

Abstract
Thyroid hormone transport into cells requires plasma membrane transport proteins. Mutations in one of these, monocarboxylate transporter 8 (MCT8), have been identified as underlying cause for the Allan–Herndon–Dudley syndrome, an X-linked mental retardation in which the patients also present with abnormally high 3′,3,5-triiodothyronine (T3) plasma levels. Mice deficient in Mct8 replicate the thyroid hormone abnormalities observed in the human condition. However, no neurological deficits have been described in mice lacking Mct8. Therefore, we subjected Mct8-deficient mice to a comprehensive immunohistochemical, neurological, and behavioral screen. Several behavioral abnormalities were found in the mutants. Interestingly, some of these behavioral changes are compatible with hypothyroidism, whereas others rather indicate hyperthyroidism. We thus hypothesized that neurons exclusively dependent on Mct8 are in a hypothyroid state, whereas neurons expressing other T3 transporters become hyperthyroid, if they are exposed directly to the high plasma T3. The majority of T3 uptake in primary cortical neurons is mediated by Mct8, but pharmacological inhibition suggested functional expression of additional T3 transporter classes. mRNAs encoding six T3 transporters, including L-type amino acid transporters (LATs), were coexpressed with Mct8 in isolated neurons. We then demonstrated Lat2 expression in cultured neurons and throughout murine brain development. In contrast, LAT2 is expressed in microglia in the developing human brain during gestation, but not in neurons. We suggest that lack of functional complementation by alternative thyroid hormone transporters in developing human neurons precipitates the devastating neurodevelopmental phenotype in MCT8-deficient patients, whereas Mct8-deficient mouse neurons are functionally complemented by other transporters, for possibly Lat2.
Introduction
Thyroid hormone signaling is critical for normal vertebrate development and controls the metabolic activity of many tissues throughout life. 3′,3,5-Triiodothyronine (T3) is the active hormone that binds to nuclear thyroid hormone receptors. Many processes during human brain development are T3 dependent (Zoeller et al., 2002; Bernal, 2005b) and the human embryo in the first weeks of gestation is dependent on sufficient maternal thyroid hormone supply. Therefore, even a transient decrease of maternal plasma T4 levels to hypothyroid levels during gestation can result in neurodevelopmental deficits in the human (Haddow et al., 1999; Bernal, 2007; Gilbert et al., 2007). Unlike other nuclear receptors, both the T3-liganded and the apo-receptors exert specific activating or repressing activities on target genes (Wallis et al., 2008).
For nuclear receptor binding, cellular entry of thyroid hormones is a prerequisite. It is now definite that carrier proteins mediate T3 and T4 transport across the plasma membrane (Hennemann et al., 2001; Visser et al., 2008). Recently, we and others demonstrated that mutations in one transporter, monocarboxylate transporter 8 (MCT8) (SLC16A2), underlie a severe form of X-linked mental retardation, the previously clinically described Allan–Herndon–Dudley syndrome (Dumitrescu et al., 2004; Friesema et al., 2004; Biebermann et al., 2005; Schwartz et al., 2005). Patients present with severe hypotonia during the first months after birth and develop a permanent severe motor and mental retardation. Most patients never attain speech or manage to walk independently. Despite abnormally high plasma T3 levels, some tissues are apparently resistant to their action.
It has been demonstrated that Mct8 mRNA is expressed during brain development and thereafter in mouse neurons and other cells (Heuer et al., 2005). Since the neurological phenotype of MCT8-deficient patients is more severe than the phenotype of patients with primary congenital hypothyroidism, it was suggested that impaired transport of maternal thyroid hormones into neurons during the first weeks of gestation may lead to neurodevelopmental defects (Bernal, 2005a). To create a model for Mct8 deficiency, two independent transgenic mouse lines were generated lacking functional MCT8 (Dumitrescu et al., 2006; Trajkovic et al., 2007). These mice closely mimic the human endocrinological phenotype. Surprisingly, however, Mct8-deficient mice do not exhibit an overt neurological impairment or locomotion deficits. Therefore, we subjected Mct8-deficient mice to an extensive battery of histochemical, neurological, and behavioral tests. Only subtle behavioral changes are observed in Mct8-deficient mice, but indicate a complex phenotype reflecting characteristics of euthyroid and dysthyroid states. We demonstrate that Mct8 accounts for most, but not all, T3 uptake in primary cortical neurons, and identify mRNAs encoding several other T3 transporters in murine neurons, including Lat2. Then, we explore the possibility whether Lat2 may compensate for the lack of Mct8 in the mouse, but not in the human brain. Unlike in the rodent, LAT2 is not coexpressed with MCT8 in human fetal neurons. Thus, we suggest that species-specific and cell type-specific differences account for the phenotypic differences between patients and mice lacking functional MCT8.
Materials and Methods
Mice.
Mct8-deficient mice on a C57BL/6 genetic background (>F8) were obtained from Deltagen. Mct8 was inactivated by insertion of a bacterial lacZ-neomycin phosphotransferase-II gene into exon 2 (Trajkovic et al., 2007). Mice were kept under standard conditions (12 h light/dark cycle) in a specific pathogen-free environment in the central animal facility of the Charité, Berlin, according to local regulations. For phenotypic assessment in the German Mouse Clinic, 15 mice each of the following genotypes were transferred to their animal house and maintained as described previously: Mct8 +/+, Mct8 +/−, Mct8 −/−, and Mct8 −/y (Gailus-Durner et al., 2005). For all phenotypic assessments, Mct8-deficient and littermate control mice were used. The dataset will be available on the EuroPhenome webpage (http://www.europhenome.org/).
Antibodies and reagents.
All reagents were of the highest chemical purity available and supplied by Sigma-Aldrich, unless otherwise indicated. Antibodies against MCT8 were raised in rabbits against an N-terminal peptide comprising amino acids 55–158 (counted from first start codon in human). Antibodies directed against the C terminus of murine LAT2 were raised in rabbits (ImmunoGlobe). Antibodies against neurochemical markers were as follows: NeuN, somatostatin-14, neuropeptide Y, GFAP, p75NTR, ChAT, MAP2, Tau (all Millipore Bioscience Research Reagents), GFAP (Dako), parvalbumin, calbindin, calretinin, GAD67 (all from Swant). Secondary antibodies and peroxidase reagents were either from Vector Laboratories (Vectastain and MOM kit; Linaris) or from Dako. Cy2- and Cy3-labeled secondary antibodies were from Jackson ImmunoResearch Laboratories and of preabsorbed quality. Human multiple tissue Western blot custom manufactured from crude membrane fractions was purchased from BioChain (BioCat).
Western blot.
Mouse organs were frozen on dry ice directly after dissection, powdered under liquid nitrogen, and cytosolic and membrane extracts were prepared as described previously (Renko et al., 2008). Neurons were cultured as described in six-well plates and scraped in ice-cold PBS. After collection of cells by centrifugation, a crude membrane fraction was prepared as described previously. Protein content was determined by the Bradford method (Bio-Rad). A total of 40 μg of protein was separated in 10% polyacrylamide gels and transblotted to nitrocellulose membrane, and even transfer was confirmed with Ponceau staining. Then the membrane was blocked with 5% milk powder and incubated overnight with antiserum (1:1000). After washing, HRP-conjugated secondary anti-rabbit antibody was applied and bands were detected using chemiluminescence (Bio-Rad) and x-ray films (Kodak). As additional loading control, blots were probed with mouse anti-transferrin receptor (TfR) antibody (Invitrogen) or rabbit anti-β-actin (Rockland).
Histology.
Mice were perfused with 0.1 m phosphate buffer (PB), pH 7.4, followed by 4% paraformaldehyde in PB and postfixed overnight at 4°C in the same fixative. After rinsing in PB, brains were either embedded in paraffin and cut on a microtome or embedded in agarose and cut at 100 μm on a vibratome; alternatively, the brains were cryoprotected in 30% sucrose in PB and cut on a cryostat at 30 μm. Blocking was performed with sera from the species from which the secondary antibodies were derived. Stainings were performed using the rabbit polyclonal antibodies against LAT2 (dilution, 1:250) and MCT8 (dilution, 1:250). Secondary antibodies and a specific streptavidin–biotin peroxidase amplification kit were from Vector Laboratories or Dako. Micrographs were taken at a Zeiss Axioscope 2mot plus equipped with an AxioCam MRc5 and Axiovision software. Alternatively, confocal images were taken at a Leica instrument at the Neuroscience Research Center core facility.
Primary cell cultures.
At embryonic day 15, brains were removed from embryos and cortical hemispheres were dissected free of meninges and other brain regions. Neurons were cultured on poly-l-lysine and collagen-treated glass coverslips or cell culture multiwell plates in Neurobasal medium (NBM) supplemented with serum-free B27 (Invitrogen) at densities of 1.5 × 105 cells/cm2 (uptake assays) and 7.5 × 104 cells/cm2 (stainings). Since B27 contains T3, we used a self-made serum-free culture supplement in those experiments in which thyroid hormone content was varied. This supplement can replace B27 for culture times up to 3 weeks (S. Roth and U. Schweizer, unpublished observations). After 7–14 d in vitro, RNA was prepared from neocortical neuron cultures, protein was prepared for Western blotting, or T3 uptake assays were performed. As judged by GFAP and NeuN staining, neuronal cultures were >90% pure. Primary fibroblasts were prepared from embryonic tissue after removal of head, heart, liver, and gastrointestinal tract and cultured on tissue culture plastic in DMEM-F12 (Invitrogen). Fibroblasts cultured in 24-well plates at passages 3–4 were used in radioactive uptake assays at 80% confluence.
T3 uptake assay.
125I-T3 solution (PerkinElmer) was liberated from iodide ions by affinity chromatography and finally resuspended in NBM for neuronal uptake assays or DMEM:F12 for MDCK1 or fibroblast uptake assays. The complete medium of neurons or fibroblasts was removed and cells were incubated for indicated times in 300 μl of prewarmed medium containing 10 nm 125I-T3. MDCK1 cells stably expressing MCT8 from pcDNA3.1 (Invitrogen) were incubated for 4 min in 500 μl of prewarmed DMEM:F12 containing radioactive T3. For inhibitor studies, probenecid (Prob) [1 mm as used by Sugiyama et al. (2003)] was dissolved in 0.1 m NaOH, rebuffered to pH 7.4, and dissolved in tracer medium. All other inhibitor substances were directly dissolved in medium containing 125I-T3. 2-Aminobicyclo-(2.2.1)-heptane-2-carboxylic acid (BCH) was used at 1 mm [three times K i (Morimoto et al., 2008)]. Inhibitors were added to the cells together with 125I-T3. At the end of the incubation, cells were placed on ice, medium was aspirated, and the cells were washed with ice-cold PBS. Cells were lysed in 500 μl of 40 mm NaOH, and the radioactivity of the lysate was measured in a gamma counter. For experiments involving transfected MDCK1 cells, background was defined as radioactivity associated with cells transfected with empty pcDNA3.1 vector.
Human fetal tissue.
Fetal brain tissues were obtained from the autopsy material archive of the Institute of Neuropathology, Charité. Local ethics committee approval for research purposes was obtained. Brains were fixed for at least 2 weeks in buffered 4% formalin before being sliced and tissue blocks embedded in paraffin. A thorough neuropathological examination and diagnosis followed. A total of 14 cases was submitted to immunohistochemical staining. The gestational age ranged from 13 to 40 weeks, and the selected cases showed no major pathology. The hippocampus was chosen for the immunohistochemical analysis, which was performed using the rabbit polyclonal antibodies against LAT2 (dilution, 1:50) and MCT8 (dilution, 1:250). Counterstaining was done according to standard protocols. The respective secondary antibody and a specific streptavidin–biotin peroxidase amplification kit were applied (Ventana). A small fragment of kidney tissue was used as a positive control on each slide.
Results
Specific pattern of thyroid hormone transporter Mct8 immunoreactivity in the brain
Using a new and very specific antiserum directed against MCT8, we show Mct8 protein expression in the murine neocortex during development from embryonic day 15 (E15) to the adult (Fig. 1 A). Specificity of the antiserum for Mct8 was demonstrated by the absence of signal in Western blot performed with membrane protein from Mct8 −/y mice (Fig. 1 B). Moreover, specific immunohistochemical staining was absent in sections from Mct8-deficient mice (Fig. 1 C). Previously, in situ hybridization had demonstrated Mct8 expression in the adult mouse brain (e.g., in dentate granule cells) (Heuer et al., 2005). Here, we show that the distribution of Mct8 protein is more complex than expected from the mRNA expression pattern. Specifically, we detected a strong signal in granule cell dendrites extending into the molecular layer (ml), whereas granule cell bodies (gc) and axons in the stratum lucidum (sl) remained mostly unstained (Fig. 1 Cb). In the cerebellar cortex, not only Purkinje cells (pc) but also bona fide stellate (arrow) and Golgi (asterisk) interneurons were stained (Fig. 1 Ce). Tanycytes also express Mct8 mRNA (Heuer et al., 2005). Here, we demonstrate that Mct8-positive processes line the wall of the third ventricle (black arrows), wrap hypothalamic blood vessels, and extend to the median eminence (Fig. 1 Cf, white arrow). From at least E15 to the adult, there is strong Mct8 expression in the choroid plexus on the apical membrane (Fig. 1 Cd). Since Mct8 protein is widely distributed to dendrites, individual Mct8-expressing cells are not easily recognized in the adult cerebral cortex (Fig. 1 Ca), whereas at postnatal day 5 (P5) single pyramidal cells expressing Mct8 immunoreactivity were easily distinguishable (Fig. 1 Cg–j).
Figure 1.
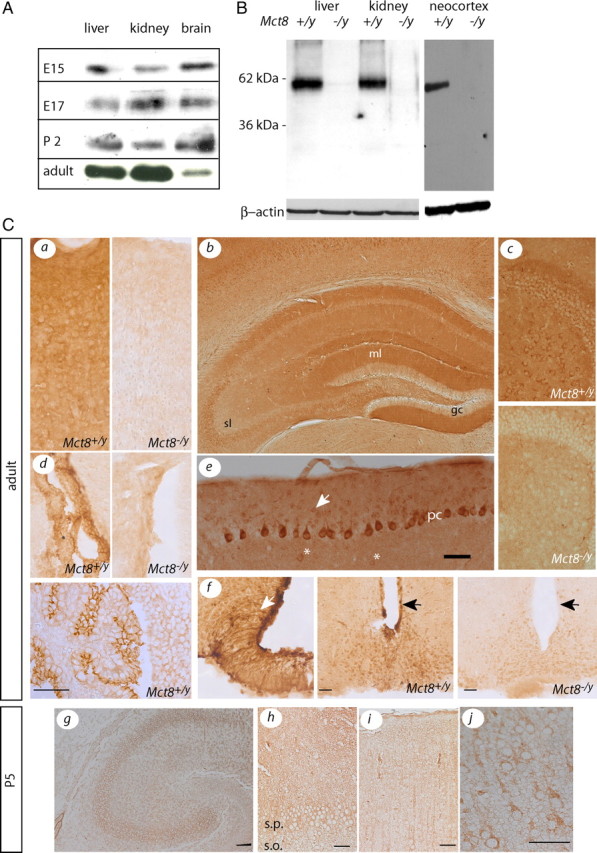
Developmental and cell type-specific expression of MCT8 immunoreactivity in the mouse brain. A, Western blots demonstrating the relative abundance of cerebrocortical MCT8 protein compared with liver and kidney at E15, E17, P2, and adult. B, Specificity of the MCT8 antiserum in Western blot. Molecular weight markers are indicated on the left. β-Actin was used as loading control. C, MCT8 immunoreactivity in Mct8 +/y brain compared with Mct8 −/y controls. a, Cerebral cortex. b, Hippocampus. c, Dentate gyrus with hilus. d, Choroid plexus. The bottom panel is from embryonic day 15. e, Cerebellar cortex. Scale bar, 50 μm. f, Hypothalamus. g, Hippocampus at P5. Scale bar, 100 μm. h, CA3 region magnified from g. s.p., Stratum pyramidale; s.o., stratum oriens. i, Somatosensory cortex at P5. j, Magnified view of i. Scale bars: h–j, 50 μm.
Gene disruption of Mct8 does not alter brain histogenesis
In search of structural or molecular alterations in Mct8-deficient mouse brains, we undertook an immunohistochemical study of Mct8-deficient brains using a panel of common informative markers (NeuN, parvalbumin, somatostatin 14, calbindin, calretinin, neuropeptide Y, p75NTR, choline acetyl transferase, microtubule associated protein 2, glutamic acid decarboxylase 67). Brain regions inspected were cerebral cortex (M1, S1BF, Pir), hippocampus, basal forebrain, amygdala, hypothalamus, and vermal cerebellar cortex. As shown in Figure 2, we did not observe a cortical layering defect in the barrel field cortex or changes in the number or distribution of parvalbumin+ interneurons, both changes that can be observed when thyroid hormone signaling is reduced during development (Lavado-Autric et al., 2003; Ausó et al., 2004; Gilbert et al., 2007; Wallis et al., 2008). Similarly, we did not observe changes in the number or distribution of the other major classes of cortical interneurons, somatostatin+ and calretinin+ cells, or in the distribution of any other marker tested. Likewise, no changes were apparent in the structure or neurochemical organization of the hippocampus (Fig. 2 B).
Figure 2.

Immunohistochemical analysis of Mct8-deficient mouse brains. A, Cerebral cortex (S1BF). No differences were observed in the staining patterns for NeuN, parvalbumin, somatostatin, calbindin, neuropeptide Y, and calretinin. Scale bar, 200 μm. B, Dorsal hippocampus. No differences were observed in the staining patterns for NeuN, parvalbumin, and calbindin. Markers tested, but not shown are MAP2, ChAT, p75NTR, GAD67, GFAP, NPY, and AChE activity.
Behavioral phenotype of mice deficient for the thyroid hormone transporter Mct8
Since we and others did not observe a clear behavioral phenotype that may have guided our analysis to specific neural circuits or transmitter systems, we subjected Mct8-deficient mice to the comprehensive neurological and behavioral screens at the German Mouse Clinic. In the neurological screen, the mice were tested according to the SHIRPA protocol, a battery of 24 independent assessments. As shown in the supplemental tables (available at www.jneurosci.org as supplemental material), the SHIRPA screen detected no differences between Mct8-deficient mice and their control littermates including normal reflexes (supplemental Tables S1–S4, available at www.jneurosci.org as supplemental material). The mice were also subjected to the modified hole-board test to reveal more subtle behavioral alterations. Unlike the situation in affected humans deficient for MCT8, Mct8-knock-out mice did not display altered locomotion (Fig. 3 A). Distances traveled and velocities were not different from control littermates. In addition, there was no difference in grip strength between Mct8-deficient mice and control littermates (control vs Mct8-deficient: males, 139 ± 7, vs 134 ± 6; females, 103 ± 1, vs 103 ± 2)—in sharp contrast to the severe hypotonia observed very early in patients with MCT8 mutations. Finally, the rotarod assay did not reveal any significant differences in movement coordination between Mct8-deficient mice and controls. However, Mct8-deficient mice exhibited decreased anxiety-related behavior (Fig. 3 B). The mutant mice entered the board significantly more often, spent more time on the board, and kept a larger mean distance to the wall of the arena. This is the opposite finding of TRα deficient mice, which exhibit increased anxiety (Guadaño-Ferraz et al., 2003; Venero et al., 2005). A notable finding was shortened latency in the hot plate test (Table 1). Such a shortened latency may indicate hyperalgesia and was described previously in hyperthyroid mice (Edmondson et al., 1990) and rats (Bruno et al., 2005, 2006). Interestingly, both hyperalgesia and reduced anxiety-related behavior are more indicative of hyperthyroidism than hypothyroidism, which would be expected from the T3 transporter deficiency. In contrast, reduced grooming and increased latency of grooming (Fig. 3 C) have been reported in hypothyroid rats (Negishi et al., 2005). Thus, certain brain regions may be exposed to the increased circulating T3 levels present in Mct8-deficient mice, whereas other neuronal systems remain functionally hypothyroid in Mct8 −/y mice. These findings and the lack of a histopathologically discernible hypothyroid phenotype in the brain, prompted us to investigate the possible presence of additional thyroid hormone transporters that may compensate for the lack of Mct8 in the mouse brain.
Figure 3.

Behavioral analysis of Mct8-deficient mice. A, Locomotion as observed in the modified hole board test. Controls are represented by black columns, and Mct8-deficient mice are represented by shaded columns. Total distance traveled and mean velocity were not different between Mct8-deficient mice and littermate controls. For rotarod test, no difference related to Mct8 genotype in the latency to fall from the rotating drum was observed. B, Anxiety-related behavior. In the modified hole board test, entries on board, time on board, mean distance to wall, and mean distance to board were assessed. All parameters suggest decreased anxiety-related behavior. C, Grooming behavior. In the modified hole board test, an increased latency to grooming was observed in mutants of both sexes, as well as reduced time spent grooming in male mutants. Error bars indicate SEM. *p < 0.05, ***p < 0.001.
Table 1.
Hot plate test
| Parameter | Latency (s) |
ANOVA, p value | |||
|---|---|---|---|---|---|
|
Mct8 control |
Mct8 mutant |
||||
| Male (n = 10) | Female (n = 13) | Male (n = 10) | Female (n = 8) | ||
| Hindpaw shaking | 11.54 ± 1.08 | 21.39 ± 1.34 | 9.31 ± 1.31 | 14.88 ± 2.02 | <0.01 |
| Hindpaw licking | 18.46 ± 2.84 | 20.15 ± 1.39 | 16.64 ± 1.60 | 21.25 ± 2.52 | NS |
| Jumping | 50.44 ± 2.97 | 49.71 ± 2.52 | 43.51 ± 3.24 | 41.00 ± 1.88 | <0.01 |
Functional characterization of thyroid hormone transporters in cortical neurons
There is no systematic data on the expression of thyroid hormone transporters in mouse neurons, but previous physiological studies have characterized several T3 transporters in brain cells (Chantoux et al., 1995; Hennemann et al., 2001). The relative importance of Mct8-mediated T3 uptake in differentiated neurons has not been demonstrated previously. To investigate Mct8-mediated T3 uptake in primary cortical neurons, we first determined expression of the protein and found almost all NeuN-positive cells stained with the Mct8 antibody (Fig. 4 A). Expression of Mct8 protein in neurons as estimated by Western blot did not depend on the presence of iodothyronines, but increased with time in culture (Fig. 4 B). To validate our T3 uptake assay, we measured 125I-T3 uptake in primary cultured fibroblasts from Mct8-deficient embryos and their wild-type littermates (Fig. 4 C). Then, we tested primary neurons for T3 uptake. Clearly, loss of Mct8 significantly diminished the rate of T3 uptake in a gene dose-dependent manner (Fig. 4 D). Still, we noted a moderate linear increase of 125I-T3 uptake even in Mct8 −/y neurons. To establish whether other thyroid hormone transporters are functional in primary neurons, we pharmacologically characterized T3 uptake in cortical neurons. To this end, we measured T3 uptake in the presence of bromosulfophtalein (BSP) (a Mct8 inhibitor), BCH (a specific Lat inhibitor), and Prob (a broad-spectrum Oatp inhibitor) (Fig. 4 E). BSP significantly inhibited T3 uptake in Mct8 +/y neurons, but the reduction was only one-half the effect in Mct8 −/y neurons, although BSP was used at five times its K i value in cortical neurons (190 μm) (data not shown). Incomplete inhibition by BSP was also observed in Mct8-transfected MDCK1 cells (Fig. 4 F). BCH and Prob inhibition was reproducible, but small, and did not reach statistical significance. However, combination of BCH and Prob exhibited an additive effect and significantly reduced T3 uptake in both Mct8 +/y and Mct8 −/y neurons. Moreover, inhibition by the combination of BSP, BCH, and Prob was larger than for BSP alone. Together, these data demonstrate the functional contribution of BCH- and Prob-sensitive T3 transporters in cortical neurons (Fig. 4 E). Moreover, overexpressed Mct8 was insensitive to BCH and Prob excluding the possibility of inadvertent inhibition of Mct8 by the Lat and Oatp inhibitors (Fig. 4 F). These data strongly suggest that Lats and/or Oatps are functionally expressed in primary cortical neurons and may partially compensate for the loss of the major T3 transporter, Mct8. The residual T3 uptake capacity may thus suffice to prevent a major neurodevelopmental defect in the Mct8-deficient mouse.
Figure 4.
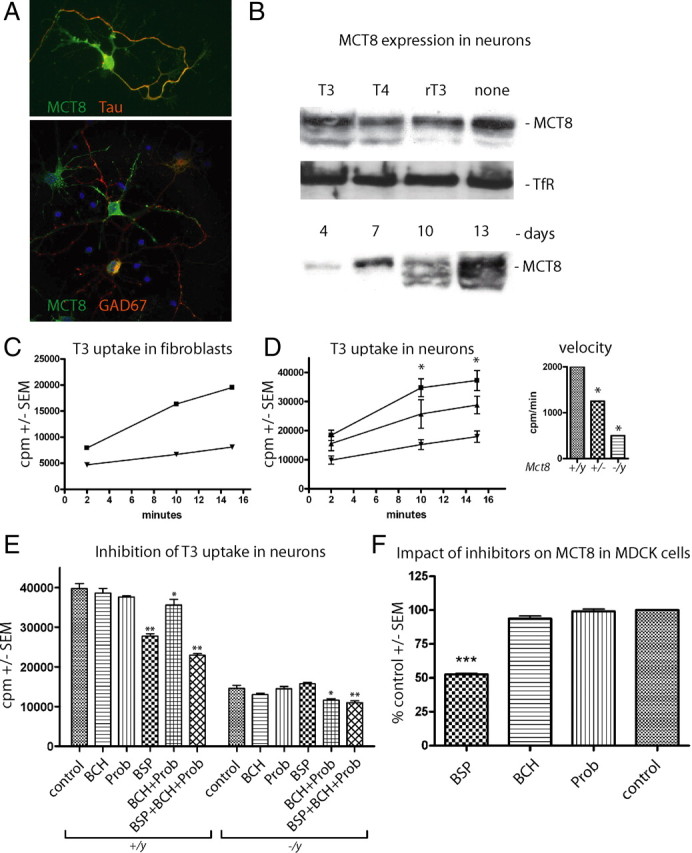
Functional characterization of T3 transporters in primary cortical neurons. A, Primary cortical neurons were cultured for 7 d in vitro and immunostained for Mct8, the axonal marker Tau, and the interneuron marker GAD67. B, Western blot for Mct8 from primary cortical neurons cultured in vitro with the indicated iodothyronines at 10 nm. TfR served as control. C, Kinetic analysis of 125I-T3 uptake in mouse embryonic fibroblasts derived from control (■) and Mct8 −/y (▼) mice. Cell-associated radioactivity (cpm) was measured in triplicate. Error bars (SEM) are smaller than the symbols. D, Kinetic analysis of 125I-T3 uptake in mouse primary cortical neurons derived from wild-type (■), Mct8 +/− (▲), and Mct8 −/y (▼) mice. Cell-associated radioactivity (cpm) was measured in triplicate from two to four independent animals. Error bars denote SEM. Inset, Initial rate kinetics of T3 uptake in relation to Mct8 genotype expressed as cpm per minute. Note that 75% of the T3 uptake rate depends on Mct8. E, Pharmacological inhibition of neuronal T3 uptake reveals Mct8-independent transport. T3 uptake assays were performed in triplicate by addition of inhibitors (1 mm) together with T3. F, BCH and Prob do not inhibit MCT8. Tracer was incubated on MDCK1 cells stably transfected with MCT8 for 4 min and background activity of empty vector-transfected MDCK1 cells was subtracted. *p < 0.05; **p < 0.01; ***p < 0.001 versus no inhibitor, one-way ANOVA followed by Dunnett's posttest.
Expression of thyroid hormone transporter mRNA in mouse neurons
Pharmacological inhibitors are useful tools for the functional characterization of T3 uptake, but do not allow the unambiguous identification of specific transporters. To determine all T3 transporters expressed in cortical neurons, we performed real-time PCR [quantitative PCR (qPCR)] for 12 described thyroid hormone transporters. mRNAs for seven transporters were detected in cultured neurons (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Among these were at lower levels Mct10 (Slc16a10), the closest relative of Mct8, which is also able to efficiently transport T3 (Friesema et al., 2008), Oatp-14 (Slco1c1), although it was mostly described in brain microcapillary endothelium (Sugiyama et al., 2003; Chu et al., 2008; Roberts et al., 2008b), and Slco4a1 and Slco4c1, which are less well described. Only Slc7a5 and Slc7a8 (L-type amino acid transporters Lat1 and Lat2, respectively) were expressed at levels comparable with Mct8 in the neurons. Both Lats have been characterized previously as T3 transporters with low K M values (Friesema et al., 2001) and have been identified as astrocytic T3 transporters (Francon et al., 1989; Blondeau et al., 1993; Chantoux et al., 1995).
Developmental expression of thyroid hormone transporters in the mouse brain
To define the developmental expression patterns of thyroid hormone transporters in the mouse brain, we performed qPCR on cortical samples in mice covering four developmental stages: E14, birth (P0), P5, and P20 (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Several transporter genes, including Lat2, were coexpressed with Mct8 at all time points tested. To gain more insight into their cell type specificity, we consulted the Allen Brain Atlas (Lein et al., 2007). In agreement with a recent report (Roberts et al., 2008b), we found that both Lat1 and Slco1c1 were clearly expressed in the choroid plexus and in brain microvessels. Strongest signals with a neuron-like distribution were observed for Lat2 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Lat2 expression in the mouse brain
To study the function of Lat2-mediated T3 transport during mouse development, we developed a specific antiserum against mouse Lat2. With this antiserum, we detected a single band of 45 kDa in mouse brain, primary neurons, and kidney (Fig. 5 A) (data not shown). Specificity of the antiserum was further assessed by recombinant expression of Lat2 in HEK293 cells and Western blotting. The 45 kDa band was the only band detected by the antiserum and was specific for Lat2-transfected cells. Lat2 protein is prominently expressed in the developing brain and spinal cord at E17, and kidney is the only organ with a similarly strong expression (Fig. 5 A). Immunocytochemical staining for Lat2 on cultured primary neurons revealed widespread expression in both the somatic and neuritic compartments (Fig. 5 B). Next, we determined the cellular expression pattern of Lat2 in the developing mouse brain by immunohistochemistry. Preabsorption of the antiserum with Lat2 peptide completely abolished immunostaining in the mouse brain (Fig. 5 C). At P5, Lat2 immunoreactivity is detected in neurons located in the cerebral cortex and hippocampus (Fig. 5 D). Similar as in the case of Mct8, Lat2 immunoreactivity is widely distributed throughout the cerebral cortex and hippocampus of the adult mouse (Fig. 5 E, F). A likely reason for the blurred immunohistochemical signal may be its dendritic localization and its reported expression in cortical astrocytes (Blondeau et al., 1993). Strikingly, the stratum lucidum was mostly devoid of Lat2 as it is devoid of Mct8 immunoreactivity. Since Lat2 is expressed in mouse cortex and hippocampus in a pattern overlapping the expression of Mct8, we speculate that Lat2 is a candidate alternative T3 transporter during neuronal development that may compensate for the lack of Mct8 in Mct8 −/y mice.
Figure 5.

Developmental expression of Lat2 in the mouse brain. A, Western blot against Lat2. Top left panel, Mouse cerebral cortex and cultured primary neurons. Lat2 protein abundance is not different between Mct8 +/y and Mct8 −/y mice. Bottom left panel, Strong Lat2 expression in brain and spinal cord at E17. Li, Liver; Ki, kidney; Br, brain; Spc, spinal cord; Lu, lung; He, heart; GI, gastrointestinal tract. Molecular weight markers are indicated on the left. TfR served as loading control. Right panel, Specificity of the Lat2 antiserum. Only one band of the appropriate size is detected in HEK cells transfected with Lat2 expression plasmid. B–G, Immunocytochemical staining for Lat2. B, Cortical neurons cultured for 7 d. C, Specificity of the antiserum. In the right panel, the antibody was preincubated with the blocking peptide before immunostaining. P15 cerebral cortex is shown. D, Hippocampus, CA3 region, P5. E, Adult cerebral cortex. F, Adult hippocampus. G, Anterior commissure in the adult mouse. Note the differential Lat2 staining in the posterior and the anterior one-half of the commissure. Scale bars: D, 50 μm; C, E–G, 100 μm.
Expression of MCT8 and LAT2 in the human brain
Mct8-deficient mice replicate the endocrine, but not the neurological phenotype of human patients suffering from mutations in MCT8. We therefore speculated that one possible explanation could be differential species-specific expression patterns of thyroid hormone transporters in humans and mice. During development, mice may express more thyroid hormone transporters than humans at their blood–brain barrier (BBB) or in neurons compared with humans and thus may be less vulnerable to the lack of one of these transporters, MCT8. To investigate which thyroid hormone transporters are expressed in the human brain, we first performed qPCR on adult human cortical cDNA and found significant expression of L-type transporters (SLC7A), SLCO4A1, and SLCO1C1 in addition to MCT8 (Fig. 6 A). Western blotting supported protein expression of LAT2 and MCT8 in brain (Fig. 6 B). We then determined the expression pattern of MCT8 in human fetal brain during development by immunohistochemistry. MCT8 expression is first detected in blood vessels at gestational week 25 (GW25) (Fig. 6 E) and maintained at GW32 and GW40 (Fig. 6 C). Later, MCT8 immunoreactivity is also found in developing neurons with punctate staining in white matter, possibly representing cortical axons. From GW32, the mature MCT8 expression pattern is established in the hippocampus and cortex, but immunoreactivity is still weak (Fig. 6 C). By GW40, prominent MCT8 staining is achieved resembling essentially the pattern observed in mice: In the dentate gyrus, granule cell dendrites are strongly labeled in the molecular layer, whereas their axons in the stratum lucidum are apparently lacking MCT8. Most hilar and pyramidal cells are strongly stained for MCT8 (Fig. 6 C). In the cerebral cortex, pyramidal cells show MCT8 immunoreactivity from GW30 with increasing intensity until birth (data not shown). Similar as in the mouse brain, MCT8 is located on the apical side of the choroid plexus (Fig. 6 E). We then tested our newly developed antibody against LAT2 and confirmed its applicability for immunohistochemistry in paraffin-embedded tissue by robust labeling of kidney tubules (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) and adult human cerebral cortex (Fig. 6 F). LAT2 immunoreactivity is clearly detectable in adult neurons. During human fetal development, microglia is clearly LAT2 positive, but not neurons (Fig. 6 D). We thus suggest that a major difference regarding T3 transporters between mice and humans is the absence of LAT2 from developing neurons in the human. Thus, human neurons may depend exclusively on MCT8 expression for T3 uptake during a critical time of development.
Figure 6.
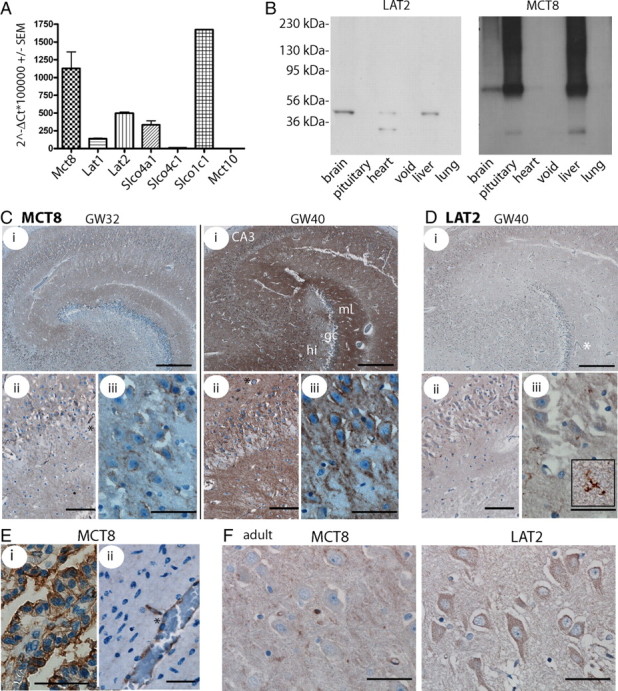
Developmental expression of thyroid hormone transporters in the human brain. A, qPCR detection of thyroid hormone transporters in adult human brain cDNA. Values are calculated according to the ΔCt method in relation to β-actin as a housekeeping gene. B, Multiple tissue Western blot on adult human membrane fractions for LAT2 and MCT8. The antibodies detect a protein of the same size as in mouse brain. Left, Molecular weight markers. C, Immunohistochemical detection of MCT8. Labeling of MCT8 increases with time in the developing human brain. i, Hippocampus. Scale bar, 500 μm. ii, CA3. Scale bar, 100 μm. iii, CA3. Scale bar, 50 μm. GW32 and GW40 are shown. D, Immunohistochemistry for LAT2 in GW40 hippocampus detects microglial, but not neuronal staining (iii, inset). Scale bars are as in C. E, MCT8 in GW25 choroid plexus (i) (scale bar, 50 μm) and cortical gray matter vessel (ii) (scale bar, 50 μm). F, Hippocampal CA3 neurons in the adult human brain stained for MCT8 and LAT2. Scale bars, 50 μm. Capillaries are indicated by asterisks.
Discussion
Despite the important role of thyroid hormones for brain development and function, we are only beginning to identify the molecules that mediate the uptake of thyroid hormones in vivo at the BBB or into neurons and astrocytes (Roberts et al., 2008a,b). Major interest in the molecular identity of brain thyroid hormone transporters was raised after the identification of mutations in MCT8 as the cause of mental retardation in human patients (Dumitrescu et al., 2004; Friesema et al., 2004; Schwartz et al., 2005). Previous reports using Mct8-deficient mice supported the observation that mutations in MCT8 cause the unusual thyroid hormone constellations in human patients (Dumitrescu et al., 2006; Trajkovic et al., 2007). However, unlike the human subjects, mice lacking functional Mct8 did not develop obvious signs of neurodevelopmental retardation. Trajkovic et al. (2007) demonstrated that cerebral uptake of 125I-T3 was greatly impaired in Mct8-deficient mice, whereas uptake of 125I-T4 was almost normal. This finding implicated a role for Mct8 in T3 uptake at the BBB, consistent with our data presented here and a recent report by Roberts et al. (2008b). Accordingly, Oatp-14, a T4-specific transporter, mediates T4 uptake at the BBB (Sugiyama et al., 2003; Roberts et al., 2008b). Moreover, type 2 deiodinase (Dio2), an astrocytic enzyme capable of converting T4 to active T3, is upregulated in Mct8-deficient mouse brain and thus indicates a relative lack of T3 behind the BBB (Dumitrescu et al., 2006; Trajkovic et al., 2007).
In the rodent brain, Mct8 mRNA is expressed in neurons (Heuer et al., 2005). Thus, it was generally concluded that Mct8 represents the neuronal T3 transporter. We set out to test this hypothesis and performed T3 uptake experiments on primary cortical neuron cultures prepared from wild-type and Mct8-deficient mice. Mct8 is quantitatively the most important T3 transporter in cultured cortical neurons. Using BCH and Prob as pharmacological inhibitors, we found that a significant fraction of neuronal T3 uptake is independent of Mct8. We may have even underestimated their quantitative contribution, if inhibition was incomplete as in the case of the Mct8 inhibitor BSP. The existence of a Mct8-independent T3 uptake mechanism may explain the general lack of an obvious hypothyroid phenotype in Mct8-deficient mice and is compatible with the normal dendritic development of Purkinje cell dendrites in Mct8-deficient cerebellar cultures (Trajkovic et al., 2007). We performed a comprehensive immunohistochemical study but were unable to identify structural defects compatible with developmental hypothyroidism in Mct8-deficient mouse brains. In addition, we subjected Mct8-deficient mice to an extensive behavioral and neurological screen. Again, no neurological defects were observed, but for the first time significant behavioral abnormalities were noted. These were, however, not consistent with changes observed in hypothyroid animals (as expected for the T3 transport deficiency into neurons) or mice deficient in nuclear TRα1 receptors, TRα+/R384C mice (Venero et al., 2005), TRα1 −/− mice (Guadaño-Ferraz et al., 2003) and TRα0/0 mice (Wilcoxon et al., 2007), since these animals display increased anxiety-related behavior. Mct8-deficient mice displayed decreased anxiety-related behavior similar to rats transiently treated with T4 after birth (Yilmazer-Hanke et al., 2004). Rather, some of our results pointed to increased T3 signaling resulting in enhanced pain perception (Edmondson et al., 1990; Bruno et al., 2005, 2006). However, in contrast to hyperthyroid rodents (Redei et al., 2001; Sala-Roca et al., 2002), there was no indication for hyperactivity in Mct8 −/y mice as shown by unaltered locomotion. We thus speculate that neurons in specific brain regions with direct access to the increased circulating T3 levels may be exposed to enhanced T3 signaling, if their T3 uptake is independent of Mct8. In contrast, other neurons may completely rely on Mct8 for T3 import and thus become hypothyroid in the absence of Mct8 (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Accordingly, we detected mRNAs encoding several potential thyroid hormone transporters in cortical neurons. Some of these were formerly known as components of the BBB (e.g., Lat1/Slc7a5 and Oatp-14/Slco1c1) (Boado et al., 1999; Sugiyama et al., 2003; Chu et al., 2008; Roberts et al., 2008b). Lat1 and Lat2 are BCH-sensitive T3 transporters and may account for one-half of Mct8-independent T3 import into neurons. The remaining T3 uptake capacity is sensitive to Prob consistent with a potential role of Slco1c1.
Why is the neurological phenotype of MCT8-deficient patients so much more severe than the phenotype of Mct8-deficient mice? One may argue that the mouse brain may not represent a good model for the human brain. Human neurons may be much more sensitive to deviations from adequate T3 supply than mouse neurons. Especially higher cortical functions like speech are simply not present in mice and cannot be studied in the rodent model. In our eyes, it is at present not possible to reject this argument in general, but there is no indication of motor deficits in Mct8-deficient mice, although this is a prominent feature of the patients.
Another question is whether a mouse with total deletion of the Mct8 gene is a model for the human patients given the large number of missense, nonsense, and splice site mutations that may allow for expression of MCT8 fragments with potential dominant-negative actions. However, patients with total or partial deletions of MCT8 or patients harboring mutations that in vitro do not allow for protein expression do not have a weaker phenotype. Conversely, the only patients who are apparently less severely affected harbor missense mutations (S105F, L434W, and L568P). Finally, the Mct8 −/y mouse is a good model for the human disease regarding the endocrine phenotypes.
Roberts et al. (2008b) reported that rodent, but not human, brain microvessels express Oatp-14. The authors propose that Oatp-14 may compensate for the loss of Mct8 in the mouse, but not in the human brain. However, Oatp-14 is significantly more active toward T4 than T3 (Sugiyama et al., 2003). If this proposed mechanism was entirely true, MCT8-deficient patients should resemble congenital hypothyroid patients, since thyroid hormone transport beyond the BBB would be generally impaired—and this is not observed. Also, this explanation does not take into account the expression of Lat1 at the BBB (Roberts et al., 2008a).
A fourth hypothetical explanation states that Mct8-deficient mice do not display neurological defects, because homeostatic mechanisms in neurons and other cells (other alternative transporters, deiodinase upregulation and downregulation) collectively succeed in establishing sufficiently high neuronal T3 levels to eventually prevent functional hypothyroidism. We have explored this possibility in more detail and therefore tested the hypothesis that differential species-specific spatiotemporal expression patterns of neuronal thyroid hormone transporters account for the more severe neurological phenotype of MCT8-deficient patients. Based on our functional and expression data in cultured neurons, we speculate that Lat2 may compensate in the mouse, but not in the human brain for the lack of Mct8. Therefore, we compared LAT2 expression in the developing human and mouse brain. Although Lat2 is significantly expressed in primary cortical neurons isolated at the time point when corticogenesis is in full progress in the mouse, there is rather low LAT2 expression in developing human neurons. At this time, it appears as if MCT8 is the critical T3 transporter during human brain development. Only in the adult, LAT2 expression increases. Our results further suggest that MCT8 is located in the dendritic compartment and we can only speculate at present whether it may be involved in synaptic function (Ruiz-Marcos et al., 1994). Such a role would be of utmost importance during the formation of functional connections. A role for thyroid hormone in these processes is well documented by the neurodevelopmental retardation occurring in severely hypothyroid children. Since thyroid hormone transporter expression is apparently developmentally and cell type-specifically regulated, it may be difficult to interpret the phenotypes associated with the lack of one T3 transporter. In the mouse, it appears as if at least a fraction of the brain gains access to the elevated plasma T3 levels in Mct8-deficient mice, and thus a mixed phenotype is created with respect to thyroid state. The same situation may occur in MCT8-deficient patients. Although their neurons may be deprived of T3 signaling during critical periods of development, they may, at least in part, be exposed to the exceedingly high T3 levels after birth, when T3 transport relies more on LAT2, as we showed for CA3 pyramidal neurons. In keeping with the proposed role of thyroid hormone signaling during development, it has recently been demonstrated that normalizing peripheral thyroid hormone levels does not improve the neurological deficits of MCT8-deficient patients but only ameliorates the peripheral hyperthyroid phenotype (Wémeau et al., 2008).
Footnotes
This work was supported by a NGFNplus grant from the Bundesministerium für Bildung und Forschung [01GS0850 (S.M.H., W.W., H.F., V.G.D., M.H.d.A.), 01GS0853 (I.R., A.Z.), 01GS0851 (L.B., T.K.)] and by European Union Grant EUMODIC LSHG-2006-037188 (German Mouse Clinic). Additional funding was provided by Deutsche Forschungsgemeinschaft Sonderforschungsbereich 665/TPA7 (A.G., U.S.) and by EnForCé, Technologiestiftung Berlin (J.K., A.G.). We thank Doreen Braun, Vartitér Seher, Anita Kinne, Antje Kretschmer, and Anja Fischbach for technical assistance. We thank the whole team of the German Mouse Clinic for helpful discussions of phenotyping results. We are grateful to Reinhard Seeliger, Regina Kneuttinger, and Bettina Sperling for expert technical help.
References
- Ausó E, Lavado-Autric R, Cuevas E, Del Rey FE, Morreale De Escobar G, Berbel P. A moderate and transient deficiency of maternal thyroid function at the beginning of fetal neocorticogenesis alters neuronal migration. Endocrinology. 2004;145:4037–4047. doi: 10.1210/en.2004-0274. [DOI] [PubMed] [Google Scholar]
- Bernal J. The significance of thyroid hormone transporters in the brain. Endocrinology. 2005a;146:1698–1700. doi: 10.1210/en.2005-0134. [DOI] [PubMed] [Google Scholar]
- Bernal J. Thyroid hormones and brain development. Vitam Horm. 2005b;71:95–122. doi: 10.1016/S0083-6729(05)71004-9. [DOI] [PubMed] [Google Scholar]
- Bernal J. Thyroid hormone receptors in brain development and function. Nat Clin Pract Endocrinol Metab. 2007;3:249–259. doi: 10.1038/ncpendmet0424. [DOI] [PubMed] [Google Scholar]
- Biebermann H, Ambrugger P, Tarnow P, von Moers A, Schweizer U, Grueters A. Extended clinical phenotype, endocrine investigations and functional studies of a loss-of-function mutation A150V in the thyroid hormone specific transporter MCT8. Eur J Endocrinol. 2005;153:359–366. doi: 10.1530/eje.1.01980. [DOI] [PubMed] [Google Scholar]
- Blondeau JP, Beslin A, Chantoux F, Francon J. Triiodothyronine is a high-affinity inhibitor of amino acid transport system L1 in cultured astrocytes. J Neurochem. 1993;60:1407–1413. doi: 10.1111/j.1471-4159.1993.tb03302.x. [DOI] [PubMed] [Google Scholar]
- Boado RJ, Li JY, Nagaya M, Zhang C, Pardridge WM. Selective expression of the large neutral amino acid transporter at the blood-brain barrier. Proc Natl Acad Sci U S A. 1999;96:12079–12084. doi: 10.1073/pnas.96.21.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno AN, Fontella FU, Crema LM, Bonan CD, Dalmaz C, Barreto-Chaves ML, Sarkis JJ. Hyperthyroidism changes nociceptive response and ecto-nucleotidase activities in synaptosomes from spinal cord of rats in different phases of development. Comp Biochem Physiol A Mol Integr Physiol. 2005;140:111–116. doi: 10.1016/j.cbpb.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Bruno AN, Fontella FU, Bonan CD, Barreto-Chaves ML, Dalmaz C, Sarkis JJ. Activation of adenosine A(1) receptors alters behavioral and biochemical parameters in hyperthyroid rats. Behav Brain Res. 2006;167:287–294. doi: 10.1016/j.bbr.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Chantoux F, Blondeau JP, Francon J. Characterization of the thyroid hormone transport system of cerebrocortical rat neurons in primary culture. J Neurochem. 1995;65:2549–2554. doi: 10.1046/j.1471-4159.1995.65062549.x. [DOI] [PubMed] [Google Scholar]
- Chu C, Li JY, Boado RJ, Pardridge WM. Blood-brain barrier genomics and cloning of a novel organic anion transporter. J Cereb Blood Flow Metab. 2008;28:291–301. doi: 10.1038/sj.jcbfm.9600538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74:168–175. doi: 10.1086/380999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology. 2006;147:4036–4043. doi: 10.1210/en.2006-0390. [DOI] [PubMed] [Google Scholar]
- Edmondson EA, Bonnet KA, Friedhoff AJ. The effect of hyperthyroidism on opiate receptor binding and pain sensitivity. Life Sci. 1990;47:2283–2289. doi: 10.1016/0024-3205(90)90160-s. [DOI] [PubMed] [Google Scholar]
- Francon J, Chantoux F, Blondeau JP. Carrier-mediated transport of thyroid hormones into rat glial cells in primary culture. J Neurochem. 1989;53:1456–1463. doi: 10.1111/j.1471-4159.1989.tb08538.x. [DOI] [PubMed] [Google Scholar]
- Friesema EC, Docter R, Moerings EP, Verrey F, Krenning EP, Hennemann G, Visser TJ. Thyroid hormone transport by the heterodimeric human system L amino acid transporter. Endocrinology. 2001;142:4339–4348. doi: 10.1210/endo.142.10.8418. [DOI] [PubMed] [Google Scholar]
- Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, Barrett TG, Mancilla EE, Svensson J, Kester MH, Kuiper GG, Balkassmi S, Uitterlinden AG, Koehrle J, Rodien P, Halestrap AP, Visser TJ. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004;364:1435–1437. doi: 10.1016/S0140-6736(04)17226-7. [DOI] [PubMed] [Google Scholar]
- Friesema EC, Jansen J, Jachtenberg JW, Visser WE, Kester MH, Visser TJ. Effective cellular uptake and efflux of thyroid hormone by human monocarboxylate transporter 10. Mol Endocrinol. 2008;22:1357–1369. doi: 10.1210/me.2007-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gailus-Durner V, Fuchs H, Becker L, Bolle I, Brielmeier M, Calzada-Wack J, Elvert R, Ehrhardt N, Dalke C, Franz TJ, Grundner-Culemann E, Hammelbacher S, Hölter SM, Hölzlwimmer G, Horsch M, Javaheri A, Kalaydjiev SV, Klempt M, Kling E, Kunder S, et al. Introducing the German Mouse Clinic: open access platform for standardized phenotyping. Nat Methods. 2005;2:403–404. doi: 10.1038/nmeth0605-403. [DOI] [PubMed] [Google Scholar]
- Gilbert ME, Sui L, Walker MJ, Anderson W, Thomas S, Smoller SN, Schon JP, Phani S, Goodman JH. Thyroid hormone insufficiency during brain development reduces parvalbumin immunoreactivity and inhibitory function in the hippocampus. Endocrinology. 2007;148:92–102. doi: 10.1210/en.2006-0164. [DOI] [PubMed] [Google Scholar]
- Guadaño-Ferraz A, Benavides-Piccione R, Venero C, Lancha C, Vennström B, Sandi C, DeFelipe J, Bernal J. Lack of thyroid hormone receptor alpha1 is associated with selective alterations in behavior and hippocampal circuits. Mol Psychiatry. 2003;8:30–38. doi: 10.1038/sj.mp.4001196. [DOI] [PubMed] [Google Scholar]
- Haddow JE, Palomaki GE, Allan WC, Williams JR, Knight GJ, Gagnon J, O'Heir CE, Mitchell ML, Hermos RJ, Waisbren SE, Faix JD, Klein RZ. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl J Med. 1999;341:549–555. doi: 10.1056/NEJM199908193410801. [DOI] [PubMed] [Google Scholar]
- Hennemann G, Docter R, Friesema EC, de Jong M, Krenning EP, Visser TJ. Plasma membrane transport of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr Rev. 2001;22:451–476. doi: 10.1210/edrv.22.4.0435. [DOI] [PubMed] [Google Scholar]
- Heuer H, Maier MK, Iden S, Mittag J, Friesema EC, Visser TJ, Bauer K. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology. 2005;146:1701–1706. doi: 10.1210/en.2004-1179. [DOI] [PubMed] [Google Scholar]
- Lavado-Autric R, Ausó E, García-Velasco JV, Arufe Mdel C, Escobar del Rey F, Berbel P, Morreale de Escobar G. Early maternal hypothyroxinemia alters histogenesis and cerebral cortex cytoarchitecture of the progeny. J Clin Invest. 2003;111:1073–1082. doi: 10.1172/JCI16262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Morimoto E, Kanai Y, Kim do K, Chairoungdua A, Choi HW, Wempe MF, Anzai N, Endou H. Establishment and characterization of mammalian cell lines stably expressing human L-type amino acid transporters. J Pharmacol Sci. 2008;108:505–516. doi: 10.1254/jphs.08232fp. [DOI] [PubMed] [Google Scholar]
- Negishi T, Kawasaki K, Sekiguchi S, Ishii Y, Kyuwa S, Kuroda Y, Yoshikawa Y. Attention-deficit and hyperactive neurobehavioural characteristics induced by perinatal hypothyroidism in rats. Behav Brain Res. 2005;159:323–331. doi: 10.1016/j.bbr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Redei EE, Solberg LC, Kluczynski JM, Pare WP. Paradoxical hormonal and behavioral responses to hypothyroid and hyperthyroid states in the Wistar-Kyoto rat. Neuropsychopharmacology. 2001;24:632–639. doi: 10.1016/S0893-133X(00)00229-3. [DOI] [PubMed] [Google Scholar]
- Renko K, Werner M, Renner-Müller I, Cooper TG, Yeung CH, Hollenbach B, Scharpf M, Köhrle J, Schomburg L, Schweizer U. Hepatic selenoprotein P (SePP) expression restores selenium transport and prevents infertility and motor-incoordination in Sepp-knockout mice. Biochem J. 2008;409:741–749. doi: 10.1042/BJ20071172. [DOI] [PubMed] [Google Scholar]
- Roberts LM, Black DS, Raman C, Woodford K, Zhou M, Haggerty JE, Yan AT, Cwirla SE, Grindstaff KK. Subcellular localization of transporters along the rat blood-brain barrier and blood-cerebral-spinal fluid barrier by in vivo biotinylation. Neuroscience. 2008a;155:423–438. doi: 10.1016/j.neuroscience.2008.06.015. [DOI] [PubMed] [Google Scholar]
- Roberts LM, Woodford K, Zhou M, Black DS, Haggerty JE, Tate EH, Grindstaff KK, Mengesha W, Raman C, Zerangue N. Expression of the thyroid hormone transporters MCT8 (SLC16A2) and OATP14 (SLCO1C1) at the blood-brain barrier. Endocrinology. 2008b;149:6251–6261. doi: 10.1210/en.2008-0378. [DOI] [PubMed] [Google Scholar]
- Ruiz-Marcos A, Cartagena-Abella P, Martinez-Galan JR, Calvo R, Morreale de Escobar G, Escobar del Rey F. Thyroxine treatment and the recovery of pyramidal cells of the cerebral cortex from changes induced by juvenile-onset hypothyroidism. J Neurobiol. 1994;25:808–818. doi: 10.1002/neu.480250706. [DOI] [PubMed] [Google Scholar]
- Sala-Roca J, Martí-Carbonell MA, Garau A, Darbra S, Balada F. Effects of chronic dysthyroidism on activity and exploration. Physiol Behav. 2002;77:125–133. doi: 10.1016/s0031-9384(02)00815-6. [DOI] [PubMed] [Google Scholar]
- Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, Ward J, Sanabria J, Marsa S, Lewis JA, Echeverri R, Lubs HA, Voeller K, Simensen RJ, Stevenson RE. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet. 2005;77:41–53. doi: 10.1086/431313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama D, Kusuhara H, Taniguchi H, Ishikawa S, Nozaki Y, Aburatani H, Sugiyama Y. Functional characterization of rat brain-specific organic anion transporter (Oatp14) at the blood-brain barrier: high affinity transporter for thyroxine. J Biol Chem. 2003;278:43489–43495. doi: 10.1074/jbc.M306933200. [DOI] [PubMed] [Google Scholar]
- Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, Raivich G, Bauer K, Heuer H. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest. 2007;117:627–635. doi: 10.1172/JCI28253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venero C, Guadaño-Ferraz A, Herrero AI, Nordström K, Manzano J, de Escobar GM, Bernal J, Vennström B. Anxiety, memory impairment, and locomotor dysfunction caused by a mutant thyroid hormone receptor alpha1 can be ameliorated by T3 treatment. Genes Dev. 2005;19:2152–2163. doi: 10.1101/gad.346105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser WE, Friesema EC, Jansen J, Visser TJ. Thyroid hormone transport in and out of cells. Trends Endocrinol Metab. 2008;19:50–56. doi: 10.1016/j.tem.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Wallis K, Sjögren M, van Hogerlinden M, Silberberg G, Fisahn A, Nordström K, Larsson L, Westerblad H, Morreale de Escobar G, Shupliakov O, Vennström B. Locomotor deficiencies and aberrant development of subtype-specific GABAergic interneurons caused by an unliganded thyroid hormone receptor alpha1. J Neurosci. 2008;28:1904–1915. doi: 10.1523/JNEUROSCI.5163-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wémeau JL, Pigeyre M, Proust-Lemoine E, d'Herbomez M, Gottrand F, Jansen J, Visser TJ, Ladsous M. Beneficial effects of propylthiouracil plus l-thyroxine treatment in a patient with a mutation in MCT8. J Clin Endocrinol Metab. 2008;93:2084–2088. doi: 10.1210/jc.2007-2719. [DOI] [PubMed] [Google Scholar]
- Wilcoxon JS, Nadolski GJ, Samarut J, Chassande O, Redei EE. Behavioral inhibition and impaired spatial learning and memory in hypothyroid mice lacking thyroid hormone receptor alpha. Behav Brain Res. 2007;177:109–116. doi: 10.1016/j.bbr.2006.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmazer-Hanke DM, Hantsch M, Hanke J, Schulz C, Faber-Zuschratter H, Schwegler H. Neonatal thyroxine treatment: changes in the number of corticotropin-releasing-factor (CRF) and neuropeptide Y (NPY) containing neurons and density of tyrosine hydroxylase positive fibers (TH) in the amygdala correlate with anxiety-related behavior of Wistar rats. Neuroscience. 2004;124:283–297. doi: 10.1016/j.neuroscience.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Zoeller TR, Dowling AL, Herzig CT, Iannacone EA, Gauger KJ, Bansal R. Thyroid hormone, brain development, and the environment. Environ Health Perspect. 2002;110(Suppl 3):355–361. doi: 10.1289/ehp.02110s3355. [DOI] [PMC free article] [PubMed] [Google Scholar]