

Abstract
Substance P is thought to play an essential role in several forms of supraspinally mediated analgesia. The actions of substance P on synaptic transmission within descending analgesic pathways, however, are largely unknown. Here, we used whole-cell recordings from rat midbrain slices to examine the effects of substance P on GABAergic and glutamatergic transmission within the periaqueductal gray (PAG), a key component of a descending analgesic pathway that projects via the rostral ventromedial medulla (RVM) to the spinal cord dorsal horn. We found that substance P reversibly decreased the amplitude and increased the paired-pulse ratio of evoked IPSCs recorded from identified PAG-RVM projection neurons and from unidentified PAG neurons. Substance P had no effect on miniature IPSCs, implying an indirect mode of action. The effects of substance P were abolished by metabotropic glutamate type 5 and cannabinoid CB1 receptor antagonists, but unaltered by NMDA, GABAB, μ,δ-opioid, adenosine A1, and 5HT1A receptor antagonists. Consistent with a role for endogenous glutamate in this process, substance P increased the frequency of action potential-dependent spontaneous EPSCs. Moreover, the effect of substance P on evoked IPSCs was mimicked and occluded by a glutamate transport inhibitor. Finally, these effects were dependent on postsynaptic G-protein activation and diacylglycerol lipase activity, suggesting the requirement for retrograde signaling by the endocannabinoid 2-arachidonoylglycerol. Thus, substance P may facilitate descending analgesia in part by enhancing glutamate-mediated excitation and endocannabinoid-mediated disinhibition of PAG-RVM projection neurons.
Introduction
Substance P plays an important role in a number of sensory, autonomic, and cognitive functions, primarily through its interaction with the neurokinin-1 (NK1) receptor. It has long been proposed that substance P contributes to nociceptive signaling within the spinal cord dorsal horn, where it is released by nociceptive primary afferent fibers and excites superficial NK1 receptor-expressing projection neurons (Morris et al., 2004). Conversely, supraspinal administration of substance P produces analgesia (Stewart et al., 1976; Frederickson et al., 1978; Malick and Goldstein, 1978; Mohrland and Gebhart, 1979; Improta and Broccardo, 2000). Indeed, behavioral studies of NK1 receptor-knock-out mice indicate that these receptors are critical for the full expression of several supraspinally mediated phenomena, including stress-induced analgesia and diffuse noxious inhibitory controls (De Felipe et al., 1998; Bester et al., 2001).
NK1 receptors are present in a number of brain regions involved in the processing of nociceptive information, including the midbrain periaqueductal gray (PAG) (Nakaya et al., 1994; Barbaresi, 1998; Commons and Valentino, 2002). The PAG is a major site of the analgesic actions of μ-opioids and cannabinoids and forms part of a descending analgesic pathway that projects via the rostral ventromedial medulla (RVM) to modulate nociceptive transmission within the spinal cord dorsal horn (Fields et al., 2006). Microinjection studies in rodents have shown that substance P produces potent analgesia when administered directly into the PAG (Stewart et al., 1976; Frederickson et al., 1978; Malick and Goldstein, 1978; Mohrland and Gebhart, 1979; Rosén et al., 2004). Furthermore, substance P is released within the PAG in response to noxious peripheral stimulation (Xin et al., 1997) and behavioral stress (Rosén et al., 1992), suggesting that endogenous substance P may contribute to PAG-mediated descending analgesia. Consistent with this notion, selective blockade of NK1 receptors in the PAG abolishes antinociception induced by stimulation of the lateral hypothalamus (Holden et al., 2009).
Exogenously applied substance P enhances the action potential frequency of PAG neurons (Ogawa et al., 1992) by activating a nonselective cation conductance and reducing a resting G-protein-coupled inwardly rectifying K+ conductance (Drew et al., 2005). However, the effects of substance P on synaptic transmission within the PAG have not previously been investigated. Here, we show that substance P suppresses GABAergic transmission onto identified PAG-RVM projection neurons by driving action potential-dependent activation of group I metabotropic glutamate receptors (mGluRs) and retrograde endocannabinoid signaling.
Materials and Methods
Retrograde tracer injections.
In some experiments, rhodamine-conjugated latex microspheres (Invitrogen) were microinjected into the RVM of 19- to 28-d-old male and female Sprague Dawley rats (n = 17), as described previously (Osborne et al., 1996). The animals were anesthetized (1–2% isoflurane in O2) and placed in a stereotaxic frame, and the dura was exposed by trephination. A glass micropipette (tip diameter 20–50 μm) was advanced into the RVM and 10 injections (9.2 nl each) were made using a calibrated injection system (Drummond Nanoject). The incision was closed and a topical antibiotic was applied. Animals were allowed to recover from anesthesia in a warmed box before being returned to holding cages. Animals were used for in vitro experiments ≥3 d postsurgery.
Slice preparation.
Rats (12–31 d old) were deeply anesthetized with isoflurane and decapitated, in accordance with protocols approved by the institutional Animal Care and Ethics Committee. Transverse midbrain slices (300 μm) containing PAG were cut using a vibratome (VT1000S, Leica Microsystems) in ice-cold artificial CSF (ACSF), as described previously (Drew et al., 2008). In animals that had received tracer injections, 500-μm-thick RVM slices were also cut and fixed (see below). PAG slices were maintained at 34°C in a submerged chamber containing ACSF equilibrated with 95% O2 and 5% CO2. The slices were then individually transferred to a chamber and superfused continuously (1.8 ml · min−1) with ACSF (34°C) of composition (in mm): 126 NaCl, 2.5 KCl, 1.4 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 11 glucose, and 25 NaHCO3.
Electrophysiology.
PAG neurons were visualized using infrared Nomarski or Dodt contrast gradient optics on an upright microscope (BX51; Olympus). Whole-cell voltage-clamp recordings (−70 mV) were made from lateral and ventrolateral PAG neurons using an Axopatch 200B (Molecular Devices) with an internal solution containing (in mm): 140 CsCl, 10 EGTA, 5 HEPES, 2 CaCl2, 2 MgATP, and 3 QX-314 (pH 7.3; osmolarity, 270–290 mOsm/L), plus 0.05% biocytin in some of the experiments. Series resistance (<20 MΩ) was compensated by 80% and continuously monitored during experiments. Liquid junction potentials of −4 mV were corrected. Electrically evoked IPSCs were elicited in neurons via bipolar tungsten-stimulating electrodes (tip separation 100 μm) placed 50–200 μm from the recording electrode (rate, 0.083 s−1; stimuli: 1–50 V, 20–400 μs), in the presence of the non-NMDA glutamate receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione disodium (CNQX) (5 μm) and the glycine receptor antagonist strychnine (5 μm). Paired-pulse ratio was calculated as the average amplitude of the second evoked IPSC divided by the average amplitude of the first evoked IPSC, measured over a period of at least 2 min during each drug or control condition. Spontaneous miniature IPSCs were recorded in the presence of CNQX (5 μm), strychnine (5 μm), and tetrodotoxin (TTX) (300 nm). IPSCs were abolished by the GABAA receptor antagonists SR95531 (10 μm, n = 3) and picrotoxin (100 μm, n = 7). Spontaneous EPSCs were recorded in the presence of picrotoxin (100 μm) and strychnine (5 μm), whereas miniature EPSCs were isolated by further addition of TTX (300 nm).
IPSCs and EPSCs were filtered (1–2 kHz low-pass filter) and sampled (5–10 kHz) for on-line and later off-line analysis (Axograph X; Axograph Scientific Software). Spontaneous IPSCs and EPSCs were sampled in 4–5 s epochs every 6 s for analysis, and PSCs above a preset threshold (4–5 SDs above baseline noise) were automatically detected by a sliding template algorithm, then manually checked off-line. All numerical data are expressed as mean ± SEM. Statistical comparisons of drug effects were made using paired Student's t test, and comparisons between treatment groups with an unpaired Student's t test or one-way ANOVA using Dunnett's correction for post hoc comparisons. Differences were considered significant if p < 0.05.
Immunohistochemistry.
Immediately after recording, slices containing biocytin-filled cells were fixed overnight in a phosphate-buffered paraformaldehyde solution. The slices were then washed six to eight times and stored in a phosphate buffer. Biocytin-filled cells were visualized by incubation with Cy5-labeled streptavidin (Jackson ImmunoResearch). Slices were mounted and coverslipped, and digital images of filled cells were collected using a Zeiss AX10 microscope with an AxioCam MRm camera. RVM sites at which the fluorescent microspheres were injected were examined using conventional fluorescence microscopy, and reconstructions of the extents of injection sites were made from digital images.
Drug solutions.
Baclofen, biocytin, CNQX, picrotoxin, strychnine hydrochloride and (−)-tetrahydrolipstatin were obtained from Sigma; substance P and methionine-enkephalin from Auspep; 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide (AM251) from Cayman Chemical; 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-4-morpholinyl-1H-pyrazole-3-carboxamide (AM281), naloxone hydrochloride, dl-2-amino-5-phosphonovaleric acid (dl-AP5), N-acetyl-l-tryptophan 3,5-bis(trifluoromethyl)benzyl ester (L732,138), 5-fluoro-3-[2-[4-methoxy-4-[[(R)-phenylsulphinyl]methyl]-1-piperidinyl]ethyl]-1H-indole (GR159897), (R)-[[2-phenyl-4-quinolinyl)carbonyl]amino]-methyl ester benzeneacetic acid (SB218795), (2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl] (phenylmethyl) phosphinic acid (CGP55845), (RS)-3,5-dihydroxyphenylglycine (DHPG), 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), 1-(1H-indol-4-yloxy)-3-[(1-methylethyl)amino]-2-propanol (pindolol), SR95531 hydrobromide, (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid (LY341495), 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP), (2S,3S,4R)-2-carboxy-4-isopropyl-3-pyrrolidineacetic acid [dihydrokainic acid (DHK)], and dl-threo-β-benzyloxyaspartic acid (TBOA) from Tocris Bioscience; and QX-314 bromide and TTX from Alomone Laboratories. Stock solutions of drugs were made in distilled water, except picrotoxin (added directly to ACSF), LY341495 (in 1N NaOH), GR159897, SB218795, AM251, AM281, THL and TBOA (in DMSO), then diluted to working concentrations using ACSF immediately before use and applied by superfusion.
Results
Substance P suppresses evoked IPSCs in identified PAG-RVM projection neurons
We first examined the effect of substance P on GABAergic synaptic transmission in PAG projection neurons retrogradely labeled from the RVM. In all animals used, injections of rhodamine microspheres were restricted to within 1 mm of the midline, directly dorsal to the pyramids (Fig. 1C). Rostrocaudally, all of the series of injections occupied a volume of nucleus raphe magnus within the RVM bounded by the caudal limit of the trapezoid body and the rostral olivary complex. After injection of the fluorescent microspheres into the RVM, many neurons in the PAG were observed to contain retrograde tracer. Post hoc imaging of the biocytin-filled neurons confirmed the presence of fluorescent microspheres within the recorded neurons (Fig. 1A–C).
Figure 1.

Substance P suppresses evoked IPSCs in retrogradely labeled PAG-RVM projection neurons. A–C, Photomicrographs showing biocytin (A) and rhodamine microspheres (B) colocalized (C; merge of A, B) in a PAG neuron after injection of microspheres into the RVM. Inset: Injection site (black) of rhodamine microspheres in the RVM. Scale bar, 20 μm. D, Averaged traces of evoked IPSCs before (Pre) and during superfusion of substance P (SubP, 300 nm). E, Averaged traces in response to identical paired stimuli (IPSC1–2 interval, 70 ms) with IPSC1 normalized (left) before and during substance P. D and E are taken from the neuron shown in A–C.
Whole-cell voltage-clamp recordings were made from retrogradely labeled PAG-RVM projection neurons of varying cell size and morphology throughout the lateral and ventrolateral PAG. Superfusion of substance P (300 nm) reduced the amplitude of electrically evoked IPSCs in all PAG-RVM projection neurons tested (74 ± 2% of control, range: 59–87% of control, p < 0.0001, n = 16) (Fig. 1D). In these neurons, paired evoked IPSCs were elicited by two stimuli of identical strength in close succession (interstimulus interval, 70 ms) to determine whether the substance P-induced inhibition of the first evoked response was associated with relative facilitation of the second evoked response. Under control conditions, the mean ratio of the amplitude of paired evoked IPSCs was 1.08 ± 0.05, with both paired-pulse depression and facilitation being observed (evoked IPSC2/IPSC1 ratio, 0.80–1.55; n = 16). Superfusion of substance P increased the mean paired-pulse ratio to 1.18 ± 0.05 (109 ± 2% of control, p < 0.001) (Fig. 1E). We also examined the effect of substance P on evoked IPSCs recorded from unidentified, randomly selected PAG neurons in these animals. Substance P reduced the amplitude (73 ± 3% of control, p < 0.01) and increased the paired-pulse ratio (110 ± 4% of control, p = 0.06) of evoked IPSCs in these neurons (n = 7) to an extent similar to that observed for labeled projection neurons (p > 0.05).
Substance P suppresses evoked IPSCs primarily through activation of NK1 receptors
Next, we investigated the pharmacology of substance P actions on GABAergic transmission by recording from unidentified PAG neurons in animals that had not received tracer injections. All subsequent results therefore refer to experiments performed using noninjected animals. In these animals, superfusion of substance P (300 nm) reversibly reduced the amplitude (63 ± 3% of control, p < 0.0001, n = 22) and increased the paired-pulse ratio (137 ± 10% of control, p < 0.01, n = 20) of evoked IPSCs (Fig. 2A–D). These effects were significantly larger than those observed for neurons recorded from tracer-injected animals (n = 23, pooled; p < 0.01), suggesting that factors associated with surgery or postsurgery recovery may have led to a reduction in substance P-mediated actions. In neurons tested, subsequent superfusion of the μ,δ-opioid receptor agonist met-enkephalin (10 μm) also reduced the amplitude (43 ± 7% of control, p < 0.0001) and increased the paired-pulse ratio (135 ± 13% of control, p < 0.05) of evoked IPSCs (n = 11). Substance P and met-enkephalin had no effect on the membrane current or the conductance of the neurons at −70 mV.
Figure 2.

Substance P suppresses evoked IPSCs primarily via activation of NK1 receptors. A, Time course of evoked IPSC amplitudes (eIPSC Ampl) during superfusion of substance P (SubP, 300 nm) and picrotoxin (Pictx, 100 μm) in an unidentified neuron. Each point is the mean of two consecutive evoked IPSCs. B, Averaged traces of evoked IPSCs before (Pre) and during substance P and picrotoxin. C, Averaged traces in response to paired stimuli (IPSC1–2 interval, 70 ms) with IPSC1 normalized (left) before and during substance P. D, Bar chart of the mean amplitude and paired-pulse ratio (PP ratio) of evoked IPSCs recorded from unidentified PAG neurons in the presence of substance P, expressed as a percentage of the predrug control (Ctl) level, in neurons preincubated in the NK1 receptor antagonist L732,138 (20 μm, NK1) and a combination of the NK1, NK2, and NK3 receptor antagonists L732,138 (20 μm), GR159897 (3 μm), and SB218795 (3 μm) (NK1–3) compared with neurons that did not undergo preincubation. **p < 0.01; ##p < 0.0001. A–C are taken from one neuron.
Preincubation of slices with the NK1 receptor-specific antagonist L732,138 (20 μm) significantly reduced (p < 0.05), but did not fully block, the effect of substance P on evoked IPSC amplitude (88 ± 7% of control, p > 0.05, n = 5) and paired-pulse ratio (108 ± 3% of control, p > 0.05, n = 5) (Fig. 2D). Because substance P may also activate NK2 and NK3 receptors located on PAG neurons (Drew et al., 2005), we examined the actions of substance P in the combined presence of L732,138 (20 μm) and the NK2 and NK3 receptor-specific antagonists GR159897 (3 μm) and SB218795 (3 μm). Under these conditions, the effect of substance P on evoked IPSC amplitude (101 ± 8% of control, p > 0.05, n = 5) and paired-pulse ratio (99 ± 7% of control, p > 0.05, n = 5) was abolished (Fig. 2D). In contrast, met-enkephalin (10 μm) reduced the amplitude (44 ± 5% of control, p < 0.05) and increased the paired-pulse ratio (121 ± 4% of control, p < 0.01) of evoked IPSCs in these neurons (n = 4).
Substance P has no effect on miniature IPSCs
The observed change in evoked IPSC paired-pulse ratio suggested that substance P was acting presynaptically to suppress GABAergic transmission. We recorded spontaneous miniature IPSCs to determine whether the effect of substance P on evoked IPSCs was attributable to a direct action at NK1 receptor-expressing GABAergic axon terminals (Barbaresi, 1998; Commons and Valentino, 2002). In the presence of TTX (300 nm), substance P (300 nm) had no significant effect on the rate (92 ± 6% of control, p > 0.05) or amplitude (103 ± 3% of control, p > 0.05) of miniature IPSCs (n = 8) (Fig. 3F). In contrast, met-enkephalin (10 μm) reduced the rate (p < 0.01) but not the amplitude (p > 0.05) of miniature IPSCs in these neurons (n = 8) (Fig. 3F).
Figure 3.

Substance P has no effect on spontaneous miniature IPSCs. A, Time course of miniature IPSC (mIPSC) rate during superfusion of substance P (SubP, 300 nm), met-enkephalin (ME, 10 μm), naloxone (NAL, 1 μm), and cadmium (Cd2+, 100 μm). B, Averaged traces of miniature IPSCs before (Pre) and during substance P and met-enkephalin. C, D, Cumulative probability (Cumul. Prob.) distribution plots of miniature IPSCs versus inter-event interval (C) and amplitude (D) before and during substance P and met-enkephalin. E, Representative raw traces before and during substance P and met-enkephalin. F, Bar chart of the mean rate and amplitude (Ampl) of miniature IPSCs in the presence of substance P, met-enkephalin, and DHPG (30 μm), expressed as a percentage of the predrug control (Ctl) level, in slices preincubated in standard (2.5 mm) and elevated (17.5 mm; Elevated [K+]o) extracellular [K+]. *p < 0.05; **p < 0.01. A–E are taken from one neuron in 17.5 mm [K+]o.
In PAG slices, the vast majority of spontaneous miniature IPSCs occur independently of extracellular Ca2+ (Vaughan and Christie, 1997). Therefore, the effect of substance P on evoked IPSCs could have been the result of inhibition of voltage-dependent Ca2+ channels in the presynaptic terminals of GABAergic neurons, which were activated during action potential-evoked release but largely inactive during TTX-resistant spontaneous release. To test this possibility, we examined the effect of substance P on miniature IPSCs during tonic activation of voltage-dependent Ca2+ channels by increasing the extracellular K+ concentration ([K+]o) from 2.5 to 17.5 mm, as described previously in the PAG (Connor et al., 2004; Hack et al., 2005). Under these conditions, substance P had no significant effect on the rate of miniature IPSCs (95 ± 5% of control, p > 0.05) (Fig. 3A,E,F), reflected as a lack of change in the cumulative inter-event interval distribution (Fig. 3C), or on the amplitude (100 ± 5% of control, p > 0.05), kinetics, or cumulative amplitude distribution of IPSCs (n = 6) (Fig. 3B,D–F). In the same neurons, met-enkephalin reduced the rate (p < 0.01) but had no effect on the amplitude (p > 0.05) of miniature IPSCs (n = 6) (Fig. 3A–F). In addition, in a separate set of experiments, superfusion of the group I mGluR agonist DHPG (30 μm) also reduced the rate (59 ± 7% of control, p < 0.05) but not the amplitude (94 ± 8% of control, p > 0.05) of miniature IPSCs recorded in 17.5 mm [K+]o (n = 5) (Fig. 3F). In all neurons tested, addition of cadmium (100 μm) robustly reduced the rate of miniature IPSCs (22 ± 3% of control, n = 9) (Fig. 3A), confirming the involvement of voltage-dependent Ca2+ channels in these experiments.
Substance P suppresses evoked IPSCs through activation of mGluRs
The above results suggested that the inhibition of evoked IPSCs by substance P was mediated by an indirect effect on GABAergic terminals. We next investigated the possibility that this inhibition was mediated via the release of other neurotransmitters, as has been described for substance P actions in the nucleus accumbens and dorsal raphe nucleus (Liu et al., 2002; Kombian et al., 2003; Valentino et al., 2003). To test this, we examined the effects of substance P on evoked IPSCs after blockade of a number of G-protein-coupled receptors that are known to presynaptically suppress GABAA receptor-mediated synaptic transmission in the PAG (Vaughan and Christie, 1997; Bagley et al., 1999; Kishimoto et al., 2001; Yang et al., 2003). Substance P produced a significant reduction in the amplitude of evoked IPSCs in the combined presence of GABAB (CGP55845, 1 μm), μ,δ-opioid (naloxone, 1 μm), adenosine A1 (DPCPX, 1 μm), and 5HT1A (pindolol, 1 μm) receptor antagonists (60 ± 5% of control, p < 0.01, n = 5) (Fig. 4A,E) that was similar to the effect of substance P alone (p > 0.05). Met-enkephalin had no effect on evoked IPSC amplitudes in these neurons (96 ± 4% of control, p > 0.05, n = 3).
Figure 4.
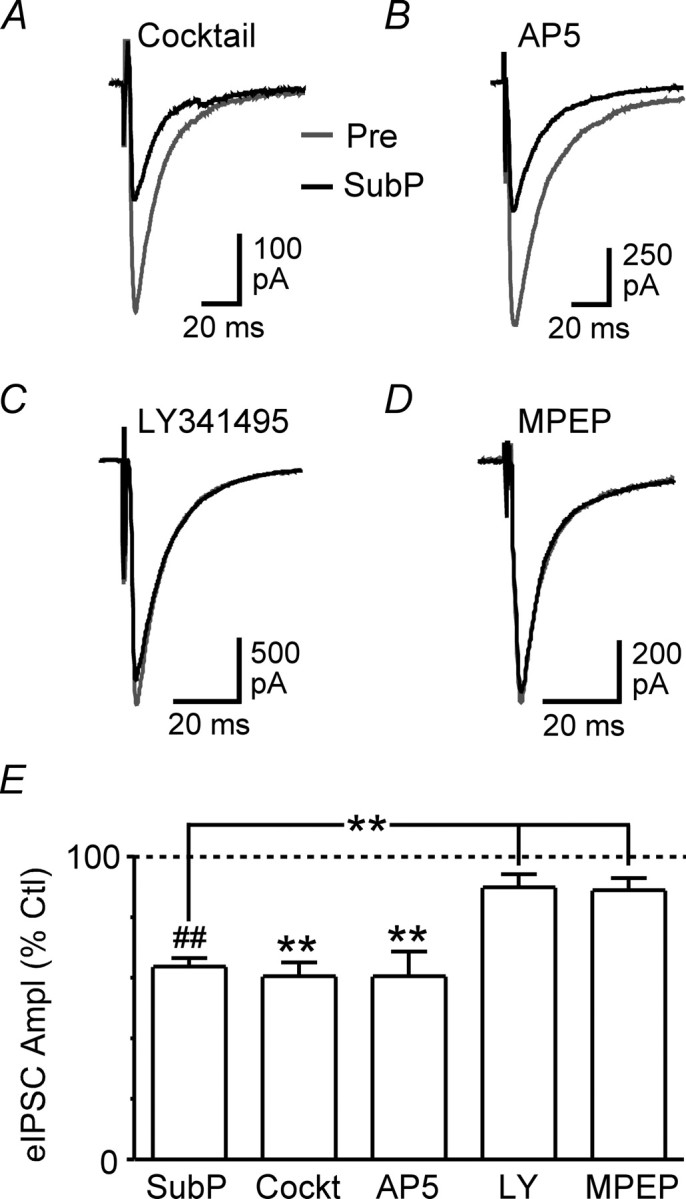
Substance P suppresses evoked IPSCs via mGluR5 activation. A–D, Averaged traces of evoked IPSCs before (Pre) and during substance P (SubP, 300 nm) in neurons preincubated in a combination of CGP55845 (1 μm), naloxone (1 μm), DPCPX (1 μm), and pindolol (1 μm) (A, Cocktail), AP5 (B, 25 μm), LY341495 (C, 100 μm), and MPEP (D, 5 μm). E, Bar chart of the mean amplitude of evoked IPSCs (eIPSC Ampl) in the presence of substance P, expressed as a percentage of the predrug control (Ctl) level, in neurons preincubated in cocktail (Cockt, see above), AP5, LY341495 (LY), and MPEP compared with neurons that did not undergo preincubation. **p < 0.01; ##p < 0.0001. A–D are taken from different neurons.
Because many NK1 receptor-expressing PAG neurons contain glutamate (Commons and Valentino, 2002), we next examined whether glutamate, acting through ionotropic or metabotropic glutamate receptors, was involved in the effect of substance P on evoked IPSCs. Substance P produced a significant reduction in the amplitude of evoked IPSCs in the presence of the NMDA receptor antagonist AP5 (25 μm) (61 ± 8% of control, p < 0.01, n = 5) (Fig. 4B,E) that was similar to the effect of substance P alone (p > 0.05). In contrast, substance P had no significant effect on the amplitude of evoked IPSCs in slices pretreated with the broad-spectrum mGluR antagonist LY341495 (100 μm) (90 ± 4% of control, p > 0.05, n = 6) (Fig. 4C,E) or the mGluR5-specific antagonist MPEP (5 μm) (89 ± 4% of control, p > 0.05, n = 4) (Fig. 4D,E). Similarly, substance P had no effect on the paired-pulse ratio of evoked IPSCs in the presence of LY341495 (99 ± 12% of control, p > 0.05, n = 6) or MPEP (99 ± 1% of control, p > 0.05, n = 4).
Substance P drives action potential-dependent glutamate release
Together, the above results indicated that substance P was reducing GABAergic transmission via an action potential- and mGluR5-dependent mechanism. A likely explanation for these observations is that substance P was activating mGluRs by eliciting action potential-dependent release of glutamate. We tested this hypothesis by examining the effect of substance P on non-NMDA glutamate receptor-mediated spontaneous EPSCs. Superfusion of substance P (300 nm) increased the rate of spontaneous EPSCs in 8 of 13 PAG neurons tested (overall mean, 322 ± 95% of control; range, 66–1035% of control; p < 0.05), which was reflected as a leftward shift in the cumulative inter-event interval distribution (Fig. 5A,C,I). In contrast, substance P had no significant effect on the amplitude (107 ± 5% of control, p > 0.05), kinetics, or cumulative amplitude distribution of spontaneous EPSCs (n = 13) (Fig. 5A,B,D,I).
Figure 5.
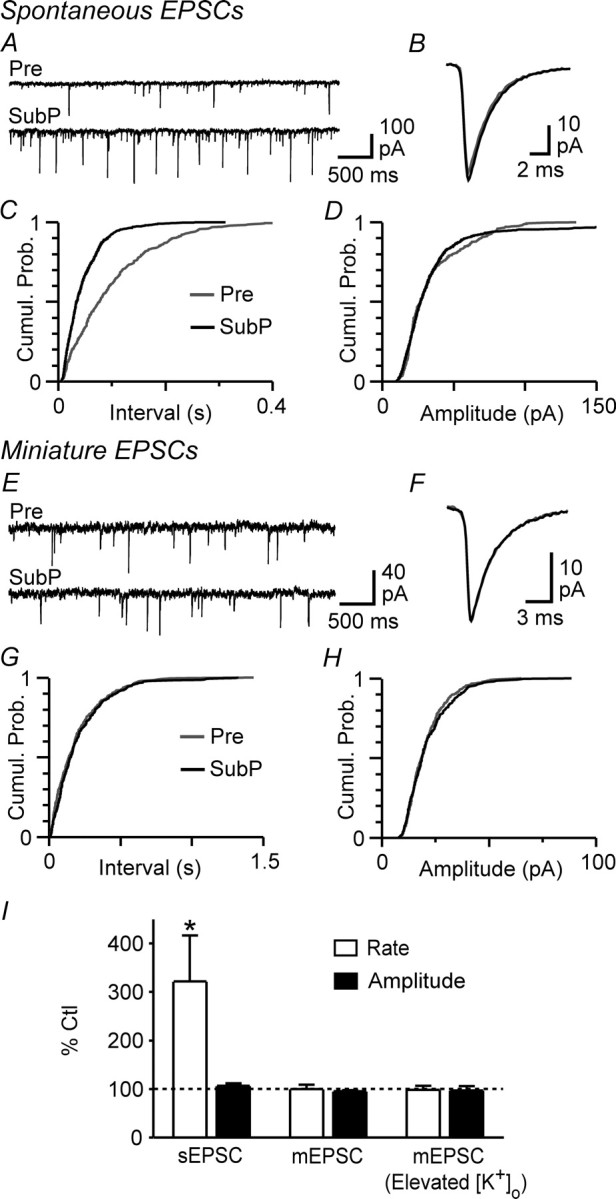
Substance P increases the frequency of action potential-dependent spontaneous EPSCs. A, Representative raw traces of spontaneous EPSCs before (Pre) and during superfusion of substance P (SubP, 300 nm). B, Averaged traces of spontaneous EPSCs before and during substance P. C, D, Cumulative probability (Cumul. Prob.) distribution plots of spontaneous EPSC versus inter-event interval (C) and amplitude (D) before (Pre) and during substance P. E, Representative raw traces of spontaneous miniature EPSCs before (Pre) and during substance P. F, Averaged traces of miniature EPSCs before and during substance P. G, H, Cumulative probability (Cumul. Prob.) distribution plots of miniature EPSC versus inter-event interval (C) and amplitude (D) before (Pre) and during substance P. I, Bar chart of the mean rate and amplitude of spontaneous EPSCs (sEPSCs) and miniature EPSCs (mEPSCs) in the presence of substance P, expressed as a percentage of the predrug control (Ctl) level, in neurons preincubated in standard (2.5 mm) and elevated (17.5 mm; Elevated [K+]o) extracellular [K+]. *p < 0.05. A–D are taken from one neuron and E–H from a different neuron in 17.5 mm [K+]o.
We next recorded miniature EPSCs to determine whether substance P was acting directly at NK1 receptor-expressing glutamatergic axon terminals (Barbaresi, 1998; Commons and Valentino, 2002). In the presence of TTX (300 nm), substance P (300 nm) had no effect on the rate (100 ± 9% of control, p > 0.05) or amplitude (96 ± 2% of control, p > 0.05) of miniature EPSCs (n = 7) (Fig. 5I). Furthermore, substance P had no effect on the rate (98 ± 9% of control, p > 0.05), amplitude (97 ± 9% of control, p > 0.05), kinetics, or cumulative distributions of miniature EPSCs recorded in 17.5 mm [K+]o (n = 4) (Fig. 5E–I).
Blocking glutamate uptake occludes substance P effects
The above findings are consistent with substance P activating mGluRs via action potential-dependent release of glutamate. To assess an involvement of endogenous glutamate in this process more directly, however, we examined the action of substance P on evoked IPSCs in the presence of TBOA, a broad-spectrum inhibitor of neuronal and glial glutamate transporters. Consistent with our previous work (Drew et al., 2008), TBOA (30 μm) reduced the amplitude of evoked IPSCs in all neurons tested (59 ± 8% of control, p < 0.01, n = 6) (Fig. 6A). In the presence of TBOA, substance P produced no further changes in evoked IPSC amplitude (99 ± 3% of the TBOA level, p > 0.05) (Fig. 6A,C) or paired-pulse ratio (95 ± 7% of the TBOA level, p > 0.05) (n = 5), whereas met-enkephalin significantly reduced the amplitude (p < 0.05) (Fig. 6A,C) and increased the paired-pulse ratio (p < 0.01) of IPSCs in these neurons (n = 4). In contrast, substance P significantly reduced the amplitude of evoked IPSCs in the presence of the GLT1-specific glial glutamate transport inhibitor DHK (200 μm) (76 ± 3% of the DHK level, p < 0.01, n = 4) (Fig. 6B,C), which is consistent with the lack of GLT1 involvement in TBOA-induced inhibition of evoked IPSCs in the PAG (Drew et al., 2008). These results confirm that substance P and endogenous glutamate inhibit GABAergic transmission in PAG neurons via a shared mechanism.
Figure 6.

The glutamate transport inhibitor TBOA occludes substance P effects on evoked IPSCs. A, Averaged traces of evoked IPSCs before (Pre) and during superfusion of TBOA (30 μm), TBOA + substance P (SubP, 300 nm), and TBOA + met-enkephalin (ME, 10 μm). B, Averaged traces of evoked IPSCs before and during DHK (200 μm) and DHK + substance P. C, Bar chart of the mean amplitude of evoked IPSCs (eIPSC Ampl) in the presence of substance P and met-enkephalin, expressed as a percentage of the predrug control (Ctl) level, in neurons preincubated in TBOA and DHK. *p < 0.05; **p < 0.01. A and B are taken from different neurons.
Substance P suppresses evoked IPSCs via retrograde endocannabinoid signaling
We have previously shown that TBOA reduces GABAergic transmission in PAG neurons primarily through mGluR5 activation and presynaptic CB1 receptor (CB1R)-mediated endocannabinoid signaling (Drew et al., 2008). To determine whether substance P also reduces GABAergic transmission via this mGluR5–CB1R pathway, we examined substance P actions in the presence of the CB1R-specific antagonists/inverse agonists AM251 and AM281. In slices preincubated in AM251 (3 μm), substance P had no significant effect on evoked IPSC amplitude (96 ± 3% of control, p > 0.05) (Fig. 7A,E) or paired-pulse ratio (101 ± 1% of control, p > 0.05) (n = 5). Similarly, substance P had no significant effect on evoked IPSC amplitude (91 ± 6% of control, p > 0.05) (Fig. 7B,E) or paired-pulse ratio (111 ± 6% of control, p > 0.05) in the presence of AM281 (3 μm) (n = 4). In contrast, met-enkephalin produced a significant reduction in the amplitude of evoked IPSCs in the presence of both AM251 (44 ± 6% of control, p < 0.01, n = 4) and AM281 (38 ± 10% of control, p < 0.01, n = 4) that was not significantly different from that produced by met-enkephalin alone (p > 0.05).
Figure 7.
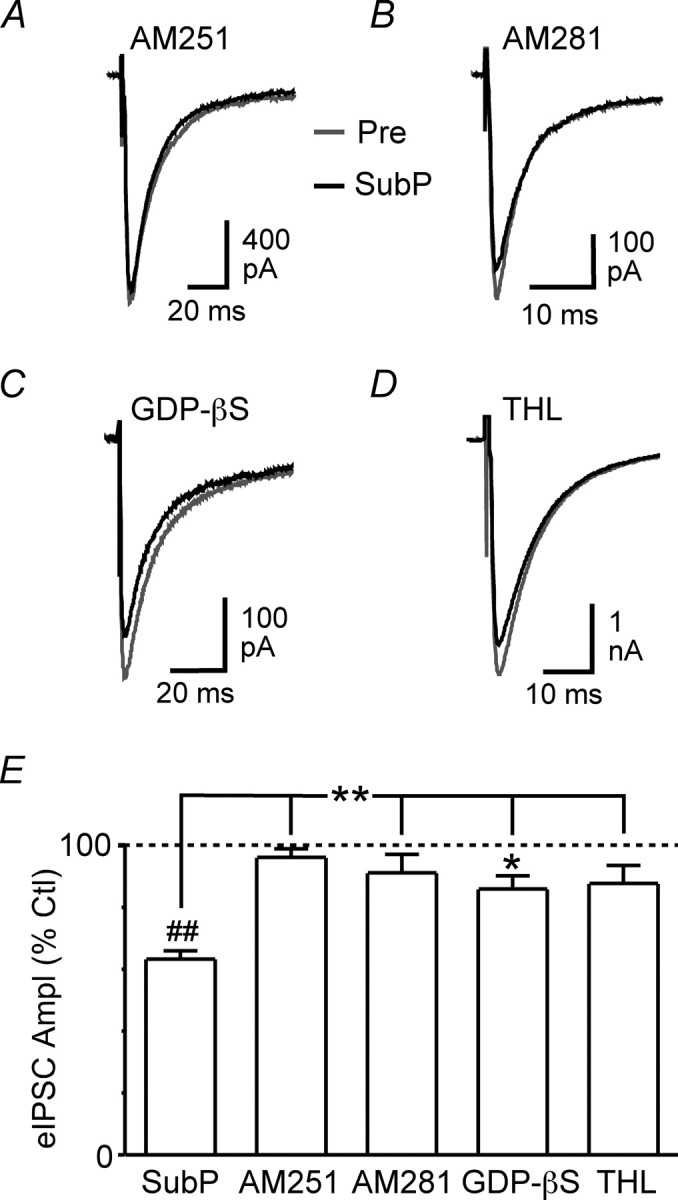
Substance P suppresses evoked IPSCs via retrograde endocannabinoid signaling. A–D, Averaged traces of evoked IPSCs before (Pre) and during substance P (SubP, 300 nm) in the presence of AM251 (A, 3 μm), AM281 (B, 3 μm), intracellular GDP-βS (C, 2 mm, added to the recording pipette solution) and THL (D, 10 μm). E, Bar chart of the mean amplitude of evoked IPSCs (eIPSC Ampl) in the presence of substance P, expressed as a percentage of the predrug control (Ctl) level, in neurons treated with AM251, AM281, GDP-βS, and THL compared with untreated control neurons. *p < 0.05; **p < 0.01; ##p < 0.0001. A–D are taken from different neurons.
Labeling for mGluR5 has been observed in the dendrites and, to a lesser extent, soma of PAG neurons (Azkue et al., 1997; de Novellis et al., 2003). To investigate a possible involvement of postsynaptic mGluR5s in substance P-induced endocannabinoid signaling, we examined the effect of substance P on evoked IPSCs after blockade of postsynaptic G-protein activity by adding the G-protein inhibitor GDP-βS (2 mm) to the recording pipette solution. In these experiments, substance P was applied at least 30 min after obtaining whole-cell configuration. Under these conditions, substance P produced a significant reduction in evoked IPSC amplitude (86 ± 4% of control, p < 0.05, n = 6); however, this was significantly less than that observed in the absence of GDP-βS (p < 0.01) (Fig. 7C,E). Substance P had no significant effect on the paired-pulse ratio of evoked IPSCs recorded from GDP-βS-filled neurons (102 ± 3% of control, p > 0.05, n = 6). In contrast, when GDP-βS was added to the pipette solution, met-enkephalin (10 μm) reduced the amplitude of evoked IPSCs (45 ± 5% of control, p < 0.01, n = 4) to an extent similar to that observed in its absence (p > 0.05).
Activation of mGluR5 stimulates biosynthesis of the endocannabinoid 2-arachidonoylglycerol (2-AG), but not anandamide, at several distinct synapses in the brain (Jung et al., 2005; Lafourcade et al., 2007; Uchigashima et al., 2007). This process is mediated by the catalytic actions of phospholipase C and 1,2-diacylglycerol (DAG) lipases (Bisogno et al., 2003; Piomelli, 2003). To test whether 2-AG was likely to be the endocannabinoid mediating the effect of substance P, we examined substance P actions in the presence of the DAG lipase inhibitor tetrahydrolipstatin (Bisogno et al., 2003; Szabo et al., 2006; Uchigashima et al., 2007). In slices preincubated in tetrahydrolipstatin (10 μm) for at least 20 min, substance P had no significant effect on evoked IPSC amplitude (88 ± 6% of control, p > 0.05) (Fig. 7D,E) or paired-pulse ratio (100 ± 7% of control, p > 0.05) (n = 7). In contrast, met-enkephalin (10 μm) reduced the amplitude of evoked IPSCs in the presence of tetrahydrolipstatin (29 ± 3% of control, p < 0.0001, n = 7) to an extent similar to that observed in its absence (p > 0.05). Together, these results suggest that substance P suppresses GABAergic transmission by driving postsynaptic mGluR5 activation and retrograde 2-AG signaling.
Discussion
This study demonstrates that substance P suppresses GABAergic synaptic transmission onto PAG neurons that form part of the PAG–RVM–spinal cord descending pathway. This effect is produced through local glutamatergic neuron-driven activation of group I mGluRs and CB1R-mediated retrograde endocannabinoid signaling. These findings may partly account for the analgesic action of substance P within the PAG and suggest a potential role for the endocannabinoid system in NK1 receptor-dependent descending analgesia.
Presynaptic inhibition mediated by local glutamatergic transmission
A number of observations indicated that substance P suppressed GABAergic transmission indirectly through activation of local glutamatergic circuits and presynaptic inhibition. First, as observed with the μ-opioid agonist met-enkephalin, inhibition of evoked IPSCs by substance P was associated with an increase in the paired-pulse ratio, indicative of a presynaptic reduction in transmitter release probability. Unlike met-enkephalin, however, substance P did not significantly affect the rate of spontaneous miniature IPSCs, suggesting that its effect on evoked IPSCs was unlikely to be the result of a direct action at GABAergic terminals. Second, substance P-induced suppression of evoked IPSCs was blocked by mGluR antagonists. This is consistent with evidence that mGluR agonists suppress IPSCs via a presynaptic mechanism(s) in PAG slices (Drew and Vaughan, 2004). Third, substance P increased the rate but not the amplitude of spontaneous EPSCs in the majority of neurons. This effect was abolished by tetrodotoxin, implying that substance P was likely to be acting on somatodendritic NK1 receptors to elicit action potential-dependent glutamate release. Indeed, in PAG slices, substance P induces a direct (tetrodotoxin-resistant) postsynaptic inward current in ∼60% of neurons (Drew et al., 2005) and immunohistochemical evidence indicates that the majority of NK1 receptor-expressing PAG neurons contain glutamate (Commons and Valentino, 2002). Finally, the actions of substance P were mimicked and occluded by a glutamate transport inhibitor, confirming that substance P and endogenous glutamate suppress evoked IPSCs in the PAG via a common mechanism.
Immunohistochemical studies have demonstrated NK1 and NK3 receptor expression in the PAG (Barbaresi, 1998; Mileusnic et al., 1999; Yip and Chahl, 2001; Commons and Valentino, 2002), and we have previously shown that the postsynaptic actions of substance P on PAG neurons are mediated in part by NK1, NK2, and NK3 receptor subtypes (Drew et al., 2005). In the present study, although an NK1 receptor-specific antagonist significantly reduced substance P-induced suppression of evoked IPSCs, this effect was completely abolished only when a combination of NK1, NK2, and NK3 antagonists was used. Collectively, these studies suggest that substance P modulates neuronal excitability and synaptic transmission in the PAG by interacting with multiple NK receptor subtypes.
Retrograde endocannabinoid signaling
In this study, substance P-induced suppression of evoked IPSCs was reduced by mGluR5 and CB1R antagonists, as well as by blocking postsynaptic G-protein activity with GDP-βS. Expression of mGluR5 in PAG neurons is restricted predominantly to the dendrites, with weaker labeling in soma (Azkue et al., 1997; de Novellis et al., 2003), although a possible presynaptic locus is not yet known. In many brain regions, mGluR5 localization is almost exclusively postsynaptic, with particularly high densities in extra- and perisynaptic regions of dendrites and dendritic spines (Shigemoto et al., 1993; Lujan et al., 1996; Négyessy et al., 1997; Kuwajima et al., 2004; Uchigashima et al., 2007). In contrast, immunohistochemical and electrophysiological studies indicate that CB1Rs are localized to the axon terminals, but not cell bodies, of GABAergic and glutamatergic PAG neurons (Tsou et al., 1998; Vaughan et al., 2000). Together with the present study, these findings are consistent with substance P driving postsynaptic group I mGluR activation to initiate retrograde endocannabinoid signaling in the PAG, as previously described for other synapses in the brain (Maejima et al., 2001; Varma et al., 2001; Ohno-Shosaku et al., 2002; Galante and Diana, 2004). In PAG slices, both the group I mGluR agonist DHPG and the glutamate transport inhibitor TBOA presynaptically suppress IPSCs via mGluR5-dependent endocannabinoid signaling (Drew et al., 2008). Importantly, mGluR5 and CB1R antagonists have little effect on IPSCs when evoked at low stimulation frequencies, but increase IPSC amplitudes during trains of high-frequency stimulation (Drew et al., 2008). This result implies that mGluR5 activation, and subsequent endocannabinoid production, requires sufficient release of glutamate to overwhelm transporters. The findings that substance P enhanced synaptic glutamate release and that its effect on evoked IPSCs was occluded by TBOA and blocked by an mGluR5 antagonist are consistent with this “glutamate spillover” model for initiating endocannabinoid signaling in the PAG (Fig. 8).
Figure 8.

Proposed model for substance P-induced suppression of GABAergic transmission in the PAG. Substance P (SubP) activates NK1, NK2, and NK3 (NK1–3) receptors located on glutamatergic (GLU) neurons to elicit action potential (AP) generation via the opening of a nonselective cation conductance (Icat) and suppression of a G-protein-coupled inwardly rectifying K+ (GIRK) conductance (Drew et al., 2005). The resultant action potential-driven glutamate release is sufficient to overwhelm neuronal glutamate transporters, presumably excitatory amino acid carrier 1 (EAAC1) (Drew et al., 2008), and engage postsynaptic mGluR5s. Synthesis of the endocannabinoid 2-AG subsequently occurs via catalysis of diacylglycerol by a 1,2-diacylglycerol lipase (DAGL). 2-AG then acts as a retrograde messenger to activate presynaptic CB1Rs located on the terminals of GABAergic neurons and to reduce GABA release. Activation of this dual excitatory and disinhibitory mechanism in PAG-RVM projection neurons would be predicted to then facilitate activation of this descending pathway.
The finding that substance P suppressed evoked but not miniature IPSCs could potentially be explained by a differential cannabinoid sensitivity of action potential-dependent and action potential-independent transmitter release. We have previously observed, however, that CB1R activation (induced by DHPG, TBOA, or the synthetic CB1R agonist WIN55,212-2) inhibits both evoked and miniature IPSCs in all PAG neurons (Vaughan et al., 2000; Drew et al., 2008). Furthermore, in this study, DHPG reduced the rate of miniature IPSCs induced by K+-stimulated voltage-dependent Ca2+ channel activation. Because CB1R activation in the PAG also reduces miniature IPSC rate in nominally Ca2+-free extracellular solution (Vaughan et al., 2000), this result suggests that substance P-induced suppression of evoked IPSCs could occur through both Ca2+-dependent and Ca2+-independent CB1R-mediated presynaptic mechanisms. Moreover, given the sensitivity of miniature IPSCs to CB1R activation, these findings also suggest that substance P has little capacity to stimulate action potential-independent endocannabinoid release.
Although mGluR and CB1R activation accounted for most of the observed effect of substance P on evoked IPSCs, a small (∼5–10%) residual inhibition persisted in the presence of antagonists for these receptors. We therefore cannot exclude the possibility of an additional mechanism for substance P-induced suppression of GABAergic transmission in the PAG. Indeed, although substance P had no effect on miniature IPSCs recorded in elevated extracellular [K+], it is possible that a non-CB1R-mediated modulation of voltage-dependent Ca2+ channels could have been missed in these experiments because strong presynaptic stimulation can overcome various receptor-mediated actions on transmitter release (Klapstein and Colmers, 1992). We can, however, exclude involvement of NMDA, GABAB, adenosine A1, 5HT1A, and μ,δ-opioid receptors because antagonists for these receptors did not alter the effects of substance P on evoked IPSCs. The lack of opioid receptor involvement, in particular, is somewhat surprising given that analgesia produced by microinjection of substance P into the PAG is naloxone sensitive (Stewart et al., 1976; Frederickson et al., 1978; Malick and Goldstein, 1978; Mohrland and Gebhart, 1979) and that substance P increases dialysate enkephalin levels in PAG slices (Del Río et al., 1983). It has been postulated that substance P elicits enkephalin release in the PAG indirectly by activating local glutamatergic interneurons (Commons and Valentino, 2002). Because in the present study IPSCs were isolated using a non-NMDA glutamate receptor antagonist, we cannot rule out the possibility that, under physiological conditions, substance P induces enkephalin release via activation of these receptors to modulate GABAergic transmission. In behavioral studies, however, naloxone was administered via the systemic route only and, therefore, the precise location and mechanism of action of opioids in substance P-induced analgesia remain to be determined.
Regardless of a potential opioidergic contribution within the PAG, the present results demonstrate that substance P is able to disinhibit PAG neurons via retrograde endocannabinoid signaling (Fig. 8). In this multistep process, we propose that substance P acts via NK1–3 receptors to elicit action potential-dependent release of glutamate from interneurons within the PAG. This endogenously released glutamate overwhelms adjacent transporters and stimulates mGluR5-mediated production of endocannabinoids in the postsynaptic neuron. This endocannabinoid then activates CB1Rs located on GABAergic axon terminals impinging on that neuron. The endocannabinoid involved in this process is likely to be 2-AG because the effect of substance P was blocked by the DAG lipase inhibitor tetrahydrolipstatin (Bisogno et al., 2003; Szabo et al., 2006; Uchigashima et al., 2007), although this requires further confirmation with the development of other more selective blockers of 2-AG synthesis.
Functional implications
The PAG is a major site of the analgesic actions of μ-opioids and cannabinoids (Martin et al., 1995; Lichtman et al., 1996; Finn et al., 2003; Fields et al., 2006). Like μ-opioids, cannabinoids are thought to produce their effects in the PAG by reducing GABAergic inhibition of output neurons that form part of a descending analgesic pathway (Osborne et al., 1996; Vaughan et al., 2000; Fields et al., 2006). In support of this hypothesis, we have shown that substance P suppresses GABAergic transmission onto PAG neurons that form part of the PAG–RVM descending pathway via the actions of endocannabinoids. In behavioral studies, noxious peripheral stimuli and psychological stressors elicit the release of substance P (Rosén et al., 1992; Xin et al., 1997), glutamate (Silva et al., 2000), and endocannabinoids (Walker et al., 1999; Hohmann et al., 2005) within the PAG. Our findings indicate that, by driving local glutamatergic transmission and group I mGluR activation, substance P represents an important upstream modulator of endocannabinoid signaling within the PAG. It has been demonstrated that NK1 and CB1 receptors are critical for the full expression of several forms of pain- and stress-induced analgesia (De Felipe et al., 1998; Bester et al., 2001; Hohmann et al., 2005). The results of the present study suggest that CB1R-mediated disinhibition of the PAG–RVM descending analgesic pathway may play an important role in NK1 receptor-dependent analgesia.
Footnotes
This work was supported by Australian National Health and Medical Research Council Grant 457563 (C.W.V.) and C. J. Martin Biomedical Fellowship 402868 (G.M.D.).
References
- Azkue JJ, Knöpfel T, Kuhn R, Mateos JM, Grandes P. Distribution of the metabotropic glutamate receptor subtype mGluR5 in rat periaqueductal grey and relationship with ascending spinofugal afferents. Neurosci Lett. 1997;228:1–4. doi: 10.1016/s0304-3940(97)00357-1. [DOI] [PubMed] [Google Scholar]
- Bagley EE, Vaughan CW, Christie MJ. Inhibition by adenosine receptor agonists of synaptic transmission in rat periaqueductal grey neurons. J Physiol. 1999;516:219–225. doi: 10.1111/j.1469-7793.1999.219aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaresi P. Immunocytochemical localization of substance P receptor in rat periaqueductal gray matter: a light and electron microscopic study. J Comp Neurol. 1998;398:473–490. doi: 10.1002/(sici)1096-9861(19980907)398:4<473::aid-cne2>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Bester H, De Felipe C, Hunt SP. The NK1 receptor is essential for the full expression of noxious inhibitory controls in the mouse. J Neurosci. 2001;21:1039–1046. doi: 10.1523/JNEUROSCI.21-03-01039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commons KG, Valentino RJ. Cellular basis for the effects of substance P in the periaqueductal gray and dorsal raphe nucleus. J Comp Neurol. 2002;447:82–97. doi: 10.1002/cne.10228. [DOI] [PubMed] [Google Scholar]
- Connor M, Bagley EE, Mitchell VA, Ingram SL, Christie MJ, Humphrey PP, Vaughan CW. Cellular actions of somatostatin on rat periaqueductal grey neurons in vitro. Br J Pharmacol. 2004;142:1273–1280. doi: 10.1038/sj.bjp.0705894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felipe C, Herrero JF, O'Brien JA, Palmer JA, Doyle CA, Smith AJ, Laird JM, Belmonte C, Cervero F, Hunt SP. Altered nociception, analgesia and aggression in mice lacking the receptor for substance P. Nature. 1998;392:394–397. doi: 10.1038/32904. [DOI] [PubMed] [Google Scholar]
- Del Río J, Naranjo JR, Yang HY, Costa E. Substance P-induced release of Met5-enkephalin from striatal and periaqueductal gray slices. Brain Res. 1983;279:121–126. doi: 10.1016/0006-8993(83)90169-5. [DOI] [PubMed] [Google Scholar]
- de Novellis V, Marabese I, Palazzo E, Rossi F, Berrino L, Rodella L, Bianchi R, Maione S. Group I metabotropic glutamate receptors modulate glutamate and gamma-aminobutyric acid release in the periaqueductal grey of rats. Eur J Pharmacol. 2003;462:73–81. doi: 10.1016/s0014-2999(03)01342-6. [DOI] [PubMed] [Google Scholar]
- Drew GM, Vaughan CW. Multiple metabotropic glutamate receptor subtypes modulate GABAergic neurotransmission in rat periaqueductal grey neurons in vitro. Neuropharmacology. 2004;46:927–934. doi: 10.1016/j.neuropharm.2004.01.015. [DOI] [PubMed] [Google Scholar]
- Drew GM, Mitchell VA, Vaughan CW. Postsynaptic actions of substance P on rat periaqueductal grey neurons in vitro. Neuropharmacology. 2005;49:587–595. doi: 10.1016/j.neuropharm.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Drew GM, Mitchell VA, Vaughan CW. Glutamate spillover modulates GABAergic synaptic transmission in the rat midbrain periaqueductal grey via metabotropic glutamate receptors and endocannabinoid signaling. J Neurosci. 2008;28:808–815. doi: 10.1523/JNEUROSCI.4876-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields HL, Basbaum AI, Heinricher MM. Central nervous systems mechanisms of pain modulation. In: McMahon SB, Koltzenburg M, editors. Textbook of pain. Ed 5. Philadelphia: Elsevier, Churchill Livingston; 2006. pp. 125–142. [Google Scholar]
- Finn DP, Jhaveri MD, Beckett SR, Roe CH, Kendall DA, Marsden CA, Chapman V. Effects of direct periaqueductal grey administration of a cannabinoid receptor agonist on nociceptive and aversive responses in rats. Neuropharmacology. 2003;45:594–604. doi: 10.1016/s0028-3908(03)00235-1. [DOI] [PubMed] [Google Scholar]
- Frederickson RC, Burgis V, Harrell CE, Edwards JD. Dual actions of substance P on nociception: possible role of endogenous opioids. Science. 1978;199:1359–1362. doi: 10.1126/science.204012. [DOI] [PubMed] [Google Scholar]
- Galante M, Diana MA. Group I metabotropic glutamate receptors inhibit GABA release at interneuron-Purkinje cell synapses through endocannabinoid production. J Neurosci. 2004;24:4865–4874. doi: 10.1523/JNEUROSCI.0403-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack SP, Bagley EE, Chieng BC, Christie MJ. Induction of delta-opioid receptor function in the midbrain after chronic morphine treatment. J Neurosci. 2005;25:3192–3198. doi: 10.1523/JNEUROSCI.4585-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- Holden JE, Pizzi JA, Jeong Y. An NK1 receptor antagonist microinjected into the periaqueductal gray blocks lateral hypothalamic-induced antinociception in rats. Neurosci Lett. 2009;453:115–119. doi: 10.1016/j.neulet.2009.01.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Improta G, Broccardo M. Effects of supraspinal administration of PG-SPI and PG-KII, two amphibian tachykinin peptides, on nociception in the rat. Peptides. 2000;21:1611–1616. doi: 10.1016/s0196-9781(00)00292-8. [DOI] [PubMed] [Google Scholar]
- Jung KM, Mangieri R, Stapleton C, Kim J, Fegley D, Wallace M, Mackie K, Piomelli D. Stimulation of endocannabinoid formation in brain slice cultures through activation of group I metabotropic glutamate receptors. Mol Pharmacol. 2005;68:1196–1202. doi: 10.1124/mol.105.013961. [DOI] [PubMed] [Google Scholar]
- Kishimoto K, Koyama S, Akaike N. Synergistic mu-opioid and 5-HT1A presynaptic inhibition of GABA release in rat periaqueductal gray neurons. Neuropharmacology. 2001;41:529–538. doi: 10.1016/s0028-3908(01)00100-9. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, Colmers WF. 4-Aminopyridine and low Ca2+ differentiate presynaptic inhibition mediated by neuropeptide Y, baclofen and 2-chloroadenosine in rat hippocampal CA1 in vitro. Br J Pharmacol. 1992;105:470–474. doi: 10.1111/j.1476-5381.1992.tb14277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kombian SB, Ananthalakshmi KV, Parvathy SS, Matowe WC. Dopamine and adenosine mediate substance P-induced depression of evoked IPSCs in the rat nucleus accumbens in vitro. Eur J Neurosci. 2003;18:303–311. doi: 10.1046/j.1460-9568.2003.02753.x. [DOI] [PubMed] [Google Scholar]
- Kuwajima M, Hall RA, Aiba A, Smith Y. Subcellular and subsynaptic localization of group I metabotropic glutamate receptors in the monkey subthalamic nucleus. J Comp Neurol. 2004;474:589–602. doi: 10.1002/cne.20158. [DOI] [PubMed] [Google Scholar]
- Lafourcade M, Elezgarai I, Mato S, Bakiri Y, Grandes P, Manzoni OJ. Molecular components and functions of the endocannabinoid system in mouse prefrontal cortex. PLoS ONE. 2007;2:e709. doi: 10.1371/journal.pone.0000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman AH, Cook SA, Martin BR. Investigation of brain sites mediating cannabinoid-induced antinociception in rats: evidence supporting periaqueductal gray involvement. J Pharmacol Exp Ther. 1996;276:585–593. [PubMed] [Google Scholar]
- Liu R, Ding Y, Aghajanian GK. Neurokinins activate local glutamatergic inputs to serotonergic neurons of the dorsal raphe nucleus. Neuropsychopharmacology. 2002;27:329–340. doi: 10.1016/S0893-133X(02)00305-6. [DOI] [PubMed] [Google Scholar]
- Lujan R, Nusser Z, Roberts JD, Shigemoto R, Somogyi P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–475. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- Malick JB, Goldstein JM. Analgesic activity of substance P following intracerebral administration in rats. Life Sci. 1978;23:835–844. doi: 10.1016/0024-3205(78)90518-0. [DOI] [PubMed] [Google Scholar]
- Martin WJ, Patrick SL, Coffin PO, Tsou K, Walker JM. An examination of the central sites of action of cannabinoid-induced antinociception in the rat. Life Sci. 1995;56:2103–2109. doi: 10.1016/0024-3205(95)00195-c. [DOI] [PubMed] [Google Scholar]
- Mileusnic D, Lee JM, Magnuson DJ, Hejna MJ, Krause JE, Lorens JB, Lorens SA. Neurokinin-3 receptor distribution in rat and human brain: an immunohistochemical study. Neuroscience. 1999;89:1269–1290. doi: 10.1016/s0306-4522(98)00349-2. [DOI] [PubMed] [Google Scholar]
- Mohrland JS, Gebhart GF. Substance P-induced analgesia in the rat. Brain Res. 1979;171:556–559. doi: 10.1016/0006-8993(79)91062-x. [DOI] [PubMed] [Google Scholar]
- Morris R, Cheunsuang O, Stewart A, Maxwell D. Spinal dorsal horn neurone targets for nociceptive primary afferents: do single neurone morphological characteristics suggest how nociceptive information is processed at the spinal level. Brain Res Rev. 2004;46:173–190. doi: 10.1016/j.brainresrev.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Nakaya Y, Kaneko T, Shigemoto R, Nakanishi S, Mizuno N. Immunohistochemical localization of substance P receptor in the central nervous system of the adult rat. J Comp Neurol. 1994;347:249–274. doi: 10.1002/cne.903470208. [DOI] [PubMed] [Google Scholar]
- Négyessy L, Vidnyánszky Z, Kuhn R, Knöpfel T, Görcs TJ, Hámori J. Light and electron microscopic demonstration of mGluR5 metabotropic glutamate receptor immunoreactive neuronal elements in the rat cerebellar cortex. J Comp Neurol. 1997;385:641–650. doi: 10.1002/(sici)1096-9861(19970908)385:4<641::aid-cne9>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Ogawa S, Kow LM, Pfaff DW. Effects of lordosis-relevant neuropeptides on midbrain periaqueductal gray neuronal activity in vitro. Peptides. 1992;13:965–975. doi: 10.1016/0196-9781(92)90058-b. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Shosaku J, Tsubokawa H, Kano M. Cooperative endocannabinoid production by neuronal depolarization and group I metabotropic glutamate receptor activation. Eur J Neurosci. 2002;15:953–961. doi: 10.1046/j.1460-9568.2002.01929.x. [DOI] [PubMed] [Google Scholar]
- Osborne PB, Vaughan CW, Wilson HI, Christie MJ. Opioid inhibition of rat periaqueductal grey neurones with identified projections to rostral ventromedial medulla in vitro. J Physiol. 1996;490:383–389. doi: 10.1113/jphysiol.1996.sp021152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Rosén A, Brodin K, Eneroth P, Brodin E. Short-term restraint stress and s.c. saline injection alter the tissue levels of substance P and cholecystokinin in the peri-aqueductal grey and limbic regions of rat brain. Acta Physiol Scand. 1992;146:341–348. doi: 10.1111/j.1748-1716.1992.tb09428.x. [DOI] [PubMed] [Google Scholar]
- Rosén A, Zhang YX, Lund I, Lundeberg T, Yu LC. Substance P microinjected into the periaqueductal gray matter induces antinociception and is released following morphine administration. Brain Res. 2004;1001:87–94. doi: 10.1016/j.brainres.2003.11.060. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Nomura S, Ohishi H, Sugihara H, Nakanishi S, Mizuno N. Immunohistochemical localization of a metabotropic glutamate receptor, mGluR5, in the rat brain. Neurosci Lett. 1993;163:53–57. doi: 10.1016/0304-3940(93)90227-c. [DOI] [PubMed] [Google Scholar]
- Silva E, Hernandez L, Contreras Q, Guerrero F, Alba G. Noxious stimulation increases glutamate and arginine in the periaqueductal gray matter in rats: a microdialysis study. Pain. 2000;87:131–135. doi: 10.1016/S0304-3959(00)00275-X. [DOI] [PubMed] [Google Scholar]
- Stewart JM, Getto CJ, Neldner K, Reeve EB, Krivoy WA, Zimmermann E. Substance P and analgesia. Nature. 1976;262:784–785. doi: 10.1038/262784a0. [DOI] [PubMed] [Google Scholar]
- Szabo B, Urbanski MJ, Bisogno T, Di Marzo V, Mendiguren A, Baer WU, Freiman I. Depolarization-induced retrograde synaptic inhibition in the mouse cerebellar cortex is mediated by 2-arachidonoylglycerol. J Physiol. 2006;577:263–280. doi: 10.1113/jphysiol.2006.119362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sañudo-Peña MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- Uchigashima M, Narushima M, Fukaya M, Katona I, Kano M, Watanabe M. Subcellular arrangement of molecules for 2-arachidonoyl-glycerol-mediated retrograde signaling and its physiological contribution to synaptic modulation in the striatum. J Neurosci. 2007;27:3663–3676. doi: 10.1523/JNEUROSCI.0448-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentino RJ, Bey V, Pernar L, Commons KG. Substance P acts through local circuits within the rat dorsal raphe nucleus to alter serotonergic neuronal activity. J Neurosci. 2003;23:7155–7159. doi: 10.1523/JNEUROSCI.23-18-07155.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE. Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci. 2001;21:RC188. doi: 10.1523/JNEUROSCI.21-24-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Christie MJ. Presynaptic inhibitory action of opioids on synaptic transmission in the rat periaqueductal grey in vitro. J Physiol. 1997;498:463–472. doi: 10.1113/jphysiol.1997.sp021872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Connor M, Bagley EE, Christie MJ. Actions of cannabinoids on membrane properties and synaptic transmission in rat periaqueductal gray neurons in vitro. Mol Pharmacol. 2000;57:288–295. [PubMed] [Google Scholar]
- Walker JM, Huang SM, Strangman NM, Tsou K, Sañudo-Peña MC. Pain modulation by release of the endogenous cannabinoid anandamide. Proc Natl Acad Sci U S A. 1999;96:12198–12203. doi: 10.1073/pnas.96.21.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin L, Geller EB, Liu-Chen LY, Chen C, Adler MW. Substance P release in the rat periaqueductal gray and preoptic anterior hypothalamus after noxious cold stimulation: effect of selective mu and kappa opioid agonists. J Pharmacol Exp Ther. 1997;282:1055–1063. [PubMed] [Google Scholar]
- Yang K, Furue H, Kumamoto E, Dong YX, Yoshimura M. Pre- and postsynaptic inhibition mediated by GABA(B) receptors in rat ventrolateral periaqueductal gray neurons. Biochem Biophys Res Commun. 2003;302:233–237. doi: 10.1016/s0006-291x(03)00156-6. [DOI] [PubMed] [Google Scholar]
- Yip J, Chahl LA. Localization of NK1 and NK3 receptors in guinea-pig brain. Regul Pept. 2001;98:55–62. doi: 10.1016/s0167-0115(00)00228-7. [DOI] [PubMed] [Google Scholar]