There is a growing appreciation that the anesthetic selectivity that is observed at the molecular level (Franks and Lieb, 1994; Rudolph and Antkowiak, 2004) may extend to the level of neuronal networks (Devor and Zalkind, 2001) and there is some evidence that anesthetics may cause a loss of consciousness through pathways that maintain cortical activation and behavioral arousal, or the sleep pathways that control them (Tung et al., 2001, 2002; Lydic and Baghdoyan, 2005; Franks, 2008).
We are pursuing the hypothesis that anesthetics inhibit pathways that maintain arousal, for example by enhancing the postsynaptic inhibitory effects of the ventrolateral preoptic nucleus (VLPO). This region increases firing before sleep onset (Szymusiak et al., 1998) causing sleep through the GABAergic inhibition of arousal nuclei (Sherin et al., 1996, 1998) such as the histaminergic tuberomammillary nucleus (TMN). This hypothesis is particularly directed toward those general anesthetics, such as propofol and etomidate, that act predominantly (Hales and Lambert, 1991; Krasowski et al., 1998; Tomlin et al., 1998) at GABAA receptors.
Both propofol and pentobarbital cause a sleep-like pattern of neuronal activity in the TMN and VLPO (Nelson et al., 2002, 2003; Ko et al., 2003; Takahashi et al., 2006), consistent with the reduction in brain histamine levels during anesthesia (Mammoto et al., 1997). Moreover, injection of the GABAA receptor agonist muscimol into the TMN caused LORR, whereas the GABAA receptor antagonist gabazine antagonized LORR (Nelson et al., 2002, 2003). However, injection of propofol itself caused sedation but not LORR, implying that other pathways must also be required. This is perhaps not surprising given the redundancy of arousal pathways: the sedative α2 adrenoceptor agonist dexmedetomidine appears to impinge upon the VLPO-TMN pathway (Nelson et al., 2003) through the locus ceruleus (LC) and orexins may cause arousal via the TMN (Huang et al., 2001). This indicates that both sleep and anesthetic-induced loss of consciousness may depend on the concerted inhibition of multiple arousal systems.
This study tests the hypothesis that specific brain nuclei might be directly inhibited by GABAergic anesthetics. We have used a mouse with a point mutation in the β3 subunit of the GABAA receptor, the β3N265M knock-in, which caused a substantial reduction in anesthetic sensitivity for propofol, pentobarbital and etomidate, but an unchanged sensitivity to the neurosteroid alphaxalone (Jurd et al., 2003; Zeller et al., 2005, 2007a,b). If a nucleus is important for anesthetic action, then in the β3N265M knock-in it should display a greatly diminished sensitivity to propofol compared with the wild type. Alphaxalone provides an important negative control; the mutation is known not to affect the alphaxalone sensitivity of GABAA receptors (Siegwart et al., 2002), so responses to the neurosteroid should be identical in the wild-type and knock-in, enabling any change in propofol sensitivity to be attributed to the β3N265M mutation per se. We apply this test to three arousal nuclei that are inhibited by GABAergic sleep pathways: the histaminergic TMN, the orexinergic perifornical area (Pef), and the noradrenergic LC.
Materials and Methods
All experiments were performed in accordance with the United Kingdom Animals (Scientific Procedures) Act of 1986 and have been approved by the Ethical Review Committee of Imperial College London. All efforts were made to minimize animal suffering and reduce the number of animals used.
Electrophysiology.
Adult male (8–12 weeks old) wild-type (WT) controls or GABAA receptor β3(N265M) mutant mice [on background 129/Sv × 129X1/SvJ (12.5%/87.5%)] were genotyped using template DNA from tail tips, which was amplified using the specific primers shown below (MWG Biotech) using the PCR: RJM-8 (5′-GTT CAG CTT CCA TTC TCA CTG-3′) and RJM-24 (5′-GCT ATG GCT TTC TGG TGG AG-3′).
Animals were deeply anesthetized with halothane and killed by cervical dislocation followed by decapitation. The tissue was immediately placed in ice-cold, carbogen (95% O2 5% CO2)-bubbled slicing solution (composition in mm: NaCl 85, KCl 2.5, NaH2PO4.H2O 1.25, NAHCO3 25, CaCl2 1, MgCl2 4, sucrose 75, glucose 25) and 200 μm coronal slices of the TMN, Pef, or the LC were taken.
All experiments were performed at room temperature (21−22°C) in external solution (composition in mm: NaCl 125, KCl 2.5, NaH2PO4.H2O 1.25, NAHCO3 25, CaCl2 2, MgCl2 1, sucrose 25, kynurenic acid 1, strychnine 1). Pipette solutions contained a CsCl internal (composition in mm: CsCl 140, NaCl 4, HEPES 10, EGTA 5, CaCl2 0.5,Mg-ATP 2) for the examination of GABAergic IPSCs. To record spontaneous action potential firing we used a KCl internal (composition in mm: KCl 130, HEPES 10, BAPTA 0.1, MgCl2 5, Na2ATP 3, Na-GTP 0.1, phosphocreatine 8) pipette solution. Pipette resistance was typically between 2 and 4 MΩ. The degree of series resistance deemed acceptable in these experiments was ∼20 MΩ Neurons that had stable series resistance and cell capacitance over the recording period (<20% change) were used for analysis. The signal was filtered at 2 kHz and sampled at 10 kHz. Recordings were made using an Axopatch 200 B amplifier and pClamp 8.2 (Molecular Devices).
Following satisfactory identification of neuronal type (see Results), the effects of propofol and alphaxalone on the GABAergic, spontaneous IPSCs were studied under voltage clamp (at −60 mV), using the standard whole-cell patch clamping technique in the WT and β3N265M TMN, Pef, and LC. IPSCs had a reversal potential close to zero. Single events with an uninterrupted rising and decaying phases were selected for analysis. The decay time constant (τ) was calculated as the IPSC integral divided by its peak.
These isolated events were used to calculate average IPSC parameters, 10–90% rise time (ms), peak amplitude (pA), and decay time (ms). These parameters were compared before and after anesthetic exposure using a two-tailed paired Student's t test (Graphpad Prism 4.03). Neuronal parameters (cell capacitance, input resistance) were analyzed using a two-tailed unpaired Student's t test (Graphpad Prism 4.03).
Drug preparation.
All chemicals were obtained from Sigma-Aldrich unless otherwise stated. Pentobarbital sodium salt, dexmedetomidine (Dex; donated by Orion Pharmaceuticals), muscimol hydrobromide, chloral hydrate, and gabazine hydrobromide (SR-95531, Tocris) were prepared in sterile saline (0.9% NaCl w/v) solution. Halothane (Fluothane, Zeneca) and isoflurane (Aerrane, Baxter) were administered through a TEC vaporizer (Ohmeda; BOC Healthcare) and anesthesia machine (Ohmeda; BOC Healthcare). Propofol for the in vitro experiments was obtained from Sigma-Aldrich and was diluted from an ethanolic stock. For the jugular vein injections, propofol was obtained as a 1% (w/v) lipid emulsion (Rapinovet; Schering-Plough Animal Health). Ketamine was obtained as Ketaset (Fort Dodge Animal Health) and diluted to 1% (v/v) in 0.9% sterile saline, and xylazine was from Millpledge Veterinary. Orexin-A and angiotensin II were diluted in 0.9% sterile saline.
Discrete administration of pharmacological probes into the Pef.
Adult male Fischer rats (250–300 g) were given access to food and water ad libitum and housed under controlled conditions (12 h of light starting at 19:00; 20−22°C). Animals were anesthetized with a ketamine/xylazine mix (70 mg/kg and 10 mg/kg respectively, i.p.) and halothane when necessary and prepared for aseptic surgery and secured in a stereotaxic frame. For bilateral Pef cannulation, two holes were drilled into the skull and filled with removable bone wax (Ethicon). Guide cannulae (22G; Tomlinson Tubes) were stereotaxically positioned [coordinates ± 1.4 mm mediolateral, −3.5 mm anteroposterior, −7.7 mm dorsoventral from Bregma (Estabrooke et al., 2001; Gerashchenko et al., 2001) based on a rat brain atlas (Paxinos and Watson, 2005)], and stabilized with dental resin (Orthoresin; Dentsply). Drugs were discretely administered into the Pef at least 4 d postsurgery using methodology similar to that previously described (Nelson et al., 2002). The GABAA receptor agonist muscimol (0.5–2 μg/0.2 μl/side) or antagonist gabazine (0.2 μg/0.2 μl/side) were microinjected using a Howard CMA microinjection pump (CMA/100; CMA/Microdialysis) at rates of 1.0 μl/min and 0.4 μl/min, respectively. The needle protruded 1 mm beyond the end of the fixed-guide cannula, and was removed 2 min after the completion of the injection. The locations of the injections were confirmed histologically at the completion of each experiment. Using dye injections into the Pef, we found that spread tended to be rather uniform rostrocaudal but more dorsal than ventral, was encompassed within the Pef and, importantly, did not extend measurably to the TMN. This is consistent with quantitative studies (Allen et al., 2008) on the spread of drugs following discrete injection. However, as with all discrete injections of drugs into brain tissue, drugs spread is complex both spatially and temporally and there will always be a degree of uncertainty about which neurons are affected and at which concentrations.
Lateral ventricle and jugular vein cannulation.
Adult male Sprague Dawley rats (300–350 g) were given access to food and water ad libitum and housed under controlled conditions (12 h of light starting at 21:00; 20−22°C). Animals were anesthetized, as above, with a ketamine/xylazine mix (intraperitoneal) and halothane when necessary and prepared for aseptic surgery. After making an incision in the skin overlying the jugular vein, the vessel was isolated by blunt dissection. Ligatures were placed at the caudal and rostral aspect of the vein, which was then punctured using a 25G needle. The catheter was inserted to a depth of ∼3.5 cm and secured to the vessel. Next, a pocket was made on the animal's back and curved tissue forceps were used to tunnel subcutaneously over the shoulder. The catheter tubing was fed through this tunnel and connected to the injection port, which was then secured within the pocket. Catheter placement was checked at various stages of the surgery by the aspiration of blood.
When cannulating the lateral ventricle, a single hole was drilled into the skull and the guide cannula (27G; Small Parts Inc., Miramar) was stereotaxically positioned [coordinates 2.0 mm mediolateral, −1.0 mm anteroposterior, −3.0 mm dorsoventral from Bregma; based on a rat brain atlas (Paxinos and Watson, 2005)], and stabilized with dental resin (Orthoresin; Dentsply). Drugs were administered via a 32G cannula (Small Parts) which protruded 1 mm beyond the end of the guide cannula. Correct placement of the cannula was assessed by an intense drinking response to angiotensin II (100 ng in 5 μl over 30 s). Orexin-A (10 nmol) was administered in 10 μl over 1 min at lights-off, 2 h before the jugular vein injection of anesthetic.
Assessment of loss of righting reflex.
Loss of righting reflex (LORR) was used as an endpoint for anesthesia, defined as the inability of an animal to right itself when placed on its back or side. Following anesthetic administration, LORR was assessed every 30 s. Sleep time was defined as the time until the animal regained its righting reflex.
Transcardial perfusion and tissue sectioning.
Animals were anesthetized with chloral hydrate (350 mg/kg, i.p.) or pentobarbital (140 mg/kg, i.p.), and transcardially perfused with normal saline (100 ml) or PBS (100 ml; 0.1 m phosphate buffer, 0.9% NaCl; pH 7.4) followed by 10% formalin (500 ml) or 4% paraformaldehyde (500 ml; BDH) in 0.1 m phosphate buffer. Whole brains were removed, postfixed in 10% formalin or 4% paraformaldehyde overnight and incubated in 20% sucrose overnight (or until tissue sank), and coronally cryosectioned (1:4 series, 30 μm).
Immunohistochemistry.
Sections were double-immunostained using previously described methods (Estabrooke et al., 2001; Nelson et al., 2002). All sections were stained for c-Fos (goat polyclonal antibody; 1:20,000; Santa Cruz, or 1:150,000; rabbit; Oncogene) using secondary donkey anti-goat IgG (1:200; Millipore Bioscience Research Reagents), VectaStain Elite ABC solution (Vector), visualized using 3,3-diaminobenzidine (DAB; Vector) with nickel ammonium sulfate for black staining. Next, they were counter-stained for orexin-A (1:5000, rabbit polyclonal; Millipore Bioscience Research Reagents) and visualized using DAB without nickel.
During experimental recordings, neurons were filled by adding 0.2% neurobiotin to the pipette solution. Slices were fixed with a freshly prepared solution of 4% 1-ethyl-3(3-dimethylamino-propyl)carbodiimide (EDAC) and 1% N-hydroxysuccinimide in 0.1 m Phosphate buffer (pH 7.4, 2–12 h at 4°C) and then stained with a rabbit anti-histamine (1:1000; Millipore Bioscience Research Reagents) polyclonal antibody (Panula et al., 1984). Slices were then incubated at room temperature with sheep anti-rabbit Cy3 (1:500) and streptavidin-Alexa 488 (1:200; Invitrogen) before mounting and visualization. Optical sections were obtained using a Zeiss LSM 510 upright confocal microscope, equipped with a 40× oil-immersion objective lens. Fluorochromes were excited with an Argon laser at 488 nm using a BP 505–550 nm emission filter (green), with a HeNe laser at 543 nm in combination with a BP 585–615 nm emission filter (red). Neurons were z-sectioned into 1 μm optical slices. The signal-to-noise ratio was improved by averaging at least two scans for each z-section.
Cell counting and analysis.
Using light microscopy, c-Fos positive neurons were identified by dense black nuclear staining, and the orexin-positive neurons in the perifornical area (Pef) of the lateral hypothalamus by brown cytoplasmic staining. Locations in the brain were confirmed by staining and reference to the literature (Estabrooke et al., 2001; Gerashchenko et al., 2001) and a rat brain atlas (Paxinos and Watson, 2005). Four sections through the middle of the Pef per animal (n = 6 per experimental group) were counted (blind to treatment) using a 500 μm counting box centered around the fornix (Estabrooke et al., 2001) and averaged. Data were analyzed using one-way ANOVA and the Bonferroni test, and are presented as means ± SEMs. Differences were considered significant at p < 0.05.
Results
Characterization of Pef, TMN and LC neurons in wild-type and β3N265M knock-in mice
The anatomical location of the Pef was identified in coronal slices of the posterior hypothalamus as being dorsal to the fornix at the level of the median eminence (Fig. 1A). The Pef is a heterogeneous region containing various cell types, including magnocellular neurons that express either orexin or melanin-concentrating hormone (MCH) (often in combination with other peptides) and other cells expressing GABA or glutamate (Bittencourt et al., 1992; Broberger et al., 1998; Peyron et al., 1998; Abrahamson and Moore, 2001). The magnocellular neurons cannot be distinguished from one another by size or morphology, however, these populations have already been well characterized (Eggermann et al., 2003; Bayer et al., 2005; Burdakov et al., 2005) as two distinct populations based upon their membrane potential and spontaneous tonic firing activity. Therefore, we used the criteria of relatively depolarized membrane potential (−45.4 ± 1.2 mV; n = 10) and tonic firing activity to distinguish orexinergic neurons from the relatively hyperpolarized (−68.0 ± 3.2 mV; n = 11) and electrically silent MCH-containing neurons. Neurons were rejected if the membrane potential was more depolarized than −40 mV. Figure 1B shows data from typical WT neurons. Orexinergic neurons from WT mice had an average capacitance of 43.8 ± 4.7 pF and an input resistance of 351 ± 68 MΩ (n = 9). Orexinergic neurons were selected under current clamp followed by examination of GABAergic IPSCs under voltage clamp (Fig. 1C). Events were sorted for an uninterrupted rising and decaying phase so that only isolated IPSCs were studied (Fig. 1D). The spread of peak amplitudes for isolated events recorded from all orexinergic neurons is shown in the inset in Figure 1E. Isolated IPSCs occurred at a frequency of 4.6 ± 1.4 Hz (n = 10) and events were bicuculline- (20 μm; n = 6) and gabazine-sensitive (20 μm; n = 4). However no change in the holding current was observed with application of either drug, indicating the absence of a basal tonic conductance in these neurons (data not shown). GABAergic IPSCs were sorted into three groups (Fig. 1D) and only isolated events were included in the final average for each neuron (Fig. 1E). Neurons from β3N265M knock-in mice had neuronal capacitance (31.3 ± 5.1 pF), input resistance (435 ± 87 MΩ), depolarized resting membrane potential (−46.0 ± 1.6 mV; n = 8) and tonic firing activity that did not differ from neurons from WT mice (unpaired Student's t test, p > 0.05; n = 8). Spontaneous IPSCs occurred at a frequency of 2.0 ± 0.5 Hz (n = 8), which was also not different from WT.
Figure 1.

Characterization of GABAergic IPSCs in identified orexinergic Pef neurons. A, The coronal slice shows the location of the perifornical region (highlighted in red) with respect to the fornix (fx). We used slices corresponding to ∼−1.7 mm from the Bregma. B, The left panel shows an example of a tonically firing (∼11 Hz) orexinergic neuron with a depolarized membrane potential (approximately −46 mV) compared with a relatively hyperpolarized (resting at approximately −60 mV), silent MCH-containing neuron shown on the right. C, Continuous current trace recorded from a WT orexinergic Pef neuron under voltage clamp (−60 mV). In this example, IPSCs occurred at an average frequency of 1.6 Hz and were confirmed as GABAergic due to their bicuculline sensitivity (data not shown; n = 10). Individual events (i–iii) highlighted in red were representative of three IPSC groups. D, Expanded trace for each example highlighted in C. The first trace (i) shows superimposition of IPSCs interrupting the decay phase. The second trace (ii) shows superimposition of IPSCs interrupting the rising phase. Finally, the third trace (iii) shows a single, isolated IPSC. To get an accurate representation of the IPSC parameters, only events falling into category (iii) were included in the analysis. E, Isolated IPSCs from a typical orexinergic neuron. The isolated events (77 events, shown in gray) were used to construct the average IPSC trace (shown in black) of the neuron. This example average IPSC trace had a 10–90% rise time of 1.3 ms, a peak amplitude of −69.2 pA and a decay time of 15.9 ms. The histogram shows the distribution of peak amplitudes recorded from all wild-type orexinergic neurons. The coefficient of variation was 59.4 ± 7.0% (n = 9).
The ventral TMN has been previously described in detail as a small, densely packed nucleus located at the level of the mammillary recess and at the edge of the tissue, ventrolateral to the fornix (Staines et al., 1987). We confirmed the localization of these neurons through immunostaining for the neurotransmitter histamine, with double-staining neurobiotin-filled neurons after electrophysiological recordings (Fig. 2C). Histaminergic neurons were also found to be scattered away from the main cluster but electrophysiological recordings were not attempted from these cells. Positively stained neurons were predominantly large, although a population of smaller neurons has been described (Staines et al., 1987). Larger neurons were selected in the present study. These cells displayed tonic firing activity in current clamp from a membrane potential of around −50 mV (Fig. 2B, left). TMN neurons from WT mice had an average capacitance of 38.7 ± 5.4 pF and input resistance of 323 ± 40 MΩ (n = 12). They were typically oval-shaped, with 2–4 primary dendrites, as visualized by filling the cell with fluorescent dye in the whole-cell configuration and subsequent image capture with confocal microscopy (Fig. 2C). This morphology is consistent with descriptions in the literature. When possible, RT-PCR was used to confirm expression of the histaminergic neuronal marker adenosine deaminase (ADA) (Fig. 2B).
Figure 2.
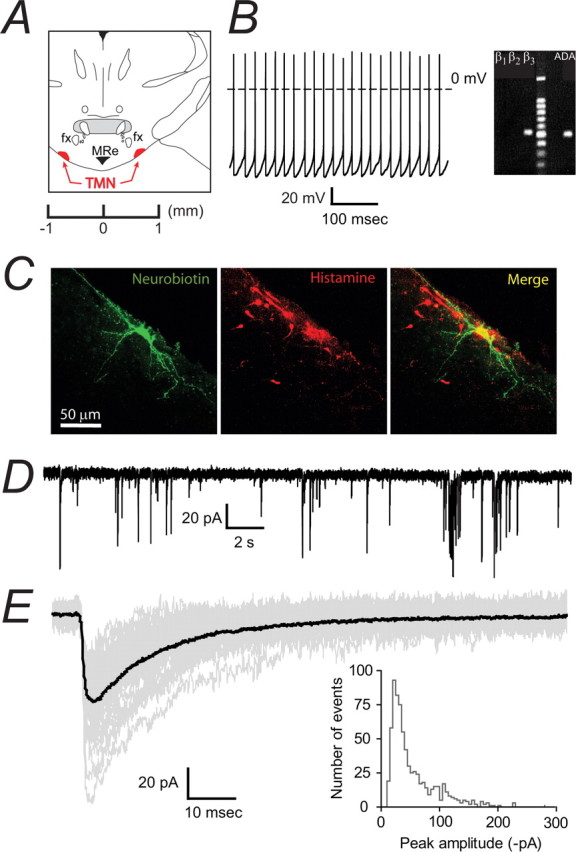
Characterization of GABAergic IPSCs in identified histaminergic neurons in the TMN. A, The coronal slice shows the location of the TMN (highlighted in red) as ventrolateral to the fornix (fx) at the level of the mammillary recess. We used slices corresponding to ∼−2.1 mm from the Bregma. B, A tonically firing (4.4 Hz) TMN neuron is shown on the left. On the right is an example of mRNA expression for the histaminergic neuronal marker ADA and β1–3 subunit of the GABAA receptor in a single neuron using single cell RT-PCR. Tonic firing and expression of ADA was typical of large cells in this region. C, A typical histaminergic TMN neuron. The left panel shows a neurobiotin-filled neuron following whole-cell recording, the center panel shows the staining pattern for histamine and the right panel is the merged image. D, GABAergic IPSCs recorded in the whole-cell configuration from a typical histaminergic TMN neuron. IPSCs occurred at a frequency of ∼1.5 Hz. E, A representative average trace (shown in black) was constructed from monotonic IPSCs (49 events, shown in gray). This example average IPSC had a 10–90% rise time of 1.8 ms, a peak amplitude of −56.0 pA and a decay time of 23.3 ms. The histogram shows the distribution of peak amplitudes recorded from all wild-type histaminergic neurons. The coefficient of variation was 60.0 ± 7.8% (n = 12).
Isolated IPSCs were recorded in voltage clamp (Figure 2D, data from a typical β3N265M knock-in neuron). As with Pef neurons, there was no change in the holding current (data not shown) in response to the GABAA receptor antagonists gabazine (20 μm; n = 6) and bicuculline (20 μm; n = 10). Spontaneous IPSCs occurred at a frequency of 2.1 ± 0.4 Hz (n = 12) in the knock-in, which was not significantly different from the WT value of 1.9 ± 0.4 Hz (n = 13). Isolated events were selected to construct the average trace for each neuron (Fig. 2E). Neurons from β3N265M knock-in mice had an average neuronal capacitance (38.6 ± 4.9 pF), input resistance (369 ± 27 MΩ) and tonic firing activity that did not differ from WT (unpaired Student's t test, p > 0.05; n = 12).
The anatomical location of the LC (Fig. 3A), which is composed entirely of noradrenergic neurons (Moore and Bloom, 1979), was identified in coronal sections of the pons by its position relative to the floor of the fourth ventricle at the level of the posterodorsal tegmental nucleus (PDTg). Individual neurons were densely clustered in this region and identified electrophysiologically by their spontaneous tonic action potential firing and hyperpolarizing response to noradrenaline (Fig. 3B, data from a typical WT neuron). This response has been characterized (Williams et al., 1985) as being an autoreceptor feedback mechanism mediated by G-protein coupled α2 adrenoceptors. LC neurons from WT mice had an average capacitance of 68.1 ± 3.8 pF and input resistance of 544 ± 37 MΩ (n = 12). Selection of noradrenergic neurons in current clamp was followed by examination of isolated GABAergic IPSCs (Fig. 3C), which occurred at a frequency of 0.9 ± 0.2 Hz (n = 12). As with neurons in the Pef and TMN, no evidence of a basal tonic conductance was found in LC neurons, although the synaptic events were reversibly inhibited by 20 μm bicuculline or gabazine (data not shown; n = 5). An average IPSC was constructed from isolated events for each neuron (Fig. 3D). Neurons from β3N265M knock-in mice had an average capacitance (60.2 ± 2.1 pF), input resistance (553 ± 40 MΩ), and noradrenaline-sensitive tonic firing activity that were not significantly different from WT neurons (unpaired Student's t test, p > 0.05; n = 10). The IPSC frequency of 0.8 ± 0.1 Hz (n = 10) was also not different to the WT value.
Figure 3.
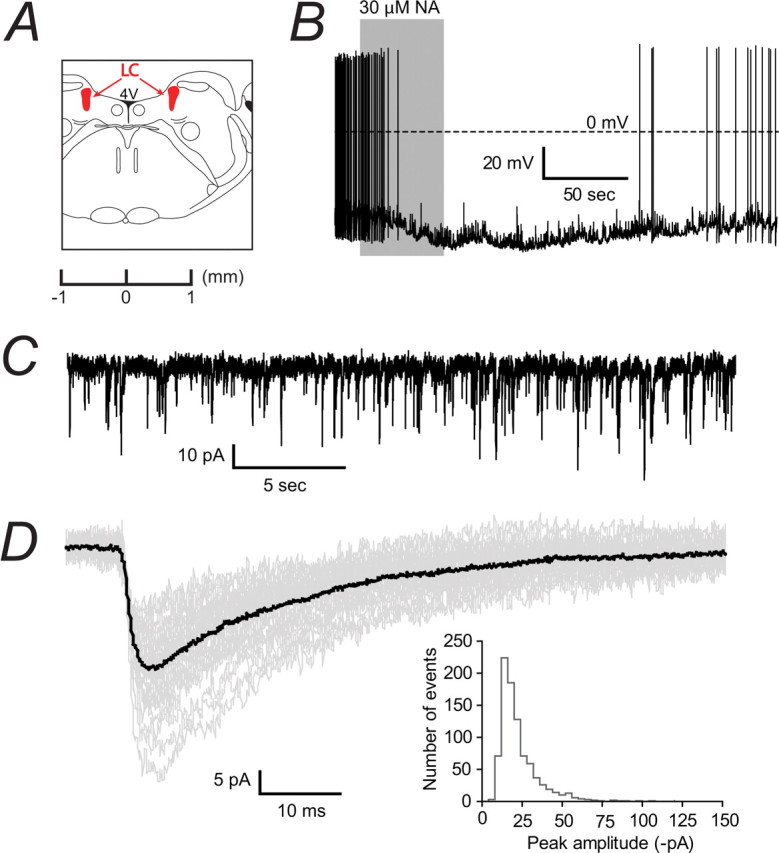
Characterization of GABAergic IPSCs in identified noradrenergic neurons in the LC. A, A coronal slice shows the LC (highlighted in red) as being at the floor of the fourth ventricle (4 V) and ventral to the cerebellum. We used slices corresponding to ∼−5.9 mm from the Bregma. B, Tonic firing (∼1.8 Hz) of an LC neuron recorded in current clamp, which hyperpolarized (from approximately −55 mV to −75 mV) and ceased firing after exposure to 30 μm noradrenaline (NA). This reversible autoreceptor response is characteristic of noradrenergic neurons and was seen in all recorded cells. C, A continuous trace showing GABAergic IPSCs recorded in voltage clamp from a typical LC neuron. D, Isolated IPSCs (37 events, shown in gray) were selected to construct a representative trace (shown in black) of individual IPSC properties. This example average IPSC had a 10–90% rise time of 3.5 ms, a peak amplitude of −28.2 pA and a decay time of 28.5 ms. The histogram shows the distribution of peak amplitudes recorded from all wild-type noradrenergic neurons. The coefficient of variation was 50.2 ± 4.3% (n = 12).
The β3N265M mutation is silent under control conditions of synaptic transmission
Spontaneous IPSCs recorded in neurons from β3N265M knock-in mice were not different from those recorded in the corresponding WT neurons in each of the brain nuclei tested. Figure 4A–C shows the average traces from all recorded β3N265M knock-in neurons (red) superimposed on average traces from all WT neurons (black) in the Pef, TMN and LC. Figure 4D–F summarizes average IPSC parameters from both genotypes: there were no difference between the WT and the knock-in parameters. Therefore, under conditions of synaptic GABA neurotransmission present in the slice preparation, the point mutation is silent and provides a clean background for the assessment of pharmacological differences.
Figure 4.

The β3N265M mutation does not affect control IPSC properties in Pef, TMN and LC neurons. A–C, The average IPSCs constructed from each recorded WT neuron (black traces) are shown together with the average β3N265M IPSCs (shown in red), the Pef (A; WT n = 9, β3N265M n = 8), the TMN (B; WT n = 12, β3N265M n = 12), and the LC (C; WT n = 12, β3N265M n = 10). D, Average 10–90% rise times for WT and knock-in Pef neurons (1.2 ± 0.1 ms; 1.3 ± 0.1 ms; means ± SEMs), TMN neurons (1.9 ± 0.2 ms; 2.1 ± 0.2 ms) and LC neurons (3.7 ± 0.4 ms; 3.3 ± 0.4 ms) respectively are shown in the top panel. E, Decay times for WT and β3N265M Pef neurons (21.3 ± 1.5 ms; 24.6 ± 2.3 ms), TMN neurons (30.3 ± 2.8 ms; 31.0 ± 2.1 ms) and LC neurons (37.1 ± 2.5 ms; 32.6 ± 2.2 ms). F, Average peak amplitudes for WT and β3N265M Pef neurons (−50.2 ± 6.6 pA; 35.3 ± 4.5 pA), TMN neurons (−37.4 ± 5.2 pA; 51.4 ± 9.2 pA), LC neurons (−19.0 ± 1.6 pA; −19.6 ± 1.5 pA). There were no differences in IPSC parameters between WT and β3N265M knock-in animals in any of the brain regions studied.
The alphaxalone-sensitivity of GABAergic IPSCs is not affected by the β3N265M mutation
The neurosteroid alphaxalone (2 μm) prolonged the IPSC decay phase regardless of brain nucleus or genotype (Fig. 5). However, it had no effect on 10–90% rise time or amplitude in any WT or β3N265M nucleus. The control decay time in WT orexinergic neurons was 22.1 ± 2.0 ms, which increased to 58.7 ± 4.7 ms (n = 5; p < 0.01) during the application of alphaxalone. Similarly, in neurons from β3N265M knock-in mice, the average IPSC decay phase increased from 21.3 ± 1.8 ms to 54.1 ± 5.2 ms (n = 4; p < 0.01). The average decay phase of IPSCs recorded from TMN neurons was equally sensitive to alphaxalone in the WT (increasing from 36.0 ± 4.0 ms to 100.4 ± 8.0 ms; n = 6; p < 0.001) and the knock-in, where the average IPSC decay time increased from 32.0 ± 4.2 ms to 84.0 ± 1.8 ms (n = 4; p < 0.01). Finally, alphaxalone also potentiated the decay phase of IPSCs recorded in LC neurons in both genotypes. In the WT the decay phase was 37.6 ± 3.3 ms and 93.2 ± 8.2 ms (n = 8; p < 0.01) before and during alphaxalone application respectively. In neurons from the knock-in mice, the corresponding values were 33.6 ± 2.4 ms and 84.6 ± 9.2 ms (n = 6; p < 0.001). Overall, these data show that the β3N265M mutation does not affect alphaxalone sensitivity of IPSCs recorded in the Pef, TMN or LC, mirroring in vivo data (Jurd et al., 2003; Zeller et al., 2005).
Figure 5.
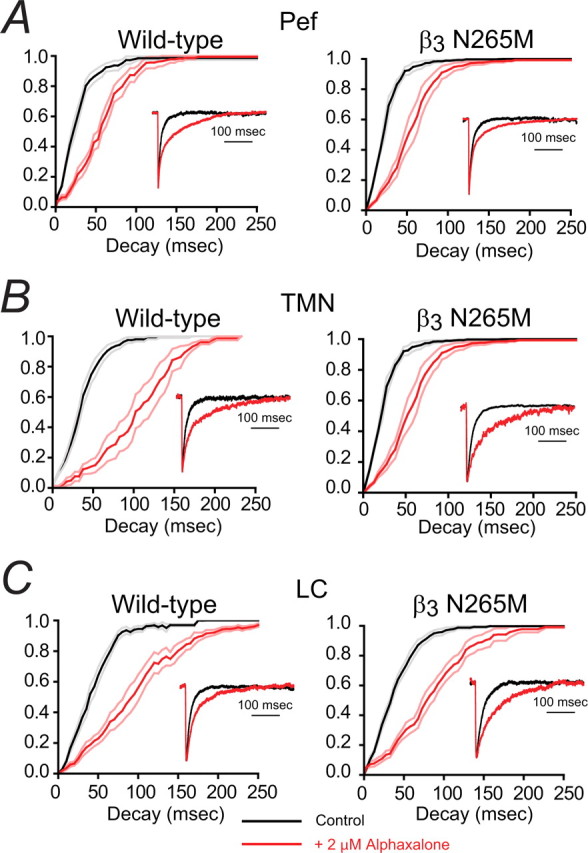
The neurosteroid alphaxalone markedly increases GABAergic IPSC decay times in both WT and β3N265M knock-in Pef, TMN, and LC neurons. The main traces in each panel shows the effects of alphaxalone on the cumulative probability distributions of IPSC decay times averaged across all recorded neurons under control conditions (black curve) and during exposure to 2 μm alphaxalone (red curve). The lighter lines show the SEMs. The insets show representative average IPSCs (normalized to peak amplitude) from individual neurons. A, The cumulative decay time distributions for Pef neurons from both WT and β3N265M knock-in mice show clear, and comparable, rightward shifts in the presence of alphaxalone (for WT neurons, τ increased by 175 ± 30%, n = 5, and for β3N265M neurons, τ increased by 159 ± 30%, n = 4). Data from a typical WT Pef neuron (left inset), shows a large increase in the average IPSC decay time from 17 ms (n = 51) for control, to 53 ms (n = 81) in the presence of alphaxalone. Data from a typical β3N265M Pef neuron (right inset) shows a similar increase from 16 ms (n = 51) to 40 ms (n = 105). B, The cumulative decay time distributions for TMN neurons from both WT and β3N265M knock-in mice show clear, and comparable, rightward shifts (for WT neurons, τ increased by 188 ± 30%, n = 5, and for β3N265M neurons, τ increased by 178 ± 41%, n = 4). Data from a typical WT TMN neuron (left inset), shows a large increase in the average IPSC decay time from 24 ms (n = 63) for control to 71 ms (n = 49) in the presence of alphaxalone. Data from a typical β3N265M TMN neuron (right inset) shows a similar increase from 21 ms (n = 50) to 82 ms (n = 48). C, The cumulative decay time distributions for LC neurons from both WT and β3N265M knock-in mice show clear, and comparable, rightward shifts (for WT neurons, τ increased by 149 ± 15%, n = 8, and for β3N265M neurons, τ increased by 154 ± 26%, n = 6). Data from a typical WT LC neuron (left inset), shows a large increase in the average IPSC decay time from 27 ms (n = 127) for control to 52 ms (n = 71) in the presence of alphaxalone. Data from a typical β3N265M LC neuron (right inset) shows a similar increase from 29 ms (n = 59) to 78 ms (n = 47).
IPSCs in β3N265M knock-in are insensitive to propofol in the Pef and TMN, but are still potentiated in the LC
Having determined, in agreement with the in vivo data, that alphaxalone-sensitivity of GABAergic IPSCs in the Pef, TMN and LC was unaffected by the β3N265M point mutation, we went on to test their sensitivities to propofol. The β3N265M knock-in mice have been shown to be markedly less affected by this anesthetic for both loss of righting reflex and loss of response to a painful stimulus (Jurd et al., 2003). Brain nuclei responsible for this loss of sensitivity should display similar phenotypes in vitro.
As expected, we found that GABAergic IPSCs recorded from WT neurons from the Pef, TMN and LC were all sensitive to propofol (Fig. 6A–C, left panels) and the average IPSC decay phases increased from 20.6 ± 2.3 ms to 41.0 ± 5.2 ms (n = 5; p < 0.01) for Pef neurons, from 25.1 ± 1.6 ms to 54.4 ± 6.2 ms (n = 7; p < 0.001) for TMN neurons and from 36.5 ± 3.6 ms to 62.4 ± 5.8 (n = 4; p < 0.05) for LC neurons.
Figure 6.
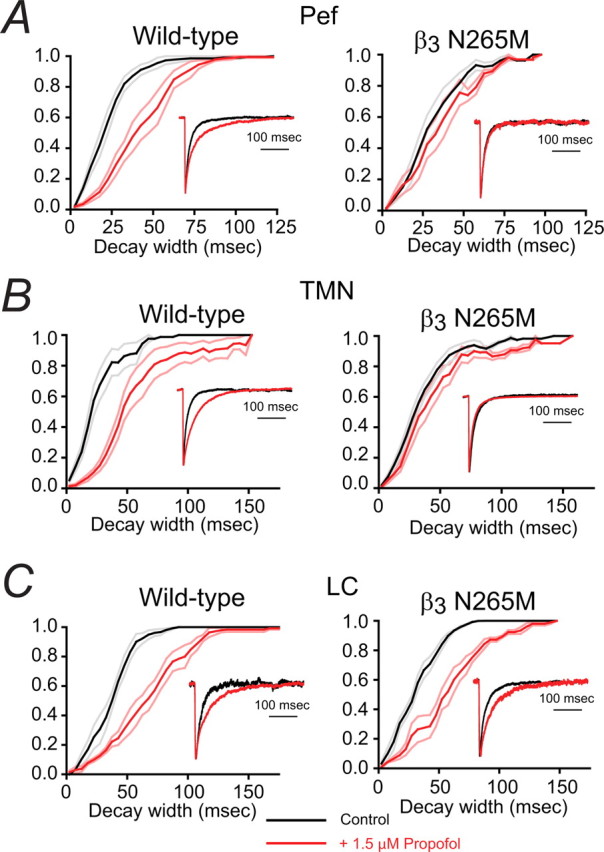
The contrasting effects of propofol on IPSC decay times in WT and β3N265M knock-in Pef, TMN and LC. Each panel shows the effect of propofol on the cumulative frequency distribution of IPSC decay times averaged across all recorded neurons before (black curve) and during exposure to 1.5 μm propofol (red curve). The lighter lines show the SEMs. The insets show the average IPSC decay phase from a representative neuron. A, For WT Pef neurons the cumulative frequency distributions show that, on average, propofol causes a 100 ± 14% (n = 5) increase in median decay time. The left inset shows data from a typical WT Pef neuron. The average IPSC decay time under control conditions was 21 ms (n = 51), which increased to 50 ms (n = 81) in the presence of propofol. In contrast, propofol caused an increase of only 16 ± 4% (n = 4) in β3N265M knock-in neurons. The right inset shows data from a typical β3N265M Pef neuron. The average IPSC decay time was 19 ms (n = 51) under control conditions and did not change significantly in the presence of propofol (20 ms; n = 105). B, For WT TMN neurons the cumulative frequency distributions show that, on average, propofol causes an 124 ± 18% (n = 7) increase in median decay time. The left inset shows data from a typical WT TMN neuron. The average decay time was 19 ms (n = 63), which increased to 48 ms (n = 49) in the presence of propofol. However, propofol caused an increase of only 23 ± 11% (n = 8) in β3N265M knock-in neurons. The right inset shows data for a typical β3N265M TMN neuron. Propofol minimally affected the IPSC decay time, changing it from 20 ms (n = 50) to 21 ms (n = 48). C, In the LC, cumulative frequency distributions show that, on average, propofol causes a similar increase in decay times for WT and β3N265M knock-in neurons (79 ± 29%, n = 4, increase in median decay time in WT LC neurons and 89 ± 23%, n = 4, in β3N265M knock-in neurons). The left inset shows data from a typical WT LC neuron. Propofol caused an increase in IPSC decay time from 27 ms (n = 127) to 57 ms (n = 71). The right inset shows data from a typical LC neuron from the β3N265M knock-in. Propofol caused an increase in IPSC decay time from 35 ms (n = 59) to 51 ms (n = 47).
In marked contrast, however, neurons in the Pef and TMN from the β3N265M knock-in mice were unaffected by propofol (Fig. 6A,B, right panels). For Pef neurons, the IPSC decay times in the presence (33.6 ± 4.4 ms) and absence (29.2 ± 4.0 ms) of propofol were not significantly different (n = 4; p > 0.05). Similarly, for TMN neurons from the β3N265M knock-in mice, the average IPSC decay time was insensitive to propofol, changing from 30.7 ± 2.3 ms to 37.3 ± 4.3 ms, which again was not significant (n = 8; p > 0.05). In the LC, in contrast, GABAergic IPSCs in β3N265M knock-in mice retained their propofol sensitivity (Fig. 6C, right panel). For these neurons the IPSC decay times increased significantly, from 30.5 ± 4.6 ms to 55.0 ± 5.0 ms (n = 4; p < 0.05).
For all nuclei, and both phenotypes, the average IPSC peak amplitudes and 10–90% rise times were not significantly affected by propofol.
Involvement of orexinergic neurons in propofol LORR
To test the involvement of Pef orexinergic neurons in propofol-induced LORR, we performed a series of in vivo experiments. We first determined whether these neurons were functionally inhibited during propofol anesthesia, as we have previously shown for histaminergic TMN neurons (Nelson et al., 2002), using c-Fos as a surrogate marker (Hoffman et al., 1993). Coronal slices of the posterior hypothalamus were taken and the Pef was identified as described above (Fig. 7A). We used immunohistochemistry to identify orexin-containing neurons and to quantify c-Fos expression following loss of righting reflex induced by a known GABA agonist (muscimol), putative GABAergic anesthetics (propofol and pentobarbital), and an anesthetic agent known to have no effects on GABAA receptors, the selective α2 adrenoceptor agonist dexmedetomidine. Approximately half (47.8 ± 2.9%) of the orexinergic neurons in the Pef stained positively for c-Fos in control awake animals (Fig. 7B). Propofol (100 mg/kg) greatly reduced the level of c-Fos expression. Similar reductions were seen with other GABAergic agents, muscimol (5 mg/kg) and pentobarbital (50 mg/kg). However, c-Fos expression did not change significantly in animals anesthetized with systemic administration of dexmedetomidine (150 μg/kg, i.p.).
Figure 7.
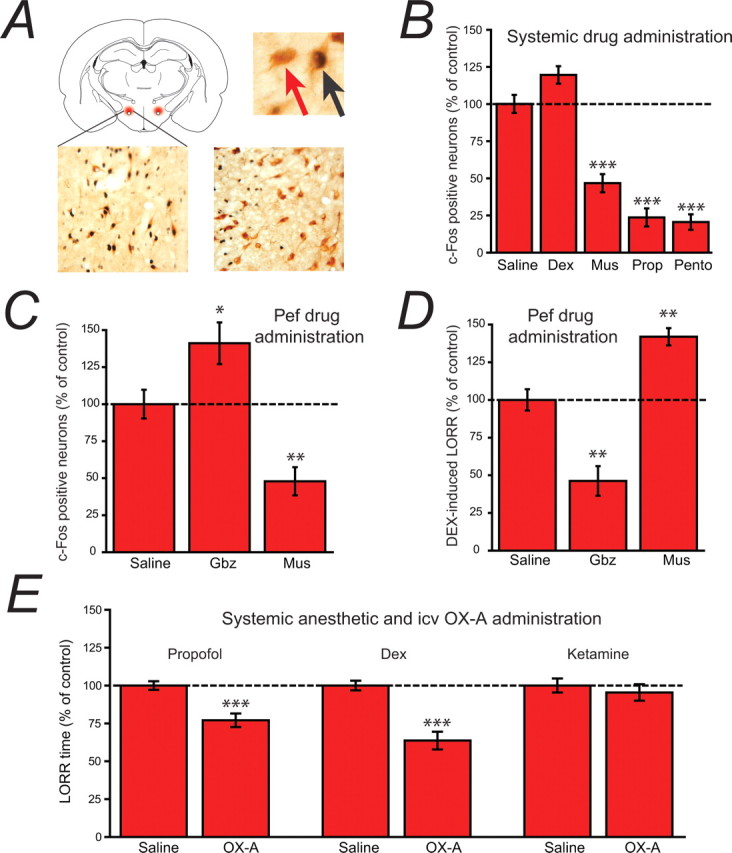
Evidence for the involvement of orexinergic neurons in anesthetic-induced LORR. A, The schematic coronal slice (top left) shows the location of Pef orexinergic neurons (red) close to the fornix. The image top right shows 3,3-diaminobenzidine tetrahydrochloride-visualized neurons, double-stained for cytosolic orexin-A (brown) and nuclear c-Fos (black). The red arrow points to a single-labeled orexinergic-positive, but c-Fos-negative neuron and the black arrow indicates a double-labeled c-Fos-positive orexinergic neuron. The lower images show (left) a level of c-Fos expression in orexinergic neurons following systemic dexmedetomidine (Dex; 150 μg/kg) that is similar to control levels, in contrast to the marked depression of c-Fos expression observed (right) following pentobarbital administration (Pento; 50 mg/kg). B, C-Fos-positive neurons following systemic administration of dexmedetomidine (Dex; 150 μg/kg, i.p.), muscimol (Mus; 5 mg/kg, i.p.), propofol (Prop; 100 mg/kg, i.p.) and pentobarbital (Pento; 50 mg/kg, i.p.) expressed as a percentage of those observed following saline administration. Following saline injection, 47.8 ± 2.9% of orexinergic neurons were c-Fos positive. C, When administered directly into the Pef (bilateral injections, 0.2 μg/0.2 μl/side), the GABAA receptor antagonist gabazine increases c-Fos expression in orexinergic neurons, whereas the GABAA receptor agonist muscimol decreases expression compared with the injection of control saline (normalized to 100%; 45.5 ± 4.4% of neurons were c-Fos positive), showing that the activity of Pef neurons can be modulated by GABAergic agents. D, When administered directly into the Pef (bilateral injections, 0.2 μg/0.2 μl/side), gabazine antagonizes Dex-induced LORR (150 μg/kg, s.c.), whereas muscimol significantly increases Dex-induced LORR compared with the injection of control saline (normalized to 100%; control sleep time was 286 min), showing that the modulation of Pef neurons can directly influence LORR. Significance was tested using an unpaired Student's t test. E, The intracerebroventricular administration of orexin-A significantly antagonizes propofol and dexmedetomidine anesthesia, but has no effect on ketamine anesthesia compared with the intracerebroventricular injection of saline. In each case the mean control sleep times have been normalized to 100% (control sleep times were 8.2 min for Prop, 26.2 min for Dex and 3.6 min for ketamine). Significance was tested using a paired Student's t test with respect to control saline injections on the same animal. The data are means ± SEMs, and the asterisks denote the level of significance (*p < 0.05, **p < 0.01, ***p < 0.001).
The expression of c-Fos in orexinergic neurons could be significantly modulated by the discrete bilateral injection of GABAergic modulators directly into the Pef. The data in Figure 7C show that bilateral injection of the GABAA receptor antagonist gabazine significantly increased c-Fos expression, whereas the injection of the GABAA receptor agonist muscimol had the opposite effect and greatly decreased c-Fos expression. Because dexmedetomidine had no effect on c-Fos expression in the Pef, it was possible to determine whether activity of orexinergic neurons in the Pef per se could affect loss of righting reflex by determining the effects of these same discrete administrations of GABAergic modulators on dexmedetomidine-induced loss of righting reflex. The data in Figure 7D show that inhibition of orexinergic neurons by muscimol caused an increase in dexmedetomidine-induced sleep time, whereas enhancing the activity of orexinergic neurons by gabazine caused a shortening of dexmedetomidine-induced sleep time.
In a parallel set of experiments (Fig. 7E) we investigated the effects of an intracerebroventricular injection of orexin A on the loss of righting reflex induced by the intravenous (jugular vein) injection of dexmedetomidine (20 μg/kg), propofol (10 mg/kg), and ketamine (10 mg/kg). The injection of orexin A caused a significant decrease in sleep time for both propofol and dexmedetomidine, but had no effect on ketamine-induced LORR.
Discussion
The principal aim of this research was to test the hypothesis that general anesthetics that are thought to act mainly at GABAA receptors (such as propofol) exert their effects in the CNS at specific hypothalamic nuclei. Our previous work has shown that these agents may act, at least in part, by potentiating the inhibitory actions of GABA on histaminergic neurons in the TMN (Nelson et al., 2002). Because the discrete application of anesthetics into the TMN caused only sedation rather than LORR, it was clear that other nuclei must be involved. The orexinergic neurons in the hypothalamus are one possible candidate (Kushikata et al., 2003; Kelz et al., 2008). These neurons provide additional excitatory drive via a dense innervation of the TMN (Peyron et al., 1998), and are also under inhibitory GABAergic control from VLPO and other sleep-active nuclei (Suntsova et al., 2002; Alam et al., 2005). Orexinergic neurons in the Pef are critically important for normal sleep architecture, and a disruption to this system is known to be responsible for the sleep disorder, narcolepsy (Mignot et al., 2002; Sakurai, 2007).
To test the direct involvement of specific brain nuclei in anesthetic-induced LORR, we have taken a novel approach which uses the β3N265M knock-in mouse. This mouse strain is markedly less sensitive to the LORR-inducing effects of propofol, but has an unchanged sensitivity to the neurosteroid alphaxalone (Jurd et al., 2003). Any neuronal target that is hypothesized to be critical for anesthetic-induced LORR should display an anesthetic sensitivity that reflects this pattern in vivo. Specifically, GABAergic IPSCs recorded in that region should be sensitive to both propofol and alphaxalone in WT mice, but when measured in β3N265M knock-in mouse should lose their sensitivity to propofol but not to alphaxalone.
In our experiments, we tested the hypothesis that the inhibition of the hypothalamic TMN and Pef are involved in propofol LORR, but we also investigated the role of the LC in the pons. Although our previous work has indicated that GABAergic inhibition of this nucleus alone is not sufficient for LORR (Nelson et al., 2002), this important noradrenergic arousal nucleus is thought to be a key target for dexmedetomidine in causing LORR (Correa-Sales et al., 1992a,b); we hypothesized that it may play an important permissive role. One caveat that should be mentioned here is that our characterizations of the neurons from which we recorded was predominantly anatomical and physiological. Although this is not necessarily as definitive as specific staining, the homogeneity of the cell populations in the TMN and the LC, together with the well characterized electrophysiological properties of the orexinergic neurons in the Pef, give us confidence that we were recording from the neurons in these nuclei that are believed to be involved in sleep pathways
The first step was to establish that the control GABAergic IPSCs in the three chosen nuclei (the Pef, TMN, and LC) were unaffected by the β3N265M mutation. Is it, in other words, a “silent” mutation? The data shown in Figure 4 establish that, for the Pef, TMN and LC, the control IPSC parameters in β3N265M knock-in mice are essentially identical to those observed in WT mice. None of the synaptic parameters (amplitude, rise time and decay time) were affected by the β3N265M mutation. The effects of this mutation on postsynaptic GABAergic currents in cortical neurons have also been investigated (Drexler et al., 2006) with the same result: the mutation appears to have no significant effect on the control synaptic parameters.
We then determined the alphaxalone sensitivity of GABAergic currents in the Pef, TMN and LC in both WT and β3N265M mice. We used an alphaxalone concentration of 2 μm because, on the basis of previous studies in hippocampal neurons (Harrison et al., 1987; Spigelman et al., 2003), this concentration might be expected to produce robust potentiations of 50% or more. In fact, we found that the decay times of GABAergic IPSCs were prolonged by between 153 and 188% in the three brain nuclei studied in both WT mice and β3N265M mice (Fig. 5). The observation that GABAergic currents in these brain nuclei appear to be significantly more sensitive than those recorded from hippocampal neurons may, or may not, be important for the in vivo actions of alphaxalone. In the context of the present study, however, what is important is the fact that alphaxalone is able to potentiate GABAergic IPSCs in the Pef, TMN and LC of the knock-in mice to the same extent as it does in the wild-type animals. This shows that the GABAA receptors in these nuclei can still be modulated by general anesthetics.
Next, we investigated the effects of propofol. For propofol we chose to use a concentration of 1.5 μm. This was based on the fact that the EC50 for LORR in rodents is ∼0.5 μm (Franks, 2008), and at the depth in the brain slice from which we make our recordings (50 – 100 μm), the concentration of propofol is between 2 – 3 times lower than that at the surface after 25–30 min of equilibration (Gredell et al., 2004). In each of the brain nuclei tested, the GABAergic currents recorded using WT mice showed prolongations of decay times of between 89 – 124%. Similar sensitivities of IPSCs to propofol have been reported for hippocampal (Orser et al., 1994; Bai et al., 2001), cortical (Kitamura et al., 2004), brainstem (McDougall et al., 2008) and hypothalamic (Sergeeva et al., 2005) neurons. Our results with the β3N265M mice were strikingly different (Fig. 6). While the LC neurons from the β3N265M mice retained a sensitivity to propofol that was comparable with the WT, the GABAergic IPSCs from TMN and Pef neurons were not significantly (p > 0.05) affected by propofol, with increases in decay times of only 23 ± 11% and 16 ± 4%, respectively.
These in vitro results rule out the LC as a major target for propofol. Although GABAergic input to the LC is clearly potentiated by propofol, and this is consistent with some reduction in LC firing (Chen et al., 1999), the identical sensitivity of the WT and β3N265M neurons is obviously inconsistent with the very different in vivo sensitivities to propofol, and so, this anesthetic cannot be exerting its principal effects via the LC. The data for the histaminergic neurons in the TMN and the orexinergic neurons in the Pef, in contrast, are consistent with propofol acting directly on these neurons to cause anesthesia. However, it is important to stress that the absence of an effect of propofol on these TMN and Pef neurons in the β3N265M knock-in mice does not prove their involvement in propofol-induced LORR. Indeed, it is likely that other brain nuclei will show similar responses to those we found in the hypothalamus, including regions that have been postulated to be involved in anesthetic action (such as the thalamus and cortex). The next challenge will be to determine which of these brain regions are causally involved, and this will, inevitably, require extensive in vivo experiments.
We have previously shown (Nelson et al., 2002) that the TMN may be causally involved, but there is little data on the role of orexinergic pathways. Recently, it has been proposed that the recovery from isoflurane anesthesia may involve the activation of orexinergic receptors (Kelz et al., 2008) and a previous study using pentobarbital has also provided evidence of their involvement in anesthesia (Kushikata et al., 2003). To explore the role of orexinergic neurons further we performed some in vivo experiments. We observed that muscimol, propofol and pentobarbital were able to markedly reduce c-Fos expression in orexinergic neurons (Fig. 1B) following LORR in rats. Comparable, although smaller, reductions have been reported with isoflurane (Kelz et al., 2008). In contrast, dexmedetomidine, a highly selective α2 adrenoceptor agonist with no GABAergic activity, had no effect on the excitability of orexinergic neurons. It has previously been established that these neurons are greatly inhibited during sleep, compared with during wakefulness, by GABAergic input from the preoptic area (Suntsova et al., 2002; Alam et al., 2005). Consistent with this, we found that when muscimol was discretely administered into the Pef during the waking state it strongly reduced c-Fos expression, whereas when gabazine was injected, c-Fos expression was elevated, the latter implying some basal GABAergic inhibition during normal wakefulness (Fig. 7C).
Because dexmedetomidine was found to have no significant effect on the expression of c-Fos in orexinergic neurons, we were able to explore the possibility that a reduction in the excitability of Pef neurons might nonetheless contribute to LORR caused by other anesthetics. The data in Figure 7D show that in the presence of dexmedetomidine, where LORR is achieved completely independently of the Pef, GABAergic modulation of this nucleus can directly affect LORR.
In a parallel set of experiments, and as further confirmation that orexin levels in the brain can influence anesthesia, we determined the effects of injecting orexin A directly (intracerebroventricularly) into the brain on the LORR induced by dexmedetomidine, propofol and ketamine. Consistent with the observation that directly exciting the Pef neurons by gabazine can antagonize dexmedetomidine anesthesia (Fig. 7D), we found that increased levels of orexin A in the brain also antagonized anesthesia, for both dexmedetomidine and propofol (Fig. 7E). A comparable antagonism has previously been reported for pentobarbital (Kushikata et al., 2003).
Interestingly, however, we observed no effect of intracerebroventricular orexin-A injection on ketamine sleep time. This important observation means that propofol and dexmedetomidine must be causing LORR by acting on neuronal pathways that are modulated by orexins. Many of these pathways are involved in wakefulness and appear to integrate at the level of the TMN because they are abolished in the H1 receptor knock-out mouse (Huang et al., 2001). Ketamine, in contrast, must be acting differently by affecting non-overlapping pathways. Consistent with this, the pattern of c-Fos during ketamine (Lu et al., 2008) and propofol anesthesia (Nelson et al., 2002) are very different, as are the patterns of neuronal activity deduced from human imaging studies (Kaisti et al., 2003; Långsjö et al., 2005) or EEG recordings (Sloan, 1998).
Thus the in vivo data implicate the neuronal pathways of sleep and arousal in the behavioral effects of propofol and, when taken together our in vitro data, suggest that these effects may be due to direct actions on histaminergic and orexinergic neurons.
Footnotes
This work was supported by grants from the Medical Research Council, UK (G0501584 and G9817980), the Chelsea and Westminster Health Care National Health Service Trust (UK; 010ANA007, 010ANA008), and the Swiss National Science Foundation (3100A0-102113). T.C.G. was a research student funded by the UK Biotechnology and Biological Sciences Research Council and Air Products and Chemicals Inc. We thank Raquel Yustos for excellent technical assistance.
References
- Abrahamson EE, Moore RY. The posterior hypothalamic area: chemoarchitecture and afferent connections. Brain Res. 2001;889:1–22. doi: 10.1016/s0006-8993(00)03015-8. [DOI] [PubMed] [Google Scholar]
- Alam MN, Kumar S, Bashir T, Suntsova N, Methippara MM, Szymusiak R, McGinty D. GABA-mediated control of hypocretin- but not melanin-concentrating hormone-immunoreactive neurones during sleep in rats. J Physiol. 2005;563:569–582. doi: 10.1113/jphysiol.2004.076927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen TA, Narayanan NS, Kholodar-Smith DB, Zhao Y, Laubach M, Brown TH. Imaging the spread of reversible brain inactivations using fluorescent muscimol. J Neurosci Methods. 2008;171:30–38. doi: 10.1016/j.jneumeth.2008.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai D, Zhu G, Pennefather P, Jackson MF, MacDonald JF, Orser BA. Distinct functional and pharmacological properties of tonic and quantal inhibitory postsynaptic currents mediated by gamma-aminobutyric acid(A) receptors in hippocampal neurons. Mol Pharmacol. 2001;59:814–824. doi: 10.1124/mol.59.4.814. [DOI] [PubMed] [Google Scholar]
- Bayer L, Eggermann E, Serafin M, Grivel J, Machard D, Muhlethaler M, Jones BE. Opposite effects of noradrenaline and acetylcholine upon hypocretin/orexin versus melanin concentrating hormone neurons in rat hypothalamic slices. Neuroscience. 2005;130:807–811. doi: 10.1016/j.neuroscience.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Bittencourt JC, Presse F, Arias C, Peto C, Vaughan J, Nahon JL, Vale W, Sawchenko PE. The melanin-concentrating hormone system of the rat brain: an immuno- and hybridization histochemical characterization. J Comp Neurol. 1992;319:218–245. doi: 10.1002/cne.903190204. [DOI] [PubMed] [Google Scholar]
- Broberger C, De Lecea L, Sutcliffe JG, Hökfelt T. Hypocretin/orexin- and melanin-concentrating hormone-expressing cells form distinct populations in the rodent lateral hypothalamus: relationship to the neuropeptide Y and agouti gene-related protein systems. J Comp Neurol. 1998;402:460–474. [PubMed] [Google Scholar]
- Burdakov D, Gerasimenko O, Verkhratsky A. Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J Neurosci. 2005;25:2429–2433. doi: 10.1523/JNEUROSCI.4925-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Yang YR, Chiu TH. Activation of rat locus coeruleus neuron GABA(A) receptors by propofol and its potentiation by pentobarbital or alphaxalone. Eur J Pharmacol. 1999;386:201–210. doi: 10.1016/s0014-2999(99)00750-5. [DOI] [PubMed] [Google Scholar]
- Correa-Sales C, Rabin BC, Maze M. A hypnotic response to dexmedetomidine, an alpha 2 agonist, is mediated in the locus coeruleus in rats. Anesthesiology. 1992a;76:948–952. doi: 10.1097/00000542-199206000-00013. [DOI] [PubMed] [Google Scholar]
- Correa-Sales C, Nacif-Coelho C, Reid K, Maze M. Inhibition of adenylate cyclase in the locus coeruleus mediates the hypnotic response to an alpha 2 agonist in the rat. J Pharmacol Exp Ther. 1992b;263:1046–1049. [PubMed] [Google Scholar]
- Devor M, Zalkind V. Reversible analgesia, atonia, and loss of consciousness on bilateral intracerebral microinjection of pentobarbital. Pain. 2001;94:101–112. doi: 10.1016/S0304-3959(01)00345-1. [DOI] [PubMed] [Google Scholar]
- Drexler B, Jurd R, Rudolph U, Antkowiak B. Dual actions of enflurane on postsynaptic currents abolished by the gamma-aminobutyric acid type A receptor beta3(N265M) point mutation. Anesthesiology. 2006;105:297–304. doi: 10.1097/00000542-200608000-00012. [DOI] [PubMed] [Google Scholar]
- Eggermann E, Bayer L, Serafin M, Saint-Mleux B, Bernheim L, Machard D, Jones BE, Mühlethaler M. The wake-promoting hypocretin-orexin neurons are in an intrinsic state of membrane depolarization. J Neurosci. 2003;23:1557–1562. doi: 10.1523/JNEUROSCI.23-05-01557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estabrooke IV, McCarthy MT, Ko E, Chou TC, Chemelli RM, Yanagisawa M, Saper CB, Scammell TE. Fos expression in orexin neurons varies with behavioral state. J Neurosci. 2001;21:1656–1662. doi: 10.1523/JNEUROSCI.21-05-01656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci. 2008;9:370–386. doi: 10.1038/nrn2372. [DOI] [PubMed] [Google Scholar]
- Franks NP, Lieb WR. Molecular and cellular mechanisms of general anaesthesia. Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- Gerashchenko D, Kohls MD, Greco M, Waleh NS, Salin-Pascual R, Kilduff TS, Lappi DA, Shiromani PJ. Hypocretin-2-saporin lesions of the lateral hypothalamus produce narcoleptic-like sleep behavior in the rat. J Neurosci. 2001;21:7273–7283. doi: 10.1523/JNEUROSCI.21-18-07273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gredell JA, Turnquist PA, Maciver MB, Pearce RA. Determination of diffusion and partition coefficients of propofol in rat brain tissue: implications for studies of drug action in vitro. Br J Anaesth. 2004;93:810–817. doi: 10.1093/bja/aeh272. [DOI] [PubMed] [Google Scholar]
- Hales TG, Lambert JJ. The actions of propofol on inhibitory amino acid receptors of bovine adrenomedullary chromaffin cells and rodent central neurones. Br J Pharmacol. 1991;104:619–628. doi: 10.1111/j.1476-5381.1991.tb12479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison NL, Vicini S, Barker JL. A steroid anesthetic prolongs inhibitory postsynaptic currents in cultured rat hippocampal neurons. J Neurosci. 1987;7:604–609. doi: 10.1523/JNEUROSCI.07-02-00604.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman GE, Smith MS, Verbalis JG. c-Fos and related immediate early gene products as markers of activity in neuroendocrine systems. Front Neuroendocrinol. 1993;14:173–213. doi: 10.1006/frne.1993.1006. [DOI] [PubMed] [Google Scholar]
- Huang ZL, Qu WM, Li WD, Mochizuki T, Eguchi N, Watanabe T, Urade Y, Hayaishi O. Arousal effect of orexin A depends on activation of the histaminergic system. Proc Natl Acad Sci U S A. 2001;98:9965–9970. doi: 10.1073/pnas.181330998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, Rudolph U. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. FASEB J. 2003;17:250–252. doi: 10.1096/fj.02-0611fje. [DOI] [PubMed] [Google Scholar]
- Kaisti KK, Långsjö JW, Aalto S, Oikonen V, Sipilä H, Teräs M, Hinkka S, Metsähonkala L, Scheinin H. Effects of sevoflurane, propofol, and adjunct nitrous oxide on regional cerebral blood flow, oxygen consumption, and blood volume in humans. Anesthesiology. 2003;99:603–613. doi: 10.1097/00000542-200309000-00015. [DOI] [PubMed] [Google Scholar]
- Kelz MB, Sun Y, Chen J, Cheng Meng Q, Moore JT, Veasey SC, Dixon S, Thornton M, Funato H, Yanagisawa M. An essential role for orexins in emergence from general anesthesia. Proc Natl Acad Sci U S A. 2008;105:1309–1314. doi: 10.1073/pnas.0707146105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura A, Sato R, Marszalec W, Yeh JZ, Ogawa R, Narahashi T. Halothane and propofol modulation of gamma-aminobutyric acidA receptor single-channel currents. Anesth Analg. 2004;99:409–415. doi: 10.1213/01.ANE.0000131969.46439.71. table of contents. [DOI] [PubMed] [Google Scholar]
- Ko EM, Estabrooke IV, McCarthy M, Scammell TE. Wake-related activity of tuberomammillary neurons in rats. Brain Res. 2003;992:220–226. doi: 10.1016/j.brainres.2003.08.044. [DOI] [PubMed] [Google Scholar]
- Krasowski MD, Koltchine VV, Rick CE, Ye Q, Finn SE, Harrison NL. Propofol and other intravenous anesthetics have sites of action on the gamma-aminobutyric acid type A receptor distinct from that for isoflurane. Mol Pharmacol. 1998;53:530–538. doi: 10.1124/mol.53.3.530. [DOI] [PubMed] [Google Scholar]
- Kushikata T, Hirota K, Yoshida H, Kudo M, Lambert DG, Smart D, Jerman JC, Matsuki A. Orexinergic neurons and barbiturate anesthesia. Neuroscience. 2003;121:855–863. doi: 10.1016/s0306-4522(03)00554-2. [DOI] [PubMed] [Google Scholar]
- Långsjö JW, Maksimow A, Salmi E, Kaisti K, Aalto S, Oikonen V, Hinkka S, Aantaa R, Sipilä H, Viljanen T, Parkkola R, Scheinin H. S-ketamine anesthesia increases cerebral blood flow in excess of the metabolic needs in humans. Anesthesiology. 2005;103:258–268. doi: 10.1097/00000542-200508000-00008. [DOI] [PubMed] [Google Scholar]
- Lu J, Nelson LE, Franks N, Maze M, Chamberlin NL, Saper CB. Role of endogenous sleep-wake and analgesic systems in anesthesia. J Comp Neurol. 2008;508:648–662. doi: 10.1002/cne.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydic R, Baghdoyan HA. Sleep, anesthesiology, and the neurobiology of arousal state control. Anesthesiology. 2005;103:1268–1295. doi: 10.1097/00000542-200512000-00024. [DOI] [PubMed] [Google Scholar]
- Mammoto T, Yamamoto Y, Kagawa K, Hayashi Y, Mashimo T, Yoshiya I, Yamatodani A. Interactions between neuronal histamine and halothane anesthesia in rats. J Neurochem. 1997;69:406–411. doi: 10.1046/j.1471-4159.1997.69010406.x. [DOI] [PubMed] [Google Scholar]
- McDougall SJ, Bailey TW, Mendelowitz D, Andresen MC. Propofol enhances both tonic and phasic inhibitory currents in second-order neurons of the solitary tract nucleus (NTS) Neuropharmacology. 2008;54:552–563. doi: 10.1016/j.neuropharm.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot E, Lammers GJ, Ripley B, Okun M, Nevsimalova S, Overeem S, Vankova J, Black J, Harsh J, Bassetti C, Schrader H, Nishino S. The role of cerebrospinal fluid hypocretin measurement in the diagnosis of narcolepsy and other hypersomnias. Arch Neurol. 2002;59:1553–1562. doi: 10.1001/archneur.59.10.1553. [DOI] [PubMed] [Google Scholar]
- Moore RY, Bloom FE. Central catecholamine neuron systems: anatomy and physiology of the norepinephrine and epinephrine systems. Annu Rev Neurosci. 1979;2:113–168. doi: 10.1146/annurev.ne.02.030179.000553. [DOI] [PubMed] [Google Scholar]
- Nelson LE, Guo TZ, Lu J, Saper CB, Franks NP, Maze M. The sedative component of anesthesia is mediated by GABA(A) receptors in an endogenous sleep pathway. Nat Neurosci. 2002;5:979–984. doi: 10.1038/nn913. [DOI] [PubMed] [Google Scholar]
- Nelson LE, Lu J, Guo T, Saper CB, Franks NP, Maze M. The alpha2-adrenoceptor agonist dexmedetomidine converges on an endogenous sleep-promoting pathway to exert its sedative effects. Anesthesiology. 2003;98:428–436. doi: 10.1097/00000542-200302000-00024. [DOI] [PubMed] [Google Scholar]
- Orser BA, Wang LY, Pennefather PS, MacDonald JF. Propofol modulates activation and desensitization of GABAA receptors in cultured murine hippocampal neurons. J Neurosci. 1994;14:7747–7760. doi: 10.1523/JNEUROSCI.14-12-07747.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panula P, Yang HY, Costa E. Histamine-containing neurons in the rat hypothalamus. Proc Natl Acad Sci U S A. 1984;81:2572–2576. doi: 10.1073/pnas.81.8.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic; 2005. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci. 2004;5:709–720. doi: 10.1038/nrn1496. [DOI] [PubMed] [Google Scholar]
- Sakurai T. The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat Rev Neurosci. 2007;8:171–181. doi: 10.1038/nrn2092. [DOI] [PubMed] [Google Scholar]
- Sergeeva OA, Andreeva N, Garret M, Scherer A, Haas HL. Pharmacological properties of GABAA receptors in rat hypothalamic neurons expressing the epsilon-subunit. J Neurosci. 2005;25:88–95. doi: 10.1523/JNEUROSCI.3209-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–219. doi: 10.1126/science.271.5246.216. [DOI] [PubMed] [Google Scholar]
- Sherin JE, Elmquist JK, Torrealba F, Saper CB. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci. 1998;18:4705–4721. doi: 10.1523/JNEUROSCI.18-12-04705.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegwart R, Jurd R, Rudolph U. Molecular determinants for the action of general anesthetics at recombinant alpha(2)beta(3)gamma(2)gamma-aminobutyric acid(A) receptors. J Neurochem. 2002;80:140–148. doi: 10.1046/j.0022-3042.2001.00682.x. [DOI] [PubMed] [Google Scholar]
- Sloan TB. Anesthetic effects on electrophysiologic recordings. J Clin Neurophysiol. 1998;15:217–226. doi: 10.1097/00004691-199805000-00005. [DOI] [PubMed] [Google Scholar]
- Spigelman I, Li Z, Liang J, Cagetti E, Samzadeh S, Mihalek RM, Homanics GE, Olsen RW. Reduced inhibition and sensitivity to neurosteroids in hippocampus of mice lacking the GABA(A) receptor delta subunit. J Neurophysiol. 2003;90:903–910. doi: 10.1152/jn.01022.2002. [DOI] [PubMed] [Google Scholar]
- Staines WA, Daddona PE, Nagy JI. The organization and hypothalamic projections of the tuberomammillary nucleus in the rat: an immunohistochemical study of adenosine deaminase-positive neurons and fibers. Neuroscience. 1987;23:571–596. doi: 10.1016/0306-4522(87)90077-7. [DOI] [PubMed] [Google Scholar]
- Suntsova N, Szymusiak R, Alam MN, Guzman-Marin R, McGinty D. Sleep-waking discharge patterns of median preoptic nucleus neurons in rats. J Physiol. 2002;543:665–677. doi: 10.1113/jphysiol.2002.023085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymusiak R, Alam N, Steininger TL, McGinty D. Sleep-waking discharge patterns of ventrolateral preoptic/anterior hypothalamic neurons in rats. Brain Res. 1998;803:178–188. doi: 10.1016/s0006-8993(98)00631-3. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Lin JS, Sakai K. Neuronal activity of histaminergic tuberomammillary neurons during wake-sleep states in the mouse. J Neurosci. 2006;26:10292–10298. doi: 10.1523/JNEUROSCI.2341-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlin SL, Jenkins A, Lieb WR, Franks NP. Stereoselective effects of etomidate optical isomers on gamma-aminobutyric acid type A receptors and animals. Anesthesiology. 1998;88:708–717. doi: 10.1097/00000542-199803000-00022. [DOI] [PubMed] [Google Scholar]
- Tung A, Lynch JP, Mendelson WB. Prolonged sedation with propofol in the rat does not result in sleep deprivation. Anesth Analg. 2001;92:1232–1236. doi: 10.1097/00000539-200105000-00028. [DOI] [PubMed] [Google Scholar]
- Tung A, Szafran MJ, Bluhm B, Mendelson WB. Sleep deprivation potentiates the onset and duration of loss of righting reflex induced by propofol and isoflurane. Anesthesiology. 2002;97:906–911. doi: 10.1097/00000542-200210000-00024. [DOI] [PubMed] [Google Scholar]
- Williams JT, Henderson G, North RA. Characterization of alpha 2-adrenoceptors which increase potassium conductance in rat locus coeruleus neurones. Neuroscience. 1985;14:95–101. doi: 10.1016/0306-4522(85)90166-6. [DOI] [PubMed] [Google Scholar]
- Zeller A, Arras M, Lazaris A, Jurd R, Rudolph U. Distinct molecular targets for the central respiratory and cardiac actions of the general anesthetics etomidate and propofol. FASEB J. 2005;19:1677–1679. doi: 10.1096/fj.04-3443fje. [DOI] [PubMed] [Google Scholar]
- Zeller A, Arras M, Jurd R, Rudolph U. Mapping the contribution of beta3-containing GABAA receptors to volatile and intravenous general anesthetic actions. BMC Pharmacol. 2007a;7:2. doi: 10.1186/1471-2210-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller A, Arras M, Jurd R, Rudolph U. Identification of a molecular target mediating the general anesthetic actions of pentobarbital. Mol Pharmacol. 2007b;71:852–859. doi: 10.1124/mol.106.030049. [DOI] [PubMed] [Google Scholar]