

Abstract
Opioid signaling in the CNS is critical for controlling cellular excitability, yet the conditions under which endogenous opioid peptides are released and the precise mechanisms by which they affect synaptic transmission remain poorly understood. The opioid peptide dynorphin is present in the soma and dendrites of vasopressin neurons in the hypothalamus and dynamically controls the excitability of these cells in vivo. Here, we show that dynorphin is released from dendritic vesicles in response to postsynaptic activity and acts in a retrograde manner to inhibit excitatory synaptic transmission. This inhibition, which requires the activation of κ-opioid receptors, results from a reduction in presynaptic release of glutamate vesicles. The opioid inhibition is downstream of Ca2+ entry and is likely mediated by a direct modulation of presynaptic fusion machinery. These findings demonstrate that neurons may self-regulate their excitability through the dendritic release of opioids to inhibit excitatory synaptic transmission.
Introduction
Neurotransmitters released from the somatodendritic compartment of the postsynaptic cell can act in a retrograde manner to induce short- or long-term changes in synaptic efficacy (Alger, 2002). These retrograde signals are chemically diverse, are recruited under different conditions, and have distinct effects on the presynaptic terminal (Ludwig and Pittman, 2003). The opioid peptide, dynorphin, powerfully inhibits excitatory transmission in the nervous system (Weisskopf et al., 1993; Castillo et al., 1996; Simmons and Chavkin, 1996; Hjelmstad and Fields, 2003; Honda et al., 2004; Ford et al., 2007). In stark contrast to the wealth of data implicating a role for exogenous dynorphin in modulating synaptic transmission, there are only a few examples, and only in the hippocampus, of dynorphin acting as a retrograde messenger (Wagner et al., 1993). Furthermore, the mechanisms responsible for the dynorphin-mediated presynaptic inhibition remain contentious.
In vivo evidence indicates that endogenous dynorphin plays a key role in regulating the excitability of neurons in the hypothalamus, which in turn control the release of neurohormones from the posterior pituitary (Wells and Forsling, 1991; Brown et al., 1998). Specifically, this work has focused on the magnocellular neurosecretory cells (MNCs) in the paraventricular (PVN) and supraoptic (SON) nuclei of the hypothalamus. The MNCs make either oxytocin (OT) or vasopressin (VP), but it is these latter cells in particular that have one of the highest concentrations of somatodendritically stored dynorphin in the brain (Watson et al., 1982; Shuster et al., 2000). Blocking κ-opioid receptors in vivo dramatically increases the excitability of MNCs (Wells and Forsling, 1991; Brown et al., 1998), indicating an ongoing and important role for dynorphin in regulating the output of this network. Since spiking in MNCs in vivo, both tonically (Nissen et al., 1995) and in response to physiological stimuli (Brown et al., 2004), is dependent on excitatory, glutamatergic synaptic inputs, we hypothesized that the retrograde modulation of glutamate neurotransmission by dynorphin would be an efficient way for MNCs to control their own activity. Since MNCs are targeted by glutamate synapses that exhibit a high rate of stochastic, action potential independent release, we hypothesized that dynorphin should inhibit both spontaneous and action potential-dependent neurotransmission.
Using whole-cell patch-clamp recordings from MNCs, we find that bath application of dynorphin A or the κ-opioid receptor agonist U69593 produces a robust and long-lasting presynaptic depression of glutamate neurotransmission. This depression appears to be mediated by a direct inhibition of presynaptic vesicle fusion. Importantly, we demonstrate that this depression is also observed in response to the release of endogenous dynorphin, specifically from VP neurons. Together, these data demonstrate that dynorphin released from the dendrites of vasopressin magnocellular neurons in an activity-dependent manner induces a novel presynaptic depression that targets both action potential-dependent and action potential-independent glutamate release.
Materials and Methods
All protocols were approved by the University of Calgary animal care and use committee in accordance with guidelines established by the Canadian Council on Animal Care.
Slice preparation.
Male wild-type Sprague Dawley [postnatal day 21 (p21)–30] or transgenic Wistar/Sprague Dawley (p21–40) rats that express enhanced green fluorescence protein (eGFP) under the vasopressin promoter were used (Ueta et al., 2005). Animals were anesthetized with sodium pentobarbital (0.1 ml/100 g body weight) and then decapitated. The brain was quickly removed and placed in ice-cold slicing solution for several minutes containing (in mm): 87 NaCl, 2.5 KCl, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 glucose, 75 sucrose; saturated with 95% O2/5% CO2. The brain was then blocked and mounted on a vibrating slicer (Leica) submerged in ice-cold slicing solution. Slices were incubated at 32.5°C in artificial CSF (ACSF) containing (in mm) 126 NaCl, 2.5 KCl, 26 NaHCO3, 2.5 CaCl2, 1.5 MgCl2, 1.25 NaH2PO4, 10 glucose; saturated with 95% O2/5% CO2, for a minimum of 60 min. When Ca2+ was decreased in the ACSF, Mg2+ was increased by the same proportion.
Electrophysiology.
Slices containing the PVN were submerged in a recording chamber and superfused with 32.5°C artificial CSF at a flow rate of 1 ml/min. Whole-cell recordings were obtained from magnocellular neurons visualized with an AxioskopII FS Plus (Zeiss) upright microscope fitted with infrared differential interference contrast optics. Recorded cells were confirmed to be MNCs based on their morphology and well defined electrophysiological characteristics (Luther and Tasker, 2000). eGFP neurons were visualized with a Zeiss AxioCam MRm camera (Zeiss).
Patch pipettes were pulled from borosilicate glass, had a resistance between 3–6 MΩ, and were filled with a solution containing (in mm) 116 potassium gluconate, 8 sodium gluconate, 2 MgCl2, 8 KCl, 1 potassium EGTA, 4 potassium ATP, and 0.3 sodium GTP, 10 HEPES, corrected to pH 7.2 with KOH. Series resistance was not compensated and recordings were accepted for analysis if changes in access resistance were <20%. The liquid junction potential was calculated to be approximately −13 mV and was not compensated for.
Glutamatergic fibers were stimulated extracellularly with a monopolar glass microelectrode (3–6 MΩ) filled with ACSF and placed either within or just outside of the PVN. Synaptic stimulation was delivered at a rate of 0.2 Hz for all experiments with the exception of endogenous dynorphin release experiments, where it was delivered at 0.4 Hz. The perfusate always contained picrotoxin (100 μm) to block GABAA-mediated conductances. We have previously reported that evoked glutamatergic currents onto MNCs have both synchronous and asynchronous components (Iremonger and Bains, 2007). In this current study, we generally selected synaptic responses that exhibited more pronounced asynchronous release. Drugs used in this study were purchased from the following locations: ω-conotoxin GVIA, ω-agatoxin IVA, tetrodotoxin (TTX), and α-dendrotoxin from Alomone labs; N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251) and 4-aminopyridine (4-AP) from Tocris Bioscience; N-[2-((p-bromocinnamyl) amino)ethyl]-5-isoquinolinesulfonamide (H-89) and botulinum toxin C (BoTC) from Calbiochem; dynorphin A(1–17) (DynA) from Bachem. All remaining drugs and chemicals were purchased from Sigma-Aldrich. In most cases, (+)-(5α,7α,8β)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]-benzeneacetamide (U69593) was applied at 1 μm for 5 min. In experiments where percentage inhibition was compared between OT and VP cells, U69593 was applied at 500 nm for 1 min.
Data collection and analysis.
Signals were amplified with the Multiclamp 700B amplifier (Molecular Devices) were low-pass filtered at 1 kHz, digitized at 10–20 kHz with the Digidata 1322 (Molecular Devices), and saved on a PC for off-line analysis. For evoked currents, analysis was performed using pClamp 9 (Molecular Devices). To better quantify synaptic efficacy at these synapses that have both fast and slow components of release, we measured charge transfer rather than peak amplitude. For all experiments [except for calculating paired-pulse ratio (PPR)], charge transfer was calculated by integrating the area under the averaged evoked EPSC from the onset of the EPSC to 100 ms. This time window generally encompasses all asynchronous release events (Iremonger and Bains, 2007). For calculating the PPR of charge transfer, the area under the averaged evoked EPSC from the onset to 45 ms was integrated. The number of individual asynchronous release events was quantified by counting the occurrence of miniature EPSCs (mEPSCs) from 5 to 100 ms after the onset of the EPSC (5 ms bins) during 30 trials. Events were not counted in the first 5 ms, because they could not be discriminated from the synchronous component. During the 5–10 ms time window (decay of the synchronous EPSC), we could clearly discern individual quanta, and these were counted as asynchronous events. Baseline spontaneous release was calculated for the 100 ms before the stimulus and subtracted. For amplitude analysis of asynchronous events, only events with a clear baseline before onset were selected, and those that were summed onto other events were rejected from analysis.
For miniature events, analysis was performed with Mini Analysis software (Synaptosoft). Miniature frequency was measured in events per minute, and amplitude was measured as the average amplitude in 1 min bins. All miniature data were normalized to the average of 5 min baseline immediately before drug application. For all data, control and treatment time points were generally taken at 5 min before drug application and 15–20 min after start of application. All data are presented as mean ± SEM, and statistical analyses were performed with Student's t test when comparing two groups and with ANOVA with a post hoc Newman–Keuls test for comparisons across multiple groups. p < 0.05 was accepted as statistically significant (*p < 0.05).
Results
To investigate the effects of both exogenous and endogenous dynorphin on synaptic transmission, we performed whole-cell recordings from MNCs in acute brain slices of the PVN (Fig. 1A). EPSCs were recorded at a holding potential of −60 mV in the presence of the GABAA receptor antagonist, picrotoxin (100 μm).
Figure 1.
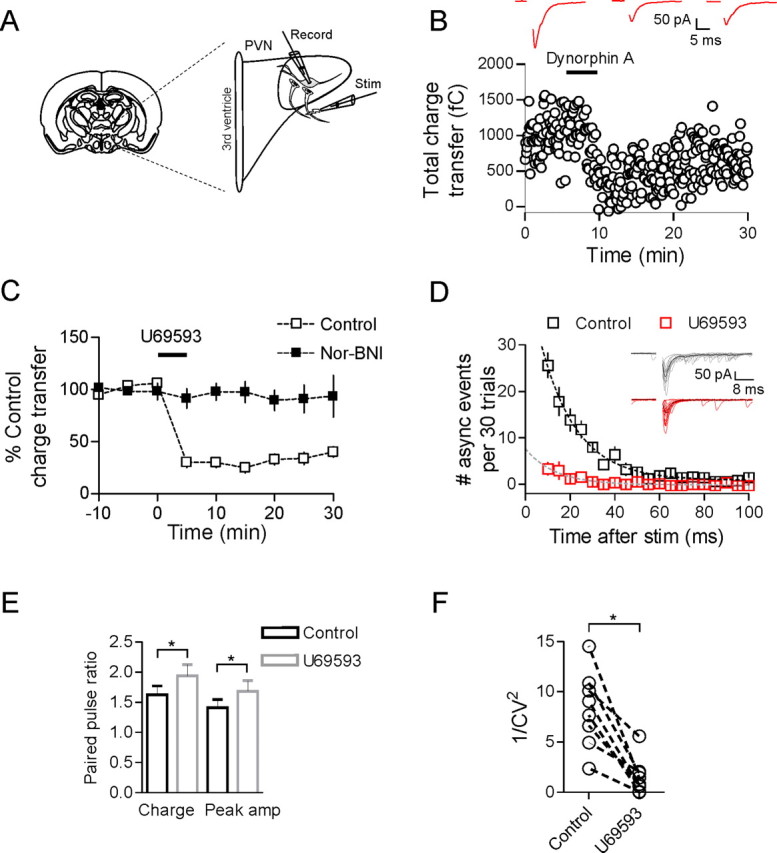
Dynorphin inhibits evoked glutamate transmission onto MNCs. A, Schematic showing a coronal brain slice at the level of the PVN. Enlarged area shows a diagram of the stimulation and recording configuration. B, Bath application of 1 μm dynorphin A(1–17) for 5 min produced a large and sustained inhibition of the EPSC (50 ± 9% inhibition of charge transfer 15 min after DynA, n = 8, p = 0.002). Sample EPSCs are the average of 30 trials from control, 10 and 25 min after DynA application. C, The κ-opioid receptor agonist U69593 (1 μm, 5 min) also produced a similar depression of the evoked EPSC (75 ± 4% inhibition, n = 9, p < 0.0001) that was also blocked by nor-BNI (2 ± 10% inhibition, n = 8, p = 0.67). D, There is a marked reduction in the number of asynchronous release events after U69593. Dashed lines are single exponential fits from the data (n = 5). Traces in D are 15 sweeps overlaid taken from control and after U69593; dark traces indicate the average EPSC. E, There is an increase in PPR measured for both amplitude and charge transfer (see Materials and Methods) after activation of κ-opioid receptors with U69593. F, The 1/CV2 of the EPSC charge transfer is decreased dramatically 15 min after U69593.
Dynorphin inhibits evoked glutamate neurotransmission
Evoked glutamate inputs onto MNCs in PVN exhibit pronounced asynchronous release, the characteristics of which have been described previously (Iremonger and Bains, 2007). To examine the effects of dynorphin on excitatory synaptic transmission, we bath applied either 1 μm DynA or 1 μm U69593, a specific agonist of κ-opioid receptors, for 5 min. DynA caused a pronounced inhibition of the evoked EPSC (percentage inhibition of charge transfer: 50 ± 9% 15 min after DynA, n = 8, p = 0.002) (Fig. 1B). This DynA-mediated inhibition was completely abolished in the presence of the κ-opioid receptor antagonist nor-binaltorphimine (nor-BNI) (100 nm, percentage inhibition in nor-BNI: 5 ± 9%, n = 7, p = 0.28), confirming that it was mediated by activation of κ-opioid receptors. Similarly, bath application of U69593 (1 μm, 5 min) produced a robust and prolonged inhibition of the EPSC (percentage inhibition: 75 ± 4%, n = 9, p < 0.0001) (Fig. 1C) that was also blocked by nor-BNI (percentage inhibition: 2 ± 10%, n = 8, p = 0.67) (Fig. 1C). Although the DynA peptide exhibits high affinity for the κ-opioid receptor (Chavkin et al., 1982), there are also reports that it binds (with lower affinity) to μ- and δ-opioid receptors (Raynor et al., 1994). Consequently, U69593 was used for the remainder of the experiments requiring exogenous ligand.
Although dynorphin is primarily released from the dendrites of VP cells (Watson et al., 1982; Shuster et al., 2000), the intermingling of processes from both OT and VP cells raises the possibility that synaptic transmission onto both OT and VP cells may be affected by dynorphin. To determine whether activation of κ-opioid receptors had differential effects depending on cell type in the PVN, we used a transgenic rat that expresses eGFP under the vasopressin promoter (Ueta et al., 2005). In these animals, the inhibitory effect of a lower concentration of U69593 (500 nm, 1 min) was found to be similar in both GFP-negative (putative OT) and GFP-positive (VP) cells (percentage inhibition VP: 40 ± 16%, n = 4; OT: 43 ± 26%, n = 5, p = 0.92). Therefore, for the remainder of the experiments (except Figs. 7, 8), wild-type rats were used, and we did not distinguish between OT and VP cells.
Figure 7.

Postsynaptic depolarization induces a presynaptic depression. Whole-cell recordings were obtained from GFP-positive (vasopressin) neurons. With repetitive postsynaptic depolarization (black bar), the evoked EPSC depresses and remains depressed for the duration of the recording (A, B; 53 ± 8% of control, p = 0.012, n = 6). Sample EPSCs are taken from control immediately after and 20 min after the end of the depolarizing protocol. When spontaneous EPSCs were analyzed, there was a decrease in sEPSC frequency after the end of the depolarizing protocol (C; 73 ± 7% of control, p = 0.024, n = 6). Finally, there was a decrease in 1/CV2 of the evoked EPSC after the postsynaptic depolarization that persisted for as long as the depression (D; p = 0.006, n = 6).
Figure 8.

Dendritic dynorphin release is responsible for retrograde depression. When the postsynaptic depolarizing protocol (black bar) was repeated in the presence of the κ-opioid receptor antagonist, nor-BNI (100 nm), it no longer induced synaptic depression (A, B; 109 ± 20% control, p = 0.389, n = 5). Sample traces are taken from control, 10 and 20 min after the end of the depolarizing protocol. Coincidentally, there was also no change in sEPSC frequency or amplitude in the presence of nor-BNI (C, D; sEPSC frequency: 99 ± 7% control, p = 0.675, n = 4; sEPSC amplitude: 103 ± 3% control, p = 0.214, n = 4). When postsynaptic syntaxin was cleaved by including botulinum toxin C in the patch pipette, the synaptic depression was also no longer observed (E; 89 ± 12% of control, p = 0.493, n = 4). Finally, eGFP-positive (putative oxytocin) neurons were patched to determine whether this dynorphin-mediated retrograde inhibition was limited to vasopressin-expressing cells. The same postsynaptic depolarizing protocol was delivered to eGFP-positive cells; however, the depression was significantly less than that in GFP plus ve cells (n = 6, p = 0.015).
To obtain insight into the mechanisms of the κ-opioid-mediated inhibition, we took advantage of the fact that these synapses exhibit pronounced asynchronous release (Iremonger and Bains, 2007) (Fig. 1B,E) and specifically analyzed the effect of κ-opioid agonist on this delayed component of the EPSC. Application of U69593 resulted in a robust decrease in the number of asynchronous events (occurring between 5–100 ms after the onset of the synchronous EPSC) (total number of asynchronous events: control, 107 ± 16; U69593, 10 ± 5; p = 0.002; n = 5) (Fig. 1D) without changing the mean amplitude of these events (control, 31.8 ± 5.3 pA; U69593, 29.6 ± 6.8 pA; p = 0.76; n = 5). This is consistent with a presynaptic reduction in transmitter release with no change in postsynaptic efficacy. To provide more evidence for this hypothesis, we measured the PPR for two EPSCs evoked 50 ms apart, both before and after U69593. PPR was calculated for both the peak amplitude and charge transfer of the EPSC (see Materials and Methods). Peak amplitude PPR increased from 1.41 ± 0.13 to 1.68 ± 0.18 after U69593 (n = 19, p = 0.049), whereas charge transfer PPR increased from 1.63 ± 0.15 to 1.94 ± 0.19 after U69593 (n = 19, p = 0.014) (Fig. 1E). In addition, when the coefficient of variation (CV) of the EPSC charge transfer was calculated and plotted as 1/CV2, there was a significant reduction after application of U69593 (p = 0.001, n = 8) (Fig. 1F).
Together, these data suggest that activation of κ-opioid receptors causes a robust inhibition of glutamate release onto both OT- and VP-expressing MNCs and that this inhibition is mediated via a presynaptic mechanism.
Activation of κ-opioid receptors inhibits action potential-independent neurotransmission
Glutamatergic synapses onto MNCs exhibit a high rate of quantal release (Gordon and Bains, 2003). The regulation of these mEPSCs is thought to be important in controlling excitability in these cells (Gordon and Bains, 2003). In the presence of TTX (1 μm), mEPSC frequency and amplitude were 45.2 ± 5.8 events/min and 25.0 ± 1.8 pA, respectively (n = 6). After the application of U69593 (1 μm, 5 min), there was a robust reduction in mEPSC frequency (49 ± 14% reduction at 15 min, p < 0.0001, n = 6) (Fig. 2A,B) with no change in mEPSC amplitude (105 ± 4% control, p = 0.29, n = 6) (Fig. 2C,D). To determine if a distinct pool of miniature events were being selectively inhibited, we analyzed both cumulative distribution plots and mEPSC rise/decay times. In the representative cell shown in Figure 2C, the mEPSC amplitude distribution was identical before and after application of U69593. In addition, there was no change in the mean rise or decay times after U69593 (rise time: control vs U69593, p = 0.83, n = 6; decay time: control vs U69593, p = 0.50, n = 6) (Fig. 2E). These data suggest that the inhibition by U69593 does not target a specific group of mEPSC events with distinct amplitudes or rise/decay times.
Figure 2.

Activation of κ-opioid receptors decreases mEPSC frequency without affecting amplitude. A, Cumulative distribution plot from a single cell showing the increase in mEPSC interevent interval after U69593 (1 μm, 5 min). B, On average, there is a 49 ± 14% reduction in mEPSC frequency after U69593 (at 15 min, p < 0.0001, n = 6). C, Cumulative distribution plot from a single cell showing no change in the mEPSC amplitude distribution after U69593. Mean data are shown in D (n = 6). Traces and cumulative distributions in A and C are taken from the same cell in control and 15 min after U69593. E, There was no change in either the 10–90 rise time or the decay tau of mEPSCs after U69593. F, The decrease in mEPSC frequency after bath application of U69593 persisted even when all Ca2+ was removed by incubating slices in 0 mm Ca2+/4 mm Mg2+, 100 μm EGTA with 50 μm BAPTA-AM.
Finally, since the frequency of mEPSCs recorded in MNCs is independent of external Ca2+ (Inenaga et al., 1998), we next tested whether the κ-opioid inhibition of mEPSC frequency persisted in the absence of Ca2+. To remove Ca2+, slices were incubated in 0 mm Ca2+/4 mm Mg2+, 100 μm EGTA, and 50 μm BAPTA-AM for 1–2 h. During whole-cell recordings, slices were perfused continually with 0 mm external Ca2+ and 100 μm EGTA. Baseline mEPSC frequency and amplitude were not different from control cells (frequency 0 Ca2+: 73.7 ± 20.1 events/min, n = 8, p = 0.26 vs control; amplitude 0 Ca2+: 33.0 ± 3.1 pA, n = 8, p = 0.06 vs control). In addition, the inhibition of mEPSC frequency by U69593 was unaffected by the removal of Ca2+ (45.4 ± 12.4% reduction in mEPSC frequency, n = 8, p = 0.85 vs control reduction) (Fig. 2F). mEPSC amplitude remained unchanged after U69593 (103 ± 4% of control amplitude, n = 8).
These data show that direct activation of κ-opioid receptors inhibits mEPSC frequency without affecting mEPSC amplitude or rise/decay kinetics. In addition, this inhibition persists in the absence of Ca2+. Together with the PPR and 1/CV2 EPSC data (Fig. 1), these data provide further support for the hypothesis that κ-opioid receptors inhibit excitatory neurotransmission via a presynaptic inhibition of glutamate release.
κ-Opioid-mediated inhibition of glutamate release is not attributable to inhibition of adenylate cyclase/protein kinase A
The κ-opioid receptor is a G-protein-coupled receptor of the Gi/o family. Once activated, the α subunit can inhibit adenylate cyclase while the liberated βγ subunit can act to inhibit Ca2+ channels (Herlitze et al., 1996) and directly interfere with vesicle fusion (Blackmer et al., 2001). To determine whether κ-opioid receptor inhibition of adenylate cyclase and protein kinase A (PKA) was responsible for the reduction in glutamate release, we bath applied the adenylate cyclase activator, forskolin (10 μm). Bath application of forskolin increased EPSC charge transfer (157 ± 28% of control after 30 min, n = 5) (Fig 3A). This, however, had no effect on the subsequent U69593-induced inhibition (80.5 ± 7.0% inhibition, p = 0.48 vs percentage control inhibition, n = 5) (Fig. 3B). We also tested whether directly inhibiting adenylate cyclase could block the effects of U69593. To test this, slices were incubated for a minimum of 40 min in the adenylate cyclase inhibitor SQ22536 (10 μm). This had no effect on the U69593-mediated inhibition (percentage inhibition in SQ22536: 87 ± 5%, n = 5, p = 0.093 vs control U69593 inhibition) (Fig. 3D). Finally, we investigated the involvement of PKA in the κ-opioid-mediated inhibition by applying U69593 to slices that had been incubated in the PKA inhibitor H-89 (100 μm) for 40–120 min. After incubation in H-89, U69593 still produced a significant inhibition of release that was similar to inhibition in control slices (H-89, n = 5: 84 ± 6% inhibition at 15 min, p = 0.25 vs control inhibition) (Fig. 3C). To ensure that H-89 was inhibiting PKA, we applied the adenylate cyclase activator, forskolin (10 μm), to either control slices or slices that had been incubated in H-89. Forskolin produced a dramatic increase in mEPSC frequency in control slices (800 ± 265% baseline frequency, n = 5), but this effect was attenuated in H-89-treated slices (154 ± 8% baseline frequency, n = 4), confirming the efficacy of this PKA inhibitor.
Figure 3.

cAMP/PKA pathways are not responsible for the κ-opioid-mediated inhibition. A, Bath application of forskolin (10 μm) increases EPSC charge transfer. Subsequent application of U69593 (1 μm, 5 min) inhibited the EPSC comparable with control (80.5 ± 7.0% inhibition, n = 5, p = 0.48 vs control inhibition; B). C, To inhibit PKA, slices were incubated in ACSF containing H-89 (100 μm) for between 40–120 min. After this incubation, U69593 was still equally effective at inhibiting evoked EPSCs (H-89: n = 5, 84 ± 6% inhibition at 15 min, p = 0.25 vs control inhibition). Sample EPSCs are from control, 15 and 30 min after U69593 application. D summarizes the ability of various pharmacological agents to prevent the dynorphin A- or U69593-mediated inhibition of the EPSC.
Together, these data demonstrate that inhibition of glutamate release mediated by activation of the κ-opioid receptor does not require the inhibition of the adenylate cyclase/PKA pathway.
κ-Opioid inhibition persists after antagonism of presynaptic Ca2+ channels
We have previously reported that glutamate release onto MNCs in the PVN is asynchronous, with transmitter release persisting for up to 100 ms after a single presynaptic action potential (Iremonger and Bains, 2007). In other systems, this form of transmitter release relies more heavily on Ca2+ influx through N-type Ca2+ channels rather than P/Q-type channels (Fedchyshyn and Wang, 2005; Hefft and Jonas, 2005). We first determined the contribution of different Ca2+ channel subtypes to transmitter release at glutamate synapses onto PVN MNCs. Then, we asked whether the depression observed in response to activation of κ-opioid receptors persisted after inhibition of these presynaptic Ca2+ channels.
The N-type Ca2+ channel antagonist ω-conotoxin GVIA (1 μm, 5 min) produced a pronounced inhibition of the EPSC (77 ± 6% inhibition at 20 min, n = 8, p = 0.01 compared with baseline charge transfer) (Fig. 4A). The P/Q-type antagonist ω-agatoxin IVA (200 nm, 10 min), however, produced only a small inhibition of evoked release (16 ± 12% inhibition at 25 min, n = 6, p = 0.21 compared with baseline) (Fig. 4B). The toxin concentrations used above have previously been shown to be saturating for both N- and P-type channels (Mintz et al., 1992; Boland et al., 1994). Based on the above data, we performed an occlusion experiment to determine if previous inhibition of N-type or P/Q-type calcium channels would attenuate the effects of U69593. Bath application of ω-conotoxin GVIA dramatically inhibited the EPSC; however, subsequent application of U69593 (n = 4) (Fig. 4C–E) still inhibited the EPSC by about the same percentage (p = 0.68, comparing percentage inhibition control vs percentage inhibition after ω-conotoxin) (Fig. 4C–E). Similarly, the inhibition in response to U69593 was not affected by previous application of ω-agatoxin IVA (n = 4, p = 0.95) (Fig. 4F). Together, these data indicate that asynchronous glutamate release is highly reliant on Ca2+ influx through N-type channels, consistent with asynchronous release in other areas of the brain. However, the inhibition of glutamate release after activation of κ-opioid receptors persists after inhibition of N- or P/Q-type Ca2+ channels. These data are in line with our observation that activation of κ-opioid receptors also reduced mEPSC frequency and that this inhibition persisted in the absence of Ca2+ (Fig. 2). Together, this suggests that the presynaptic inhibition mediated by activation of κ-opioid receptors is downstream of Ca2+ entry.
Figure 4.

κ-Opioid inhibition persists after antagonism of presynaptic Ca2+ channels. A, Evoked glutamate release onto MNCs is mediated mostly by N-type Ca2+ channels as bath application of the N-type Ca2+ channel blocker ω-conotoxin GVIA (1 μm, 5 min) reduced the charge transfer of the EPSC by 77 ± 6% (n = 8, p = 0.01). Sample EPSCs are from control, 15 and 30 min after ω-conotoxin. B, Blocking P/Q-type channels with ω-agatoxin IVA (200 nm, 10 min) reduced the EPSC charge transfer by only 16 ± 12% (n = 6, p = 0.21). Sample EPSCs are from control, 15 and 30 min after ω-agatoxin. C–F, When U69593 was applied to cells that had previously had either N- or P/Q-type channels blocked, there was no occlusion of the inhibitory effect of the κ-opioid agonist. C and D are data from the same cell. Traces in C were taken in control, 15 min after ω-conotoxin and 10 min after subsequent application of U69593. The inset in E shows the charge transfer after ω-conotoxin normalized to 100%.
Blocking potassium channels attenuates κ-opioid-mediated inhibition by increasing presynaptic Ca2+
In a number of different brain areas, the activation of μ- or κ-opioid receptors has been shown to inhibit transmitter release via activation of presynaptic potassium channels (Simmons and Chavkin, 1996; Vaughan et al., 1997; Finnegan et al., 2006). To determine if activation of a presynaptic potassium conductance is responsible for the inhibition of glutamate release observed in this current study, we applied the nonspecific potassium channel antagonist 4-AP. Bath application of 4-AP (2 mm) resulted in a large increase in the EPSC charge transfer (250 ± 74% at 20 min, n = 7, p = 0.028) (Fig. 5A). In the presence of 4-AP, the inhibition mediated by U69593 was significantly attenuated (28 ± 8% inhibition, n = 6, p < 0.001 compared with control inhibition) (Fig. 5A,B). Since high concentrations of 4-AP can have effects that are not specific to voltage-gated K+ channels (Solis and Nicoll, 1992), we repeated these experiments using 100 μm 4-AP. The effects of U69593 were attenuated under these conditions as well (U69593 inhibition in 100 μm 4-AP: 32 ± 13%, p = 0.003, n = 6) (Fig. 5B). This reduction in the κ-opioid inhibition could not be replicated with other potassium channel blockers such as tetraethylammonium, α-dendrotoxin, or Ba2+ (data not shown). To determine whether the effects of 4-AP are solely attributable to block of potassium channels or result from a secondary increase in presynaptic Ca2+ influx from a broader presynaptic action potential, we performed additional experiments in low external Ca2+. We again bath applied 100 μm 4-AP but then washed in an ACSF containing low Ca2+ (0.3 mm Ca2+/3.7 mm Mg2+), which subsequently decreased EPSC amplitude. Under these conditions, U69593 caused a robust inhibition of glutamate release that was not significantly different to control inhibition (79 ± 5% inhibition, n = 3, p = 0.61) (Fig. 5C,D). To confirm that potassium channels were not directly responsible for mediating the κ-opioid inhibition, we bath applied 100 μm 4-AP in the presence of TTX. 4-AP alone had no effect on mEPSC frequency (101 ± 21% control frequency after 15 min, n = 6, p = 0.87 compared with baseline) (Fig. 5E), and subsequent application of U69593 decreased mEPSC frequency to the same extent as in control (43 ± 9% reduction at 15 min, n = 9, p < 0.0001, compared with baseline) (Fig. 5F). These data indicate that activation of presynaptic potassium channels do not contribute directly to the κ-opioid inhibition. Rather, they hint at a process whereby glutamate neurotransmission can be restored under conditions that elevate presynaptic Ca2+.
Figure 5.

κ-Opioid-mediated inhibition does not require presynaptic potassium channels. A, Application of 2 mm 4-AP produces a robust increase in the evoked EPSC charge transfer (250 ± 74% at 20 min, n = 7, p = 0.028) and prevents U69593-mediated inhibition. EPSCs in A are taken from time points 10 min after 4-AP, 15 and 30 min after U69593 application. A lower concentration of 4-AP (100 μm) is also effective at preventing the U69593 inhibition (U69593 inhibition in 100 μm 4-AP: 32 ± 13%, p = 0.003 compared with control inhibition, n = 6). C, D, If a low Ca2+ containing ACSF is washed in 15 min after application of 100 μm 4-AP and followed by U69593, the κ-opioid-mediated inhibition is again unmasked (79 ± 5% inhibition, n = 3, p = 0.61). In C, low Ca2+ refers to 0.3 mm Ca2+/3.7 mm Mg2+ containing ACSF. E, There is no change in mEPSC frequency after bath application of 2 mm 4-AP (101 ± 21% control frequency after 15 min, n = 6, p = 0.87). In addition, after a minimum of 15 min application of 2 mm 4-AP, U69593 is still equally effective at decreasing mEPSC frequency (43 ± 9% reduction at 15 min, n = 9, p < 0.0001; F).
Increasing presynaptic Ca2+ overcomes κ-opioid-mediated inhibition
To test this idea directly, we used the Ca2+ ionophore, ionomycin (1 μm), to increase presynaptic Ca2+ independently of voltage-gated calcium channels. For these experiments, 10 mm BAPTA was included in the patch pipette to buffer postsynaptic Ca2+. Bath application of ionomycin for 4 min (in the presence of 1 μm TTX) produced a robust increase in mEPSC frequency in control cells in 2.5 mm external Ca2+. Cells that had been previously treated with U69593 (1 μm 5 min, followed by 10 min washout) exhibited an increase in mEPSC frequency in response to ionomycin that was not significantly different from the control response (peak increase in absolute number of events: control, 686 ± 260 events, n = 6; U69593, 649 ± 102 events, n = 5; p = 0.91) (Fig. 6). In low external Ca2+ (0.3 mm), control cells still showed an increase in mEPSC frequency in response to bath applied ionomycin; however, cells previously treated with U69593 now showed a significantly attenuated response (peak increase in absolute number of events: control, 165 ± 31 events, n = 6; U69593, 35 ± 6 events, n = 4, p = 0.01) (Fig. 6). Together, all these data suggest that activation of κ-opioid receptors inhibits vesicle fusion and that this inhibition can be overcome with higher presynaptic Ca2+ concentrations.
Figure 6.

Increasing presynaptic Ca2+ overcomes the κ-opioid-mediated inhibition. In normal ACSF (2.5 mm Ca2+/1.5 mm Mg2+), bath application of ionomycin (1 μm, 4 min) induced a large increase in mEPSC frequency in control cells as well as in cells previously treated with U69593 (peak increase in absolute number of events: control, 686 ± 260 events, n = 6; U69593, 649 ± 102 events, n = 5, p = 0.91). In low Ca2+ ACSF (0.3 mm Ca2+/3.7 mm Mg2+), ionomycin evoked a much smaller increase in mEPSC frequency in U69593-treated cells compared with control cells (peak increase in absolute number of events: control, 165 ± 31 events, n = 6; U69593, 35 ± 6 events, n = 4, p = 0.01). Representative cells and responses for each condition are shown in A and B (control, black; U69593, red). Each trace is from a different cell.
Somatodendritic dynorphin release inhibits excitatory synaptic transmission
Somatodendritic dynorphin/VP release modulates the postsynaptic firing properties of VP MNCs through a direct action on postsynaptic channels (Brown et al., 1998; Brown and Bourque, 2004), but there are no reports demonstrating the effects of dynorphin, released from a single neuron, on synaptic transmission. To evoke dendritic release, vasopressin MNCs (GFP-positive cells) were repetitively depolarized in voltage clamp from −60 to 0 mV for 100 ms at a frequency of 2 Hz, 240 times. Single synaptic stimuli were administered after every fourth depolarization to assay synaptic strength. This protocol was performed in the presence of the CB1 antagonist AM251 (5 μm) and the aminopeptidase inhibitor, amastatin (10 μm).
The evoked EPSC depressed quickly during the postsynaptic depolarizing protocol and remained depressed for the duration of the recording (53 ± 8% of control, p = 0.012, n = 6) (Fig. 7A,B). To determine the locus of this inhibition, we analyzed spontaneous EPSCs (sEPSCs) before and after the depolarizing protocol. There was no change in the amplitude of sEPSCs (98 ± 4% control, p = 0.711, n = 6), but there was a significant decrease in the frequency of sEPSCs after the depolarizing protocol (73 ± 7% of control, p = 0.024, n = 6) (Fig. 7C). In addition, we analyzed the coefficient of variation of the evoked EPSC over the course of the synaptic depression. We found that there was a significant reduction in 1/CV2 after the depolarizing protocol that persisted for the duration of the recordings (p = 0.006, n = 6) (Fig. 7D). Together, these data suggest that repetitive postsynaptic depolarization induces a presynaptically mediated depression of glutamate release onto VP neurons, likely mediated by a retrograde transmitter.
To determine that the retrograde transmitter responsible for the synaptic depression was indeed dynorphin, we repeated the depolarizing protocol in the presence of the κ-opioid receptor antagonist nor-BNI (100 nm). Figure 8A shows that with κ-opioid receptors blocked, repetitive depolarizations no longer induced a prolonged depression of the evoked EPSC (109 ± 20% control, p = 0.389, n = 5) (Fig. 8A,B). In addition, sEPSC frequency was no longer depressed after the depolarizing protocol in the presence of nor-BNI (99 ± 7% control, p = 0.675, n = 4) (Fig. 8C). If dynorphin is released from the postsynaptic cell by the exocytosis of somatodendritic large dense core vesicles, then the dynorphin-dependent synaptic depression should be blocked by preventing SNARE-dependent exocytosis in the postsynaptic cell. To test this, we included 5 μg/ml BoTC in the patch pipette to cleave syntaxin only in the recorded cell. A minimum of 15 min was allowed for BoTC to dialyze the postsynaptic cell before control EPSCs were recorded. In the presence of BoTC, the repetitive postsynaptic depolarizations no longer induced a depression of the evoked EPSC (89 ± 12% of control, p = 0.493, n = 4) (Fig. 8E), suggesting that dynorphin is released via SNARE-dependent exocytosis. Finally, to test the specificity of the retrograde depression, we repeated the depolarizing protocol in neighboring eGFP-negative neurons (putative oxytocin cells) that do not produce dynorphin (Watson et al., 1982). The depression observed in these cells (19 ± 5%, n = 6) (Fig. 8F) was significantly less than that observed in eGFP-positive cells (p = 0.015) and not significantly different from depression in the presence of nor-BNI (p = 0.131). Together, these data suggest that dendritically released dynorphin from vasopressin neurons can induce a presynaptic depression of glutamate release.
Discussion
In this study, we have shown that dynorphin acts as a retrograde transmitter to inhibit excitatory synaptic transmission through the activation of presynaptic κ-opioid receptors. This inhibition is attributable to a presynaptic modulation of vesicle fusion that can, in part, be overcome by increasing presynaptic Ca2+.
Retrograde signaling is now accepted as a fundamental property of synaptic transmission in the brain. Although the most commonly examined retrograde signaling molecules are the endocannabinoids, many different areas of the brain also employ additional retrograde transmitters, such as GABA, glutamate, nitric oxide, and dopamine (Ludwig and Pittman, 2003). Such a variety of retrograde transmitters not only allows for many different forms of plasticity but also allows specificity of retrograde signaling. In this study, we have shown for the first time that the opioid peptide, dynorphin, also acts as a retrograde messenger to modulate excitatory neurotransmission onto VP-expressing MNCs. Dynorphin is released in an activity- and SNARE-dependent manner from the postsynaptic cell to induce a presynaptic depression of glutamate release. Importantly and consistent with its described localization with VP in large dense core vesicles (Watson et al., 1982), dynorphin only acts as a retrograde transmitter in VP-expressing neurons and not in neighboring OT-expressing cells. VP itself has been shown to promote dendritic release (Ludwig et al., 2005), suggesting that initial dendritic secretion of dynorphin and VP may act to prime further dendritic release.
Although the long-lasting depression of EPSCs seen after repetitive activation of VP neurons was blocked by the κ-opioid receptor antagonist nor-BNI, a transient depression that manifests only during the depolarizing protocol persisted. This transient depression was unaffected either by the inclusion of BoTC in the patch pipette or by bath application of the CB1 receptor antagonist AM251. These data rule out both the vesicular release of a dendritic transmitter and the dendritic release of endocannabinoid. Currently, the identity of the signal responsible for this transient depression in response to repetitive depolarization is unknown.
Opioids depress transmission at a number of central synapses including those onto MNCs (Honda et al., 2004). Many of these studies have implicated an opioid-mediated inhibition of presynaptic Ca2+ channels (Rusin et al., 1997; Hjelmstad and Fields, 2003) or the activation of presynaptic K+ channels (Simmons and Chavkin, 1996; Vaughan et al., 1997). Other studies, however, rule out Ca2+ channels and/or K+ channels (Castillo et al., 1996; Ford et al., 2007), indicating that the nature of a unifying mechanism remains unresolved. Our data provide evidence for a novel form of inhibition, whereby neither of these mechanisms contribute to the decrease in glutamate release after activation of κ-opioid receptors. Instead, we propose that once activated, κ-opioid receptors directly modulate the presynaptic release machinery, to reduce vesicular glutamate release.
We have provided several pieces of evidence consistent with the idea that κ-opioid inhibition is mediated by direct modulation of vesicle release machinery. First, the κ-opioid receptor agonist, U69593, produces a large reduction in the frequency of both asynchronous release events and mEPSCs but has no effect on their mean amplitude. Second, mEPSCs in MNCs are independent of external Ca2+ (Inenaga et al., 1998), and since the inhibition of mEPSC frequency persisted when all Ca2+ was removed/buffered, this suggests that the target of inhibition is downstream of Ca2+ entry. Third, blocking either N- or P/Q-type Ca2+ channels failed to reduce the relative magnitude of the inhibition. It should be noted, however, that because of the nonlinear relationship between presynaptic Ca2+ and transmitter release (Dodge and Rahamimoff, 1967), interpreting changes in the magnitude of opioid inhibition after blocking a single subtype of Ca2+ channels is problematic. However, when taken in concert with other pieces of data, the most parsimonious explanation is that κ-opioid inhibition results from a direct modulation of the vesicle fusion apparatus at a site downstream of Ca2+ entry.
Although the molecular target of the inhibition by dynorphin is unresolved, our results are consistent with recent work implicating direct modulation of release machinery by the βγ subunit of Gi/o-coupled receptors (Blackmer et al., 2001; Delaney et al., 2007). Consistent with our data, this βγ-mediated inhibition can be overcome by elevating presynaptic Ca2+ levels (Yoon et al., 2007). In previous studies, βγ interactions either decreased the size of the fusion pore (Photowala et al., 2006) or decreased the number of active release sites (Delaney et al., 2007). We do not observe any changes in quantal size after dynorphin, indicating that fusion pore size is not changing. And although we cannot completely rule out a decrease in the number of release sites, our data demonstrating that there is an increase in PPR as well as an increase in the coefficient of variation of the EPSC argue against this idea. Rather, our data indicate that activation of presynaptic κ-opioid receptors reduces the probability of vesicle fusion, an effect distinct from that reported in the above studies.
Glutamate synapses onto MNCs also exhibit a unique release profile that is worth considering when interpreting our findings. Specifically, these synapses exhibit asynchronous transmitter release (Iremonger and Bains, 2007). Unlike fast neurotransmission at most central synapses, glutamate transmission at synapses onto MNCs is sensitive to the slow calcium buffer EGTA-AM. This finding is in line with the properties of other central synapses that display pronounced asynchronous release (Atluri and Regehr, 1998; Hefft and Jonas, 2005). Interestingly, many of the synapses that show asynchronous release (or ectopic release) rely more heavily on presynaptic N-type calcium channels than other subtypes (Matsui and Jahr, 2004; Fedchyshyn and Wang, 2005; Hefft and Jonas, 2005). This is consistent with the observation, at some synapses, that P/Q-type channels are more tightly localized in the active zone, whereas N-type channels are more evenly dispersed throughout the terminal (Wu et al., 1999). In this current study, we find that asynchronous release onto MNCs in the PVN is also mainly reliant on N-type calcium channels, with only a small contribution from P/Q-type channels. Similar observations have been made at glutamate synapses onto MNCs in the SON (Hirasawa et al., 2001).
Physiological significance
In vivo application of κ-opioid receptor antagonists into the magnocellular region of the hypothalamus increases the excitability of vasopressin MNCs (Brown et al., 1998). Dendritically released dynorphin has previously been shown to act as an autocrine messenger in these cells (Brown and Bourque, 2004). Specifically, during a burst of activity, dynorphin is released, which in turn binds to κ-opioid receptors on the same postsynaptic cell to transiently inhibit a depolarizing after potential and help terminate a burst. The current data suggest that dendritically released dynorphin also curtails MNC excitability by inhibiting both spontaneous and action potential-dependent glutamate release. Since bursting in these cells requires excitatory synaptic drive (Nissen et al., 1995; Brown et al., 2004), this would be a very efficient mechanism to control firing and hence hormone secretion into the blood.
Footnotes
This work was supported by operating grants from the Canadian Institutes of Health Research (J.S.B.) and the Heart and Stroke Foundation of Alberta, Yukon, and Northwest Territories (J.S.B.). K.J.I. is supported by studentships from the Heart and Stroke Foundation of Canada and the Alberta Heritage Foundation for Medical Research (AHFMR). J.S.B. is an AHFMR Senior Scholar. We thank Dr. G. W. Zamponi for comments on this manuscript and members of the Bains laboratory for fruitful discussions.
References
- Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68:247–286. doi: 10.1016/s0301-0082(02)00080-1. [DOI] [PubMed] [Google Scholar]
- Atluri PP, Regehr WG. Delayed release of neurotransmitter from cerebellar granule cells. J Neurosci. 1998;18:8214–8227. doi: 10.1523/JNEUROSCI.18-20-08214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmer T, Larsen EC, Takahashi M, Martin TF, Alford S, Hamm HE. G protein betagamma subunit-mediated presynaptic inhibition: regulation of exocytotic fusion downstream of Ca2+ entry. Science. 2001;292:293–297. doi: 10.1126/science.1058803. [DOI] [PubMed] [Google Scholar]
- Boland LM, Morrill JA, Bean BP. ω-Conotoxin block of N-type calcium channels in frog and rat sympathetic neurons. J Neurosci. 1994;14:5011–5027. doi: 10.1523/JNEUROSCI.14-08-05011.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CH, Bourque CW. Autocrine feedback inhibition of plateau potentials terminates phasic bursts in magnocellular neurosecretory cells of the rat supraoptic nucleus. J Physiol. 2004;557:949–960. doi: 10.1113/jphysiol.2004.063818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CH, Ludwig M, Leng G. κ-Opioid regulation of neuronal activity in the rat supraoptic nucleus in vivo. J Neurosci. 1998;18:9480–9488. doi: 10.1523/JNEUROSCI.18-22-09480.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CH, Bull PM, Bourque CW. Phasic bursts in rat magnocellular neurosecretory cells are not intrinsically regenerative in vivo. Eur J Neurosci. 2004;19:2977–2983. doi: 10.1111/j.0953-816X.2004.03408.x. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Salin PA, Weisskopf MG, Nicoll RA. Characterizing the site and mode of action of dynorphin at hippocampal mossy fiber synapses in the guinea pig. J Neurosci. 1996;16:5942–5950. doi: 10.1523/JNEUROSCI.16-19-05942.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- Delaney AJ, Crane JW, Sah P. Noradrenaline modulates transmission at a central synapse by a presynaptic mechanism. Neuron. 2007;56:880–892. doi: 10.1016/j.neuron.2007.10.022. [DOI] [PubMed] [Google Scholar]
- Dodge FA, Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedchyshyn MJ, Wang LY. Developmental transformation of the release modality at the calyx of Held synapse. J Neurosci. 2005;25:4131–4140. doi: 10.1523/JNEUROSCI.0350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan TF, Chen SR, Pan HL. Mu opioid receptor activation inhibits GABAergic inputs to basolateral amygdala neurons through Kv1.1/1.2 channels. J Neurophysiol. 2006;95:2032–2041. doi: 10.1152/jn.01004.2005. [DOI] [PubMed] [Google Scholar]
- Ford CP, Beckstead MJ, Williams JT. Kappa opioid inhibition of somatodendritic dopamine inhibitory postsynaptic currents. J Neurophysiol. 2007;97:883–891. doi: 10.1152/jn.00963.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GR, Bains JS. Priming of excitatory synapses by α1 adrenoceptor-mediated inhibition of group III metabotropic glutamate receptors. J Neurosci. 2003;23:6223–6231. doi: 10.1523/JNEUROSCI.23-15-06223.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Hirasawa M, Kombian SB, Pittman QJ. Oxytocin retrogradely inhibits evoked, but not miniature, EPSCs in the rat supraoptic nucleus: role of N- and P/Q-type calcium channels. J Physiol. 2001;532:595–607. doi: 10.1111/j.1469-7793.2001.0595e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmstad GO, Fields HL. Kappa opioid receptor activation in the nucleus accumbens inhibits glutamate and GABA release through different mechanisms. J Neurophysiol. 2003;89:2389–2395. doi: 10.1152/jn.01115.2002. [DOI] [PubMed] [Google Scholar]
- Honda E, Ono K, Inenaga K. DAMGO suppresses both excitatory and inhibitory synaptic transmission in supraoptic neurones of mouse hypothalamic slice preparations. J Neuroendocrinol. 2004;16:198–207. doi: 10.1111/j.0953-8194.2004.01151.x. [DOI] [PubMed] [Google Scholar]
- Inenaga K, Honda E, Hirakawa T, Nakamura S, Yamashita H. Glutamatergic synaptic inputs to mouse supraoptic neurons in calcium-free medium in vitro. J Neuroendocrinol. 1998;10:1–7. doi: 10.1046/j.1365-2826.1998.00662.x. [DOI] [PubMed] [Google Scholar]
- Iremonger KJ, Bains JS. Integration of asynchronously released quanta prolongs the postsynaptic spike window. J Neurosci. 2007;27:6684–6691. doi: 10.1523/JNEUROSCI.0934-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig M, Pittman QJ. Talking back: dendritic neurotransmitter release. Trends Neurosci. 2003;26:255–261. doi: 10.1016/S0166-2236(03)00072-9. [DOI] [PubMed] [Google Scholar]
- Ludwig M, Bull PM, Tobin VA, Sabatier N, Landgraf R, Dayanithi G, Leng G. Regulation of activity-dependent dendritic vasopressin release from rat supraoptic neurones. J Physiol. 2005;564:515–522. doi: 10.1113/jphysiol.2005.083931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther JA, Tasker JG. Voltage-gated currents distinguish parvocellular from magnocellular neurones in the rat hypothalamic paraventricular nucleus. J Physiol. 2000;523:193–209. doi: 10.1111/j.1469-7793.2000.t01-1-00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui K, Jahr CE. Differential control of synaptic and ectopic vesicular release of glutamate. J Neurosci. 2004;24:8932–8939. doi: 10.1523/JNEUROSCI.2650-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz IM, Venema VJ, Swiderek KM, Lee TD, Bean BP, Adams ME. P-type calcium channels blocked by the spider toxin omega-Aga-IVA. Nature. 1992;355:827–829. doi: 10.1038/355827a0. [DOI] [PubMed] [Google Scholar]
- Nissen R, Hu B, Renaud LP. Regulation of spontaneous phasic firing of rat supraoptic vasopressin neurones in vivo by glutamate receptors. J Physiol. 1995;484:415–424. doi: 10.1113/jphysiol.1995.sp020674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Photowala H, Blackmer T, Schwartz E, Hamm HE, Alford S. G protein betagamma-subunits activated by serotonin mediate presynaptic inhibition by regulating vesicle fusion properties. Proc Natl Acad Sci U S A. 2006;103:4281–4286. doi: 10.1073/pnas.0600509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynor K, Kong H, Chen Y, Yasuda K, Yu L, Bell GI, Reisine T. Pharmacological characterization of the cloned kappa-, delta-, and mu-opioid receptors. Mol Pharmacol. 1994;45:330–334. [PubMed] [Google Scholar]
- Rusin KI, Giovannucci DR, Stuenkel EL, Moises HC. κ-Opioid receptor activation modulates Ca2+ currents and secretion in isolated neuroendocrine nerve terminals. J Neurosci. 1997;17:6565–6574. doi: 10.1523/JNEUROSCI.17-17-06565.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuster SJ, Riedl M, Li X, Vulchanova L, Elde R. The kappa opioid receptor and dynorphin co-localize in vasopressin magnocellular neurosecretory neurons in guinea-pig hypothalamus. Neuroscience. 2000;96:373–383. doi: 10.1016/s0306-4522(99)00472-8. [DOI] [PubMed] [Google Scholar]
- Simmons ML, Chavkin C. k-Opioid receptor activation of a dendrotoxin-sensitive potassium channel mediates presynaptic inhibition of mossy fiber neurotransmitter release. Mol Pharmacol. 1996;50:80–85. [PubMed] [Google Scholar]
- Solis JM, Nicoll RA. Pharmacological characterization of GABAB-mediated responses in the CA1 region of the rat hippocampal slice. J Neurosci. 1992;12:3466–3472. doi: 10.1523/JNEUROSCI.12-09-03466.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueta Y, Fujihara H, Serino R, Dayanithi G, Ozawa H, Matsuda K, Kawata M, Yamada J, Ueno S, Fukuda A, Murphy D. Transgenic expression of enhanced green fluorescent protein enables direct visualization for physiological studies of vasopressin neurons and isolated nerve terminals of the rat. Endocrinology. 2005;146:406–413. doi: 10.1210/en.2004-0830. [DOI] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Connor MA, Christie MJ. How opioids inhibit GABA-mediated neurotransmission. Nature. 1997;390:611–614. doi: 10.1038/37610. [DOI] [PubMed] [Google Scholar]
- Wagner JJ, Terman GW, Chavkin C. Endogenous dynorphins inhibit excitatory neurotransmission and block LTP induction in the hippocampus. Nature. 1993;363:451–454. doi: 10.1038/363451a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson SJ, Akil H, Fischli W, Goldstein A, Zimmerman E, Nilaver G, van wimersma Griedanus TB. Dynorphin and vasopressin: common localization in magnocellular neurons. Science. 1982;216:85–87. doi: 10.1126/science.6121376. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Zalutsky RA, Nicoll RA. The opioid peptide dynorphin mediates heterosynaptic depression of hippocampal mossy fibre synapses and modulates long-term potentiation. Nature. 1993;362:423–427. doi: 10.1038/362423a0. [DOI] [PubMed] [Google Scholar]
- Wells T, Forsling ML. Kappa-opioid modulation of vasopressin secretion in conscious rats. J Endocrinol. 1991;129:411–416. doi: 10.1677/joe.0.1290411. [DOI] [PubMed] [Google Scholar]
- Wu LG, Westenbroek RE, Borst JG, Catterall WA, Sakmann B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J Neurosci. 1999;19:726–736. doi: 10.1523/JNEUROSCI.19-02-00726.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon EJ, Gerachshenko T, Spiegelberg BD, Alford S, Hamm HE. Gbetagamma interferes with Ca2+-dependent binding of synaptotagmin to the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex. Mol Pharmacol. 2007;72:1210–1219. doi: 10.1124/mol.107.039446. [DOI] [PubMed] [Google Scholar]