

Abstract
Objective:
There is considerable uncertainty regarding the optimal use of rifampicin for the treatment of tuberculous (TB) meningitis. A pharmacokinetic modeling and simulation study of rifampicin concentrations in cerebrospinal fluid (CSF) during TB meningitis treatment was performed in this study.
Methods:
Parameters for rifampicin pharmacokinetics in CSF were estimated using individual-level rifampicin pharmacokinetic data, and the model was externally validated in three separate patient cohorts. Monte Carlo simulations of rifampicin serum and CSF concentrations were performed. The area under the rifampicin CSF concentration-versus-time curve during 24 h (AUC0–24) relative to the minimum inhibitory concentration (MIC) served as the pharmacodynamic target.
Results:
Across all simulated patients on the first treatment day, 85% attained the target AUC0–24/MIC ratio of 30 under a weight-based dosing scheme approximating 10 mg/kg. At the rifampicin MIC of 0.5 mg/l, the probability of AUC0–24/MIC target attainment was 26%. With an intensified dosing strategy corresponding to 20 mg/kg, target attainment increased to 99%, including 93% with a MIC of 0.5 mg/l.
Conclusions:
Under standard dosing guidelines, few TB meningitis patients would be expected to attain therapeutic rifampicin exposures in CSF when the MIC is ≤0.5 mg/l. Either downward adjustment of the rifampicin MIC breakpoint in the context of TB meningitis, or intensified rifampicin dosing upwards of 20 mg/kg/day, would reflect the likelihood of pharmacodynamic target attainment in CSF.
Keywords: Tuberculosis, Meningitis, Tuberculous meningitis, Pharmacokinetics, Pharmacodynamics
Introduction
Meningitis is the most devastating clinical manifestation of active tuberculosis (TB) disease (Thwaites et al., 2013; Gu et al., 2015), and disproportionately affects children and individuals with impaired immune response (Leeds et al., 2012; Centers for Disease Control and Prevention, 2003). As there have been few trials of antimicrobial treatment for TB meningitis, treatment regimens have largely been extrapolated from principles of treatment of pulmonary TB. Among TB meningitis patients without suspected drug-resistant disease, the first-line anti-TB drugs (rifampicin, isoniazid, pyrazinamide, and ethambutol) are administered according to weight-based dosing guidelines that are identical to those for the treatment of pulmonary TB, with treatment duration extended from 6 months to 9–12 months (Centers for Disease Control and Prevention, 2003).
The rupture of tuberculomas adjacent to the meninges, with release of Mycobacterium tuberculosis bacilli directly into the cerebrospinal fluid (CSF), is essential to the pathogenesis of TB meningitis (Rock et al., 2008). In the treatment of bacterial meningitis, rapid sterilization of the CSF is critical for survival without neurological disability (Nau et al., 1998). One goal of therapy in TB meningitis is to achieve antimicrobial concentrations in the CSF that are sufficient to achieve bacterial sterilization, which depends on the intrinsic susceptibility of the infecting M. tuberculosis strain to the antimicrobial agent, as defined by the minimum inhibitory concentration (MIC). Thus, attaining sufficient drug exposures in the CSF during TB meningitis therapy depends both on the pharmacokinetic variability of the drug and the underlying variability in the susceptibility of the M. tuberculosis strain. Drug susceptibility testing for M. tuberculosis is typically performed by public health laboratories at a single drug concentration (the ‘breakpoint’), which defines the isolate as resistant or sensitive to the tested drug, and identical breakpoints are used regardless of the site of disease.
Historically, the mortality rate of TB meningitis began to decline several decades ago with the introduction of rifampicin into the first-line treatment regimen. The importance of rifampicin has been demonstrated by the significant increase in TB meningitis mortality conferred by rifampicin-resistant strains, despite the pharmacological properties of rifampicin (including protein-binding) that limit its distribution into the CSF (Tho et al., 2012; Vinnard et al., 2011; Donald, 2016). Intensified rifampicin dosing regimens have gained increasing focus in the treatment of TB disease. Yet, two recent clinical trials of increased rifampicin dosing for TB meningitis demonstrated mixed results. Among adult TB meningitis patients in Indonesia, high-dose intravenous rifampicin improved survival, with or without the inclusion of a fluoroquinolone (Ruslami et al., 2013). In contrast, among adult TB meningitis patients in Vietnam, intensified oral rifampicin dosing (approximating 15 mg/kg in weight-based dosing bands) accompanied by a fluoroquinolone failed to demonstrate a mortality benefit (Heemskerk et al., 2016).
The optimal initial treatment regimen for TB meningitis remains uncertain, including the role for intensified rifampicin dosing strategies. The objective of the present study was to evaluate rifampicin pharmacodynamic target attainment in the CSF under standard and intensified weight-based dosing strategies, considering variability in both the pharmacokinetics of rifampicin distribution into the CSF and the variability in rifampicin MIC levels among infecting M. tuberculosis strains. It was hypothesized that some M. tuberculosis strains classified as ‘rifampicin-susceptible’ based on the standard MIC breakpoint of 1.0 mg/l would demonstrate de facto resistance when evaluated from the perspective of pharmacodynamic target attainment in the CSF.
Materials and methods
Semi-mechanistic model of serum rifampicin with auto-induction and saturation of hepatic extraction
Pharmacokinetic model-building was started with a simplified model of first-order oral absorption from the gastrointestinal tract and a one-compartment distributional model. Chirehwa et al. analyzed rifampicin pharmacokinetic data from 61 TB patients, with samples collected on days 1, 8, 15, and 29 of TB treatment (Chirehwa et al., 2015). After first-pass metabolism in the liver, rifampicin transfers between the liver compartment and the central compartment. Hepatic clearance (CLH) is defined as the product of hepatic blood flow (QH) and hepatic extraction (EH):
EH is determined by the unbound fraction of rifampicin (fu) and the intrinsic hepatic clearance of rifampicin (CLint):
To include the saturation effects of hepatic extraction, CLint is defined by the Michaelis–Menten relationship between the maximum hepatic clearance (CLint,max) and the Michaelis constant (Km), according to the relationship:
The intrinsic clearance of rifampicin at any time (t) during treatment is characterized by an exponential relationship between the maximum intrinsic clearance at baseline () and the maximum intrinsic clearance under steady-state conditions (), capturing the effect of rifampicin auto-induction of clearance, according to the relationship:
CSF rifampicin concentrations linked to serum concentrations
CSF concentrations of rifampicin (CCSF) were determined by the rate constant of transfer between central (serum) and CSF compartments (Ke0), and the penetration coefficient (PC) (Savic et al., 2015):
The CSF compartment was joined to the mechanistic model of serum rifampicin concentrations, incorporating both saturation of hepatic extraction and auto-induction of systemic clearance, as shown in Figure 1.
Figure 1.
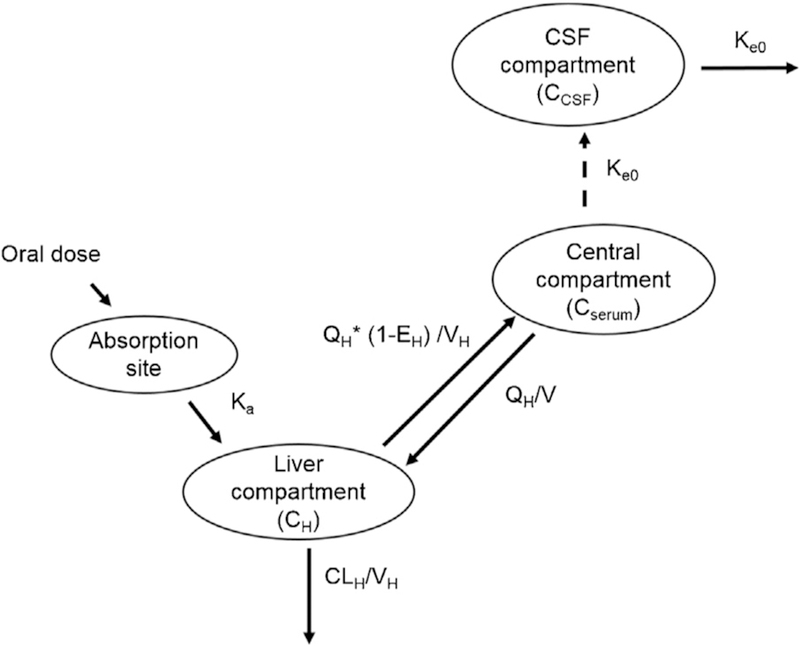
Pharmacokinetic model of rifampicin in the treatment of TB meningitis, including serum and CSF compartments. First-pass metabolism occurs in the liver compartment, followed by distribution into the central compartment. Rifampicin distribution into the CSF is determined by the penetration coefficient and the rate constant between the central and CSF compartments. Abbreviations: QH, hepatic flow rate (liters/hour); VH, volume of hepatic compartment (liters); Ka, absorption rate constant (hour—1); Ke0, rate constant for rifampicin transfer between serum and CSF (hour—1); V, volume of the central compartment (liters); EH, hepatic extraction ratio.
Comparison of model predicted and observed rifampicin serum and CSF concentrations among TB meningitis patients
D’Oliveira (D’Oliveira (1972)) reported a study of 10 TB meningitis patients treated with oral rifampicin (300 mg twice daily, approximating 10 mg/kg per day), with serum and CSF samples taken at various time points during the first 3 days of treatment. This pharmacokinetic dataset included a total of 80 rifampicin CSF concentrations and 80 rifampicin serum concentrations. These intensive pharmacokinetic data were fitted to the model described above, which included saturation of hepatic extraction and auto-induction of systemic clearance. Volume and clearance parameters were estimated for the central compartment, and PC and Ke0 for the CSF compartment, while fixing all other model parameters at literature estimates (Chirehwa et al., 2015). Reasonable levels were set for between-subject variability in the central volume of distribution (20%), systemic clearance (20%), and the penetration coefficient PC (30%). Rifampicin serum concentrations included a combined additive and proportional error model, and CSF concentrations included a proportional error model. Visual predictive checks of the observed data and model-predicted concentrations in both serum and CSF were plotted.
External validation of the rifampicin pharmacokinetic model during TB meningitis treatment using independent external datasets
The rifampicin pharmacokinetic model was validated using rifampicin serum and CSF pharmacokinetic data from two independent cohorts of TB meningitis patients in Indonesia (Dian et al., 2018) and Thailand (Kaojarern et al., 1991), with n = 1000 patients per simulation run. The Indonesian study included 60 adult TB meningitis patients assigned randomly to daily administration of 10 mg/kg, 20 mg/kg, or 30 mg/kg oral rifampicin dosing, with intensive serum sampling (0, 1, 2, 4, 8, and 12 h post-dosing) and sparse CSF sampling (between 3 and 6 h) performed on day 1 and day 10 of TB meningitis treatment. The Thailand study included eight TB meningitis patients administered 10 mg/kg oral rifampicin, with serum and CSF rifampicin concentrations obtained weekly during the first 6 weeks of TB meningitis treatment. In addition, the rifampicin serum and CSF pharmacokinetic model was evaluated using a cohort of seven patients with intraventricular shunt placement and intensive sampling of blood and CSF following rifampicin administration (600 mg intravenously), with CSF samples obtained 0.5, 1, 2, 4, 6, 8, 10, 12, and 14 h post-dosing (Nau et al., 1992). Overall, the performance of the pharmacokinetic model in this validation step was used to guide the selection of dosing regimens and observation time periods for the Monte Carlo simulations.
Monte Carlo simulation of rifampicin CSF concentrations during the first 8 weeks of treatment
Population simulations of the rifampicin CSF pharmacokinetic model were performed with 1000 virtual TB meningitis patients under each scenario, varying the rifampicin dosing regimen and observation time-point during treatment. Body weight was sampled from a normal distribution with a mean of 55 kg and a standard deviation (SD) of 8 kg. Rifampicin dose was first assigned according to World Health Organization (WHO) guidelines by weight bands, approximating 10 mg/kg (World Health Organization, 2010). Next, an intensified weight-based oral rifampicin dosing strategy corresponding to a doubling of rifampicin dose size for each weight stratum (approximating 20 mg/kg) was evaluated, as shown in Table 1.
Table 1.
Standard and intensified weight-based dosing bands for oral rifampicin therapy in TB meningitis treatment.
| Weight band | WHO guidelines | Intensified oral therapy |
|---|---|---|
| <41 kg | 300 mg oral | 600 mg oral |
| 41–55.9 kg | 450 mg oral | 900 mg oral |
| 56–70.9 kg | 600 mg oral | 1200 mg oral |
| ≥71 kg | 750 mg oral | 1500 mg oral |
TB, tuberculosis; WHO, World Health Organization.
Pharmacodynamic target attainment in CSF
A literature review was performed to identify the efficacy thresholds for CSF rifampicin concentrations. For the primary analysis, the area under the rifampicin CSF concentration-versus-time curve during 24 h (AUC0–24)/MIC ratio of 30 was selected as the minimum pharmacodynamic target, which was associated with a 1-log10 fall in colony-forming units for extracellular M. tuberculosis (Jayaram et al., 2003). As a secondary target, the rifampicin CSF AUC0–24/MIC ratio of 297 was examined as the optimal pharmacodynamic target, which corresponded to 50% of the maximal effect of rifampicin efficacy against extracellular M. tuberculosis (EC50). For each population simulation, rifampicin MICs were sampled from a wild-type distribution of M. tuberculosis isolates with rifampicin MICs below the clinical breakpoint of 1.0 mg/l (Schön et al., 2009), thus defined as rifampicin-susceptible, and the AUC0–24/MIC ratio in CSF was determined. For each scenario in the population simulation, the proportion of TB meningitis patients that successfully achieved the pharmacodynamic target across the range of wild-type MIC values was plotted. An additional plasma pharmacokinetic target predictive of a favorable outcome, independent of rifampicin MIC, identified in a cohort of adult Indonesian TB meningitis patients treated with rifampicin according to standard (10 mg/kg oral) or high-dose (13.5 mg/kg intravenous) dosing regimens, was examined (te Brake et al., 2015). All pharmacokinetic modeling and simulation was performed using Phoenix NLME 7.0 (Certara USA, Inc., Princeton, NJ, USA), and figures were generated using GraphPad Prism 6 (GraphPad Software, La Jolla, CA, USA). Rifampicin pharmacokinetic data were extracted from published figures using WebPlot-Digitizer V3.9 (https://automeris.io/WebPlotDigitizer/).
Results
Estimation of pharmacokinetic parameters for the CSF compartment
Using the intensive serum and CSF pharmacokinetic data reported by D’Oliveira et al., key pharmacokinetic parameters for serum () and CSF (PC, Ke0) were estimated, as shown in Table 2, while fixing other pharmacokinetic parameters at previously reported population values (Chirehwa et al., 2015). The visual predictive checks for the pharmacokinetic model are shown in Figure 2, including comparisons of the serum (Figure 2a) and CSF (Figure 2b) predicted concentrations with the observed values in the estimation cohort. Given the sparse nature of the pharmacokinetic data used in model estimation, it was not possible to include covariate effects on the pharmacokinetic parameters, aside from allometric scaling of weight and clearance parameters.
Table 2.
Rifampicin pharmacokinetic model of serum and CSF concentrations during TB meningitis treatment.
| Pharmacokinetic parameter | Typical value for a 55 kg adult |
|---|---|
| Volume of the central compartmenta,c (V, liters) | 51.1 |
| Baseline value of intrinsic maximum clearanceb,c (, liters/hour) | 53.9 |
| Steady-state value of intrinsic maximum clearanceb,c (, liters/hour) | 149.5 |
| Hepatic flow rateb (QH, liters/hour) | 50 |
| Volume of hepatic compartmenta (VH, liters) | 1 |
| Absorption rate constant (Ka, hour−1) | 1.15 |
| Bioavailability (F, %) | 100 |
| Fraction of unbound rifampicin (fu) | 0.2 |
| Half-life of the rifampicin clearance induction process (t1/2, days) | 4.5 |
| Michaelis–Menten constant for the saturation of rifampicin hepatic clearance (Km, mg/l) | 3.35 |
| Rate constant for rifampicin transfer between serum and CSF (Ke0, hour−1)c | 0.081 |
| Penetration coefficientc | 0.18 |
| Additive error in serum | 0.2 |
| Proportional error in serum | 0.5 |
| Proportional error in CSF | 0.5 |
| Between-subject variability in clearance (%) | 20% |
| Between-subject variability in volume of central compartment (%) | 20% |
| Between-subject variability in the partition coefficient (%) | 30% |
CSF, cerebrospinal fluid.
Allometrically scaled according to (weight/55 kg).
Allometrically scaled according to (weight/55 kg)0.75.
Estimated from rifampicin pharmacokinetic data in TB meningitis patients (D’Oliveira, 1972).
Figure 2.
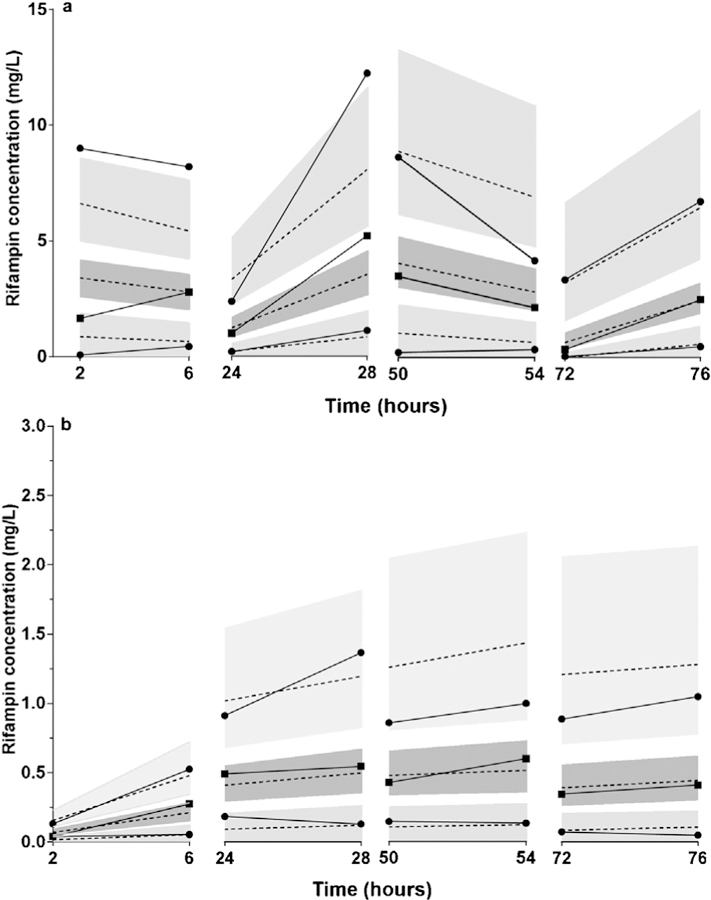
Visual predictive check of the rifampicin pharmacokinetic model. Rifampicin was administered as 300 mg orally every 12 h. (a) Model-predicted and observed serum rifampicin concentrations during the first 3 days of TB meningitis treatment. Solid lines represent the 10th, 50th, and 90th percentiles of observed serum rifampicin concentrations; dashed lines represent the 10th, 50th, and 90th percentiles of predicted serum rifampicin concentrations. Shaded regions correspond to the 90% confidence intervals for each percentile. Squares represent the median observed serum rifampicin concentrations; circles represent the 10th and 90th percentiles of observed serum rifampicin concentrations. (b) Model-predicted and observed CSF rifampicin concentrations during the first 3 days of TB meningitis treatment. Solid lines represent the 10th, 50th, and 90th percentiles of observed CSF rifampicin concentrations; dashed lines represent the 10th, 50th, and 90th percentiles of predicted CSF rifampicin concentrations. Shaded regions correspond to the 90% confidence intervals for each percentile. Squares represent the median observed CSF rifampicin concentrations; circles represent the 10th and 90th percentiles of observed CSF rifampicin concentrations.
Validation of the rifampicin pharmacokinetic model of serum and CSF concentrations
The comparisons of rifampicin concentrations predicted by the pharmacokinetic model with the observed distributions are shown in Figure 3, including the serum rifampicin AUC0–24 at each dose size (Figure 3a) and sparse CSF samples obtained between 3 and 6 h post-dosing (Figure 3b). As shown, substantial overlap was observed between model-predicted and observed serum AUC0–24 values across all dose groups, at each time-point (days 1 and 10). While the 10 mg/kg dosing group demonstrated agreement between model-predicted and observed CSF concentrations at both time-points (days 1 and 10), the 20 mg/kg dosing group demonstrated agreement only on day 1 of treatment. In the 30 mg/ kg dose group, the pharmacokinetic model over-predicted CSF concentrations, most notably on day 10 of treatment.
Figure 3.
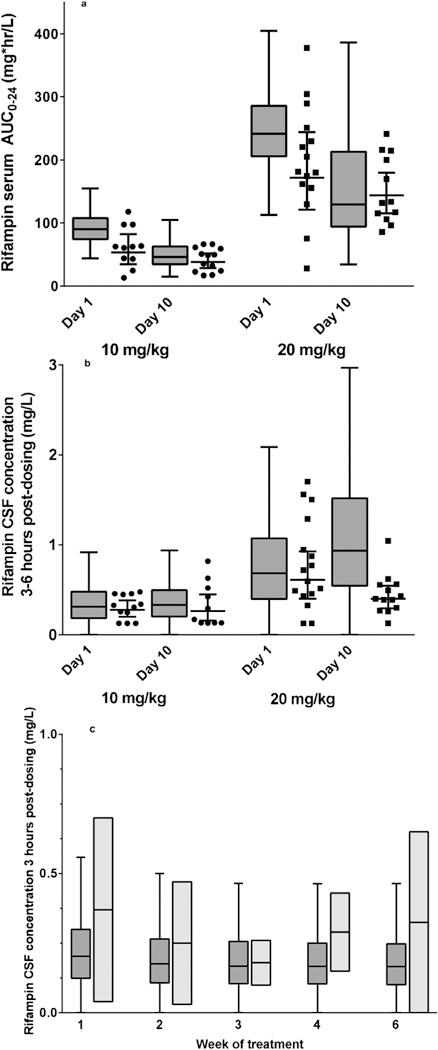
External validation of the rifampicin pharmacokinetic model among adult TB meningitis patients in Indonesia and Thailand. (a) Model-predicted and observed rifampicin serum AUC0–24 on day 1 of treatment of adult TB meningitis patients in Indonesia (Dian et al., 2018). Tukey box plots of predicted rifampicin concentrations, with boxes corresponding to median and interquartile range (IQR) and whiskers corresponding to T ±.5 × IQR. Circles represent the observed rifampicin serum AUC0–24 at 10 mg/kg dosing; squares represent the observed rifampicin serum AUC0–24 at 20 mg/kg dosing. Line and whiskers correspond to the geometric mean and associated 95% confidence interval of observed rifampicin serum AUC0–24 at each dose size and time-point. (b) Model-predicted and observed rifampicin CSF concentrations on day 1 of treatment, between 3 h and 9 h post dosing (Dian et al., 2018). Tukey box plots of predicted rifampicin concentrations, with boxes corresponding to the median and interquartile range (IQR) and whiskers corresponding to ± 1.5 × IQR. Circles represent the observed rifampicin CSF concentrations at 10 mg/kg dosing; squares represent the observed rifampicin concentrations at 20 mg/kg dosing. Line and whiskers correspond to the geometric mean and associated 95% confidence interval of observed rifampicin CSF concentrations at each dose size. (c) Model-predicted and observed rifampicin CSF concentrations during the first 6 weeks of TB meningitis treatment (Kaojarern et al., 1991). Tukey box plots of predicted rifampicin concentrations, with dark grey shaded boxes corresponding to the median and interquartile range (IQR) and whiskers corresponding to ± 1.5 × IQR; light grey shaded rectangles correspond to the range of observed CSF rifampicin concentrations between 3 h and 6 h post-dosing, and the line corresponds to the geometric mean of the observations at each time-point.
Next, the rifampicin CSF pharmacokinetic model was evaluated in an additional cohort of eight Thai TB meningitis patients (Kaojarern et al., 1991) with sparse sampling of rifampicin CSF concentrations obtained 3 h post-dosing during the first 6 weeks of treatment (600 mg dose size, patient weight not reported). A pharmacokinetic simulation of CSF rifampicin concentrations during the first 6 weeks of treatment with 10 mg/kg dosing was performed, and the 3–6 h CSF concentrations were compared between model-predicted and observed distributions (Figure 3c). As shown, overlap was observed between model-predicted and observed rifampicin CSF concentrations throughout the 6-week period of observation.
Finally, the rifampicin CSF pharmacokinetic model was evaluated in a cohort of seven patients with ventricular shunt placement and intensive rifampicin sampling from serum and CSF following 600 mg dose sizes (Figure 4). Agreement with model-predicted and observed rifampicin concentrations was observed in both serum and CSF over a period of 20 h following intravenous dosing in this cohort.
Figure 4.
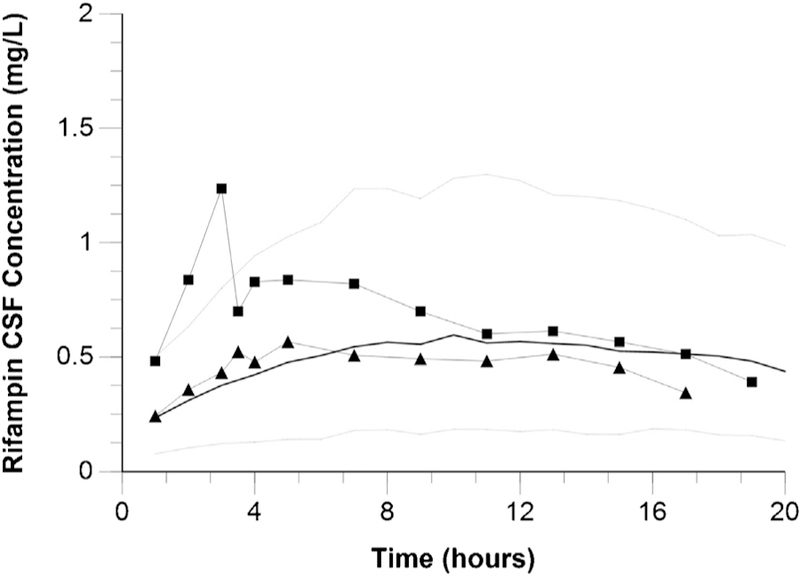
Model-predicted and observed rifampicin CSF concentrations during 24 h following intravenous administration to seven patients post intraventricular shunt placement (Kaojarern et al., 1991). Squares represent the highest observed rifampicin CSF concentrations among study subjects; triangles represent the lowest observed rifampicin CSF concentrations among study subjects. Black lines represent the median predicted rifampicin CSF concentrations; grey dotted lines represent the 10th and 90th percentiles of predicted rifampicin CSF concentrations.
Comparison of target attainment between rifampicin dosing strategies
Based on the performance of the pharmacokinetic model in the evaluation step, Monte Carlo simulations of standard and intensified rifampicin dosing strategies, corresponding to 10 mg/ kg and 20 mg/kg, administered as weight-based dosing bands, were performed. Although clinical data support rifampicin dosing upwards of 20 mg/kg, simulations of target attainment beyond 20 mg/kg were not performed because of the poor agreement of predicted and observed CSF concentrations in model evaluation at the 30 mg/kg dose size. Along with rifampicin population pharmacokinetic variability, rifampicin MIC variability in the infecting M. tuberculosis strain was incorporated, with each virtual patient assigned an infecting M. tuberculosis strain with rifampicin MIC randomly sampled from the distribution observed among rifampicin-susceptible strains (i.e., rifampicin MIC below the breakpoint of 1.0 mg/l). Under each scenario, a population (n = 1000) of TB meningitis patients was simulated, and oral rifampicin was administered according to standard and alternative weight-based dosing strategies (Table 1). In these simulations, we examined pharmacodynamic target attainment with the rifampicin dose size of 10 mg/kg and 20 mg/kg, with a rifampicin AUC0–24/ MIC ratio of 30 in the primary analysis, and a higher rifampicin AUC0–24/MIC ratio of 297 in the secondary analysis (World Health Organization, 2010).
The pharmacodynamic target attainment probabilities on the first day of treatment, according to M. tuberculosis rifampicin MIC levels, are shown in Figure 5, with separate plots for the lower AUC0–24/MIC target ratio of 30 (Figure 5a) and the higher AUC0–24/ MIC target ratio of 297 (Figure 5b). Under standard WHO guidelines using weight-based dosing bands that approximate 10 mg/kg, an 84% overall probability of attaining rifampicin AUC0– 24/MIC ratio of 30 was observed, with a probability of 24% at the MIC level of 0.5 mg/l, and a 1% overall probability of attaining an AUC0–24/MIC ratio of 297. Next, the performance of an intensified weight-based oral rifampicin dosing strategy was examined, using double dose sizes for each WHO weight-based dosing band, beginning on day 1 of treatment (corresponding to approximately 20 mg/kg). The overall probability of pharmacodynamic target attainment in CSF was 99% for the lower target (AUC0–24/MIC ratio of 30) and 25% for the higher target (AUC0–24/MIC ratio of 297). Yet even under the intensified dosing strategy, there were no virtual patients that attained this higher AUC0–24/MIC target at the M. tuberculosis MIC level of 0.5 mg/l (Figure 5b).
Figure 5.
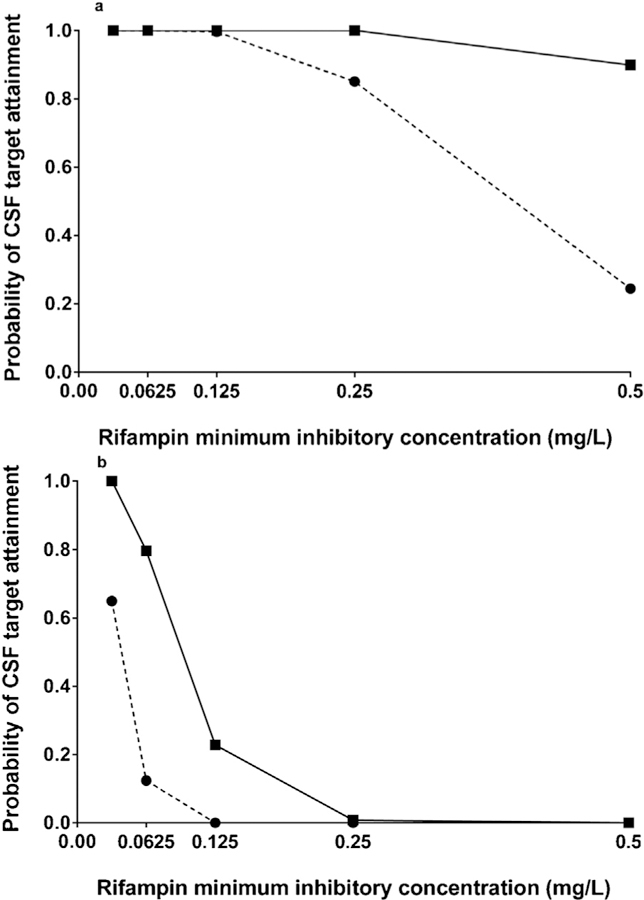
Monte Carlo simulation of CSF rifampicin target attainment according to rifampicin MIC level, comparing standard (10 mg/kg) and intensified (20 mg/kg) dosing strategies. (a) Rifampicin CSF AUC0–24/MIC ratio of 30, corresponding to a 1-log10 fall in colony-forming units for extracellular Mycobacterium tuberculosis. The dashed line and circles represent the standard weight-based dosing bands, approximating 10 mg/kg. The solid line and squares represent the intensive weight-based dosing bands, approximating 20 mg/kg. (b) Optimal rifampicin CSF AUC0–24/ MIC ratio of 297, corresponding to rifampicin EC50 against extracellular M. tuberculosis. The dashed line and circles represent the standard weight-based dosing bands, approximating 10 mg/kg. The solid line and squares represent the intensive weight-based dosing bands, approximating 20 mg/kg.
Finally, we determined the probabilities of attaining a plasma pharmacokinetic target (AUC0–6 of 70 mg*h/l) linked to a favorable outcome among Indonesian TB meningitis patients treated with rifampicin under standard (10 mg/kg orally administered) and high-dose (13.5 mg/kg intravenously administered) regimens (te Brake et al., 2015). On the first day of treatment, it was found that 2% of 1000 virtual patients administered rifampicin orally according to WHO guidelines (approximating 10 mg/kg) achieved this threshold (median AUC0–6 36.2 mg*h/l, 90% confidence interval (CI) 21.4–54.8 mg*h/l), compared with 59% of patients administered rifampicin according to an intensified regimen that approximated 20 mg/kg (median AUC0–6 76.7 mg*h/l, 90% CI 45.2–116.9 mg*h/l). By day 10 of treatment, the proportion of patients achieving the plasma target under the intensified regimen had decreased to 42% (median AUC0–6 64.9 mg*h/l, 90% CI 35.9–105.2 mg*hr/l), reflecting the effects of rifampicin auto-induction of hepatic clearance.
Discussion
There is a critical need to identify interventions that can reduce early mortality in TB meningitis (Vinnard et al., 2017). In this modeling and simulation study, it was found that doubling the rifampicin dose sizes for each WHO weight band, approximating an increase from 10 mg/kg to 20 mg/kg, led to marked improvements in the probability of pharmacodynamic target attainment during TB meningitis treatment, whether those targets were defined according to plasma rifampicin AUC0–6 levels, or CSF rifampicin AUC0–24/MIC ratios.
It has been suggested that the failure of a recent randomized clinical trial of intensified rifampicin dosing among Vietnamese TB meningitis patients could be attributed to an insufficiently intensive dosing regimen, with an average oral daily dose size of 15 mg/kg in the intensive treatment arm (Boeree et al., 2016). In this context, it is noteworthy that pediatric outcomes of TB meningitis are often better than reports from adult patients, and the higher rifampicin dose that is used in treating children with TB meningitis (up to 20 mg/kg) may contribute to this difference, although this practice is not universally followed (van Well et al., 2009). Ongoing clinical trials of intensive rifampicin dosing in TB meningitis, including intensive pharmacokinetic sampling, are also essential to refine our understanding of the ‘minimal’ and ‘optimal’ pharmacodynamic targets in the treatment of TB meningitis.
There is increasing recognition that the susceptibility break-point for rifampicin during pulmonary TB treatment should reflect the probability of pharmacodynamic target attainment (Gumbo, 2010). In this study, it was found that the MIC value of 0.5 mg/l served as a de facto breakpoint under standard dosing guidelines (approximating 10 mg/kg daily dosing). We propose that the definition of rifampicin susceptibility or resistance (based on the critical MIC threshold, or ‘breakpoint’) during TB meningitis treatment should be informed by the likelihood of pharmacodynamic target attainment in the CSF. Notably, this approach is used for the treatment of meningitis caused by Streptococcus pneumoniae, where the penicillin breakpoints published by the Clinical and Laboratory Standards Institute (CLSI) are lower for meningeal isolates compared with non-meningeal isolates (Weinstein et al., 2009). Applied to TB meningitis, this paradigm shift would recognize that sophisticated clinical assessments such as CSF pharmacokinetic monitoring or phenotypic MIC values are infeasible in many high-burden TB settings with limited resources. The use of a single, lower MIC breakpoint for M. tuberculosis in the context of TB meningitis, redefining M. tuberculosis isolates with a rifampicin MIC of 0.5 mg/l as ‘resistant’ when isolated from the CSF, would address the reality of limited rifampicin CSF penetration. Alternatively, more aggressive adoption by TB control programs of intensified rifampicin dosing strategies, at 20 mg/kg and beyond, for the treatment of TB meningitis would provide improvement in target attainment among patients at this critical MIC value of 0.5 mg/l, thereby limiting the use of more costly and toxic second-line treatment regimens.
One surprising finding was the failure of the pharmacokinetic model to predict CSF rifampicin exposures at 20 mg/kg and 30 mg/ kg at 10 days of treatment, with over-prediction of rifampicin CSF concentrations despite agreement between predicted and observed rifampicin serum concentrations at these dose sizes. One explanation for this finding would be that rifampicin distribution into the CSF is determined by active transport mechanisms along the choroid plexus epithelium, the anatomical site of the blood–CSF barrier, which could become saturated at high substrate concentrations (Sanchez-Covarrubias et al., 2014). Alternatively, increasing rifampicin exposures may induce the expression of membrane efflux transporters that remove substrate from CSF into blood (such as P-glycoprotein) (Spector, 2010). In either scenario, involvement of host drug transporters along the blood–CSF barrier would explain the observation that non-linear dose–exposure relationships for rifampicin were observed in serum but not CSF among TB meningitis patients (Dian et al., 2018). Physiologically based pharmacokinetic modeling may provide one avenue towards dissecting these potential mechanisms limiting CSF rifampicin exposures under intensive dosing strategies (Cresswell et al., 2018).
This pharmacokinetic modeling and simulation study had several limitations. There is considerable uncertainty regarding the relative contributions of M. tuberculosis killing and the modulation of the host inflammatory response in determining the clinical response during TB meningitis treatment (Thwaites et al., 2013). Furthermore, we evaluated MIC-based pharmacodynamic targets in CSF (rifampicin CSF AUC0–24/MIC ratios of 30 and 297) that were not established in the context of TB meningitis. Although a negligible role of protein-binding in CSF was assumed, the blood–CSF barrier disruption that is characteristic of TB meningitis may lead to non-negligible contributions of protein-bound rifampicin, which requires further investigation (Shen et al., 2004; Tang et al., 2014). Finally, the oral bioavailability of rifampicin is influenced by a single nucleotide polymorphism in a host gene, SLCO1B1 (Weiner et al., 2010; Kwara et al., 2014), which encodes a membrane transporter mediating biliary uptake and hepatic clearance, and limited data regarding the distribution of SLCO1B1 alleles in populations of TB meningitis patients prevented the inclusion of this variability in the pharmacokinetic model.
In summary, when rifampicin is administered according to standard weight-based dosing guidelines, few adult TB meningitis patients would be expected to achieve therapeutic rifampicin exposures in CSF when the rifampicin MIC is ≥0.5 mg/l. Until high-dose strategies for rifampicin become widely adopted in the treatment of TB meningitis, particularly during the early phase, a downward adjustment of the rifampicin MIC breakpoint, from 1.0 mg/l to 0.5 mg/l, is supported by pharmacokinetic modeling and simulation of CSF target attainment probabilities. The optimal approach for TB meningitis patients with a rifampicin MIC of 0.5 mg/l – whether to add second-line drugs and/or intensify rifampicin dosing – would be clarified with formal pharmacokinetic/pharmacodynamics studies of CSF rifampicin exposures and clinical outcomes.
Funding
This work was supported by the National Institutes of Health (K23AI102639, R01AI137080 to CV) NICHD/NIH1R01HD074944–01, NINDS/NIH K23NS105935–01 to KTT. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Corresponding Editor: Eskild Petersen, Aarhus, Denmark
Ethical approval
Human subjects research was not performed. Animal research was not performed.
Conflict of interest
All authors report no conflicts of interest.
References
- Boeree MJ, Gillespie SH, Hoelscher M, PanACEA core team. Therapy for tuberculous meningitis. N Engl J Med 2016;374:2187–8. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. Treatment of tuberculosis, American Thoracic Society, CDC, and Infectious Diseases Society of America. MMWR 2003;52: RR-11. [Google Scholar]
- Chirehwa MT, Rustomjee R, Mthiyane T, Onyebujoh P, Smth P, McIlleron H, et al. Model-based evaluation of higher doses of rifampin using a semimechanistic model incorporating autoinduction and saturation of heaptic extraction. Antimicrob Agents Chemother 2015;60:487–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cresswell FV, Te Brake L, Atherton R, Ruslami R, Dooley KE, Aarnoutse R, et al. Intensified antibiotic treatment of tuberculous meningitis. Expert Rev Clin Pharmacol 2018; (November), doi: 10.1080/17512433.2019.1552831. [DOI] [PubMed]
- D’Oliveira JJG. Cerebrospinal fluid concentrations of rifampin in meningeal tuberculosis. Am Rev Respir Dis 1972;106:432–7. [DOI] [PubMed] [Google Scholar]
- Dian S, Yunivita V, Ganiem AR, Pramaesya T, Chaidir L, Wahyudi K, et al. Double-blind, randomized, placebo-controlled phase ii dose-finding study to evaluate high-dose rifampin for tuberculous meningitis. Antimicrob Agents Chemother 2018;62(November (12)), doi: 10.1128/AAC.01014-18 pii: e01014–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald PR. Chemotherapy for tuberculous meningitis. N Engl J Med 2016;374:179–81. [DOI] [PubMed] [Google Scholar]
- Gu J, Xiao H, Wu F, Ge Y, Ma J, Sun W. Prognostic factors of tuberculous meningitis: a single-center study. Int J Clin Exp Med 2015;8:4487–93. [PMC free article] [PubMed] [Google Scholar]
- Gumbo T New susceptibility breakpoints for first-line antituberculosis drugs based on antimicrobial pharmacokinetic/pharmacodynamic science and population pharmacokinetic variability. Antimicrob Agents Chemother 2010;54:1484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk AD, Bang ND, Mai NT, Chau TT, Phu NH, Loc PP, et al. Intensified antituberculosis therapy in adults with tuberculous meningitis. N Engl J Med 2016;374:124–34. [DOI] [PubMed] [Google Scholar]
- Jayaram R, Gaonkar S, Kaur P, Suresh BL, Mahesh BN, Jayashree R, et al. Pharmacokinetics-pharmacodynamics of rifampin in an aerosol infection model of tuberculosis. Antimicrob Agents Chemother 2003;47:2118–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaojarern S, Supmonchai K, Phuapradit P, Mokkhavesa C, Krittiyanunt S. Effect of steroids on cerebrospinal fluid penetration of antituberculous drugs in tuberculous meningitis. Clin Pharmacol Ther 1991;49:6–12. [DOI] [PubMed] [Google Scholar]
- Kwara A, Cao L, Yang H, Poethke P, Kurpewski J, Tashima KT, et al. Factors associated with variability in rifampin plasma pharmacokinetics and the relationship between rifampin concentrations and the induction of efavirenz clearance. Pharmacother 2014;34:265–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeds IL, Magee MJ, Kurbatova EV, del Rio C, Blumberg HM, Leonard MK, et al. Site of extrapulmonary tuberculosis is associated with HIV infection. Clin Infect Dis 2012;55:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nau R, Prange HW, Menk S, Kolenda H, Visser K, Seydel JK. Penetration of rifampicin into the cerebrospinal fluid of adults with uninflamed meninges. J Antimicrob Chemother 1992;29:719–24. [DOI] [PubMed] [Google Scholar]
- Nau R, Sörgel F, Prange HW. Pharmacokineticoptimisation of the treatment of bacterial central nervous system infections. Clin Pharmacokinet 1998;35:223–46. [DOI] [PubMed] [Google Scholar]
- Rock RB, Olin M, Baker CA, Molitor TW, Peterson PK. Central nervous system tuberculosis: pathogenesis and clinical aspects. Clin Microbiol Rev 2008;21:243–61 pmid:18400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruslami R, Ganiem AR, Dian S, Apriani L, Achmad TH, van der Ven AJ, et al. Intensified regimen containing rifampicin and moxifloxacin for tuberculous meningitis: an open-label, randomised controlled phase 2 trial. Lancet Infect Dis 2013;13:27–35. [DOI] [PubMed] [Google Scholar]
- Sanchez-Covarrubias L, Slosky LM, Thompson BJ, Davis TP, Ronaldson PT. Transporters at CNS barrier sites: obstacles or opportunities for drug delivery?. Curr Pharm Des 2014;20:1422–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savic RM, Ruslami R, Hibma JE, Hesseling A, Ramachandran G, Ganiem AR, et al. Pediatric tuberculous meningitis: model-based approach to determining optimal doses of the anti-tuberculosis drugs rifampin and levofloxacin for children. Clin Pharmacol Ther 2015;98:622–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schön T, Juréen P, Giske CG, Chryssanthou E, Sturegård E, Werngren J, et al. Evaluation of wild-type MIC distributions as a tool for determination of clinical breakpoints for Mycobacterium tuberculosis. J Antimicrob Chemother 2009;64:786–93. [DOI] [PubMed] [Google Scholar]
- Shen DD, Artru AA, Adkinson KA. Principles and applicability of CSF sampling for the assessment of CNS drug delivery and pharmacodynamics. Adv Drug Deliv Rev 2004;56:1825–57. [DOI] [PubMed] [Google Scholar]
- Spector R Nature and consequences of mammalian brain and CSF efflux transporters: four decades of progress. J Neurochem 2010;112:13–23. [DOI] [PubMed] [Google Scholar]
- Tang J, An Y, Liao Y, Li Y, Li L, Wang L. The association between blood-cerebrospinal fluid barrier dysfunction and the therapeutic effect in tuberculous meningitis patients. Eur Neurol 2014;71:331–6. [DOI] [PubMed] [Google Scholar]
- te Brake L, Dian S, Ganiem AR, Ruesen C, Burger D, Donders R, et al. Pharmacokinetic/pharmacodynamics analysis of an intensified regimen containing rifampicin and moxifloxacin for tuberculous meningitis. Int J Antimicrob Agents 2015;45:496–503. [DOI] [PubMed] [Google Scholar]
- Tho DQ, Török ME, Yen NT, Bang ND, Lan NT, Kiet VS, et al. Influence of antituberculosis drug resistance and Mycobacterium tuberculosis lineage on outcome in HIV-associated tuberculous meningitis. Antimicrob Agents Chemother 2012;56:3074–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thwaites GE, van Toorn R, Schoeman J. Tuberculous meningitis: more questions, still too few answers. Lancet Neurol 2013;12:999–1010. [DOI] [PubMed] [Google Scholar]
- van Well GT, Paes BF, Terwee CB, Springer P, Roord JJ, Donald PR, et al. Twenty years of pediatric tuberculous meningitis: a retrospective cohort study in the western cape of South Africa. Pediatrics 2009;123:e1–8. [DOI] [PubMed] [Google Scholar]
- Vinnard C, Winston CA, Wileyto EP, MacGregor RR, Bisson GP. Multidrug resistant tuberculous meningitis in the United States, 1993–2005. J Infect 2011;63:240–2. [DOI] [PubMed] [Google Scholar]
- Vinnard C, King L, Munsiff S, Crossa A, Iwata K, Pasipanodya J, et al. The long term mortality of tuberculosis meningitis in New York City. Clin Infect Dis 2017;64:401–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner M, Peloquin C, Burman W, Luo CC, Engle M, Prihoda TJ, et al. Effects of tuberculosis, race, and human gene SLCO1B1 polymorphisms on rifampin concentrations. Antimicrob Agents Chemother 2010;54:4192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein MP, Klugman KP, Jones RN. Rationale for revised penicillin susceptibility breakpoints versus Streptococcus pneumoniae: coping with antimicrobial susceptibility in an era of resistance. Clin Infect Dis 2009;48:1596–600. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Guidelines for treatment of tuberculosis 2010. … [Accessed 7 December 2016] http://www.who.int/tb/publications/2010/9789241547833/en/.