

Abstract
Fetal nicotine exposure blunts hypoxia-induced catecholamine secretion from neonatal adrenomedullary chromaffin cells (AMCs), providing a link between maternal smoking, abnormal arousal responses, and risk of sudden infant death syndrome. Here, we show that the mechanism is attributable to upregulation of KATP channels via stimulation of α7 nicotinic ACh receptors (AChRs). These KATP channels open during hypoxia, thereby suppressing membrane excitability. After in utero exposure to chronic nicotine, neonatal AMCs show a blunted hypoxic sensitivity as determined by inhibition of outward K+ current, membrane depolarization, rise in cytosolic Ca2+, and catecholamine secretion. However, hypoxic sensitivity could be unmasked in nicotine-exposed AMCs when glibenclamide, a blocker of KATP channels, was present. Both KATP current density and KATP channel subunit (Kir 6.2) expression were significantly enhanced in nicotine-exposed cells relative to controls. The entire sequence could be reproduced in culture by exposing neonatal rat AMCs or immortalized fetal chromaffin (MAH) cells to nicotine for ∼1 week, and was prevented by coincubation with selective blockers of α7 nicotinic AChRs. Additionally, coincubation with inhibitors of protein kinase C and CaM kinase, but not protein kinase A, prevented the effects of chronic nicotine in vitro. Interestingly, chronic nicotine failed to blunt hypoxia-evoked responses in MAH cells bearing short hairpin knockdown (>90%) of the transcription factor, hypoxia-inducible factor-2α (HIF-2α), suggesting involvement of the HIF pathway. The therapeutic potential of KATP channel blockers was validated in experiments in which hypoxia-induced neonatal mortality in nicotine-exposed pups was significantly reduced after pretreatment with glibenclamide.
Introduction
Exposure of the neonate to episodes of asphyxia during birth results in a variety of adaptive changes that include fluid reabsorption and surfactant secretion in the lungs so as to facilitate air breathing, as well as improved cardiac conductance (Slotkin and Seidler, 1988). These physiological responses depend critically on catecholamine (CAT) secretion from adrenomedullary chromaffin cells (AMCs), triggered by asphyxial stressors such as low O2 (hypoxia), elevated CO2 (hypercapnia), and low pH (acidosis). Neonatal AMCs express a direct hypoxia-sensing mechanism, independent of the nervous system, that is lost or suppressed postnatally along a time course that approximately parallels maturation of the splanchnic cholinergic innervation (Seidler and Slotkin, 1985, 1986; Lagercrantz and Slotkin, 1986; Slotkin and Seidler, 1988). These findings raised the possibility that activation of nicotinic acetylcholine receptors (nAChRs) on chromaffin cells may be involved in the postnatal loss of direct chemosensing properties in these cells. Indeed, prenatal nicotine exposure is known to cause a loss of hypoxia tolerance, decreased arousal responses in the neonate, and impaired hypoxia-induced CAT secretion (Slotkin et al., 1995; Slotkin, 1998; Cohen et al., 2005).
We recently demonstrated that chronic nicotine in utero and in vitro resulted in the selective loss of direct hypoxic sensitivity in perinatal rat AMCs (Buttigieg et al., 2008b). The signaling mechanisms underlying this loss of hypoxic sensitivity in nicotine-treated perinatal AMCs have been mostly unexplored, although activation of neuronal nAChRs appears to be involved (Cohen et al., 2005; Buttigieg et al., 2008b). This effect of chronic nicotine may be the result of alterations at any one or more of several steps in the signal transduction pathway. For example, it may occur at more upstream sites such as the proposed PO2 sensor (Thompson et al., 2007; Buttigieg et al., 2008a) or, alternatively, at more downstream sites involving O2-regulated K+ channels. The hypoxic sensitivity in neonatal AMCs depends ultimately on the depolarizing effects of K+ channel inhibition, of which several subtypes including large-conductance (BK) and small-conductance (SK) Ca2+-dependent K+ channels appear to be involved (Thompson and Nurse, 1998; Thompson et al., 2002; Bournaud et al., 2007). In contrast, the plasma membrane ATP-sensitive K+ channel (KATP) is actually activated by hypoxia, thereby favoring membrane hyperpolarization and suppression of voltage-gated Ca2+ entry (Thompson and Nurse, 1998; Bournaud et al., 2007). Therefore, nicotine-induced alteration in expression of any one or more of these K+ channels provides a potential mechanism for suppressing hypoxic sensitivity in chromaffin cells. Indeed, such a mechanism seems to promote hypoxia-evoked CAT secretion in fetal adrenal chromaffin cells, in which reduced KATP combined with enhanced BK channel expression at late gestation favors membrane depolarization during hypoxia (Bournaud et al., 2007). We therefore compared K+ channel expression in nicotine- versus saline-treated AMCs to test this possibility and obtained strong evidence for KATP channel upregulation as a key contributor to nicotine-induced loss of hypoxic sensitivity. In addition, we probed the signaling pathway mediating the effects of nicotine, including the particular subtype(s) of nAChRs involved as well as the potential roles of protein kinases such as protein kinase C (PKC), protein kinase A (PKA), and Ca2+/calmodulin-dependent protein kinase (CaM kinase). Finally, because the transcription factor hypoxia-inducible factor (HIF) has previously been implicated in the effects of chronic nicotine exposure (Zhang et al., 2007), we used a model chromaffin cell line to investigate the potential role of HIF-2α in mediating the effects of chronic nicotine on KATP channel expression and hypoxic sensitivity.
Materials and Methods
Animal preparation
All animal experiments were approved by the Animal Research and Ethics Board at McMaster University, in accordance with the guidelines of the Canadian Council for Animal Care. Female Wistar rats (Harlan) were maintained under controlled lighting (12 h light/dark) and temperature (22°C) with ad libitum access to food and water. Dams were randomly assigned to receive either saline (vehicle) or nicotine bitartrate (1 mg · kg body weight−1 · d−1; Sigma-Aldrich) daily by subcutaneous injection for 14 d before mating, and then during pregnancy until parturition as previously described (Holloway et al., 2005; Buttigieg et al., 2008b). Dams were allowed to deliver naturally, and pups were collected soon after birth [postnatal day 0 (P0)]. Before removal of the adrenal glands, P0 pups were first rendered unconscious by a blow to the head and then immediately killed by decapitation. Isolated adrenal glands were kept in sterile medium, in which most of the outer cortex was removed before enzymatic digestion of the medullary tissue.
Cell culture
Primary chromaffin cells.
Primary cultures enriched in adrenal chromaffin cells were prepared from P0 rat pups by combined enzymatic and mechanical dissociation as described in detail previously (Thompson et al., 1997, 2007; Thompson and Nurse, 1998). After preplating for ∼2 h to remove most of the cortical cells, the nonadherent chromaffin cells were plated on modified culture wells coated with Matrigel (Collaborative Research). Cells were grown at 37°C in a humidified atmosphere of 95% air–5% CO2 for 18–36 h before use. The growth medium consisted of F-12 nutrient medium (Invitrogen) supplemented with 10% fetal bovine serum and other additives as previously described (Thompson et al., 1997).
Immortalized chromaffin (MAH) cells.
Immortalized wild-type MAH cells (wt MAH) (a generous gift from Dr. Laurie Doering, McMaster University, Hamilton, ON, Canada), derived from fetal rat adrenal medulla, were grown in modified L-15/CO2 medium supplemented with 0.6% glucose, 1% penicillin/streptomycin, 10% fetal bovine serum, and 5 μm dexamethasone (Fearon et al., 2002). All cultures were grown in a humidified atmosphere of 95% air–5% CO2 at 37°C. Cultures were fed every 1–2 d and split every 3–4 d. To passage cells, the culture medium was removed, and 0.25% trypsin was added to detach cells from the culture substrate. The resulting cell suspension was pelleted by centrifugation, the supernatant discarded, and the pellet resuspended in fresh medium. Cells were then plated onto standard 35 mm culture dishes, which had been previously coated with poly-d-lysine and laminin to promote cell adhesion.
Drugs
All solutions containing drugs were made fresh on the day of the experiment. Drugs were obtained from Sigma-Aldrich unless otherwise stated. In cases in which cells were exposed chronically to a drug for 7 d, the medium was replaced every 2 d and fresh drug was introduced.
Electrophysiology
Voltage-clamp data were obtained using the nystatin perforated-patch technique as previously described (Thompson and Nurse, 1998; Thompson et al., 2002, 2007; Buttigieg et al., 2008a). The pipette solution contained the following (in mm): 110 K-gluconate, 25 KCl, 5 NaCl, 2 CaCl2, 10 HEPES, at pH 7.2, and nystatin (300–450 μg ml−1). Experiments were performed at 37°C in HEPES-buffered extracellular medium that contained the following (in mm): 135 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 10 glucose, and 10 HEPES, at pH 7.4, with or without 0.5 μm tetrodotoxin. Hypoxic solutions (PO2, 5–15 mmHg) were generated by bubbling N2 gas and were applied to the cells by gravity flow or by a rapid perfusion system (Thompson et al., 1997; Zhang et al., 2000). In voltage-clamp experiments, cells were held at −60 mV and step depolarized to the indicated test potential (between −100 and +80 mV in 10 mV increments) for 100 ms at a frequency of 0.1 Hz. In the majority of experiments, cells were studied within 24 h of isolation. In cases in which cultures were treated chronically with vehicle or drugs, recordings were performed 1 week after cell isolation. All electrophysiological data are expressed as mean ± SEM and compared using the paired or independent Student's t tests (Microcal Origin, version 7.0).
Intracellular Ca2+measurements
Intracellular Ca2+ was monitored using the fluorescent Ca2+ indicator fura-2 AM. Cells were loaded with 5 μm fura-2 AM for 30 min at 37°C, and then rinsed (three times) before use (∼30 min later). Ratiometric Ca2+ imaging was performed using a Nikon Eclipse TE2000-U inverted microscope equipped with a Lambda DG-4 ultra high-speed wavelength changer (Sutter Instrument; exposure time, 100 ms), a Hamamatsu OCRCAT-ET digital CCD camera, and a Nikon S-Fluor 40× oil-immersion objective lens with a numerical aperture of 1.3. Dual images (340 and 380 nm excitation; 510 nm emission) were collected, and pseudocolor ratiometric data were obtained using Simple PCI software, version 5.3 (Compix). The imaging system was standardized with a two-point calibration, using Ca2+-free solution and Ca2+ solution (39 μm) from Invitrogen (F-6774). The parameters used for the two-point calibration included the dissociation constant of fura-2 (K D = 224 nm), the ratio values for the (−) and (+) concentration standards (R min = 0.026 and R max = 4.4) and β value of 5.6. [Ca2+]i (in nanomolar) was calculated according to the equation described previously (Grynkiewicz et al., 1985). All experiments were performed at ∼34°C, and cells were continuously perfused with bicarbonate/CO2-buffered extracellular medium as previously described (Buttigieg et al., 2008a). The switch between control and test solutions was aided with a double-barrel fast perfusion system (Zhang et al., 2000). Ca2+ levels were measured by averaging peak values of the data points collected for the duration of each treatment.
Carbon fiber amperometry
CAT secretion from chromaffin cells was monitored using carbon fiber amperometry after the culture dish was placed on the stage of a Zeiss Axioskop 2 upright microscope equipped with a 40× water-immersion objective. The culture was perfused under gravity with extracellular solution containing the following (in mm): 135 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 10 glucose, and 10 HEPES, at pH 7.4 and 37°C. In some experiments, high K+ (30 mm) solutions were used after equimolar substitution for NaCl. Hypoxic solution (PO2, 15–20 mmHg) was obtained by continuously bubbling with N2. Catecholamine secretion was monitored from single cells that were usually part of a cell cluster with ProCFE low noise carbon fiber electrodes (5 μm diameter tip; Dagan) connected to a CV 23BU headstage and an Axopatch 200B amplifier set at 800 mV. Data acquisition and analysis were performed using Clampfit 9.2 (Molecular Devices); currents were filtered at 100 Hz, digitized at 250 Hz, and stored on a personal computer. Events smaller than 3 pA were excluded from the analysis and spike frequency was calculated as the number of spike events per minute. Samples were compared using Student's t test, and level of significance was set at p < 0.05. Unless otherwise noted, the data are expressed as mean ± SEM.
Western blot
Protein was extracted from enriched chromaffin cell cultures in lysis buffer A containing the following (in mm): 10 HEPES, pH 7.6, 10 KCl, 0.1 EDTA, pH 8, 0.1 EGTA, pH 8, 1 DTT). Thirty micrograms of protein was loaded onto an 8% SDS-polyacrylamide gel and run at 120 V for 2 h. For HIF-2α determination, nuclear lysate was obtained from cells lysed in buffer A, and NP-40 was then added to a final concentration of 0.6% and vortexed for 1 min. The lysate was centrifuged at 13,000 rpm for 30 s, the supernatant removed, and the pellet was resuspended in 50 μl of buffer C (20 mm HEPES, pH 7.6, 0.4 m NaCl, 1 mm EDTA, pH 8, 1 mm EGTA, pH 8, 1 mm DTT, 5% glycerol) containing protein inhibitors, before freezing at −80°C. Protein was transferred from the gel onto a PVDF (polyvinylidene difluoride) membrane (Millipore) and incubated in either sheep polyclonal antibody against Kir 6.2 (Alomone Labs), rabbit polyclonal antibody against HIF-2α (Novus Biologicals), rabbit polyclonal antibody against TATA binding protein (Santa Cruz Biotechnology), or mouse monoclonal antibody against β-actin (Santa Cruz Biotechnology) at 4°C overnight. The membrane was washed with PBS, incubated for 1 h at room temperature with an HRP-linked secondary antibody, and rewashed in PBS. The blot was visualized using Immobilon Western Chemiluminescent HRP substrate (Millipore) and autoradiography.
Immunocytochemistry
Neonatal AMCs were grown in the central wells of modified 35 mm Nunc dishes as previously described (Fearon et al., 2002). The well was formed by drilling a central hole (∼1 cm in diameter) in the dish and attaching a glass coverslip to the underside. Medium was removed, and the cells were washed two times in 3 ml of PBS. Cells were then fixed with 3 ml of 5% acetic acid and 95% methanol at −20°C for 60 min, and the solution was replaced with 2 ml of PBS. Samples were then washed three times with PBS, before addition of 30 μl of primary antibody followed by incubation for 24 h at 4°C. The following primary antibodies were used at the dilutions indicated: anti-SK2, 1:50; anti-Kv1.2, 1:50; anti-Kv1.5, 1:50; and anti-Ca2+-dependent K+ or BK, 1:100 (Alomone Labs). After incubation, the primary antibody solution was removed and samples were washed three times in PBS. Secondary antibody, conjugated with FITC (Jackson ImmunoResearch) was diluted in PBS (1:50) and incubated with the cells for 1 h at room temperature in the dark. After removing the solution, the samples were washed three times in PBS. Vectashield was then added to the dishes to prevent photobleaching. Control experiments were performed for SK2, BK, Kv1.2, and Kv1.5 channel antibodies, in which the primary antibody was preincubated with excess blocking peptide overnight at 4°C (at 3 μg of fusion peptide per 1 μg of antibody). The samples were visualized with a Zeiss inverted microscope (IM 35) equipped with epifluorescence, and fluorescein and rhodamine filter sets. Images were acquired using a digital camera with Northern Eclipse software program and saved in TIFF format.
RNA interference knockdown of HIF-2α in MAH cells
Oligonucleotides containing the short hairpin RNA interference (RNAi) sequence for HIF-2α (5′-GCC AGA ACT TTG ATG AAT CTT CATA GAG AGA TTC ATC AAA GTT CTG GC-3′) and a scrambled negative control (scControl) (5′-tag cga cta aac aca tca att caa gag att gat gtg ttt agt cgc ta-3′) were cloned into the pSuper Retroviral vector (Brummelkamp et al., 2002). The resulting plasmids were transfected into the Phoenix packaging cell line and selected under puromycin for 1–2 weeks. Subsequently, cell culture medium containing virus was collected and filtered, and the resulting viral supernatant was used to infect dividing MAH cells.
Quantitative reverse transcription–PCR
RNA from chromaffin cell cultures was extracted using the RNeasy Mini Kit (QIAGEN; 74104) following the manufacturer's protocol. RNA was quantified in an Eppendorf Biophotometer and 500 ng was treated with DNase I (Invitrogen; 18068-015) to remove any contaminating DNA. Reverse transcription (RT) was performed on 100 ng of DNase-treated RNA using Superscript III (Invitrogen; 18080-044) and random primers (100 ng). A no-RT control was also run to test for the presence of DNA contamination (data not shown). Quantitative PCR (QPCR) was performed and data analyzed using the Absolute QPCR SYBR Green Mix (ABgene; AB-1166/A) and a Stratagene MX3000P machine. Gene-specific primers were designed using GeneFisher (Giegerich et al., 1996) and synthesized by a local facility (MOBIX; McMaster University). The following primers were used and listed as gene amplified, sequence (forward, reverse), and annealing temperature: Lamin A/C: 5′-GCATGTACATAGAAGGAGCTA-3′ and 5′-CATGCATATTCCTGGTACTCAT-3′, 55°C; HIF-2α: 5′-CCAT GTG TAT CATT GTG TGT CAT-3′ and 5′-CATA AGT TCT GGC TTC CG AA-3′, 55°C. Verification of the PCR products was done using the QIAquick Gel Extraction kit (QIAGEN; 28704) to extract PCR fragments from a 2% agarose gel. The DNA sample was then sequenced (at MOBIX) using an ABI Prism automated Sequencer (with T7 polymerase). The sequencing results were analyzed by BLAST and the sequences were matched to the Rattus norvegicus Lamin A/C (GenBank accession number BC062018.1 and X99257.1), HIF-2α (GenBank accession number NM_023090).
Results
Normal expression of hypoxia-inhibitable K+ channels in nicotine-treated P0 adrenal chromaffin cells
Several K+ channel subtypes contribute to the O2-sensing properties of perinatal AMCs. In particular, hypoxic inhibition of large-conductance (BK) and small-conductance (SK) Ca2+-activated K+, and of delayed rectifier (Kv) K+ channels are thought to be the major mechanisms that facilitate membrane depolarization, voltage-gated Ca2+ entry, and catecholamine secretion (CAT) (Mochizuki-Oda et al., 1997; Thompson and Nurse, 1998; Fearon et al., 2002; Thompson et al., 2002; Keating et al., 2005; Bournaud et al., 2007). To confirm that the loss of O2 sensitivity in AMCs derived from neonatal pups born to nicotine-treated dams was not attributable to the lack of expression of these O2-sensitive K+ channels, we used pharmacological blockers and immunocytochemistry. As shown in Figure 1, outward K+ currents in neonatal AMCs obtained from nicotine-treated P0 pups were inhibited by the selective BK and SK channel blockers iberiotoxin (IbTx) (100 nm) and apamin (Apa) (100 nm), respectively, as well as by the general Ca2+ channel blocker cadmium (50 μm), which indirectly blocks Ca2+-dependent K+ channels. In general, these results were not significantly different from those obtained for control AMCs derived from pups born to saline-treated dams. For a voltage step to +30 mV, inhibition by IbTx was 34 ± 5.4% (n = 10) for saline-exposed (Fig. 1 A1) versus 32 ± 4.9% (n = 10) for nicotine-exposed cells (Fig. 1 B1). In the case of apamin, the inhibition was 20 ± 5.7% (n = 10) for saline-exposed P0 AMCs (Fig. 1 A2), versus 21 ± 6.9% (n = 10) for nicotine-exposed P0 AMCs (Fig. 1 A2). The percentage inhibition by 50 μm Cd2+ was 47 ± 6.7% (n = 10) for saline-exposed P0 AMCs (Fig. 1 A3) versus 53 ± 6.2% (n = 10) for nicotine-exposed P0 AMCs (Fig. 1 B3). Additionally, we used immunofluorescence to confirm the presence of BK and SK2 channels in nicotine-exposed P0 AMCs (Fig. 1 C). These data indicate that AMCs obtained from pups exposed to nicotine in utero express functional Ca2+-dependent K+ channels previously shown to mediate at least part of the O2-sensing properties of chromaffin cells. Moreover, because delayed-rectifier K+ channels consisting of Kv1.2 and Kv1.5 subunits have also been implicated in O2 sensing in chromaffin cells (Conforti and Millhorn, 2000; Fearon et al., 2002), we used immunocytochemistry to test for their expression as well. As shown in Figure 1 C, positive immunoreactivity for Kv1.2 and Kv1.5 subunits was obtained in nicotine-exposed P0 AMCs. In control experiments for all immunofluorescence studies, positive immunostaining was abolished after preincubation with the corresponding blocking peptide (data not shown). Thus, the previously reported loss of hypoxia sensing in nicotine-exposed AMCs (Buttigieg et al., 2008b) was not attributable to an overt loss of expression of K+ channel proteins that mediate O2 sensitivity.
Figure 1.
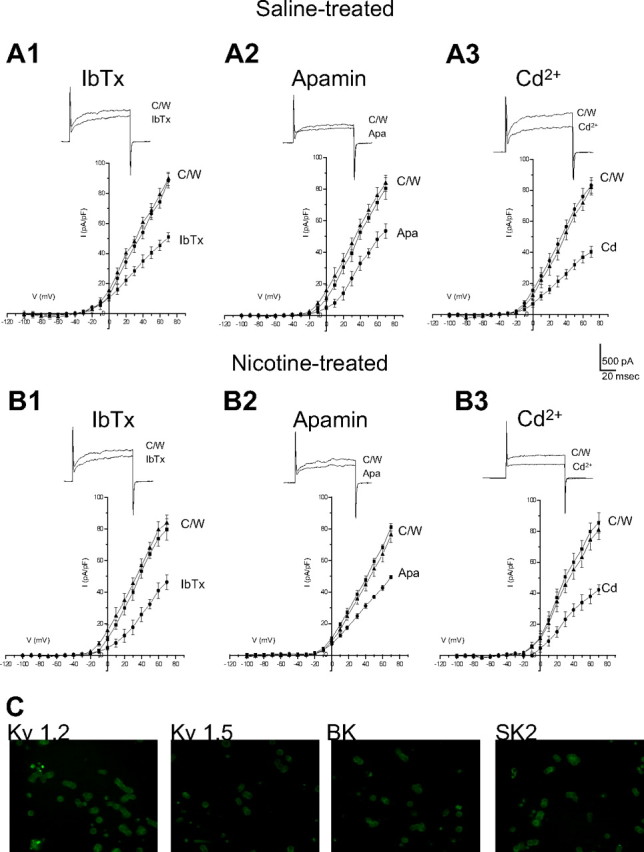
K+ channel expression in AMCs derived from P0 pups born to saline-treated versus nicotine-treated dams. In A1–A3, mean (±SEM) current density versus voltage plots are shown for saline-treated AMCs exposed to control (C) and either the BK channel blocker IbTx (100 nm; n = 10) (A1), the SK channel blocker Apa (100 nm; n = 10) (A2), or the nonspecific blocker of Ca2+ channels cadmium (Cd2+) (50 μm; n = 10) (A3), which indirectly blocks Ca2+-dependent K+ channels. The insets show sample recordings at +30 mV; holding potential was −60 mV. Corresponding data for P0 nicotine-treated AMCs are shown in B1–B3 (n = 10). Note nicotine-treated AMCs expressed similar Ca2+-dependent K+ currents as saline-treated AMCs. Isolated AMCs from pups exposed to nicotine in utero also showed expression of the K+ channel subunits Kv1.2 and Kv1.5, BK (α-subunit), and SK2, as determined by immunofluorescence (C).
Functional enhancement of hypoxia-activated KATP currents in nicotine-exposed P0 adrenal chromaffin cells
In contrast to the hypoxia-inhibitable K+ channels described above, KATP channels in neonatal AMCs are actually activated by hypoxia, thereby favoring membrane hyperpolarization (Mochizuki-Oda et al., 1997; Thompson and Nurse, 1998; Bournaud et al., 2007). Therefore, it was plausible that an increased expression of functional KATP channels in nicotine-exposed AMCs could contribute to the blunting of hypoxic sensitivity. To test this hypothesis, we monitored K+ currents and membrane potential in the presence of glibenclamide, a potent inhibitor of KATP channels.
Consistent with previous studies (Thompson and Nurse, 1998), glibenclamide (50 μm) significantly enhanced the inhibitory effect of hypoxia on outward K+ currents at more positive potentials in control, saline-exposed AMCs (Fig. 2 A). For a voltage step to +30 mV, hypoxia caused a 27 ± 5.1% inhibition of outward current (Fig. 2 A) (n = 12), whereas coapplication of hypoxia and glibenclamide caused an additional inhibition of outward current (57.8 ± 6.9% inhibition; n = 12; p < 0.05) (Fig. 2 A). Notably, glibenclamide alone had no significant effect on outward K+ current in normoxia (Fig. 2 A), suggesting that KATP channel opening occurs primarily during exposure to hypoxia. Consistent with our previous studies, hypoxia had a negligible effect on outward K+ current in neonatal AMCs from pups born to nicotine-exposed dams (Buttigieg et al., 2008b). However, when these cells were exposed to glibenclamide, hypoxia induced a pronounced inhibition of outward current (Fig. 2 B). These data are consistent with an upregulation of functional KATP channels in nicotine-treated AMCs. To obtain a quantitative estimate of the contribution of KATP current (IKATP), the glibenclamide-sensitive difference current density during hypoxia was calculated by subtracting the current recorded in hypoxia alone from that in the presence of hypoxia plus glibenclamide. IKATP current density versus voltage plots for both saline- and nicotine-exposed AMCs are shown in Figure 2 E, in which it is evident that IKATP is significantly augmented in nicotine-exposed AMCs. For example, at a voltage step to +30 mV, the saline-exposed cells had a KATP difference current density of 16.7 ± 2.8 pA/pF compared with 24.9 ± 3.1 pA/pF for nicotine-exposed cells, corresponding to an increase of ∼57% (Fig. 2 E) (p < 0.05). The reversal potential obtained for this glibenclamide-sensitive difference current was −76 ± 4.2 mV (n = 12) (Fig. 2 E), close to the Nernst potential for K+ (E K = −80 mV) in these experiments. Additional experiments based on membrane potential measurements under current clamp confirmed that the contribution of IKATP was significantly increased in nicotine-exposed AMCs. As exemplified in Figure 2 D, although hypoxia alone had a negligible effect on the resting membrane potential of nicotine-exposed neonatal AMCs (Buttigieg et al. 2008b), it strongly depolarized and increased excitability in the same cells when combined with glibenclamide (n = 10). This effect of glibenclamide appeared less dramatic in control saline-exposed AMCs, which often displayed depolarizing responses during hypoxia alone (Fig. 2 C) (Thompson and Nurse, 1998; Buttigieg et al. 2008b).
Figure 2.

Upregulation of glibenclamide-sensitive KATP channels in AMCs derived from P0 pups born to saline-treated versus nicotine-treated dams. Although glibenclamide (glib) had no effect on its own, it potentiated the hypoxia-induced (hox) inhibition of outward K+ current (A) and membrane depolarization (C) in P0 saline-treated AMCs. In contrast, P0 nicotine-treated AMCs did not respond to hypoxia alone (B); however, the combination of hypoxia plus glibenclamide resulted in a marked inhibition of outward K+ current (B), as well as membrane depolarization accompanied by spike activity (D). The glibenclamide-sensitive difference current density, which provides an estimate of IKATP current density, is plotted against voltage for the two conditions in E. Note the significantly larger IKATP density in nicotine-treated AMCs suggesting upregulation of KATP channels, and the reversal potential of IKATP near K+ equilibrium potential (E K = −80 mV) in E. Error bars indicate SEM. In F, Western blot analysis demonstrated a significant upregulation of the Kir 6.2 subunit of the KATP channel in P0 nicotine-treated AMCs compared with saline-treated AMCs, with β-actin as control.
To test whether the proposed functional upregulation of KATP channels in nicotine-exposed cells was associated with an increase in protein levels, Western blot analysis was used to probe for KATP channel subunits. The KATP channel consists of a pore-forming Kir subunit and a regulatory Sur subunit, and in neonatal AMCs the Kir 6.2 and Sur 1 isoforms appear to be major subunits expressed (our unpublished RT-PCR data) (Bournaud et al. 2007). As illustrated in Figure 2 F, Western blot analysis confirmed the upregulation of Kir 6.2 subunits in nicotine-exposed neonatal AMCs relative to saline controls. Interestingly, we did not observe any significant change in expression of the SUR subunit (data not shown).
Effects of hypoxia and glibenclamide on intracellular [Ca2+] in control saline- versus nicotine-exposed P0 adrenal chromaffin cells
Elevation in intracellular calcium (Cai), arising principally from voltage-gated Ca2+ entry, represents a key step leading to catecholamine secretion from neonatal AMCs after exposure to hypoxia and hypercapnia (Mochizuki-Oda et al., 1997; Thompson et al., 1997, 2002; Fearon et al., 2002; Muñoz-Cabello et al., 2005; Buttigieg et al., 2008a,b). We used fura-2 spectrofluorimetry to monitor Cai in P0 AMCs obtained from pups born to saline- and nicotine-exposed dams, after exposure to hypoxia and/or glibenclamide. Hypoxia caused a significant rise in cytosolic Cai in saline-exposed P0 AMCs, and consistent with our electrophysiological results, glibenclamide (50 μm) on its own had no effect on Cai (Fig. 3 A). However, combined application of hypoxia and glibenclamide resulted in a significant increase in Cai compared with hypoxia alone. The nicotine-exposed P0 AMCs failed to respond to hypoxia (Fig. 3 B), as previously demonstrated (Buttigieg et al., 2008b). However, consistent with our electrophysiological results, combined application of hypoxia and glibenclamide caused a significant rise in Cai (Fig. 3 B), further suggesting that the nicotine-induced loss of hypoxia sensing in neonatal AMCs is attributable to upregulation of glibenclamide-sensitive KATP channels.
Figure 3.

Fura-2 spectrofluorimetric determination of intracellular calcium (Cai) levels in AMCs derived from P0 pups born to saline-treated versus nicotine-treated dams. In P0 saline-treated AMCs, significant increases in Cai relative to normoxic (Nox) control (ANOVA; *p < 0.001) occurred during exposure to hypoxia (Hox) (PO2 ∼ 15 mmHg) and the depolarizing stimulus, high extracellular K+ (30 mm). Although the KATP channel blocker glibenclamide (glib) had no effect on its own, it significantly potentiated the hypoxia-induced rise in Cai seen in saline-treated cells as illustrated in the histogram in A (significance compared with hypoxia alone, **p < 0.001, n = 42). Comparative data are shown for nicotine-treated AMCs in B (n = 35). Note lack of effect of hypoxia alone on Cai levels in nicotine-treated AMCs, although coapplication of glibenclamide and hypoxia resulted in a significant increase in Cai (ANOVA; *p < 0.001). Error bars indicate SEM. Sample recordings are shown in the top traces.
Stimulation of α7 nAChRs is responsible for suppressing hypoxic sensitivity in nicotine-treated P0 adrenal chromaffin cells
Although direct stimulation of nAChRs on neonatal AMCs during chronic nicotine exposure in vitro is known to cause a loss of hypoxia sensing, the signaling pathway remains to be elucidated (Buttigieg et al., 2008b). As a first step to address this, we investigated whether specific subtypes of nAChRs on chromaffin cells needed to be activated. Therefore, neonatal AMCs were cultured for 7 d in the presence chronic nicotine base (50 μm), with or without mecamylamine (a nonspecific nAChR inhibitor), hexamethonium (an inhibitor of most nAChRs except homomeric α7), or α-bungarotoxin (a potent α7 blocker). Additionally, to address whether the effects of nAChR stimulation were simply mediated via membrane depolarization, cells were also cultured in the presence of a depolarizing stimulus, high K+ (30 mm). Hypoxic sensitivity, as determined by inhibition of outward K+ current at more positive potentials under voltage clamp (+30 mV), was retained in saline-exposed P0 AMCs cultured in control (nicotine-free) medium for 7 d (Fig. 4 A). In contrast, hypoxic sensitivity was lost when such cells were cultured for 7 d in medium supplemented with 50 μm nicotine base (Fig. 4 A), and similar to our previous findings (Buttigieg et al., 2008b), this blunting effect of nicotine was prevented by the continuous presence of mecamylamine (100 μm) (Fig. 4 A). Interestingly, hexamethonium (100 μm), which is an effective blocker of most nAChR α subunits except for homomeric α7 (Martin et al., 2005), did not prevent the blunting effect of chronic nicotine on hypoxic sensitivity (Fig. 4 A). To confirm that nAChRs on neonatal chromaffin cells had the expected pharmacological profile, we examined the effects of hexamethonium and mecamylamine on nicotine-induced inward currents. As expected, the currents evoked by 50 μm nicotine at −60 mV were inhibited almost completely by 100 μm mecamylamine (mean, 98.4 ± 3.6% inhibition; n = 4) and only partially by 100 μm hexamethonium (mean, 44.1 ± 6.8%; n = 4). The implication that activation of α7 nAChRs played a key role in the blunting of hypoxic sensitivity was confirmed in experiments in which neonatal AMCs were cultured in the presence of both nicotine and the potent α7 blocker α-bungarotoxin (100 nm). As illustrated in Figure 4 A, the ability of nicotine to blunt hypoxic sensitivity in neonatal AMCs was prevented by the presence of α-bungarotoxin, suggesting a critical role for α7 homomeric nAChRs. Because α7 receptors have high Ca2+ permeability, we investigated whether their effect could be mimicked by conditions that promote a sustained Ca2+ entry through voltage-gated Ca2+ channels. This was not the case since neonatal AMCs cultured in the presence of a depolarizing stimulus (30 mm K+) were still responsive to hypoxia (Fig. 4 A), thus confirming our hypothesis that nicotine exerted its effects via stimulation of α7 nAChRs.
Figure 4.
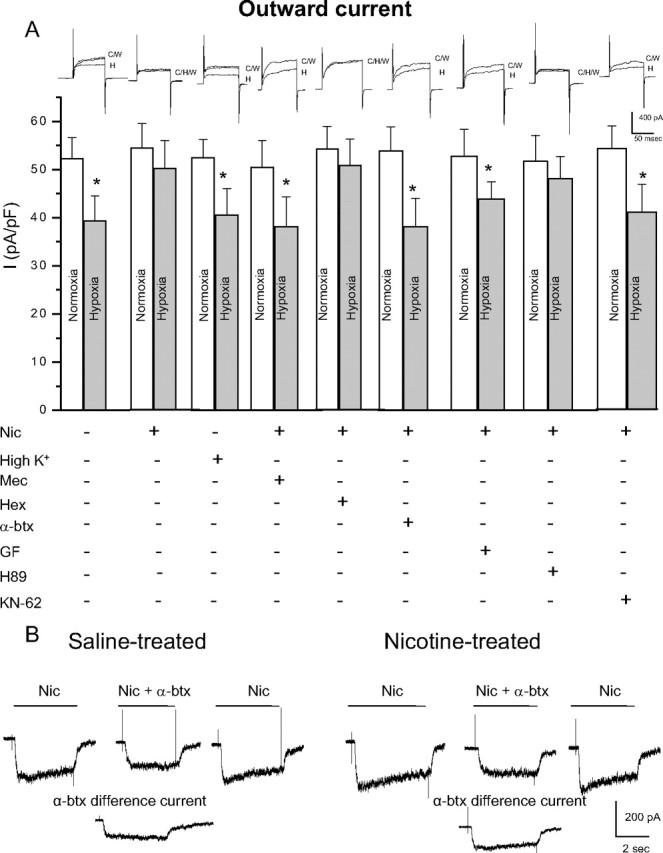
Role of nAChR subtypes and intracellular signaling pathways in the nicotine-mediated loss of hypoxic sensitivity in neonatal AMCs. Dissociated AMCs from saline-treated P0 pups were grown for ∼1 week in culture under control conditions, or in presence of nicotine base (Nic) (50 μm) with either mecamylamine (Mec) (100 μm; general nAChR blocker), hexamethonium (Hex) (100 μm; blocker of most nAChRs except α7), α-bungarotoxin (α-btx) (100 nm; specific α7 nAChR blocker), or the depolarizing stimulus high K+ (30 mm) as indicated. To probe the signaling pathways, neonatal AMCs were cultured with nicotine and one of the following: the PKC blocker GF109203X (GF) (2 μm), CaM kinase inhibitor KN-62 (3 μm), or the PKA inhibitor H-89 (20 μm). Histogram indicates (mean ± SEM) outward current density (in picoamperes per picofarad) at +30 mV during normoxia or hypoxia (A). Note that hypoxic sensitivity, indicated by inhibition of outward current, was lost after chronic nicotine treatment in culture and that this effect was prevented by α7 nAChR antagonists Mec and α-btx only. Also, hypoxia sensitivity was detectable in nicotine-treated AMCs after coincubation with the PKC inhibitor GF, the CaM kinase inhibitor KN-62, but not the PKA inhibitor H-89. Representative traces for each treatment are located above each pair of bins (C, control; H, hypoxia; W, wash). Sample size was n = 7 cells for each treatment, asterisk (*) denotes significantly different from normoxia, p < 0.01. In B, fast application of nicotine (10 μm) induced an inward current at −60 mV (holding potential) that was partially inhibited by the α7 nAChR blocker α-btx (100 nm) applied to the same saline-treated (left traces) or nicotine-treated (right traces) cell; the α-btx-sensitive difference current for each of the two cells is shown in the bottom traces.
To confirm the presence of α7 nAChRs, we acutely isolated neonatal AMCs and examined nicotine-induced current responses under voltage clamp. Fast application of nicotine (10 μm) induced a robust inward current at −60 mV in P0 AMCs obtained from pups born to saline-exposed dams (Fig. 4 B). This inward current was inhibited 27.1 ± 3.8% (n = 10) in the presence of α-bungarotoxin (100 nm) (Fig. 4 B), suggesting that neonatal AMCs do indeed express functional α7 nAChRs. At the concentration (50 μm) of nicotine used in our chronic in vitro experiments, 100 nm α-bungarotoxin caused a similar inhibition of nicotine-evoked currents (mean, 26.8 ± 2.1% inhibition; n = 6). Examination of the α-bungarotoxin-sensitive difference current revealed it accounted for 38 ± 5.4% of the nicotine-evoked current in nicotine-exposed AMCs compared with ∼27% in saline-exposed AMCs, suggesting an increased contribution of α7 nAChRs with nicotine exposure (Fig. 4 B). In preliminary Western blot analyses, it appears that both α3 and α7 subunit protein expression is increased during chronic nicotine (data not shown); however, this point requires validation in future studies.
Catecholamine secretion
Using carbon fiber amperometry, we monitored CAT secretion from chromaffin cells to obtain independent confirmation of the key role of α7 nAChRs in mediating the blunting effect of nicotine on hypoxia sensing. Cells cultured for 7 d in control media still retained hypoxic sensitivity as revealed by hypoxia-induced CAT secretion, whereas cells cultured for the same period in chronic nicotine lacked hypoxic sensitivity (Fig. 5). In concert with the electrophysiological studies reported above, the blunting effect of chronic nicotine in vitro on hypoxia-induced CAT secretion from AMCs was prevented by coincubation with either the specific α7 blocker, α-bungarotoxin, or the general nAChR blocker, mecamylamine, but not by hexamethonium (100 μm), which is ineffective at α7 nAChRs on these cells (Martin et al., 2005). Similarly, a chronic depolarizing stimulus high K+ (30 mm) was ineffective in blunting the hypoxia-induced CAT secretion from cultured AMCs (Fig. 5), indicating a specific requirement for the activation of α7 nAChRs.
Figure 5.

Effects of hypoxia on catecholamine secretion from AMCs after various chronic treatments in culture. Carbon fiber amperometry was used to detect stimulus-evoked release of CATs from AMCs cultured for 7 d in the presence of nicotine (Nic) (50 μm), with or without mecamylamine (Mec) (100 μm), hexamethonium (Hex) (100 μm), and α-bungarotoxin (α-btx) (100 nm), or in the presence of the depolarizing stimulus high K+ (30 mm). Hypoxia stimulated quantal CAT release as indicated by an increase in event frequency (events per minute) in control AMCs and AMCs grown chronically in high K+. In contrast, hypoxia failed to stimulate CAT secretion from AMCs cultured in the presence of nicotine; however, this blunting effect of nicotine was prevented after coincubation with Mec or α-btx, but not hex. Data were obtained from seven cells for each treatment [the asterisk (*) indicates significant difference, p < 0.01, from normoxic (Nox) control]. Error bars indicate SEM. The top traces show sample recordings of quantal CAT release from AMCs under the conditions indicated.
Role of PKC and CaM kinase in mediating the loss of hypoxic sensitivity in nicotine-treated chromaffin cells
Given that activation of α7 nAChRs appears to be a key initial step, we next investigated the downstream second messenger pathways that participate in the nicotine-induced upregulation of KATP channels in chromaffin cells. Potential roles of PKC, CaM kinase, and PKA were investigated after coincubation with the blockers 2-[1-(3-dimethylaminopropyl)-1H-indol-3-yl]-3-(1H-indol-3-yl)maleimide (GF109203X) (2 μm), N-[2-((o-bromocinamyl)amino)ethyl]-5-isoquinolinesulfonamide (H-89) (20 μm), [4(2S)-2-[(5-isoquinolinylsulfonyl)methylamino]-3-oxo-3-(4-phenyl-1-piperazinyl)propyl]phenyl isoquinolinesulfonic acid ester (KN-62) (3 μm), respectively. In voltage-clamp experiments, neonatal AMCs cultured in the presence of nicotine plus the PKC blocker GF109203X retained hypoxic sensitivity as measured by the inhibition of outward current at +30 mV (Fig. 4 A). Similar results were obtained with the CaM kinase blocker KN-62 (Fig. 4 A), but coincubation with the PKA blocker H-89 was ineffective in reversing the blunting effect of nicotine on hypoxic sensitivity (Fig. 4 A). These data support the idea that loss of hypoxia sensing in nicotine-treated neonatal AMCs involves activation of both PKC and CaM kinase, but not PKA.
We tested whether a similar pathway for nicotine-induced loss of hypoxic sensitivity was present in the v-myc adrenal-derived HNK1+ immortalized chromaffin cell line (MAH cells) (Birren and Anderson, 1990). This line was chosen because previous studies from this laboratory have demonstrated that MAH cells express O2-sensing properties similar to those of neonatal rat AMCs, and moreover, compared with primary cells, this cell line is easier to manipulate using RNA interference techniques (Fearon et al. 2002; Brown and Nurse, 2008; Buttigieg et al., 2008a). Indeed, Western blot analysis of protein extracts from MAH cells cultured for 7 d in the presence of chronic nicotine (50 μm) also demonstrated an upregulation of the KATP channel subunit Kir 6.2 relative to controls (Fig. 6 A,B) (see also Fig. 2 F). Moreover, similar to the results on primary neonatal AMCs, the presence of the specific α7 nAChR blocker α-bungarotoxin or the general blocker mecamylamine prevented the nicotine-induced upregulation of Kir 6.2 subunit in MAH cells, whereas the non-α7 nAChR blocker hexamethonium was ineffective (Fig. 6 A).
Figure 6.

Nicotinic AChR-mediated regulation of the expression of the KATP channel subunit Kir 6.2 in cultured immortalized chromaffin (MAH) cells. In Western blots, MAH cells cultured for 7 d in the presence of nicotine (Nic) (50 μm) showed an increased expression of the Kir 6.2 subunit, relative to control untreated cultures. This effect of nicotine persisted during coincubation with hexamethonium (Hex) (100 μm), but was blocked during coincubation with the α7 nAChR antagonist α-bungarotoxin (α-btx) (100 nm) or mecamylamine (Mec) (100 μm). Chronic membrane depolarization with 30 mm K+ did not result in upregulation of Kir 6.2 (B). Upregulation of Kir 6.2 was also prevented when MAH cells were cultured with nicotine and either the PKC blocker GF or the CaM kinase blocker KN62; the PKA inhibitor H-89 was ineffective. Relative staining intensities (compared with control) are summarized for four Western blots in the corresponding histograms below. *p < 0.005. Error bars indicate SEM.
We next investigated whether the upregulation of Kir 6.2 subunit in MAH cells by chronic nicotine involved the PKC and CaM kinase pathways, as demonstrated above for primary neonatal AMCs. As illustrated in Figure 6 B, coincubation with either the PKC blocker GF109203X or the CaM kinase blocker KN-62 prevented the upregulation of Kir 6.2 subunit by chronic nicotine in MAH cells. In contrast, the PKA blocker H-89 failed to prevent upregulation of the Kir 6.2 subunit by nicotine (Fig. 6 B). Furthermore, the chronic depolarizing stimulus high K+ (30 mm) failed to mimic the effects of nicotine on Kir 6.2 subunit expression (Fig. 6 B), in agreement with our electrophysiological studies on native AMCs (Figs. 3 A, 4 B). These results suggest that during chronic nicotine exposure, stimulation of α7 nAChRs on both neonatal and immortalized fetal-derived chromaffin cells activates a signaling cascade involving PKC and CaM kinase, but not PKA, leading to an upregulation of KATP channels and, consequently a suppression of hypoxic sensitivity.
HIF-2α activation is involved in the nicotine-mediated loss of hypoxia sensing in chromaffin cells
It has been shown that activation of nAChRs in human lung cancer cells induces expression of HIF, resulting in altered gene regulation (Zhang et al., 2007). Because HIF-2α is the predominant form present in MAH cells (Brown and Nurse, 2008), we tested the effects of chronic nicotine on MAH cells bearing a short hairpin RNAi sequence that resulted in >90% knockdown of HIF-2α (shMAH cells) as confirmed by RT-PCR and Western blot analysis (Fig. 7 F). HIF-2α mRNA was expressed in scrambled control cells (scMAH) but was considerably reduced in the knockdown shMAH cells. To validate that HIF-2α protein was also deficient in shMAH cells, we exposed cells to chronic hypoxia (2% O2) for 2 h, a stimulus that normally induces HIF-2α protein in control MAH cells (Brown and Nurse, 2008). As illustrated in Figure 7 F, Western blot analysis confirmed that, although hypoxia induced HIF-2α protein in scMAH cells, it failed to do so in shMAH cells.
Figure 7.
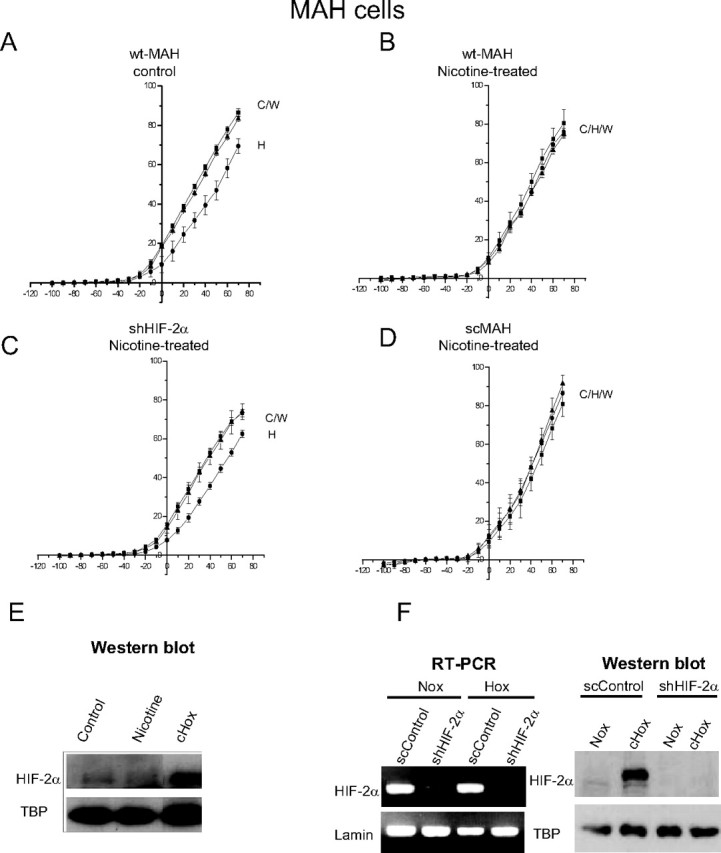
Role of HIF-2α in mediating the nicotine-induced loss of hypoxia sensitivity. MAH cells deficient in HIF-2α (>90% knockdown) (shMAH) were cultured with or without nicotine, and hypoxic sensitivity was determined based on inhibition of outward K+ current. Control, wild-type MAH cells (wtMAH) were hypoxia-sensitive (A) but became hypoxia-insensitive when cultured with chronic nicotine (B). In contrast, nicotine treatment failed to affect the hypoxic sensitivity of MAH cells deficient in HIF-2α (shMAH) (C); however, in scrambled control MAH cells (scMAH), nicotine exposure still resulted in a loss of hypoxic sensitivity (D). Error bars indicate SEM. In E, the Western blots show that HIF-2α protein was induced in MAH cells by chronic hypoxia (CHox) (2% O2 for 24 h), but not by chronic nicotine; HIF-2α induction by chronic hypoxia was also present in scMAH cells but was absent in shMAH cells (F). RT-PCR analysis in F shows downregulation of HIF-2α mRNA in shMAH cells, but not in scMAH cells.
We next investigated the potential role of HIF-2α in mediating the blunting effect of nicotine on hypoxia sensing in MAH cells. Similar to primary neonatal AMCs, hypoxic sensitivity was lost in control wild-type MAH cells (wtMAH) exposed to chronic nicotine in vitro, as measured by the inhibition of outward current (Fig. 7 A,B). However, this nicotine-induced loss of hypoxia sensing appeared to require HIF-2α function, because shMAH cells that lack HIF-2α expression showed hypoxic inhibition of outward current even after culture in chronic nicotine (Fig. 7 C). This sensitivity to hypoxia was also present in shMAH cells grown in control media; moreover, these cells still showed a hypoxia-inhibitable outward current that was enhanced by glibenclamide (data not shown). In contrast, the hypoxic response in scMAH cells was blunted after exposure to chronic nicotine in culture (Fig. 7 D).
Western blot analysis was done to determine whether or not HIF-2α protein levels were altered during chronic exposure to nicotine. This was not the case because no significant change in HIF-2α protein level was observed in MAH cells cultured with nicotine (Fig. 7 E). Thus, although our electrophysiological studies indicated that the nicotine-induced loss in hypoxia sensing was dependent on HIF-2α expression, the exact mechanism by which this occurs requires additional study.
Effect of glibenclamide pretreatment on hypoxia tolerance in nicotine-exposed pups
The above results suggest that chronic nicotine exposure in utero leads to an upregulation of KATP channels that open during acute hypoxia, causing cell hyperpolarization and blunting of hypoxic sensitivity in neonatal chromaffin cells. If so, administration of glibenclamide, a drug that inhibits KATP channels and commonly used as a diabetic medication, should reverse the blunting effect of hypoxia and conceivably remedy the loss of hypoxia tolerance in nicotine-treated pups. To test this possibility, pups were placed into three groups. Group 1 consisted of pups born to saline-treated mothers, whereas groups 2 and 3 were matched litter mates from nicotine-treated mothers. Group 2 pups were injected with vehicle (PBS and 0.1% DMSO) subcutaneously, and group 3 pups were injected with glibenclamide (90 mg/kg plus 0.1% DMSO). Group 1 pups experienced a mortality of 17.3 ± 7.6% when exposed to 10% O2 for 60 min (n = 40) (Fig. 8). Similar to previous findings (Slotkin et al., 1995), group 2 pups experienced a significantly higher mortality in which 38.6 ± 4.1% of them died after a similar hypoxic exposure (n = 40) (Fig. 8). Interestingly, group 3 pups obtained from nicotine-treated dams, but injected with glibenclamide, had a significantly lower mortality (p < 0.003) when exposed to the same hypoxic challenge (Fig. 8). These findings strongly suggest that injection of glibenclamide may ameliorate the adverse effects of hypoxia on the survival of neonates born to nicotine-exposed mothers and that this is attributable to its blocking action on KATP channels.
Figure 8.

Effect of prenatal nicotine exposure and acute glibenclamide injection on neonatal (postnatal day 1) mortality during a 60 min exposure to hypoxia (10% O2). Compared with rat pups born to saline-treated dams (control), those born to nicotine-treated dams had a higher percentage of mortality after exposure to hypoxia; both groups received a saline injection (saline inj) just before exposure to hypoxia. In contrast, neonatal nicotine-treated pups that were injected with glibenclamide (glib inj) just before exposure to hypoxia, had a percentage of mortality that was not significantly different from control. N = 40 for each bin. *p < 0.009. Error bars indicate SEM.
Discussion
In this study, we demonstrate that maternal nicotine exposure had a dramatic effect in upregulating the expression of KATP channels in AMCs of the offspring. This mechanism appears central to the nicotine-induced loss of hypoxia sensing in neonatal AMCs, with the resulting failure of these cells to initiate the critical catecholamine surge during hypoxic stress. Catecholamine release in the neonate is required for redistribution of blood flow to the brain and heart, for maintenance of cardiac contractility during O2 deprivation, and for transformation of the lung into an air breathing organ (Lagercrantz and Slotkin, 1986; Slotkin et al., 1995). Although inhibition of certain classes of K+ channels (e.g., the Ca2+-dependent BK and SK channels) during hypoxia is the major mechanism that facilitates membrane depolarization, Ca2+ influx, and CAT secretion, the activation of KATP channels has an antagonistic effect in neonatal chromaffin cells (Mochizuki-Oda et al., 1997; Thompson et al., 1997; Thompson and Nurse, 1998). This mechanism appears to be a protective one that tends to hyperpolarize the cell during hypoxia, thereby reducing the intracellular Ca2+ load, as well as limiting excitability and conserving energy stores required for the maintenance of ion gradients (Thompson and Nurse, 1998; Bournaud et al., 2007). Therefore, the action of nicotine, by selectively upregulating KATP channels, leads to a blunting of hypoxic sensitivity in these cells, and consequently loss of hypoxic tolerance in the neonate. Interestingly, in late fetal stages, there is a normal developmental switch such that KATP channel expression decreases while Ca2+-dependent BK channel expression increases in rat adrenal chromaffin cells (Bournaud et al., 2007). Thus, chronic nicotine exposure during fetal development may simply delay KATP channel downregulation that normally occurs and/or promote an additional upregulation of these KATP channels. The end result is that the neonate is born with a higher density of KATP channels in adrenal chromaffin cells and this diminishes the animal's ability to evoke the critical catecholamine surge in response to hypoxic stress.
It is possible that chronic nicotine in utero may also affect other targets in the hypoxia signaling pathway. For example, it was recently proposed that postnatal downregulation of T-type calcium channels may contribute to the developmental loss of hypoxic sensitivity in chromaffin cells (Levitsky and López-Barneo, 2009). It remains to be determined whether or not chronic nicotine exposure can regulate T-type calcium channels in neonatal AMCs.
Signaling pathways mediating KATP channel upregulation
Our results favor a model in which the effects of nicotine in utero are mediated via direct activation of neuronal nicotinic α7 ACh receptors (nAChRs), known to be present on embryonic/neonatal adrenal chromaffin cells (Mousavi et al., 2001; Martin et al., 2005; this study). We found that neonatal chromaffin cells cultured in the presence of chronic nicotine at subsaturating doses (50 μm) for ∼1 week lost hypoxic sensitivity via an upregulation of KATP channels, similar to acutely isolated chromaffin cells derived from pups born to nicotine-exposed dams. Both the loss of hypoxic sensitivity and upregulation of KATP channels seen in cultured cells were prevented when mecamylamine, a general blocker of nicotinic nAChRs, was coincubated with nicotine throughout the culture period. Significantly, this effect of nicotine was not inhibited by hexamethonium, at doses expected to block α3-containing nAChRs on neonatal chromaffin cells (Sala et al., 2008), but was blocked by the specific α7 nAChR blocker α-bungarotoxin. These data strongly suggest that the nicotine-induced loss of hypoxic sensitivity was mediated via direct stimulation of α7 nAChRs on chromaffin cells. Because α7 nAChRs are highly Ca2+-permeable, it is likely that Ca2+ entry through these receptors initiated a signaling cascade that ultimately resulted in KATP channel upregulation. This particular route of Ca2+ entry appeared critical since an alternative route via voltage-gated Ca2+ channels, mediated by chronic exposure of the cultures to the depolarizing stimulus high K+ (30 mm), was ineffective.
Other intracellular signaling pathways also appeared to be involved in the effects of chronic nicotine. In particular, key roles for PKC and CaM kinase activation (but not PKA) were demonstrated by the observation that coincubation with specific blockers of these kinases (i.e., GF109203X and KN-62, respectively) prevented the nicotine-induced loss of hypoxic sensitivity. The identity of the proteins that are presumed to be phosphorylated by these kinases during the signal transduction cascade is currently unknown. In this regard, however, it was of interest that the transcription factor HIF-2α was required for the nicotine-induced blunting of hypoxic sensitivity. This was demonstrated in a chromaffin cell line (MAH cells) bearing >90% knockdown of HIF-2α using short hairpin RNAi, in which chronic nicotine in vitro did not cause suppression of hypoxic sensitivity as it did in control MAH cells or MAH cells treated with scrambled RNA. The fact that, in previous studies, both PKC and CaM kinase, as well as increases in intracellular Ca2+, were able to stabilize HIF and/or increase HIF transcriptional activity (Yuan et al., 2005; Lee et al., 2007), raises the possibility of a similar role for these intracellular signals in mediating the KATP upregulation via changes in HIF-2α activity. In the present study, chronic nicotine did not cause any detectable upregulation of HIF-2α protein in Western blot assays, although changes in phosphorylation state of HIF-2α could not be excluded. Because HIFs do not appear to have much, if any, transcriptional activity on their own and require other cofactors for functionality, we can only speculate on the role of HIF-2α in the effects of nicotine on chromaffin cells. It is possible that nicotine acts so as to modulate HIF-2α activity by some posttranslational modification (e.g., phosphorylation), or alternatively, nicotine acts on some other factor that uses HIF-2α as a cofactor. Additional experiments are necessary to distinguish between these two possibilities.
Clinical significance
These results open up a new avenue for possible pharmacological intervention in infants that have been exposed to nicotine in utero. Maternal smoking is correlated with poor pregnancy outcome, premature delivery, and a higher incidence of sudden infant death syndrome (SIDS), and this is attributed, at least in part, to deficient adrenal catecholamine secretion during hypoxic stress at birth (Slotkin et al., 1995; Cohen et al., 2005). We identified the upregulation of KATP channels as a key factor contributing to the apparent loss of hypoxic sensitivity in nicotine-exposed adrenal chromaffin cells. Our data provide compelling evidence that blockade of these channels in vivo using the drug glibenclamide can offset or reverse the loss of hypoxia tolerance because of prenatal nicotine exposure. Glibenclamide is currently used as a treatment in individuals with certain types of diabetes and has even been shown to be effective in certain clinical cases of Kir 6.2 mutations (Mlynarski et al., 2007). Conceivably, infants exposed to nicotine in utero might experience a similar upregulation of KATP channel expression as observed in this study, resulting in impaired responses to hypoxia as a result of excessive KATP channel activation. We propose that KATP channel blockers such as glibenclamide might have therapeutic potential in reducing mortality in newborn infants who are prone to SIDS as a result of maternal exposure to cigarette smoke.
Footnotes
This work was supported by operating and equipment grants from the Heart and Stroke Foundation (HSF) of Ontario (T-4641) (C.A.N.) and Canadian Institutes of Health Research Grant MOP 69025 (A.C.H.). J.B. and S.B. were supported by Focus on Stroke awards from HSF of Canada. We thank Cathy Vollmer and Min Zhang for their expert technical assistance.
References
- Birren SJ, Anderson DJ. A v-myc-immortalized sympathoadrenal progenitor cell line in which neuronal differentiation is initiated by FGF but not NGF. Neuron. 1990;4:189–201. doi: 10.1016/0896-6273(90)90094-v. [DOI] [PubMed] [Google Scholar]
- Bournaud R, Hidalgo J, Yu H, Girard E, Shimahara T. Catecholamine secretion from rat foetal adrenal chromaffin cells and hypoxia sensitivity. Pflugers Arch. 2007;454:83–92. doi: 10.1007/s00424-006-0185-z. [DOI] [PubMed] [Google Scholar]
- Brown ST, Nurse CA. Induction of HIF-2alpha is dependent on mitochondrial O2 consumption in an O2-sensitive adrenomedullary chromaffin cell line. Am J Physiol Cell Physiol. 2008;294:C1305–C1312. doi: 10.1152/ajpcell.00007.2008. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell. 2002;2:243–247. doi: 10.1016/s1535-6108(02)00122-8. [DOI] [PubMed] [Google Scholar]
- Buttigieg J, Brown ST, Lowe M, Zhang M, Nurse CA. Functional mitochondria are required for O2 but not CO2 sensing in immortalized adrenomedullary chromaffin cells. Am J Physiol Cell Physiol. 2008a;294:C945–C956. doi: 10.1152/ajpcell.00495.2007. [DOI] [PubMed] [Google Scholar]
- Buttigieg J, Brown S, Zhang M, Lowe M, Holloway AC, Nurse CA. Chronic nicotine in utero selectively suppresses hypoxic sensitivity in neonatal rat adrenal chromaffin cells. FASEB J. 2008b;22:1317–1326. doi: 10.1096/fj.07-9194com. [DOI] [PubMed] [Google Scholar]
- Cohen G, Roux JC, Grailhe R, Malcolm G, Changeux JP, Lagercrantz H. Perinatal exposure to nicotine causes deficits associated with a loss of nicotinic receptor function. Proc Natl Acad Sci U S A. 2005;102:3817–3821. doi: 10.1073/pnas.0409782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Millhorn DE. Regulation of Shaker-type potassium channels by hypoxia. Oxygen-sensitive K+ channels in PC12 cells. Adv Exp Med Biol. 2000;475:265–274. doi: 10.1007/0-306-46825-5_25. [DOI] [PubMed] [Google Scholar]
- Fearon IM, Thompson RJ, Samjoo I, Vollmer C, Doering LC, Nurse CA. O2-sensitive K+ channels in immortalised rat chromaffin-cell-derived MAH cells. J Physiol. 2002;545:807–818. doi: 10.1113/jphysiol.2002.028415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giegerich R, Meyer F, Schleiermacher C. GeneFisher—software support for the detection of postulated genes. Proc Int Conf Intell Syst Mol Biol. 1996;4:68–77. [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Holloway AC, Lim GE, Petrik JJ, Foster WG, Morrison KM, Gerstein HC. Fetal and neonatal exposure to nicotine in Wistar rats results in increased beta cell apoptosis at birth and postnatal endocrine and metabolic changes associated with type 2 diabetes. Diabetologia. 2005;48:2661–2666. doi: 10.1007/s00125-005-0022-5. [DOI] [PubMed] [Google Scholar]
- Keating DJ, Rychkov GY, Giacomin P, Roberts ML. Oxygen-sensing pathway for SK channels in the ovine adrenal medulla. Clin Exp Pharmacol Physiol. 2005;32:882–887. doi: 10.1111/j.1440-1681.2010.04279.x. [DOI] [PubMed] [Google Scholar]
- Lagercrantz H, Slotkin TA. The “stress” of being born. Sci Am. 1986;254:100–107. doi: 10.1038/scientificamerican0486-100. [DOI] [PubMed] [Google Scholar]
- Lee JW, Park JA, Kim SH, Seo JH, Lim KJ, Jeong JW, Jeong CH, Chun KH, Lee SK, Kwon YG, Kim KW. Protein kinase C-delta regulates the stability of hypoxia-inducible factor-1 alpha under hypoxia. Cancer Sci. 2007;98:1476–1481. doi: 10.1111/j.1349-7006.2007.00535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitsky KL, López-Barneo J. Developmental change of T-type Ca2+ channel expression and its role in rat chromaffin cell responsiveness to acute hypoxia. J Physiol. 2009;587:1917–1929. doi: 10.1113/jphysiol.2009.168989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AO, Alonso G, Guérineau NC. Agrin mediates a rapid switch from electrical coupling to chemical neurotransmission during synaptogenesis. J Cell Biol. 2005;169:503–514. doi: 10.1083/jcb.200411054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlynarski W, Tarasov AI, Gach A, Girard CA, Pietrzak I, Zubcevic L, Kusmierek J, Klupa T, Malecki MT, Ashcroft FM. Sulfonylurea improves CNS function in a case of intermediate DEND syndrome caused by a mutation in KCNJ11. Nat Clin Pract Neurol. 2007;3:640–645. doi: 10.1038/ncpneuro0640. [DOI] [PubMed] [Google Scholar]
- Mochizuki-Oda N, Takeuchi Y, Matsumura K, Oosawa Y, Watanabe Y. Hypoxia-induced catecholamine release and intracellular Ca2+ increase via suppression of K+ channels in cultured rat adrenal chromaffin cells. J Neurochem. 1997;69:377–387. doi: 10.1046/j.1471-4159.1997.69010377.x. [DOI] [PubMed] [Google Scholar]
- Mousavi M, Hellström-Lindahl E, Guan ZZ, Bednar I, Nordberg A. Expression of nicotinic acetylcholine receptors in human and rat adrenal medulla. Life Sci. 2001;70:577–590. doi: 10.1016/s0024-3205(01)01427-8. [DOI] [PubMed] [Google Scholar]
- Muñoz-Cabello AM, Toledo-Aral JJ, López-Barneo J, Echevarría M. Rat adrenal chromaffin cells are neonatal CO2 sensors. J Neurosci. 2005;25:6631–6640. doi: 10.1523/JNEUROSCI.1139-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala F, Nistri A, Criado M. Nicotinic acetylcholine receptors of adrenal chromaffin cells. Acta Physiol (Oxf) 2008;192:203–212. doi: 10.1111/j.1748-1716.2007.01804.x. [DOI] [PubMed] [Google Scholar]
- Seidler FJ, Slotkin TA. Adrenomedullary function in the neonatal rat: responses to acute hypoxia. J Physiol. 1985;358:1–16. doi: 10.1113/jphysiol.1985.sp015536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidler FJ, Slotkin TA. Ontogeny of adrenomedullary responses to hypoxia and hypoglycemia: role of splanchnic innervation. Brain Res Bull. 1986;16:11–14. doi: 10.1016/0361-9230(86)90005-5. [DOI] [PubMed] [Google Scholar]
- Slotkin TA. Fetal nicotine or cocaine exposure: which one is worse? J Pharmacol Exp Ther. 1998;285:931–945. [PubMed] [Google Scholar]
- Slotkin TA, Seidler FJ. Adrenomedullary catecholamine release in the fetus and newborn: secretory mechanisms and their role in stress and survival. J Dev Physiol. 1988;10:1–16. [PubMed] [Google Scholar]
- Slotkin TA, Lappi SE, McCook EC, Lorber BA, Seidler FJ. Loss of neonatal hypoxia tolerance after prenatal nicotine exposure: implications for sudden infant death syndrome. Brain Res Bull. 1995;38:69–75. doi: 10.1016/0361-9230(95)00073-n. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Nurse CA. Anoxia differentially modulates multiple K+ currents and depolarizes neonatal rat adrenal chromaffin cells. J Physiol. 1998;512:421–434. doi: 10.1111/j.1469-7793.1998.421be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RJ, Jackson A, Nurse CA. Developmental loss of hypoxic chemosensitivity in rat adrenomedullary chromaffin cells. J Physiol. 1997;498:503–510. doi: 10.1113/jphysiol.1997.sp021876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RJ, Farragher SM, Cutz E, Nurse CA. Developmental regulation of O2 sensing in neonatal adrenal chromaffin cells from wild-type and NADPH-oxidase-deficient mice. Pflugers Arch. 2002;444:539–548. doi: 10.1007/s00424-002-0853-6. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Buttigieg J, Zhang M, Nurse CA. A rotenone-sensitive site and H2O2 are key components of hypoxia-sensing in neonatal rat adrenomedullary chromaffin cells. Neuroscience. 2007;145:130–141. doi: 10.1016/j.neuroscience.2006.11.040. [DOI] [PubMed] [Google Scholar]
- Yuan G, Nanduri J, Bhasker CR, Semenza GL, Prabhakar NR. Ca2+/calmodulin kinase-dependent activation of hypoxia inducible factor 1 transcriptional activity in cells subjected to intermittent hypoxia. J Biol Chem. 2005;280:4321–4328. doi: 10.1074/jbc.M407706200. [DOI] [PubMed] [Google Scholar]
- Zhang M, Zhong H, Vollmer C, Nurse CA. Co-release of ATP and ACh mediates hypoxic signalling at rat carotid body chemoreceptors. J Physiol. 2000;525:143–158. doi: 10.1111/j.1469-7793.2000.t01-1-00143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Tang X, Zhang ZF, Velikina R, Shi S, Le AD. Nicotine induces hypoxia-inducible factor-1alpha expression in human lung cancer cells via nicotinic acetylcholine receptor-mediated signaling pathways. Clin Cancer Res. 2007;13:4686–4694. doi: 10.1158/1078-0432.CCR-06-2898. [DOI] [PMC free article] [PubMed] [Google Scholar]