

Abstract
PURPOSE:
Germline variants in double-strand DNA damage repair (dsDDR) genes (e.g., BRCA1/2;) predispose to pancreatic adenocarcinoma (PDAC) and may predict sensitivity to platinum-based chemotherapy and PARP inhibitors. We sought to determine the prevalence and significance of germline cancer susceptibility gene variants in PDAC with paired somatic and survival analyses.
METHODS:
Using a customized next-generation sequencing panel, germline/somatic DNA was analyzed from 289 patients with resected PDAC ascertained without preselection for high-risk features (e.g., young age, personal/family history). All identified variants were assessed for pathogenicity. Outcomes were analyzed using multivariable-adjusted Cox proportional hazards regression.
RESULTS:
28/289 (9.7%; 95% CI 6.5%−13.7%) patients carried pathogenic/likely pathogenic germline variants, including 21 (7.3%) dsDDR gene variants (3 BRCA1, 4 BRCA2, 14 other dsDDR genes [ATM, BRIP1, CHEK2, NBN, PALB2, RAD50, RAD51C)], 3 Lynch syndrome, and 4 other genes (APC p.I1307K, CDKN2A, TP53). Somatic sequencing and immunohistochemistry identified second hits in the tumor in 12/27 (44.4%) patients with germline variants (1 failed sequencing). Compared to non-carriers, patients with germline dsDDR gene variants had superior overall survival (HR 0.54; 95% CI 0.30–0.99; P=0.05).
CONCLUSION:
Nearly 10% of PDAC patients harbor germline variants, although the majority lack somatic second hits, the therapeutic significance of which warrants further study.
Keywords: PARP inhibitors, familial pancreatic cancer, Lynch syndrome, hereditary breast and ovarian cancer
INTRODUCTION:
As the third leading cause of cancer-related death in the United States,1 pancreatic ductal adenocarcinoma (PDAC) is a disease for which novel approaches to risk assessment, early detection, and treatment are critically needed. Deleterious germline variants in double-strand DNA damage repair (dsDDR) genes, including BRCA1, BRCA2, ATM, and PALB2, have been linked to inherited risks of PDAC.2 In addition, several other high-penetrance cancer susceptibility genes/syndromes have also been linked to increased lifetime risks of PDAC: APC in familial adenomatous polyposis, CDKN2A in familial atypical multiple mole/melanoma syndrome, STK11 in Peutz-Jeghers syndrome, TP53 in Li-Fraumeni syndrome, and MLH1, MSH2, MSH6, PMS2, and EPCAM in Lynch syndrome.2
Recent data3–7 have demonstrated that 3.8–7.4% of PDAC cases harbor germline cancer susceptibility gene variants and that classic high-risk features (e.g., young age at diagnosis, family history of PDAC or other cancers)5–8 have poor sensitivity in identifying PDAC patients with inherited risk. Diagnosing such germline variants can facilitate cancer prevention and increase early detection by signaling the need for enhanced surveillance and risk-reducing interventions in mutation carriers and their healthy relatives.9 Furthermore, germline testing has growing therapeutic implications for individuals with advanced cancer, given the effectiveness of poly(ADP) ribose polymerase (PARP) inhibitors in patients with inherited mutations in dsDDR genes10–12 and anti-PD-1 antibodies in patients with Lynch syndrome-associated cancers.13,14
The preventive benefits for at-risk relatives and suspected therapeutic implications of these germline variants have prompted calls for systematic germline testing in all individuals with PDAC, regardless of age at diagnosis or personal/family cancer history.5,9,14 For PDAC patients with germline cancer susceptibility gene variants, however, the fate of the wild-type allele within the tumor has not been rigorously evaluated. The status of this allele has great importance in determining the functional impact of an inherited susceptibility variant within the tumor and therefore is highly relevant to determining the causality of germline variants and in guiding therapeutic decision-making. To address these important knowledge gaps, we used a customized next-generation sequencing panel to identify germline variants among 24 cancer susceptibility genes with paired somatic sequencing and survival analyses in nearly 300 PDAC patients.
MATERIALS AND METHODS:
Study population
The study population was comprised of 289 patients with resected PDAC seen at Dana-Farber/Brigham and Women’s Cancer Center (DF/BWCC; Boston, MA; n=93) between 10/26/2002–5/21/2012; at the University of Rochester Medical Center (URMC; Rochester, NY; n=80) between 3/1/2006–11/1/2013; or Stanford Cancer Institute (SCI; Stanford, CA; n=116) between 9/26/1995–5/22/2013. IRB approval was granted at each institution and all participants provided written informed consent for participation. Clinicopathologic data were collected from medical records, including sex, age at PDAC resection, race, perioperative chemotherapy/radiotherapy, tumor location, stage, histologic grade, lymphovascular invasion, resection margin status, recurrence pattern, and personal/family cancer history.
Germline DNA sequencing and interpretation
To study genes known or suspected to play a role in PDAC biology, we built a customized next-generation sequencing panel (Supplemental Methods). Analysis of germline variants was restricted to 24 genes linked to inherited cancer risk, including those related to dsDDR (ATM, BRCA1, BRCA2, BRIP1, CHEK2, NBN, PALB2, RAD50, RAD51C, and RAD51D), Lynch syndrome (MLH1, MSH2, MSH6, and PMS2), and other cancer susceptibility pathways (APC, CDH1, CDK4, CDKN2A, POLE, PRSS1, PTEN, SMAD4, STK11, and TP53). Detection of SNVs and small insertions/deletions in these genes was performed using the GATK HaplotypeCaller. Only variants assigned a quality score of ≥30 were included. Germline variants were annotated using Variant Effect Predictor and filtered by the “Best Effect” classification to include only variants of the following types: missense, frameshift, splice_donor, nonsense, splice_acceptor, inframe_del, inframe_ins, and initiator_codon. Variants present in the NHLBI GO Exome Sequencing Project or the Exome Aggregation Consortium at >1% overall population frequency were excluded. A molecular genetic pathologist (J.A.N.) manually reviewed and confirmed all germline variants by direct inspection of sequencing data using Integrative Genomics Viewer.15 All germline variants were confirmed in the matched somatic DNA sample when analyzed in an unpaired (tumor-only) mode against an unrelated reference sample.
The pathogenicity of all identified germline variants was evaluated with Alamut® Visual (http://www.interactive-biosoftware.com/alamut-visual/) and according to published guidelines from the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology.16 All null (nonsense, frameshift, canonical +/− 1 or 2 splice sites, initiation codon, single- or multi-exon deletion) variants and missense variants with supporting evidence as described by the 2015 ACMG guidelines were considered pathogenic or likely pathogenic (P/LP). Supporting evidence for pathogenicity of specific germline variants was assessed by two cancer genetics experts (M.B.Y. and A.B.C.), who evaluated each variant against available data in ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) and the published literature as standards for interpretation. Three well-described low-penetrance variants – the APC p.I1307K Ashkenazi founder variant17 (associated with modestly increased lifetime risks of colorectal cancer) and the CHEK2 p.I157T and p.S428F variants18 (associated with modestly increased lifetime risks of breast cancer) – were classified as pathogenic for the purposes of this study (Table S1). For all other variants with conflicting pathogenicity interpretations, the most conservative pathogenicity classification was assigned (i.e. variants categorized as both likely pathogenic and variant of uncertain significance on ClinVar were considered variants of uncertain significance in this analysis). All germline variants considered pathogenic or likely pathogenic (P/LP) were reported.
Somatic sequencing and tumor testing
Somatic analyses19 (Supplemental Methods) were performed to determine whether tumors with P/LP germline variants harbored a somatic alteration (second hit) in the affected gene. Prior to analysis for somatic second hits, all somatic samples were confirmed to contain an adequate tumor content for reliable detection of additional somatic mutations in the gene(s) in which a germline variant was detected. Patients with P/LP germline variants were considered to have a second hit if (i) somatic sequencing detected a deleterious alteration (e.g., nonsense or frameshift sequence alterations, single copy deletions, or copy-neutral loss-of-heterozygosity) in the gene altered on germline sequencing, (ii) immunohistochemistry (IHC) showed absent/abnormal expression of the protein produced by the gene altered on germline sequencing (Lynch syndrome, CDKN2A, and TP53 carriers), or (iii) the tumor was found to have high-level MSI (MSI-H) or mismatch repair deficiency (MMR-D) by IHC for MLH1, MSH2, MSH6, and PMS2 (Lynch syndrome carriers). For microsatellite instability analysis, DNA from paired tumor and normal tissue was analyzed using five markers (D2S123, D5S346, D17S250, BAT25, BAT26).20 We also classified tumors by status of four driver genes commonly altered in PDAC (KRAS, TP53, SMAD4, CDKN2A) by somatic sequencing and IHC (Supplemental Methods).
Outcome measures
Disease-free survival (DFS) was assessed using the time elapsed between surgery and the date of first PDAC recurrence. Patients who died without definitive evidence of recurrent disease were censored for DFS analyses on the date of last clinical contact. Overall survival (OS) was defined as time between surgery and date of death. Follow-up continued through 6/28/2016 for DF/BWCC, 3/17/2016 for URMC, and 3/11/2016 for SCI. Participants found to have metastatic disease at the time of PDAC resection (n=7) and those with 30-day and/or in-hospital post-operative mortality (n=10) were excluded from DFS and OS analyses (Figure S1). Details on adjuvant therapy, palliative therapy, and disease progression were obtained from medical record review (Figure S2).
Statistical analysis
Associations of P/LP germline variants with clinicopathological characteristics were analyzed using the Fisher exact test and the Wilcoxon rank-sum test for categorical and continuous variables, respectively. We evaluated the associated of P/LP germline variants with DFS and OS using multivariable-adjusted Cox proportional hazards regression, calculating hazard ratios (HR) and 95% confidence intervals (CI). Cox regression models were adjusted for prognostic factors and potential confounding covariates, including age at surgery, sex, tumor location (head/uncinate, body, tail, other), resection margin status (R0, R1, R2, Rx), perioperative chemotherapy (yes, no), perioperative radiotherapy (yes, no), institution (DF/BWCC, URMC, SCI), and year of surgery. We verified the proportionality of hazards assumption by evaluating time-dependent variables of the cross-product of each exposure of interest and time. DFS and OS were presented using Kaplan-Meier curves, from which median and 2-year survival rates were calculated. All P values were two-sided and considered statistically significant at <0.05. Statistical analyses were performed using SAS software (version 9.4, SAS Institute).
RESULTS:
Twenty-eight (9.7%; 95% CI 6.5%−13.7%) of the 289 study patients carried P/LP germline variants in ≥1 of the 24 genes analyzed (Table 1). Median patient age at surgery was similar among patients with and without germline variants (66 years versus 67 years, respectively; P=0.77). Individuals with P/LP germline variants were more likely than those without germline variants to have a primary tumor in the pancreatic tail (39% versus 12%, respectively; P=0.01). There was no significant difference in the presence of somatic alterations in KRAS, CDKN2A, SMAD4, or TP53 between individuals with and without germline cancer susceptibility gene variants (all P≥0.13; Tables S2 and S3).
Table 1:
Clinical and pathologic characteristics of 289 individuals with resected pancreatic adenocarcinoma
| Overall | Pathogenic or likely pathogenic germline variant | |||
|---|---|---|---|---|
| Yes | No | P-valuea | ||
| Number of subjects | 289 | 28 | 261 | |
| Women (n, %) | 138 (48%) | 14 (50%) | 124 (48%) | 0.84 |
| Median age at diagnosis, years, (IQR) | 67 (15) | 66 (17) | 67 (14) | 0.77b |
| Center (n, %) | ||||
| DF/BWCC | 93 (32%) | 13 (46%) | 80 (31%) | 0.22 |
| URMC | 80 (28%) | 8 (29%) | 72 (27%) | |
| SCI | 116 (40%) | 7 (25%) | 109 (42%) | |
| Racial background (n, %) | ||||
| White | 220 (76%) | 23 (82%) | 197 (75%) | 0.66 |
| Black | 4 (1%) | - | 4 (2%) | |
| Asian | 28 (10%) | 1 (4%) | 27 (10%) | |
| Unknown | 37 (13%) | 4 (14%) | 33 (13%) | |
| Tumor location (n, %) | ||||
| Head/Uncinate | 213 (74%) | 14 (50%) | 199 (76%) | 0.01 |
| Body | 28 (10%) | 2 (7%) | 26 (10%) | |
| Tail | 41 (14%) | 11 (39%) | 30 (12%) | |
| Overlapping sites | 7 (2%) | 1 (4%) | 6 (2%) | |
| pT stage (n, %) | ||||
| T1–T2 | 46 (16%) | 4 (14%) | 42 (16%) | 1.00 |
| T3–T4 | 242 (83%) | 24 (86%) | 218 (83%) | |
| Tx | 1 (1%) | - | 1 (1%) | |
| pN stage (n, %) | ||||
| N0 | 75 (26%) | 9 (32%) | 66 (25%) | 0.50 |
| N1 | 213 (73%) | 19 (68%) | 194 (74%) | |
| Nx | 1 (1%) | - | 1 (1%) | |
| Tumor differentiation (n, %) | ||||
| Well/Moderately | 160 (55%) | 16 (57%) | 144 (55%) | 0.84 |
| Poorly/Undifferentiated | 123 (43%) | 11 (39%) | 112 (43%) | |
| Unknown | 6 (2%) | 1 (4%) | 5 (2%) | |
| Lymphovascular invasion (n, %) | ||||
| Present | 137 (47%) | 11 (39%) | 126 (48%) | 0.23 |
| Absent | 126 (44%) | 16 (57%) | 110 (42%) | |
| Unknown | 26 (9%) | 1 (4%) | 25 (10%) | |
| Resection margin status (n, %) | ||||
| R0 | 138 (48%) | 12 (43%) | 126 (48%) | 0.73 |
| R1 | 146 (50%) | 16 (57%) | 130 (50%) | |
| R2 | 3 (1%) | - | 3 (1%) | |
| Rx (not evaluable) | 2 (1%) | - | 2 (1%) | |
| First site of recurrence (n, %) | ||||
| Local only | 44 (15%) | 3 (11%) | 41 (16%) | 0.71 |
| Distant only | 108 (37%) | 10 (35%) | 98 (38%) | |
| Synchronous local and distant | 42 (15%) | 5 (18%) | 37 (14%) | |
| No known recurrence | 68 (24%) | 7 (25%) | 61 (23%) | |
| Unknown | 27 (9%) | 3 (11%) | 24 (9%) | |
Abbreviations: DF/BWCC, Dana-Farber/Brigham and Women’s Cancer Center; URMC, University of Rochester Medical Center; SCI, Stanford Cancer Institute.
Calculated using Fisher exact test unless otherwise specified.
Calculated using Wilcoxon rank sum test.
Twenty-one (7.3%) of the 289 study participants carried P/LP germline variants in dsDDR genes (Figure 1), including 7 (2.4%) with BRCA1 or BRCA2 variants (one with a concurrent APC variant) and 14 (4.8%) with other dsDDR gene variants, including ATM (N=4), BRIP1 (N=3), CHEK2 (N=3), NBN (N=1), PALB2 (N=1), RAD50 (N=1), and RAD51C (N=1). Three (1.0%) patients carried variants consistent with Lynch syndrome (1 MSH2, 2 MSH6), and 4 (1.4%) had variants identified in other cancer susceptibility genes (1 APC p.I1307K, 2 CDKN2A, 1 TP53).
Figure 1:
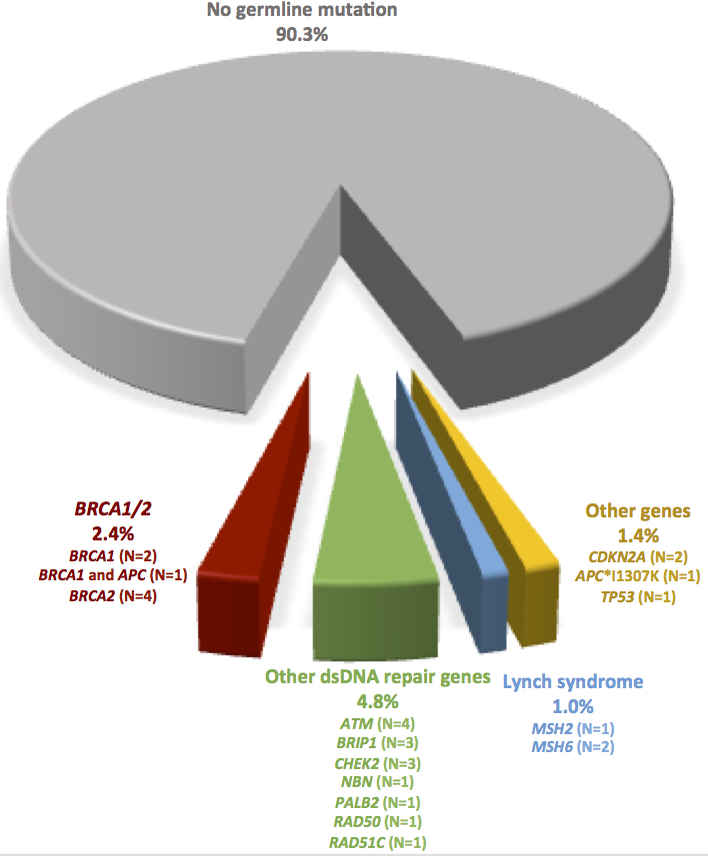
Pathogenic and likely pathogenic germline variants identified among 289 individuals with resected pancreatic adenocarcinoma. Abbreviation: dsDNA, double-strand DNA.
Of the 28 patients with P/LP germline variants, 6 (21.4%) had a family history of PDAC and 16 (57.1%) had a personal history of another cancer, including 5 (17.9%) each with colorectal and breast cancer (Tables 2–3). However, of the 7 patients with P/LP BRCA1/2 variants, none had a personal history of BRCA1/2-associated (breast, ovarian, or prostate) cancer and only 4 (57.1%) had a family history of these cancers in first- or second-degree relatives. Only one (33%) of the three Lynch syndrome carriers had a personal/family history of Lynch syndrome-associated cancer.
Table 2:
Characteristics of pancreatic adenocarcinoma patients with pathogenic or likely pathogenic germline variants in double strand DNA repair genes
| ID # | Age/Sex | Pathogenic/likely pathogenic germline alteration(s) | Somatic sequencing and tumor testing data | Other personal history of cancer* (age at diagnosis, if known) | PDAC Location | Family history of cancer (relation, age if known) |
|---|---|---|---|---|---|---|
| BRCA1/2 mutation carriers | ||||||
| 1 | 38/M | BRCA1 c.5444G>A (p.W1815*) and APC c.694C>T (p.R232*) | CN-LOH of BRCA1; somatic c.4611_4612delAG (p.E1538Ifs*5) APC alteration | REC (29) | Tail | LG (pat uncle, 65), CV (pat aunt, 64), BR (pat cousin), CNS (pat cousin) |
| 2 | 68/F | BRCA1 c.427G>T (p.E143*) | Somatic c.376C>T (p.Q126*) BRCA1 alteration | None | Head/Uncinate | BR (mother, 40; sister), OV (sister, 53; sister) |
| 3 | 75/M | BRCA1 c.5266dupC (p.Q1756Pfs*74) | No somatic BRCA1 alteration identified | CO (62) | Head/Uncinate | BR (daughter, 39; maternal aunt, 33) |
| 4 | 29/F | BRCA2 c.5946delT (p.S1982Rfs*22) | No somatic BRCA2 alteration identified | None | Head/Uncinate | BR (pat grandmother, 42), SKIN (pat grandmother) PR (mat grandfather), OV (pat great grandmother, 52) |
| 5 | 55/F | BRCA2 c.4634delT (p.F1546Lfs*22) | Inadequate somatic NGS coverage | None | Tail | CNS (sister, 49); LG (father, 68) |
| 6 | 62/M | BRCA2 c.1189_1190insTTAG (p.Q397Lfs*25) | Single copy deletion of BRCA2 | None | Head/Uncinate | BR (mother) |
| 7 | 71/F | BRCA2 c.5946delT (p.S1982Rfs*22) | No somatic BRCA2 alteration identified | None | Tail | None |
| Other germline double strand DNA repair gene mutation carriers | ||||||
| 8 | 65/M | ATM c.6843C>G (p.Y2281*) | Single copy deletion of ATM | BR | Tail | CNS (niece, 40s) |
| 9 | 66/M | ATM c.3802delG (p.V1268*) | No somatic ATM alteration identified | REC (52) | Head/Uncinate | BR (mother), BL (mother), PAN (mat uncle) |
| 10 | 66/F | ATM c.5931delT (p.F1977Lfs*13) | CN-LOH of ATM | BR (52), SAC (57) | Head/Uncinate | BR (mother, 62; pat aunt, 67), SKIN (daughter, 26; father, 65; mat uncle), PAN (mat uncle, 80), ESO (pat uncle, 58) |
| 11 | 77/F | ATM c.3023delC (p.S1008Lfs*14) | Single copy deletion of ATM | None | Overlapping Sites | None |
| 12 | 65/M | BRIP1 c.440dupA (p.Y147*) | Single copy deletion of BRIP1 | None | Tail | BR (mother) |
| 13 | 67/M | BRIP1 c.2684_2687delCCAT (p.S895*) | No somatic BRIP1 alteration identified | GIST (67) | Tail | PAN (brother, 57), DCIS (sister, 58), SKIN (sister; mat half-cousin), OV (pat grandmother, 75), BR (mat grandmother, 55), LG (mat half-sister, 81; pat cousin, 85), HN (pat cousin, 72), LYM (mat cousin, 62), KID (mat cousin, 55), CO (mat half-cousin, 85), LK (mat half-cousin, 85) |
| 14 | 80/F | BRIP1 c.2108delAinsTCC (p.K703Ifs*) | No somatic BRIP1 alteration identified | BR (37, 52) | Head/Uncinate | BR (sister; daughter), HN (brother) |
| 15 | 61/F | CHEK2 c.470T>C (p.I157T) | No somatic CHEK2 alteration identified | None | Head/Uncinate | CV (sister), MEL (father) |
| 16 | 68/M | CHEK2 c.1392delT (p.S465Vfs*15) | No somatic CHEK2 alteration identified | BL (68) | Tail | BR (sister, 45), STO (brother), UNK (father; brother; brother; sister) |
| 17 | 86/M | CHEK2 c.1283C>T (p.S428F) | No somatic CHEK2 alteration identified | None | Tail | None |
| 18 | 78/F | NBN c.698_701delAACA (p.K233Sfs*5) | No somatic NBN alteration identified | MEL | Head/Uncinate | None |
| 19 | 74/F | PALB2 c.3113G>A (p.W1038*) | Somatic c.2323C>T (p.Q775*) PALB2 alteration | BR (59) | Head/Uncinate | BR (sister, 29; sister, 42; sister, 68; mat aunt, 70) |
| 20 | 77/F | RAD50 c.1875C>G (p.Y625*) | No somatic RAD50 alteration identified | NHL (73) | Tail | None |
| 21 | 47/F | RAD51C c.706–2A>G | Single copy deletion of RAD51C | None | Head/Uncinate | OV (mat grandmother) |
Abbreviations: BL, bladder cancer; BR, breast cancer; CO, colon cancer; CNS, brain tumor; CV, cervical cancer; DCIS, ductal carcinoma in situ of the breast; ESO, esophageal cancer; GIST, gastrointestinal stromal tumor; HN, head and neck cancer not otherwise specified; KID, kidney cancer; LG, lung cancer; LK, leukemia; LYM, lymphoma not otherwise specified; MEL, melanoma; NHL, non Hodgkin lymphoma; OV, ovarian cancer; PAN, pancreatic cancer; REC, rectal cancer; SAC, sarcoma not otherwise specified; SKIN, skin cancer not otherwise specified; STO, stomach cancer; UNK, cancer with unknown primary site. CN-LOH = copy-neutral loss of heterozygosity
Excludes non-melanoma skin cancers.
Table 3:
Characteristics of pancreatic adenocarcinoma patients with pathogenic or likely pathogenic germline variants in other cancer susceptibility gene variants
| ID # | Age/Sex | Pathogenic/likely pathogenic germline alteration(s) | Somatic sequencing and tumor testing data | Other personal history of cancer* (age at diagnosis, if known) | PDAC Location | Family history of cancer (relation, age if known) |
|---|---|---|---|---|---|---|
| Lynch syndrome | ||||||
| 22 | 53/F | MSH2 c.1906G>C (p.A636P) | No somatic MSH2 alteration identified; absent MSH2/MSH6 by IHC; MSI-H | CO (46, 46) | Tail | PAN (mother, 65; mat grandmother, 68), CO (sister, 57; mat uncle, 65; mat grandmother), ENDO (sister, 52; mother, 56; mat grandmother, 50; mat cousin, 47; mat cousin, 54), MEL (niece, 26), EW (pat uncle) |
| 23 | 72/M | MSH6 c.125_126insT (p.S43Ffs*47) | No somatic MSH6 alteration identified; intact MMR IHC; MSS | None | Head/Uncinate | None |
| 24 | 77/M | MSH6 c.3968_3969insTGAGAAGATGAATC (p.Q1328Lfs*4) | No somatic MSH6 alteration identified; intact MMR IHC; MSS | PAN (second primary 85), LG | Body | None |
| Other pathogenic cancer susceptibility gene mutation carriers | ||||||
| 25 | 60/M | APC c.3920T>A (p.I1307K) | Single copy deletion of wild type APC allele | MEL (60) | Head/Uncinate | HN (father, 90), BR (mat grandmother, 65) |
| 26 | 46/M | CDKN2A c.225_243delCGCCACTCTCACCCGACCC (p.A76Cfs*64) | No somatic CDKN2A alteration identified | None | Head/Uncinate | MEL (brother, 41), PAN (mat great aunt, 81; pat great aunt, 65) |
| 27 | 65/F | CDKN2A c.44G>A (p.W15*) | No somatic CDKN2A alteration identified | BR (33), MEL (45, 55), CO (62), DES (65) | Body | MEL (brother, 24; daughter; mat uncle; mat cousin), PAN (mother, 89), LG (father, 63; pat grandfather, 58), DN (niece; nephew), BR (mat aunt, 35), CO (mat grandmother, 65), STO (mat grandfather, 65) |
| 28 | 48/M | TP53 c.742C>T (p.R248W) | Inadequate somatic NGS coverage; mutant TP53 IHC pattern | AMP (38), STO (48) | Tail | CNS (brother, 17), STO (brother, 45), BR (mother, late 30s; pat aunt, 40s), SAC (nephew, 17), ESO (mat uncle, late 40s), UNK (mat aunt, 40s; mat aunt, 40s; mat cousin, 20s; mat cousin, 30s) |
Abbreviations: AMP, ampullary adenocarcinoma; BR, breast cancer; CO, colon cancer; CNS, brain tumor; DES, desmoid tumor; DN, dysplastic nevus; ESO, esophageal cancer; EW, Ewing sarcoma; HN, head and neck cancer not otherwise specified; LG, lung cancer; MEL, melanoma; PAN, pancreatic cancer; SAC, sarcoma not otherwise specified; STO, stomach cancer; UNK, cancer with unknown primary site.
IHC = immunohistochemistry; MMR = mismatch repair protein; MSI-H = high-level microsatellite instability by PCR; MSS = microsatellite stable by PCR
Excludes non-melanoma skin cancers.
Somatic sequencing and IHC/MSI tumor testing was performed to evaluate for a second hit in the tumors of patients with P/LP germline variants. Somatic sequencing data were adequate for evaluation of second hits for all patients with P/LP germline variants, except for one individual with a P/LP germline BRCA2 variant. Twelve (44.4%) of 27 patients’ tumors had evidence of a second hit, including 3/6 (50.0%) with BRCA1/2 variants, 6/14 (42.9%) with other dsDDR gene variants (3/4 ATM, 1/3 BRIP1, 0/3 CHEK2, 0/1 NBN, 1/1 PALB2, 0/1 RAD50, 1/1 RAD51C), 1/3 (33.3%) with Lynch syndrome, and 2/4 (50.0%) with other germline variants (1/1 APC p.I1307K, 0/2 CDKN2A, 1/1 TP53; Figure S3).
In multivariable adjusted Cox proportional hazards models, individuals with any P/LP germline variant had superior OS (adjusted HR 0.54; 95% CI 0.32–0.91; P=0.02), compared to non-carriers (Figure 2). This association appeared to be driven primarily by individuals carrying P/LP germline dsDDR gene variants, who had significantly longer OS compared to non-carriers (HR 0.54; 95% CI 0.30–0.99; P=0.05) with a median OS of 34.4 months versus 19.1 months for non-carriers, and a 2-year OS rate of 65.0% versus 39.5% for non-carriers. No significant difference was identified in DFS for those with P/LP germline dsDDR gene variants, compared to non-carriers (HR 0.78; 95% CI 0.42–1.44; P=0.43). Among the subset of 13 individuals with P/LP germline dsDDR gene variants who developed recurrent/metastatic disease, there was a non-significant trend towards improved OS among the 5 who received oxaliplatin-based chemotherapy versus the 8 who did not (median OS 20.9 months versus 14.4 months; HR 0.59; 95% CI 0.17–2.13); none received other platinum agents. Two of the 3 Lynch syndrome probands were alive >90 months after PDAC resection, including one MSH2 proband with prolonged disease control for >7 years after developing distant metastases. None of the individuals with P/LP germline variants received treatment with PARP inhibitors or anti-PD-1 antibodies during the study period.
Figure 2:

Kaplan-Meier survival curves by germline status: A. Disease-free survival, and B. Overall survival for PDAC patients with any pathogenic/likely pathogenic germline cancer susceptibility gene variant; C. Disease-free survival, and D. Overall survival for PDAC patients with pathogenic/likely pathogenic double-stranded DNA damage repair gene variants. Abbreviations: dsDDR, double-stranded DNA damage repair gene; HR, adjusted hazard ratio; CI, confidence interval.
DISCUSSION:
In this multicenter study of 289 patients with resected PDAC who were not preselected for age or personal/family cancer history, targeted germline analysis of 24 genes related to inherited cancer predisposition revealed P/LP germline variants in nearly 10% of patients, including 7.3% of patients with variants in genes related to dsDDR. Compared to non-carriers, individuals with germline dsDDR gene variants in this study had superior overall survival after PDAC resection. Intriguingly, however, fewer than half of the PDAC probands with germline cancer susceptibility gene variants in this study had an identified somatic second hit in the wild-type allele of the tumor.
While other recent studies have similarly examined the prevalence and spectrum of germline variants in PDAC patients, our findings provide novel insight on the somatic, therapeutic, and prognostic consequences of such germline alterations. Importantly, unlike most other forms of cancer, the vast majority of PDAC patients, even those who present with early-stage disease, will ultimately require palliative systemic therapy, and knowledge of germline status increasingly has the potential to guide treatment decisions. Building upon successes in BRCA1/2-associated breast, ovarian, and prostate cancer,11,21,22 early-phase studies23,24 of single-agent PARP inhibitors in BRCA1/2-associated PDAC have shown durable responses, and an ongoing large, randomized trial is evaluating maintenance olaparib in PDAC patients with germline BRCA1/2 variants. PARP inhibition has also shown benefit in advanced prostate cancers in patients with germline ATM variants,22 suggesting that alterations in other dsDDR genes beyond BRCA1/2 may predict for PARP inhibitor sensitivity. Additionally, anti-PD-1 anitbodies can benefit patients with Lynch syndrome-associated cancers (including PDAC),13 and pembrolizumab was recently FDA-approved for treating patients with advanced MSI-H/MMR-D cancers. Intriguingly, the identification of somatic BRCA2 alterations in MSI-H cancers,25 as well as data demonstrating increased immune activation and high neoantigen loads in PDAC with defective dsDDR26 have raised speculation about therapeutic synergy for PARP and PD-1 inhibition in both Lynch- and dsDDR-associated cancers.
Although most of the P/LP germline variants identified in this study were in genes previously linked to inherited PDAC susceptibility (e.g., BRCA1/2, ATM, PALB2, DNA mismatch repair genes), only about half of such carriers had a confirmed second hit on paired somatic analyses. While some somatic second hits may have occurred through other forms of allelic inactivation that would have gone undetected in this analysis (e.g., hypermethylation), this finding raises critical questions about whether each of these germline variants were truly causative of the individual’s PDAC. Furthermore, many PDAC patients with germline BRCA1/2 variants have not had tumor responses to PARP inhibitors in early-phase studies.23,24 These modest response rates could potentially be due to tumors lacking somatic second hits and thus having proficient homologous recombination (HR) machinery, such that germline status alone may be insufficient for determining sensitivity to PARP inhibitors in PDAC
While none of the carriers identified in this study underwent treatment with therapies specifically tailored towards their germline status (e.g., PARP inhibitors or anti-PD-1 antibodies), we observed a non-significant trend towards superior OS for individuals with dsDDR variants who received oxaliplatin-based chemotherapy after disease recurrence. Such findings complement those from a recent single-institution analysis of 29 stage III/IV PDAC patients with germline BRCA1, BRCA2, or PALB2 variants and 58 matched controls which demonstrated that such germline variants significantly predicted for improved OS among individuals treated with oxaliplatin- or cisplatin-based chemotherapy.27 Another study of 24 BRCA1/2 carriers and 49 matched controls with early-stage/resected PDAC28 likewise reported a non-significant trend towards superior DFS in BRCA1/2 carriers versus controls among those who received platinum-based adjuvant/neoadjuvant chemotherapy. A recent single-institution analysis of 91 stage III/IV PDAC patients similarly found a non-significant trend towards improved progression-free survival (but not OS) among individuals with high HR deficiency scores (as determined by a commercial functional assay) treated with oxaliplatin-based chemotherapy (FOLFIRINOX) versus those with low HR deficiency scores, regardless of whether or not they harbored a known germline BRCA1/2 variant.29 Our findings in conjunction with these prior data support the notion that germline dsDDR variants may predict for improved response to platinum-based chemotherapy, although the small number of carriers and heterogeneous treatment regimens in each of these studies have resulted in limited power to define this predictive benefit conclusively.
Beyond the potentially predictive impact of germline status in PDAC, however, our data demonstrate the prognostic value of germline dsDDR variants in PDAC, since such carriers had significantly superior OS versus non-carriers, regardless of treatment status. Further clinical trial outcomes examining whether PARP inhibitors, PD-1 inhibitors, and other agents targeted towards germline status can further improve these outcomes are eagerly anticipated.
In addition to the therapeutic and prognostic considerations for PDAC probands themselves, identifying germline variants is also critically important for at-risk relatives, who can undergo genetic testing followed by appropriate cancer risk-reducing interventions (e.g., colonoscopies in Lynch syndrome; salpingo-oophorectomy in BRCA1/2 carriers).9 Recent data have also suggested potential benefit to regular PDAC screening with endoscopic ultrasound and MRI in such families.30,31
The overall 9.7% prevalence of P/LP germline cancer susceptibility gene variants in our study is higher than the 3.8–7.4% range observed in prior studies of systematic germline testing in other cohorts of PDAC patients.3–7 The variable mutation prevalence seen across these studies is likely due in part to differences in the number of genes analyzed as well as potential founder effects, which may be particularly noticeable in single-institution studies where the study cohorts are presumably of more limited geographic and ethnic diversity.5,7,32 Recent studies, for instance, have described a 5.3% and 10.0–15.6% prevalence of founder mutations, respectively, among French Canadian and Ashkenazi Jewish pancreatic cancer patients, respectively, regardless of age or clinical history.8,32 Our findings build on these prior studies by exploring the superior survival outcomes among PDAC patients with P/LP germline variants, and also by examining paired somatic data, which may be particularly important in understanding the functional and therapeutic significance of germline variants in dsDDR genes.
A common finding among prior studies5–8 and verified in this analysis is that many PDAC patients with germline variants lack personal/family histories suggestive of inherited cancer risk. Although national guidelines for BRCA1/2 and Lynch syndrome testing mention PDAC as a component cancer,2,33,34 our findings demonstrate that these syndromes only account for about one-third of germline alterations found in PDAC patients. These results suggest that the current standard of care of using high-risk clinical features to guide the use of syndrome-specific germline evaluation will fail to identify many PDAC patients and families with inherited risk. Assessing for hereditary cancer risk in PDAC patients is inherently different from doing so in individuals with breast, colorectal, and other types of cancer, since the aggressive natural history of PDAC may lead many patients to become seriously ill or die before they undergo genetic evaluation, unless testing is done promptly after diagnosis.9 These findings support the notion that systematic multigene germline testing,5 performed promptly at the time of initial diagnosis,9 may indeed be the optimal means of identifying PDAC patients with inherited cancer susceptibility.
Our findings include the novel observation that PDACs from patients with germline P/LP variants, particularly those with germline dsDDR gene variants, were significantly more likely to arise in the tail of the pancreas, compared to individuals lacking germline variants. This observation should be assessed in other cohorts of patients with resected PDAC since primary tumor location would be important for surveillance strategies in at-risk individuals with germline dsDDR gene variants.
This study has several important strengths. This was a large, multicenter cohort of patients with resected PDAC, for whom extensive clinical data were collected and without preselection by age of diagnosis or personal/family cancer history. Furthermore, in contrast to other recent studies of multigene germline analysis in PDAC, we conducted paired somatic analyses which may be critical in predicting efficacy of agents such as PARP inhibitors in individuals with germline dsDDR variants. Somatic second hits were identified in tumors from probands with germline alterations in genes not previously linked to PDAC risk (e.g., BRIP1, RAD51C), raising intriguing questions about whether such genes may play an etiologic role in PDAC. We also performed detailed survival analyses on individuals with and without germline cancer susceptibility gene variants, demonstrating germline status as a favorable prognostic factor in resected PDAC.
Limitations of this study also require consideration. All participants underwent PDAC resection and were recruited through academic medical centers. Therefore, our findings may not be fully generalizable to individuals with advanced disease or those seen in community practices. The heterogeneity of therapeutic regimens received by patients in this study limits the ability to draw firm conclusions about the predictive value of P/LP germline variants in the setting of particular treatment regimens (Figure S2). Evaluation of germline copy number changes was not performed and thus a small number of patients with germline deletion or duplication events may have gone undetected. Although we were conservative with germline variant classification and adhered to ACMG guidelines, we acknowledge that some subjectivity is inherent to pathogenicity assessments, such as with the low-penetrance CHEK2 p.I157T alteration, which is classified as pathogenic in this analysis and by most commercial genetic testing companies.35 Although one prior case-control study described a modest association between the CHEK2 p.I157T founder variant and likelihood of familial pancreatic cancer in Polish individuals, few data are available linking the common low-penetrance variants identified in this study cohort (APC p.I1307K, CHEK2 p.I157T, and CHEK2 p.S428F) to PDAC risk, and whether such variants are causative of increased PDAC risk is unclear.36
Furthermore, the absence of an identifiable somatic second hit in the PDAC tumor of several patients with P/LP germline variants and strong family cancer histories is somewhat surprising. Nevertheless, although other recent studies have described similar phenomena across a spectrum of tumor types from individuals with P/LP germline variants.37,38 While our sequencing platform was specifically designed to maximize sensitivity for somatic mutations and copy number changes, we cannot rule out the possibility that some somatic second hits may have gone undetected due in part to the abundant stroma and low neoplastic cell content that is characteristic of most primary PDAC tumor specimens. For the current study, when feasible, we have bolstered our somatic sequence analyses with orthogonal testing methods (eg IHC). For example, Patient #22 with a germline MSH2 variant and a striking personal and family history of Lynch-associated cancers had a PDAC that demonstrated MMR-D by IHC and MSI-H by PCR, providing compelling evidence for the presence of a somatic “second hit” even in the absence of a definitively identified somatic sequence alteration or copy number change. On the other hand, Patient #26 with a germline frameshift CDKN2A variant and a family history of melanoma and PDAC had a tumor with intact IHC for CDKN2A (Figure S3), strongly suggesting preserved CDKN2A expression. Comprehensive functional and multi-omic analyses, including methylation, RNA, and protein studies,3 would be ideal for reconciling such discordances in PDAC patients with P/LP germline variants lacking obvious somatic second hits.
Germline testing costs continue to rapidly decrease, and data have demonstrated a greater than expected yield to systematic multigene testing in various clinical situations.39,40 The most prominent such scenario is in women with ovarian cancer, where the high prevalence of germline variants, the poor predictive value of age and personal/family history at identifying carriers, the disease’s high lethality, and emerging therapeutic implications (e.g., platinum, PARP inhibitors) have prompted guidelines33 to recommend germline testing of all women with ovarian cancer. This study demonstrates how germline cancer risk in PDAC closely mirrors these same features of ovarian cancer, supporting the recent assertion5 that systematic multigene germline testing should be considered for all PDAC patients, regardless of age or family history.9,14 To maximize the potential value of such a practice, however, it will be critical to better understand how germline variants correspond with somatic and functional data (e.g., HR deficiency assays) in predicting therapeutic response to agents such PARP inhibitors, immune checkpoint inhibitors, and other novel agents.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Footnotes
Supplementary information is available at the Genetics in Medicine website.
An abstract from the data in this manuscript was presented as an oral presentation at the Annual Meeting of the Collaborative Group of the Americas on Inherited Gastrointestinal Cancer in October, 2017.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. January 2017;67(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Syngal S, Brand RE, Church JM, et al. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. February 2015;110(2):223–262;quiz 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. August 14 2017;32(2):185–203 e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johns AL, McKay SH, Humphris JL, et al. Lost in translation: returning germline genetic results in genome-scale cancer research. Genome Med. April 28 2017;9(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shindo K, Yu J, Suenaga M, et al. Deleterious Germline Mutations in Patients With Apparently Sporadic Pancreatic Adenocarcinoma. J Clin Oncol. August 02 2017:JCO2017723502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grant RC, Selander I, Connor AA, et al. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology. March 2015;148(3):556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holter S, Borgida A, Dodd A, et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. J Clin Oncol. October 01 2015;33(28):3124–3129. [DOI] [PubMed] [Google Scholar]
- 8.Salo-Mullen EE, O’Reilly EM, Kelsen DP, et al. Identification of germline genetic mutations in patients with pancreatic cancer. Cancer. December 15 2015;121(24):4382–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yurgelun MB. Germline Testing for Individuals With Pancreatic Cancer: The Benefits and Challenges to Casting a Wider Net. J Clin Oncol. August 23 2017:JCO2017747535. [DOI] [PubMed] [Google Scholar]
- 10.Robson M, Im SA, Senkus E, et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med. August 10 2017;377(6):523–533. [DOI] [PubMed] [Google Scholar]
- 11.Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med. December 01 2016;375(22):2154–2164. [DOI] [PubMed] [Google Scholar]
- 12.Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. July 24 2010;376(9737):245–251. [DOI] [PubMed] [Google Scholar]
- 13.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. July 28 2017;357(6349):409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwark AL, Stadler ZK. Should We Lower Our Threshold for Germline Genetic Assessment in Pancreatic Adenocarcinoma? JCO Precis Oncol. 2018. [DOI] [PubMed] [Google Scholar]
- 15.Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. January 2011;29(1):24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. May 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma X, Zhang B, Zheng W. Genetic variants associated with colorectal cancer risk: comprehensive research synopsis, meta-analysis, and epidemiological evidence. Gut. February 2014;63(2):326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tung N, Domchek SM, Stadler Z, et al. Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat Rev Clin Oncol. September 2016;13(9):581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qian ZR, Rubinson DA, Nowak JA, et al. Association of Alterations in Main Driver Genes With Outcomes of Patients With Resected Pancreatic Ductal Adenocarcinoma. JAMA Oncol. November 2 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao X, Lochhead P, Nishihara R, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. October 25 2012;367(17):1596–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. July 09 2009;361(2):123–134. [DOI] [PubMed] [Google Scholar]
- 22.Mateo J, Carreira S, Sandhu S, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. October 29 2015;373(18):1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Bono J, Ramanathan RK, Mina L, et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. June 2017;7(6):620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. January 20 2015;33(3):244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deihimi S, Lev A, Slifker M, et al. BRCA2, EGFR, and NTRK mutations in mismatch repair-deficient colorectal cancers with MSH2 or MLH1 mutations. Oncotarget. June 20 2017;8(25):39945–39962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Connor AA, Denroche RE, Jang GH, et al. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol. June 01 2017;3(6):774–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reiss KA, Yu S, Judy R, Symecko H, Nathanson KL, Domchek SM. Retrospective Survival Analysis of Patients with Advanced Pancreatic Ductal Adenocarcinoma and Germline BRCA or PALB2 Mutations. JCO Precis Oncol. 2018. [DOI] [PubMed] [Google Scholar]
- 28.Golan T, Sella T, O’Reilly EM, et al. Overall survival and clinical characteristics of BRCA mutation carriers with stage I/II pancreatic cancer. Br J Cancer. March 14 2017;116(6):697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shahda S, Timms KM, Ibrahim AA, et al. Homologous Recombination Deficiency in Patients with Pancreatic Ductal Adenocarcinoma and Response to Chemotherapy. JCO Precis Oncol. 2018. [DOI] [PubMed] [Google Scholar]
- 30.Vasen H, Ibrahim I, Ponce CG, et al. Benefit of Surveillance for Pancreatic Cancer in High-Risk Individuals: Outcome of Long-Term Prospective Follow-Up Studies From Three European Expert Centers. J Clin Oncol. June 10 2016;34(17):2010–2019. [DOI] [PubMed] [Google Scholar]
- 31.Canto MI, Hruban RH, Fishman EK, et al. Frequent detection of pancreatic lesions in asymptomatic high-risk individuals. Gastroenterology. April 2012;142(4):796–804; quiz e714–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith AL, Wong C, Cuggia A, et al. Reflex Testing for Germline BRCA1, BRCA2, PALB2, and ATM Mutations in Pancreatic Cancer: Mutation Prevalence and Clinical Outcomes From Two Canadian Research Registries. JCO Precis Oncol. 2018. [DOI] [PubMed] [Google Scholar]
- 33.NCCN Clinical Practice Guidelines in Oncology. Genetic/Familial High-Risk Assessment: Breast and Ovarian. Version 2.2017. http://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf.Accessed September 2, 2017.
- 34.NCCN Clinical Practice Guidelines in Oncology. Genetic/Familial High-Risk Assessment: Colorectal. Version 2.2017. http://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf. Accessed September 2, 2017..
- 35.Balmana J, Digiovanni L, Gaddam P, et al. Conflicting Interpretation of Genetic Variants and Cancer Risk by Commercial Laboratories as Assessed by the Prospective Registry of Multiplex Testing. J Clin Oncol. December 2016;34(34):4071–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lener MR, Kashyap A, Kluzniak W, et al. The Prevalence of Founder Mutations among Individuals from Families with Familial Pancreatic Cancer Syndrome. Cancer Res Treat. April 2017;49(2):430–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mandelker D, Zhang L, Kemel Y, et al. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs Guideline-Based Germline Testing. JAMA. September 5 2017;318(9):825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schrader KA, Cheng DT, Joseph V, et al. Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNA. JAMA Oncol. January 2016;2(1):104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yurgelun MB, Kulke MH, Fuchs CS, et al. Cancer Susceptibility Gene Mutations in Individuals With Colorectal Cancer. J Clin Oncol. April 01 2017;35(10):1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tung N, Lin NU, Kidd J, et al. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J Clin Oncol. May 01 2016;34(13):1460–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.