

Abstract
Isoflurane is a widely used anesthetic which safely and reversibly induces deep coma and associated burst suppression (BS) electroencephalographic patterns. Here we investigate possible underlying causes for the state of cortical hyperexcitability which was recently shown to be one of the characteristics of BS. Our hypothesis was that cortical inhibition is diminished during isoflurane-induced BS. Experiments were performed in vivo using intracellular recordings of cortical neurons to assess their responsiveness to stimulations of connected thalamic nuclei. We demonstrate that during BS EPSPs were diminished by 44%, whereas inhibitory potentials were completely suppressed. This finding was supported by additional results indicating that a decrease in neuronal input resistance normally found during inhibitory responses under low isoflurane conditions was abolished in the BS condition. Moreover, removal of inhibition occasionally revealed excitatory components which were absent during recordings before the induction of BS. We also show that the absence of inhibition during BS is not caused by a blockage of GABA receptors, since iontophoretically applied GABA shows receptor availability. Moreover, the concentration of extracellular chloride was increased during BS, as would be expected after reduced flow of chloride through GABAA receptors. Also inhibitory responses were reinstated by selective blockage of glial glutamate transporters with dihydrokainate. These results suggest that the lack of inhibition during BS is caused by reduced excitation, probably resulting from increased glial uptake of glutamate stimulated by isoflurane, which creates a diminished activation of cortical interneurons. Thus cortical hyperexcitability during BS is favored by suppressed inhibition.
Introduction
It is commonly believed that general anesthetics act by shifting the excitatory-inhibitory balance of brain signaling toward inhibition. This assumption is mainly supported by investigations into molecular mechanism of anesthesia suggesting that most of these agents act on GABA receptors (for review, see Franks, 2006). Within the large variety of anesthetics, inhalation agents such as isoflurane are widely used in clinical practice. Isoflurane appears to target several membrane proteins: glycine and GABAA receptors (Harrison et al., 1993; Nishikawa et al., 2002), as well as two-pore-domain K+ channels (Franks and Lieb, 1988; Patel et al., 1999). In addition, isoflurane has been shown to stimulate the activity of the GLT1/EAAT2 glial glutamate transporter (Larsen et al., 1997; Zuo, 2001).
A great wealth of data has been produced relating to the targets and mechanisms of anesthesia in general, and of isoflurane in particular. Interestingly, most of these studies were conducted in vitro or in cultures and despite the simplified preparation most still did not lead to definitive conclusions. The few in vivo studies (Vahle-Hinz et al., 2001, 2007; Detsch et al., 2002) used extracellular recordings which render the interpretation of excitatory versus inhibitory activities uncertain. In slices, isoflurane reduced excitatory synaptic transmission in the hippocampus (Berg-Johnsen and Langmoen, 1986; Miu and Puil, 1989) and amygdala (Ranft et al., 2004). Moreover, at concentrations of ∼0.2–0.3 mm isoflurane increased the duration of GABAA inhibitory synaptic currents without affecting their amplitude (Ranft et al., 2004). This result contrasts somewhat with a series of studies showing that similar concentrations of volatile anesthetics decreased the current amplitude of GABAA IPSCs in hippocampal neurons (Jones and Harrison, 1993; Pearce, 1996; Banks and Pearce, 1999) as well as in cerebellar neurons (Antkowiak and Heck, 1997), while confirming the prolonged decay time. These effects are believed to result from postsynaptic binding of the agent (Banks and Pearce, 1999; Westphalen and Hemmings, 2003a).
Recently, we have used a variety of anesthetic agents to demonstrate that a deep level of anesthesia, producing burst suppression (BS), generates a state of unstable cortical hyperexcitability (Kroeger and Amzica, 2007). This finding prompted us to question the strength of the associated inhibitory mechanisms within states of anesthesia-induced unconsciousness.
BS is an electroencephalographic (EEG) pattern consisting of an alternating pattern of (1) epochs with silenced cortical activity and associated EEG isoelectric line, and (2) periods displaying high-amplitude slow and sharp waves (Swank and Watson, 1949). BS is generally developed during coma (Brenner, 1985) with various etiologies ranging from anesthesia (safe, fully controlled and reversible) to hypoxia (Silverman, 1975), drug-related intoxications (Weissenborn et al., 1991; Ostermann et al., 2000; De Rubeis and Young, 2001) and hypothermia (Pagni and Courjon, 1964; Michenfelder and Milde, 1991; Nakashima et al., 1995). The common EEG patterns of BS, regardless of the underlying etiology (Chatrian, 1990), suggest common cellular mechanisms. For instance, the hyperexcitable responses evoked during BS were elicited using various anesthetic agents (Kroeger and Amzica, 2007).
For our present investigation we began with the assumption that the hyperexcitability seen during BS results from diminished or blocked inhibition.
Materials and Methods
Animal preparation.
All experimental procedures were performed in acute preparations (cats) and were approved by the committee for animal care of Laval University, Quebec City, QC, Canada. They fully comply with the principles of responsible conduct toward animals of the National Institutes of Health. Experiments started with an initial application of ketamine-xylazine anesthesia (10 mg/kg, i.m. and 2 mg/kg, i.m., respectively). Once a painless state was achieved (verified through absence of withdrawal reflex), all future pressure and incision points were infiltrated with lidocaine and the animal was tracheotomized, artificially ventilated (respiratory rate ∼30 strokes/min) and paralyzed with gallamine triethiodide (20 mg/kg) through a cannula inserted in the cephalic vein. These procedures lasted for ∼15 min. From this point on, the animal was continuously monitored to maintain vital signs within a physiological range. Body temperature was controlled through a heating pad (38 ± 1°C), end-tidal CO2 concentrations were kept at 3.7% (±0.2%) by adjusting the O2 in the inhaled airflow. Deep anesthesia was verified by continuous patterns of slow and ample EEG waves. We also monitored the heart rate throughout the experiments (<110 beats/min). Before the onset of surgical procedures we gradually switched anesthesia to isoflurane (1–1.5%) to compensate for the vanishing initial dose of ketamine-xylazine. From that point onwards the animal was kept only under isoflurane anesthesia. After the isoflurane induction, the animal underwent craniotomy exposing the suprasylvian gyrus. First we lowered bipolar tungsten stimulation electrodes into the centrolateral (CL) and lateroposterior (LP) nuclei of the thalamus (respective stereotaxic coordinates: anterior 10 mm, lateral 3.5 mm and vertical 4 mm above the ear bar plane; and anterior 10 mm, lateral 6 mm and vertical 4 mm). Next we placed intracellular microelectrodes as well as field potential electrodes and Cl−-sensitive electrodes into the cortex. The stability of the recordings was enhanced by bilateral pneumothorax, hip suspension, drainage of the atlanto-occipital space and filling of the hole in the calvarium with a 4% agar solution. Fluid loss during the experiment was compensated for by intravenous injections of saline (20–30 ml/experiment). Unless otherwise indicated, all chemicals used in these experiments were purchased from Sigma. At the end of the experiments, the animals received a lethal dose of sodium pentobarbital.
Electrode preparation and recordings.
Intracellular recordings were obtained from the association areas 5 and 7 of the suprasylvian gyrus using glass micropipettes (tip diameter <0.5 μm) filled with a 3 m solution of potassium acetate (in situ impedance 30–50 MΩ). Field potential electrodes were similarly shaped, except that the tip was widened under the microscope to achieve in situ impedance of ∼10 MΩ. These field potential electrodes were filled with a 0.2 m NaCl solution. The EEG was recorded monopolarly with stainless steel screws implanted into the skull in the immediate vicinity (∼2 mm) of the cellular recordings. All electrodes shared a chlorided silver reference placed in the paralyzed neck muscles. The EEG potentials were bandpass filtered between 0.3 Hz and 1 kHz. Thalamic stimulation was elicited with current impulses of 0.1–0.2 ms duration and 0.05–0.8 mA intensity. Under BS conditions, the electrical stimuli were generally delivered during the suppression periods to avoid the interference of spontaneous (bursting) activities.
Our Cl−-sensitive microelectrodes were manufactured according to procedures described in other studies (Massimini and Amzica, 2001). We used double-barrel pipettes in which the Cl−-sensitive electrode was pretreated with dimethylchlorosilane, dried at 120°C for 2 h, and the tip filled with the Cl− ionophore I-cocktail A (Fluka). This provides a rapid electrode (see below) and logarithmic selectivity against bicarbonate of 1.5 (Kondo et al., 1989) corresponding to a 3% selectivity. The rest of the barrel was loaded with NaCl (0.1 m), while the other barrel contained NaCl (0.2 m). The Cl−-sensitive electrodes were calibrated in two solutions containing, respectively, (1) NaCl 100 mm, and (2) NaCl 10 mm and NaGlu 90 mm, pH 7.4. To ascertain the relationship between concentration and voltage we used the Nicolsky-Eisenmann equation (Ammann, 1986).
We measured the time course of the response of Cl−-sensitive microelectrodes by stepping the electrodes through 2 drops containing different Cl− concentrations (10 and 100 mm). The drops were held at close distance by silver rings, which were individually grounded. Only electrodes reaching 90% of the response in <20 ms were selected for recording, ensuring that the electrode response was far faster than the phenomena under investigation. To preclude the possible contamination of ion potentials through capacitive coupling by field potentials, the latter were measured with the pair electrode and subtracted from the former. The resulting signal was linearized and transposed into concentration values using the parameters extracted from the logarithmic fitting of the calibration points. The headstage amplifier for Cl−-sensitive electrodes was modified with an ultra ultra-low-input current (<25 fA) amplifier (National Semiconductor). Intracellular, ion-sensitive and field potential signals were passed through a high-impedance amplifier with active bridge circuitry (Neurodata). All signals were digitally converted (20 kHz sampling rate) and stored on a computer for off-line analysis.
Iontophoretic application of GABA was performed by means of double barrel glass electrodes which were broken to a diameter of 7–15 μm. One of the barrels was loaded with GABA (0.25 m in distilled water, pH 4), while the other barrel contained NaCl (0.2 m) and served as a blank electrode. The double barrel electrode was then glued to an intracellular microelectrode whose recording tip protruded by 40–60 μm beyond the iontophoretic tip. GABA was retained in its barrel by negative current (8–10 nA) and was ejected by positive current (3–70 nA) by means of an electrophoresis device (Dagan).
Dihydrokainate is an antagonist of the glutamate transporter GLT1/EAAT2 (Arriza et al., 1994). In our experiments a 9 mm solution of dihydrokainate in lactate ringer was applied topically using a Hamilton syringe on a small filter paper (3 × 3 mm) placed over the suprasylvian gyrus. The paper had a central hole through which the recording intracellular microelectrode was lowered into the cortex.
Analysis.
All of our analysis was performed with software from WaveMetrics, and relies on time relationships between the recorded voltage (concentration) time series. Most of the results are presented as stimulus-triggered averages (evoked potentials). These were obtained through extracting sweeps of the intracellular or extracellular field/EEG potentials and synchronization with the triggering stimulus. Statistical analysis was performed with XLSTAT.
Results
Database
Fifty-six neurons were recorded intracellularly in 24 cats. Only recordings satisfying high quality criteria were retained for this study: stable membrane potentials more negative than −60 mV without holding current and overshooting action potentials. The average resting membrane potential recorded under low isoflurane conditions, immediately after impaling and after stabilization of the membrane potential was −72.4 ± 3.2 mV (average ± SD). We also recorded the cell's input resistance: 23.6 ± 9.7 MΩ (tested with 0.5 nA negative current pulses). These parameters were modulated during the induction of BS (see below). All recorded neurons displayed regular discharge patterns according to the established criteria (Connors et al., 1982; Nuñez et al., 1993). All intraneuronal recordings were held for >30 min to include at least one recording of a change of state (e.g., control to BS)—although some recordings lasted for up to 4 such cycles.
Patterns of activity related to the induction of BS
All recording sessions started under 1.5% isoflurane anesthesia, which produced activity patterns comparable to those recorded during slow-wave sleep (SWS) and under ketamine-xylazine anesthesia (Fig. 1A) (for further examples, see Amzica and Steriade, 1995; Contreras and Steriade, 1995). Briefly, these patterns consist of a quasi regular slow oscillation with a frequency range just below 1 Hz. The oscillation is the result of membrane potential alternations in the recorded neurons between depolarized (“up”) states and hyperpolarized (“down”) states. This oscillation is reflected in an in-phase lock of EEG and field potential signals. The up-states correspond to field negative potentials, whereas down-states match field positive potentials (Contreras and Steriade, 1995).
Figure 1.

Typical progression toward burst suppression (BS) pattern, recorded at the EEG and field potential level as well as intracellularly in a cortical neuron in the suprasylvian gyrus (areas 5–7). A, Under light anesthesia (1.5% isoflurane) neuronal activity is mainly characterized by slow-wave sleep-like (SWS) oscillation dominated by a quasi regular slow rhythm of <1 Hz. B, Application of higher concentrations of isoflurane (2.5%) hyperpolarizes the neuronal membrane potential and accelerates cortical oscillations. Occasionally, this transitory activity is interrupted by short periods of flat EEG (*) that increase in duration and become more frequent. C, The transition period is followed by a stable pattern of BS, which is characterized by long silent periods of flat EEG and occasional bursts of activity. In this and subsequent figures, signals are presented with positive polarity upward.
BS was induced by raising isoflurane in the inhaled airflow to 2.5%. The first change in activity patterns was characterized by an accelerated and spikier EEG as well as by a hyperpolarization of the neuronal membrane (Fig. 1B). With a delay of ∼5 min (for details, see Tétrault et al., 2008) the first episodes of isoelectric lines (Fig. 1B, asterisks) appeared and soon afterward a stable pattern of BS was established (Fig. 1C). The EEG patterns during BS consisted of periods of bursting and isoelectric lines. During the latter, all recorded cortical neurons displayed a hyperpolarized membrane potential devoid of any synaptic activity, whereas bursts consisted of depolarizing triangular-shaped potentials, synchronous with corresponding field and EEG waves.
The distribution of membrane potential values differed between low isoflurane conditions and BS (Fig. 2). SWS patterns always produced a bimodal distribution illustrating the preference for either the “up” state or the “down” state (Fig. 2A, left). In contrast, the bursting activity displayed a unimodal distribution (Fig. 2A, right) even though this activity was also characterized by depolarizing events— albeit exhibiting prevalence for hyperpolarized values and an absence of peaks in the depolarizing range. This aspect reflects the short duration of the depolarizing potentials during the bursting phase of BS. Under low isoflurane anesthesia depolarizing phases of the slow oscillation also reflect the activity of cortical interneurons, thus the above observation is a first indication of the possible reduction of inhibitory activity during BS (see Discussion).
Figure 2.

Comparison of slow oscillatory activity of intracellular potentials during sleep-like patterns and bursting during BS. A1, Intracellular activity of the same neuron recorded under lighter (1.5%) isoflurane anesthesia (left) and during BS (right). The latter trace was obtained by artificially eliminating all isoelectric phases, which served the purpose of producing an oscillatory period of equivalent duration to the trace depicting activity under light anesthesia. During the low isoflurane recording the neuron was injected with hyperpolarizing current (−0.5 nA) to prevent firing of action potentials and to reach comparative membrane potentials as during BS. A2, Histograms of membrane potentials during recording periods displayed above (20 s) gave raise to different patterns: sleep-like activity under light anesthesia resulted in a biphasic oscillatory pattern with clear up-and-down states, whereas BS activity produced a unimodal distribution reflecting a prevalence of the down state and a shortened up state. The ordinate represents the incidence of a particular membrane potential value during the analyzed period. B, C, Pairs of histograms (gray for light anesthesia, white for BS) from two other neurons recorded under the same conditions as in A, showing consistently similar distributions of the membrane potentials.
Modification of inhibition during BS
Under low isoflurane conditions cortical neurons respond to volleys from functionally related thalamic nuclei with an initial, short-latency EPSP (Fig. 3A), followed by an inhibitory potential (a), a long-lasting hyperpolarization (b), and a rebound depolarization (c). At hyperpolarized membrane potentials, such as during spontaneously occurring “down” states, the initial IPSP (Fig. 3Aa) appeared reverted and added on top of the initial EPSP. On the other hand, the secondary hyperpolarization reached a similar membrane potential (Fig. 3A, second trace). As proposed in other reports with similar responses (Connors et al., 1988; Berman et al., 1991; Contreras et al., 1997), we suggest that the initial hyperpolarization might represent a GABAA IPSP, while the second one could be GABAB mediated. During the low isoflurane condition we observed no membrane potential dependence of the rebound effect in all tested neurons (n = 37). However, after the induction of BS (third trace) the rebound excitation (Fig. 3Ac) was virtually abolished, despite the more hyperpolarized membrane potential. In addition, the reverted (Fig. 3Aa) component was decreased. The comparison of the amplitudes of the initial EPSPs between low isoflurane conditions and BS shows a reduction of the EPSP by 44% (n = 12). This comparison was performed with each neuron kept at the same membrane potential (±0.5 mV) during the two conditions (this implied depolarization of the membrane through intracellular current injection during BS). No analysis of the duration the EPSPs was made, because of the variability in resting membrane potential among the recorded population of neurons, which could have been responsible for voltage-dependent components such as NMDA, INa(p), etc. Additionally, BS unmasked occasionally excitatory components (see below) that would have rendered the task unrealistic. Furthermore, no inhibitory responses were observed during BS even when depolarizing the neuronal membrane at values close to firing threshold (Fig. 3B, −60 mV).
Figure 3.
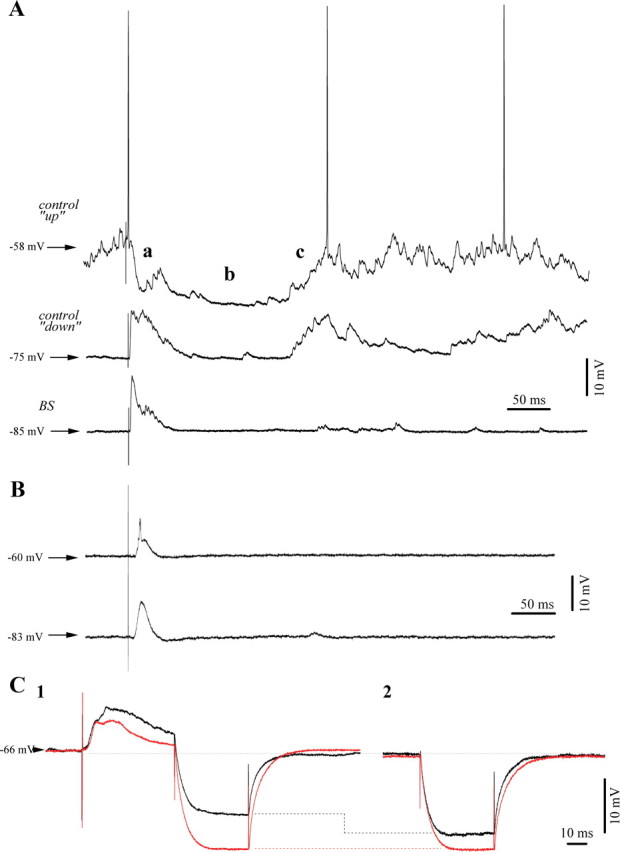
Typical responses of cortical neurons to thalamic (CL) electrical stimuli. A, Responses of a cortical neuron (area 5) to electrical stimuli delivered to the thalamic LP nucleus. During spontaneous depolarized membrane potentials of the “up-state” of the slow oscillation (upper trace) under low isoflurane anesthesia, we observed the following response sequence: an initial EPSP, a fast IPSP (a), a long lasting hyperpolarization (b), and a sustained rebound depolarization (c). The same stimulus was applied during the hyperpolarized “down-state” (middle trace) and we recorded a very similar response with the exception of a modified short IPSP (a). After BS induction with 3% IF, the initial excitatory response diminished in amplitude despite a further hyperpolarized membrane potential. Moreover, the rebound component (c) appeared mostly abolished (lower trace). B, Absence of inhibitory components in the response of another cortical neuron to thalamic stimulation even under steady depolarization through current injection (+1 nA) to bring the membrane potential at −60 mV. Average of 20 responses. C, Assessment of input resistance: responses under low isoflurane in black, BS responses in red recorded 9 min later (average traces from the same neuron, n = 25). Note the overall increase in resistance during BS (C2). Resistance tests during the time frame of the inhibitory responses after stimulation displayed a lack of resistance modification under BS and an 18% reduced resistance during the low isoflurane condition (C1).
We suggest that the a component in the response represents an active inhibition. Therefore we measured the input resistance of the neuron during the respective time frame (Fig. 3C). Under low isoflurane conditions the input resistance was decreased during the period after the initial EPSP, compared with periods without electrical stimulation (Fig. 3C1,C2, compare black traces), as expected if an IPSP would have occurred. In the depicted neuron, the initial IPSP was associated with a 23% drop in resistance, whereas at the extent of the complete population of tested neurons (n = 37) the resistance drop was of 27%. During BS on the other hand, a similar comparison, at the same membrane potential, showed no overt modification of the input resistance and indeed only displayed the overall increased resistance during BS unaffected by the presence or absence of preceding stimulation (Fig. 3C, red traces). The lack of resistance modification during the time frame of the IPSP under BS conditions was a consistent finding in all tested neurons (n = 37).
Furthermore, we could demonstrate that late inhibitory potentials evoked by thalamic electrical stimulation during low isoflurane conditions were voltage-dependent, as would be expected from a phenomenon that opens membrane conductances. Thus, the long-lasting inhibition was enhanced by depolarization under low isoflurane conditions (Fig. 4A, black trace) compared with those at resting membrane potential (Fig. 4B). In contrast, responses to the same stimuli were markedly reduced by steady hyperpolarization of the membrane potential (Fig. 4C). The fact that this component was absent at a very hyperpolarized level (−107 mV in this case) suggests that it might be generated by K+ currents. This hyperpolarizing component was canceled at a membrane potential of −93 mV (±2.1 mV; n = 16). We were, however, unable to reverse this putative GABAB IPSP to provide a clear reversal potential. During BS overt inhibitory potentials were completely abolished at any levels of membrane polarization (Fig. 4, red traces).
Figure 4.

Voltage dependence of cortical responses to thalamic (CL) electrical stimuli. A–C, Intraneuronal recording in cortical area 5 at three levels of polarization: under depolarizing current injection of 1 nA (A), rest (B), and under hyperpolarizing current application of −1.5 nA (C). Note: light anesthesia (black traces) and BS (red traces). Each trace is the result of an average of 10 individual responses, which were selected among those occurring at comparable membrane potentials between the two states. Depicted above are the averaged traces of A with two examples for individual responses from each state. Note the presence of an inhibitory response after the initial excitation with a consecutive rebound excitation during light anesthesia, as well as the absence of the inhibition and rebound during BS.
In several neurons (n = 14, 25% of the recorded neurons) the thalamic response during BS consisted of the initial EPSP and a secondary excitation. They occurred with variable delay within the time frame of the control inhibition (Fig. 5). In these cases, responses under low isoflurane exhibited the usual excitation-inhibition-rebound sequences (Fig. 5A1). The BS responses, however, were devoid of the rebound phase, in a similar way as in the overall majority of the recorded cells. Moreover, we observed that the secondary excitation displayed a clear voltage dependence (Fig. 5A2), but found it unlikely that this may represent a reversal of the IPSP because this response was also present at rather depolarized membrane potentials (Fig. 5A2, e.g., −65 mV). In addition, the latency of this component was variable and was expressed at varying points during the inhibitory window (Fig. 5B). We suggest that the observed excitatory potential may represent a secondary synaptic excitatory event unmasked by the absence of inhibition during BS. Under low isoflurane conditions, this response is shunted by inhibitory potentials.
Figure 5.

Unmasking of excitatory components during BS responses matching the timing of inhibitory components during low isoflurane conditions. Averaged evoked responses of a cortical neuron (area 5) to thalamic (LP) stimulation (n = 25). A1, Responses of the same neuron: before BS induction (under low isoflurane), during BS (high isoflurane), and after recovery from BS (return to low isoflurane). During the BS period a depolarizing potential replaces the inhibitory component of the control period. This change is also apparent in the nearby recorded field potential (FP). All stimuli were applied during the suppression period. A2, The BS-related depolarizing event displayed voltage dependence and increased with membrane hyperpolarization. B, Similar patterns in another neuron exhibiting an excitatory BS component expressed early after the initial excitatory response.
Since Cl− inhibition is achieved by opening of the GABAA receptors and entrance of Cl− ions in the cell, we investigated whether the observed absence of inhibition during BS might have consequences on the extracellular distribution of various ions, particularly those involved in inhibitory processes (Cl− and K+). Our results suggest that during low isoflurane conditions ongoing inhibitory activities establish a given extracellular Cl− concentration ([Cl−]o). Application of high levels of isoflurane (3%) caused an increase of [Cl−]o in parallel with the onset of BS. The extracellular concentration returned to control values after the supply of isoflurane was discontinued. We observed an average increase in [Cl−]o by 27.3 mm (±7.4, n = 6) with an onset latency of 112 s (±21). Considering that the average [Cl−]o is around 150 mm (Dietzel et al., 1982), our recorded difference thus represents a 18% increase. Within seconds of the onset of this increase we recorded the appearance of the first isoelectric episode (Fig. 6, marking the onset of BS, asterisk in the inset), suggesting that the reduction of the inhibitory activity in the cortex might create a surplus of Cl− ions in the extracellular space.
Figure 6.

Extracellular Cl− concentration ([Cl−]o) during BS induction and recovery. Note progressively increased Cl− values after application of increased (3%) isoflurane (high IF) and return to control values after the withdrawal of the additional dose. The dotted horizontal line indicates the average Cl− concentration under low isoflurane, while the continuous horizontal line represents the summation of the average and SD values. The time delay of the Cl− increase was determined as the first value crossing the SD line without subsequently returning (vertical arrow). The EEG period within the square is expanded below to show the appearance of the first isoelectric episode within seconds after the onset of the Cl− increase (vertical arrow).
Pharmacological correlates of inhibition during BS
Thus far, our data suggest that inhibition is strongly impaired during isoflurane-induced BS. One reason for this impairment may be the action of isoflurane on an enhancement of glutamate uptake by specific glial GLT1/EAAT2 transporters (Larsen et al., 1997; Zuo, 2001) and the consequent reduction of excitatory neuronal inputs to inhibitory interneurons. The idea behind this reasoning is that a reduction in excitation would result in a diminished inhibitory drive. If this was the case, then blockage of glial glutamate transporters should reinstate inhibition even during BS.
To test this hypothesis we applied dihydrokainate (9 mm) topically onto the neocortex during isoflurane-induced BS and performed intracellular recordings from neurons situated in the dihydrokainate affected area (Fig. 7). In all cases (n = 12), we observed an overall depolarized membrane potential in these neurons compared with recordings without dihydrokainate. The average membrane potential was –62.3 mV (±3.7; statistically different from controls, p = 0.02, signed-rank Wilcoxon test). We observed a continuation of neuronal bursting activity in synchrony with the EEG. However, neurons exhibited continuous synaptic potentials during the contralaterally recorded EEG isoelectric line (Fig. 7A). These potentials might in part also be the result of a direct enhancement of the excitatory activity by dihydrokainate. The amplitude of this activity during isoelectric episodes was clearly increased compared with recordings without dihydrokainate (Fig. 7B). The width of the Gaussian fit of the membrane potential distribution measured at half amplitude was 3.3 mV (±1.2), and was statistically different (p < 0.001, signed-rank Wilcoxon test) from the corresponding values of neurons without dihydrokainate (0.4 ± 0.09 mV). By itself, this result provides evidence that our topical application of dihydrokainate was indeed effective.
Figure 7.

Blockage of glial glutamate uptake restores inhibition during BS. A, Intracellular recording in the suprasylvian gyrus during topical application of dihydrokainate. Neurons discharged synchronously during the bursting phase of the BS pattern. However, the membrane potential displayed an unusually increased background activity during the isoelectric line (see inset). B, Histograms of membrane potentials of two neurons during 20 s periods of isoelectric activity. The upper histogram corresponds to a neuron recorded under low isoflurane conditions, while the lower histogram reflects the neuron at left. C, Neuronal response to CL thalamic stimulation during BS shows a similar excitatory-inhibitory pattern as during light anesthesia. Averaged responses (n = 15) are shown on the left, whereas individual responses during BS (top) and during low isoflurane conditions (bottom) are displayed on the right.
Moreover, dihydrokainate application during BS reinstated the inhibitory responses to thalamic stimulations, which were in fact comparable to those obtained in the same neuron before the induction of BS (Fig. 7C). This suggests that the diminished inhibition observed during isoflurane-induced BS mainly relies on a reduction of glutamate availability within cortical networks.
Further investigation into the status of interneurons focused on verifying the availability of GABAergic receptors for ligand binding during BS. We therefore used an iontophoretic application of GABA directly onto intracellularly recorded neurons. The effectiveness of this method of GABA application was confirmed by an observed amplitude reduction of synaptic intracellular activity (Fig. 8). Assessment of the distribution of membrane potential values shows a progressive reduction of the width of the Gaussian fit measured at half amplitude by 42% (n = 7) and a return to control values after the discontinuation of iontophoresis. Similarly, GABA application also reduced the amplitude of cortical responses to thalamic stimuli. During low isoflurane conditions the amplitude of EPSPs was reduced by 66% whereas IPSP amplitudes were reduced by 82% (Fig. 9A1). During BS on the other hand no inhibitory component could be elicited and the observed EPSPs were reduced by 59% (Fig. 9A2). Additionally, we recorded a reduction in input resistance of neurons undergoing GABA iontophoresis (Fig. 9B), a finding that meshes well with the known GABA shunting of neuronal membranes. During low isoflurane conditions and during BS input resistance values dropped by 70% and 68%, respectively. These virtually equal values imply that the availability of the GABA receptors was similar in the two states.
Figure 8.

Effect of iontophoretic application of GABA on a cortical neuron from area 5. A, Spontaneous activity under light (1.5%) isoflurane anesthesia displaying a slow oscillatory activity. GABA application reduced the amplitude of this oscillation, which subsequent recovered after the discontinuation of GABA. B, We quantified the amplitude of membrane potential variations of the five 17 s epochs indicated in A. The histograms (bars) were fitted with Gaussian functions (lines) and indicate the narrowing of amplitude ranges during the GABA application, suggesting a decrease in amplitude variations. The histogram widths (at half amplitude) for the five selected epochs are: 19, 12.8, 11.1, 11.5, and 18.3 mV, respectively.
Figure 9.

Effect of iontophoretic application of GABA on cortical responsiveness to thalamic stimulation. A1, Averaged (n = 15) evoked membrane potentials during low isoflurane conditions (light 1.5% isoflurane anesthesia), before and after the application of GABA (black traces), and during GABA iontophoresis (red traces). Note the diminished amplitude of both excitatory and inhibitory components. A2, Similar sequence of procedures in the same neuron recorded during BS. The application of GABA resulted in diminished excitatory responses, whereas inhibitory components were completely absent. B, Effect of GABA application on membrane input resistance during low isoflurane conditions (1) and during BS (2). In both cases GABA reduced the input resistance of the neuron.
We also investigated the effect of increasing doses of isoflurane on neuronal membranes during BS induction. As described above, we measured the input resistance of neurons with intracellularly injected current pulses. A minimum of 5 pulses were applied during each state (low isoflurane and BS) and we averaged the ensuing responses. Overall, neurons (n = 35 tested) displayed a slight increase in resistance from 23.6 MΩ (±9.7) to 26.8 MΩ (±11.7), representing a 14% increase. This increase was, however, not statistically significant (signed-pair Wilcoxon test). On the other hand, we noticed a clear increase of input resistance in a subset of neurons (Fig. 10A, n = 24), whereas others displayed a decrease (n = 11). This suggests the presence of two populations which are modulated differently by isoflurane application. Thus, we recalculated the response averages for each group: neurons with increased input resistance displayed an increase of 32% (from 20.2 MΩ to 26.7 MΩ), whereas the other group displayed a drop of 16% (from 27.9 MΩ to 23.3 MΩ). The resistance changes within these separated groups were indeed statistically significant (p < 0.01, signed-pair Wilcoxon test). We could not, however, correlate these to neuronal populations with any particular location within the suprasylvian gyrus or with a particular recording depth.
Figure 10.

Effect of isoflurane-induced BS on the neuronal input resistance. A, Hyperpolarizing pulses during low isoflurane (1) and BS (2) as used in B, demonstrating increased resistance during BS. B, Input resistance test (above) and EPSP amplitudes during three successive inductions of BS (periods marked with continuous lines), by high levels of isoflurane (3%) (periods indicated by dotted lines). Note the systematically increased resistance and diminished EPSP amplitude during BS.
Nevertheless, since the majority of the recorded neurons displayed a clearly increased input resistance during BS induced by isoflurane, our results are at least partially in agreement with the reduced inhibition measured in this study and with the hypothesized effect of isoflurane on the increased transglial transport of glutamate. To further clarify this point we monitored the dynamic evolution of the input resistance during the induction of BS in several neurons (n = 5) (Fig. 10B). During these procedures we ensured constant membrane potential values with current injection to avoid the contribution of voltage-dependent currents. We observed a clear increase of input resistance with each induction of BS. Occasionally we applied thalamic stimuli to assess the amplitude of the initial EPSP. Our results suggest a clear reduction in EPSP amplitudes during BS (Fig. 10B).
Last, we studied the modulation of inhibitory processes during BS induced with a short-acting barbiturate (thiopental, 32 mg/kg applied systemically). Since it is known that the primary action of barbiturates is an enhancement of GABAA receptor activity, we would have expected that control values of inhibition would be maintained or even increased during barbiturate-induced BS. However, all recorded neurons (n = 5) displayed a reduction of inhibitory potentials during BS, although some inhibitory components were maintained (Fig. 11). Therefore our results raise the possibility that the reduced inhibition reported in this study might not result from a specific anesthetic action on GABAergic receptors, but rather from a more general mechanism particular to the state of BS. Two facts support this idea: (1) the periodic variations of extracellular Ca2+ and its overall reduced availability, especially during the suppression phase, decreases the synaptic efficacy in cortical networks (Kroeger and Amzica, 2007), and (2) thalamic neurons display rhythmic bursting activities during the suppression phase of the BS (Steriade et al., 1994) creating an excitatory pressure on cortical elements.
Figure 11.

Reduction of cortical inhibition under barbiturate (thiopental) anesthesia. A, Control conditions under ketamine-xylazine anesthesia with slow sleep-like oscillations in the EEG (left panel) and voltage-dependent responses to thalamic stimulations (right panel; average of 15 sweeps). Note enhanced EPSPs and reduced inhibition with hyperpolarization. B, Systemic application of thiopental (32 mg/kg) induces BS in EEG signals (left) and decreasing amplitudes and durations of inhibitory responses (right).
Discussion
The results presented above show a significant reduction of cortical inhibition during isoflurane-induced BS and thus support earlier reports of a hyperexcitable state during this level of anesthesia-induced coma (Kroeger and Amzica, 2007). Several facts highlight this conclusion. (1) The composition and timing of our recorded responses to thalamic stimulation during low isoflurane conditions were similar to responses observed in other studies (Connors et al., 1988; Berman et al., 1991; Contreras et al., 1997). Both inhibitory components were absent during isoflurane induced BS. (2) The decreased input resistance measured during inhibitory responses under low isoflurane conditions is abolished under BS conditions. Moreover, as observed in some neurons, excitatory potentials replaced inhibitory responses during stimulation, which is probably the result of a lifted shunting effect. (3) During BS only the initial excitatory response to thalamic stimuli was preserved. All subsequent components did not display voltage dependence, as would be expected if receptor channels were open. (4) We observed a steady and significant rise in the extracellular Cl− concentration during BS induction emphasizing the possibility that inhibitory activity through GABAA receptors may have been absent.
At a first glance, it may surprise to learn that inhibition is abolished during a state that is associated with deep coma (Brenner, 1985), especially if the coma is provoked by a drug that (1) occupies a specific binding site on GABAA receptors (Mihic et al., 1997; Jenkins et al., 2001) and (2) enhances GABAergic transmission (Harrison et al., 1993; Banks and Pearce, 1999; Nishikawa et al., 2002). However, a blocking effect of volatile anesthetics on GABAA receptors has been described in vitro by Banks and Pearce (1999), who found that this block was most prominent using enflurane. However, isoflurane only produced a 20% reduction in the permeability of the receptor at aqueous concentrations of 0.6 mm (Banks and Pearce, 1999, their Fig. 3). This concentration corresponds to ∼2 MAC and is thus within the range used in the present experiments Moreover, in our experiments GABA application still elicited an effect on neuronal membranes even under high isoflurane concentration. This result was probably mediated by extrasynaptic receptors (Salin and Prince, 1996) and possibly reflecting a shunting effect (reduced amplitude of the synaptic potentials and increased input conductance). Therefore, the direct effect of isoflurane on GABA receptors might represent only a part of the mechanisms of isoflurane action.
Further membrane targets of isoflurane are two-pore-domain K+ channels (Franks and Lieb, 1988; Patel et al., 1999). These channels have been shown to underlie isoflurane resistance in transgenic mice with deficient TREK-1 proteins (Heurteaux et al., 2004). However, isoflurane action on these channels cannot fully account for the reduced inhibition seen in our study, but could possibly explain the decreased input resistance of a subpopulation of our recorded neurons. On the other hand, results from the neuron population displaying increased resistance during isoflurane application support the suggestion that reduced GABAergic influence could cause the loss of inhibition during BS.
In our view, the main effect of isoflurane in these experiments was a reduction in excitation, which in turn resulted in a reduced activation of interneurons, and thus diminished inhibition. This result may be explained by modulation of: (1) neurotransmitter release, (2) blockage of postsynaptic targets, and/or (3) glutamate uptake by glial cells. The first explanation receives support from findings indicating a reduced synaptic release of glutamate under isoflurane resulting from various presynaptic mechanisms (MacIver et al., 1996; Westphalen and Hemmings, 2003b; Wu et al., 2004). The most appealing target of presynaptic depression, found in the calyx of Held, appears to be a reduction in amplitude of action potentials invading the axon terminal, resulting in a 50% reduction of glutamate release (Wu et al., 2004). This value compares well with our 44% reduction in the amplitude of the thalamically evoked EPSPs.
A similar presynaptic effect might exist also at the level of GABA release, even though experimental evidence is so far lacking. Isoflurane has little effect at the glutamatergic postsynaptic site (Perouansky and Antognini, 2003), although a moderate inhibition of AMPA receptors has been reported at concentrations of 2 MAC (Dildy-Mayfield et al., 1996), which are sufficient to induce BS.
Glial cells play an important role in regulating the neuronal excitability by several mechanisms, e.g., glutamate uptake via specific transporters. This process is enhanced by volatile anesthetics such as isoflurane (Larsen et al., 1997; Zuo, 2001; Huang and Zuo, 2003) and halothane (Miyazaki et al., 1997). Blockage of glutamate uptake leads to an increased demand of anesthesia (Cechova and Zuo, 2006), which is used to counteract increasing excitatory signaling. Therefore, the net result of isoflurane is a diminished availability of glutamate in the synaptic cleft, as glial glutamate uptake is enhanced. Moreover, the isoflurane-induced reduction in glutamatergic signaling reduces the activation of inhibitory cells, and thus inhibition declines. With this scenario in mind, we investigated the effect of a blockage of the glial transporters. Our results indicate that blocking glial transporters does indeed restore inhibitory responses. However, since dihydrokainate also acts directly on glutamatergic receptors, it may be conceivable that this result could be obtained by dihydrokainate itself. Indeed, during the EEG isoelectric line the membrane potential displayed increased synaptic noise suggesting local excitatory activity. On the other hand, dihydrokainate restored phasic inhibition related to the stimulation of ascending excitatory pathways. This suggests an increased availability of glutamate in the synapse which could then have produced this response.
Increasing doses of isoflurane also result in a diminished duration of the depolarizing phases during spontaneous bursting phases of BS with respect to the slow oscillations expressed under low isoflurane anesthesia. Indeed, all excitatory and inhibitory cortical neurons discharge during the depolarizing phase (“up-state”) of the control slow (<1 Hz) sleep-like oscillation (Steriade et al., 1993). The measured reduction in duration of the “up state” during BS could be caused by increased inhibitory activity. However, considering that inhibitory responses are absent during BS, we conclude that the shape of the recurrent depolarizations during bursting activity would rather lack a contribution of local inhibitory interneurons.
A further deepening of the comatose state with higher doses of isoflurane results in progressively shorter EEG bursts and longer isoelectric episodes until a constant cortical silence is achieved. During this state cortical excitability is probably suppressed altogether.
From the above-discussed mechanisms it appears that both excitatory and inhibitory pathways are affected by isoflurane—at least during concentrations eliciting BS. Inhibition is more drastically affected, which in turn leads to a shift in the excitation/inhibition balance toward excitation, even though excitation is reduced. The abolishing of inhibition leads to a hyperexcitable state permitting the genesis of EEG bursts occasionally interrupting the cortical silence of the EEG isoelectric line (Kroeger and Amzica, 2007). EEG bursts may then be elicited either by subliminal stimuli (Kroeger and Amzica, 2007) or by ongoing subcortical activities: thalamic (Steriade et al., 1994) or hippocampal (D. Kroeger and F. Amzica, unpublished observation).
In clinical settings, this hyperexcitability, and the associated bursting activity, may occasionally be mistaken for symptoms of epileptic fits (Dan and Boyd, 2006). This idea receives further support from the abolishment of inhibition shown in the present study. Both results are surprising findings especially since isoflurane is frequently used for the treatment of status epilepticus at concentrations which induce BS (Kofke et al., 1985, 1989; Meeke et al., 1989). This therapeutic effect probably relies on the ability of isoflurane to block gap junctions (Mantz et al., 1993), a primary instrument in the generalization of spike-wave seizures (Amzica et al., 2002). The above described paradox underlines the complexity, and probably the heterogeneity, of the epileptic syndromes.
Footnotes
This work was supported by grants from the Canadian Institutes for Health Research to F.A. (MSH-69176 and MOP-67175). J.-F.F is a M.Sc. student in the laboratory, while D.K. and O.C. are Ph.D. students. We are grateful to Pierre Giguère for technical assistance.
References
- Ammann D. Ion sensitive microelectrodes. Berlin: Springer; 1986. [Google Scholar]
- Amzica F, Steriade M. Short- and long-range neuronal synchronization of the slow (<1 Hz) cortical oscillation. J Neurophysiol. 1995;73:20–38. doi: 10.1152/jn.1995.73.1.20. [DOI] [PubMed] [Google Scholar]
- Amzica F, Massimini M, Manfridi A. Spatial buffering during slow and paroxysmal sleep oscillations in cortical networks of glial cells in vivo. J Neurosci. 2002;22:1042–1053. doi: 10.1523/JNEUROSCI.22-03-01042.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antkowiak B, Heck D. Effects of the volatile anesthetic enflurane on spontaneous discharge rate and GABA(A)-mediated inhibition of Purkinje cells in rat cerebellar slices. J Neurophysiol. 1997;77:2525–2538. doi: 10.1152/jn.1997.77.5.2525. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks MI, Pearce RA. Dual actions of volatile anesthetics on GABA(A) IPSCs: dissociation of blocking and prolonging effects. Anesthesiology. 1999;90:120–134. doi: 10.1097/00000542-199901000-00018. [DOI] [PubMed] [Google Scholar]
- Berg-Johnsen J, Langmoen IA. Isoflurane effects in rat hippocampal cortex: a quantitative evaluation of different cellular sites of action. Acta Physiol Scand. 1986;128:613–618. doi: 10.1111/j.1748-1716.1986.tb08019.x. [DOI] [PubMed] [Google Scholar]
- Berman NJ, Douglas RJ, Martin KA, Whitteridge D. Mechanisms of inhibition in cat visual cortex. J Physiol. 1991;440:697–722. doi: 10.1113/jphysiol.1991.sp018731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner RP. The electroencephalogram in altered states of consciousness. Neurol Clin. 1985;3:615–631. [PubMed] [Google Scholar]
- Cechova S, Zuo Z. Inhibition of glutamate transporters increases the minimum alveolar concentration for isoflurane in rats. Br J Anaesth. 2006;97:192–195. doi: 10.1093/bja/ael152. [DOI] [PubMed] [Google Scholar]
- Chatrian GE. Coma, other states of altered responsiveness, and brain death. In: Daly DD, Pedley TA, editors. Current practice in clinical electroencephalography. New York: Raven; 1990. pp. 425–487. [Google Scholar]
- Connors BW, Gutnick MJ, Prince DA. Electrophysiological properties of neocortical neurons in vitro. J Neurophysiol. 1982;48:1302–1320. doi: 10.1152/jn.1982.48.6.1302. [DOI] [PubMed] [Google Scholar]
- Connors BW, Malenka RC, Silva LR. Two inhibitory postsynaptic potentials, and GABAA and GABAB receptor-mediated responses in neocortex of rat and cat. J Physiol. 1988;406:443–468. doi: 10.1113/jphysiol.1988.sp017390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras D, Steriade M. Cellular basis of EEG slow rhythms: a study of dynamic corticothalamic relationships. J Neurosci. 1995;15:604–622. doi: 10.1523/JNEUROSCI.15-01-00604.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras D, Dürmüller N, Steriade M. Absence of a prevalent laminar distribution of IPSPs in association cortical neurons of cat. J Neurophysiol. 1997;78:2742–2753. doi: 10.1152/jn.1997.78.5.2742. [DOI] [PubMed] [Google Scholar]
- Dan B, Boyd SG. Stimulus-sensitive burst-spiking in burst-suppression in children: implications for management of refractory status epilepticus. Epileptic Disord. 2006;8:143–150. [PubMed] [Google Scholar]
- De Rubeis DA, Young GB. Continuous EEG monitoring in a patient with massive carbamazepine overdose. J Clin Neurophysiol. 2001;18:166–168. doi: 10.1097/00004691-200103000-00008. [DOI] [PubMed] [Google Scholar]
- Detsch O, Kochs E, Siemers M, Bromm B, Vahle-Hinz C. Differential effects of isoflurane on excitatory and inhibitory synaptic inputs to thalamic neurones in vivo. Br J Anaesth. 2002;89:294–300. doi: 10.1093/bja/aef170. [DOI] [PubMed] [Google Scholar]
- Dietzel I, Heinemann U, Hofmeier G, Lux HD. Stimulus-induced changes in extracellular Na+ and Cl− concentration in relation to changes in the size of the extracellular space. Exp Brain Res. 1982;46:73–84. doi: 10.1007/BF00238100. [DOI] [PubMed] [Google Scholar]
- Dildy-Mayfield JE, Eger EI, 2nd, Harris RA. Anesthetics produce subunit-selective actions on glutamate receptors. J Pharmacol Exp Ther. 1996;276:1058–1065. [PubMed] [Google Scholar]
- Franks NP. Molecular targets underlying general anaesthesia. Br J Pharmacol. 2006;147(Suppl 1):S72–S81. doi: 10.1038/sj.bjp.0706441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franks NP, Lieb WR. Volatile general anaesthetics activate a novel neuronal K+ current. Nature. 1988;333:662–664. doi: 10.1038/333662a0. [DOI] [PubMed] [Google Scholar]
- Harrison NL, Kugler JL, Jones MV, Greenblatt EP, Pritchett DB. Positive modulation of human gamma-aminobutyric acid type A and glycine receptors by the inhalation anesthetic isoflurane. Mol Pharmacol. 1993;44:628–632. [PubMed] [Google Scholar]
- Heurteaux C, Guy N, Laigle C, Blondeau N, Duprat F, Mazzuca M, Lang-Lazdunski L, Widmann C, Zanzouri M, Romey G, Lazdunski M. TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J. 2004;23:2684–2695. doi: 10.1038/sj.emboj.7600234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Zuo Z. Isoflurane enhances the expression and activity of glutamate transporter type 3 in C6 glioma cells. Anesthesiology. 2003;99:1346–1353. doi: 10.1097/00000542-200312000-00016. [DOI] [PubMed] [Google Scholar]
- Jenkins A, Greenblatt EP, Faulkner HJ, Bertaccini E, Light A, Lin A, Andreasen A, Viner A, Trudell JR, Harrison NL. Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J Neurosci. 2001;21(RC136):1–4. doi: 10.1523/JNEUROSCI.21-06-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Harrison NL. Effects of volatile anesthetics on the kinetics of inhibitory postsynaptic currents in cultured rat hippocampal neurons. J Neurophysiol. 1993;70:1339–1349. doi: 10.1152/jn.1993.70.4.1339. [DOI] [PubMed] [Google Scholar]
- Kofke WA, Snider MT, Young RS, Ramer JC. Prolonged low flow isoflurane anesthesia for status epilepticus. Anesthesiology. 1985;62:653–656. doi: 10.1097/00000542-198505000-00023. [DOI] [PubMed] [Google Scholar]
- Kofke WA, Young RS, Davis P, Woelfel SK, Gray L, Johnson D, Gelb A, Meeke R, Warner DS, Pearson KS. Isoflurane for refractory status epilepticus: a clinical series. Anesthesiology. 1989;71:653–659. doi: 10.1097/00000542-198911000-00005. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Bührer T, Seiler K, Frömter E, Simon W. A new double-barrelled, ionophore-based microelectrode for chloride ions. Pflugers Arch. 1989;414:663–668. doi: 10.1007/BF00582133. [DOI] [PubMed] [Google Scholar]
- Kroeger D, Amzica F. Hypersensitivity of the anesthesia-induced comatose brain. J Neurosci. 2007;27:10597–10607. doi: 10.1523/JNEUROSCI.3440-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M, Hegstad E, Berg-Johnsen J, Langmoen IA. Isoflurane increases the uptake of glutamate in synaptosomes from rat cerebral cortex. Br J Anaesth. 1997;78:55–59. doi: 10.1093/bja/78.1.55. [DOI] [PubMed] [Google Scholar]
- MacIver MB, Mikulec AA, Amagasu SM, Monroe FA. Volatile anesthetics depress glutamate transmission via presynaptic actions. Anesthesiology. 1996;85:823–834. doi: 10.1097/00000542-199610000-00018. [DOI] [PubMed] [Google Scholar]
- Mantz J, Cordier J, Giaume C. Effects of general anesthetics on intercellular communications mediated by gap junctions between astrocytes in primary culture. Anesthesiology. 1993;78:892–901. doi: 10.1097/00000542-199305000-00014. [DOI] [PubMed] [Google Scholar]
- Massimini M, Amzica F. Extracellular calcium fluctuations and intracellular potentials in the cortex during the slow sleep oscillation. J Neurophysiol. 2001;85:1346–1350. doi: 10.1152/jn.2001.85.3.1346. [DOI] [PubMed] [Google Scholar]
- Meeke RI, Soifer BE, Gelb AW. Isoflurane for the management of status epilepticus. DICP. 1989;23:579–581. doi: 10.1177/1060028089023007-809. [DOI] [PubMed] [Google Scholar]
- Michenfelder JD, Milde JH. The relationship among canine brain temperature, metabolism, and function during hypothermia. Anesthesiology. 1991;75:130–136. doi: 10.1097/00000542-199107000-00021. [DOI] [PubMed] [Google Scholar]
- Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, Mascia MP, Valenzuela CF, Hanson KK, Greenblatt EP, Harris RA, Harrison NL. Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature. 1997;389:385–389. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- Miu P, Puil E. Isoflurane-induced impairment of synaptic transmission in hippocampal neurons. Exp Brain Res. 1989;75:354–360. doi: 10.1007/BF00247941. [DOI] [PubMed] [Google Scholar]
- Miyazaki H, Nakamura Y, Arai T, Kataoka K. Increase of glutamate uptake in astrocytes: a possible mechanism of action of volatile anesthetics. Anesthesiology. 1997;86:1359–1366. doi: 10.1097/00000542-199706000-00018. [DOI] [PubMed] [Google Scholar]
- Nakashima K, Todd MM, Warner DS. The relation between cerebral metabolic rate and ischemic depolarization. A comparison of the effects of hypothermia, pentobarbital, and isoflurane. Anesthesiology. 1995;82:1199–1208. doi: 10.1097/00000542-199505000-00015. [DOI] [PubMed] [Google Scholar]
- Nishikawa K, Jenkins A, Paraskevakis I, Harrison NL. Volatile anesthetic actions on the GABAA receptors: contrasting effects of alpha 1(S270) and beta 2(N265) point mutations. Neuropharmacology. 2002;42:337–345. doi: 10.1016/s0028-3908(01)00189-7. [DOI] [PubMed] [Google Scholar]
- Nuñez A, Amzica F, Steriade M. Electrophysiology of association cortical cells in vivo: intrinsic properties and synaptic responses. J Neurophysiol. 1993;70:418–430. doi: 10.1152/jn.1993.70.1.418. [DOI] [PubMed] [Google Scholar]
- Ostermann ME, Young B, Sibbald WJ, Nicolle MW. Coma mimicking brain death following baclofen overdose. Intensive Care Med. 2000;26:1144–1146. doi: 10.1007/s001340051330. [DOI] [PubMed] [Google Scholar]
- Pagni CA, Courjon J. Electroencephalographic modifications induced by moderate and deep hypothermia in man. Acta Neurochir. 1964;14(Suppl 13):35–49. [PubMed] [Google Scholar]
- Patel AJ, Honoré E, Lesage F, Fink M, Romey G, Lazdunski M. Inhalational anesthetics activate two-pore-domain background K+ channels. Nat Neurosci. 1999;2:422–426. doi: 10.1038/8084. [DOI] [PubMed] [Google Scholar]
- Pearce RA. Volatile anaesthetic enhancement of paired-pulse depression investigated in the rat hippocampus in vitro. J Physiol. 1996;492:823–840. doi: 10.1113/jphysiol.1996.sp021349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perouansky M, Antognini JF. Glutamate receptors. In: Antognini JF, Carstens EE, Raines DE, editors. Neural mechanisms of anesthesia. Totowa, NJ: Humana; 2003. pp. 319–332. [Google Scholar]
- Ranft A, Kurz J, Deuringer M, Haseneder R, Dodt HU, Zieglgänsberger W, Kochs E, Eder M, Hapfelmeier G. Isoflurane modulates glutamatergic and GABAergic neurotransmission in the amygdala. Eur J Neurosci. 2004;20:1276–1280. doi: 10.1111/j.1460-9568.2004.03603.x. [DOI] [PubMed] [Google Scholar]
- Salin PA, Prince DA. Spontaneous GABAA receptor-mediated inhibitory currents in adult rat somatosensory cortex. J Neurophysiol. 1996;75:1573–1588. doi: 10.1152/jn.1996.75.4.1573. [DOI] [PubMed] [Google Scholar]
- Silverman D. The electroencephalogram in anoxic coma. In: Rémond A, editor. Handbook of electroencephalography and clinical neurophysiology. Amsterdam: Elsevier; 1975. pp. 81–94. [Google Scholar]
- Steriade M, Nuñez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Amzica F, Contreras D. Cortical and thalamic cellular correlates of electroencephalographic burst-suppression. Electroencephalogr Clin Neurophysiol. 1994;90:1–16. doi: 10.1016/0013-4694(94)90108-2. [DOI] [PubMed] [Google Scholar]
- Swank RL, Watson CW. Effects of barbiturates and ether on spontaneous electrical activity of dog brain. J Neurophysiol. 1949;12:137–160. doi: 10.1152/jn.1949.12.2.137. [DOI] [PubMed] [Google Scholar]
- Tétrault S, Chever O, Sik A, Amzica F. Opening of the blood-brain barrier during isoflurane anaesthesia. Eur J Neurosci. 2008;28:1330–1341. doi: 10.1111/j.1460-9568.2008.06443.x. [DOI] [PubMed] [Google Scholar]
- Vahle-Hinz C, Detsch O, Siemers M, Kochs E, Bromm B. Local GABA(A) receptor blockade reverses isoflurane's suppressive effects on thalamic neurons in vivo. Anesth Analg. 2001;92:1578–1584. doi: 10.1097/00000539-200106000-00046. [DOI] [PubMed] [Google Scholar]
- Vahle-Hinz C, Detsch O, Siemers M, Kochs E. Contributions of GABAergic and glutamatergic mechanisms to isoflurane-induced suppression of thalamic somatosensory information transfer. Exp Brain Res. 2007;176:159–172. doi: 10.1007/s00221-006-0604-6. [DOI] [PubMed] [Google Scholar]
- Weissenborn K, Wilkens H, Hausmann E, Degen PH. Burst suppression EEG with baclofen overdose. Clin Neurol Neurosurg. 1991;93:77–80. doi: 10.1016/0303-8467(91)90015-h. [DOI] [PubMed] [Google Scholar]
- Westphalen RI, Hemmings HC., Jr Effects of isoflurane and propofol on glutamate and GABA transporters in isolated cortical nerve terminals. Anesthesiology. 2003a;98:364–372. doi: 10.1097/00000542-200302000-00016. [DOI] [PubMed] [Google Scholar]
- Westphalen RI, Hemmings HC., Jr Selective depression by general anesthetics of glutamate versus GABA release from isolated cortical nerve terminals. J Pharmacol Exp Ther. 2003b;304:1188–1196. doi: 10.1124/jpet.102.044685. [DOI] [PubMed] [Google Scholar]
- Wu XS, Sun JY, Evers AS, Crowder M, Wu LG. Isoflurane inhibits transmitter release and the presynaptic action potential. Anesthesiology. 2004;100:663–670. doi: 10.1097/00000542-200403000-00029. [DOI] [PubMed] [Google Scholar]
- Zuo Z. Isoflurane enhances glutamate uptake via glutamate transporters in rat glial cells. Neuroreport. 2001;12:1077–1080. doi: 10.1097/00001756-200104170-00042. [DOI] [PubMed] [Google Scholar]