

Abstract
Annotation and identification of metabolite biomarkers is critical for their biological interpretation in metabolic phenotyping studies, presenting a significant bottleneck in the successful implementation of untargeted metabolomics. Here, a systematic multistep protocol was developed for the purification and de novo structural elucidation of urinary metabolites. The protocol is most suited for instances where structure elucidation and metabolite annotation are critical for the downstream biological interpretation of metabolic phenotyping studies. First, a bulk urine pool was desalted using ion-exchange resins enabling large-scale fractionation using precise iterations of analytical scale chromatography. Primary urine fractions were collected and assembled into a “fraction bank” suitable for long-term laboratory storage. Secondary and tertiary fractionations exploited differences in selectivity across a range of reversed-phase chemistries, achieving the purification of metabolites of interest yielding an amount of material suitable for chemical characterization. To exemplify the application of the systematic workflow in a diverse set of cases, four metabolites with a range of physicochemical properties were selected and purified from urine and subjected to chemical formula and structure elucidation by respective magnetic resonance mass spectrometry (MRMS) and NMR analyses. Their structures were fully assigned as tetrahydropentoxyline, indole-3-acetic-acid-O-glucuronide, p-cresol glucuronide, and pregnanediol-3-glucuronide. Unused effluent was collected, dried, and returned to the fraction bank, demonstrating the viability of the system for repeat use in metabolite annotation with a high degree of efficiency.
Metabolic profiling of human biofluids by liquid chromatography–mass spectrometry (LC-MS) is widely used in clinical and epidemiological studies. Improvements in analytical technologies and automation of data processing have made it possible to increase both the number and throughput of sample analysis. In addition, technological advancements allowing greater analytical sensitivity, precision, and selectivity have led to the increase of the number of the detectable chemicals providing greater metabolome coverage. The extensive breadth of metabolic profiling data ensures that metabolite annotation and identification remain major bottlenecks in the metabolic phenotyping workflow, with low abundance metabolites, gut microbial cometabolites, secondary metabolites (glucuronides and sulfates), and chemically modified drug and diet related metabolites representing specific challenges. Growth of publicly available databases containing mass spectral reference data for thousands of chemical species1−3 is limited by the availability of authentic chemical standards, and the value of establishing method-specific in-house databases (e.g., complete with chromatographic retention time measurement) is further hampered by the lack of consistency in standard chromatographic profiling methods employed. Some in silico spectra prediction computational tools and pipelines4,5 are being developed,5−8 as well as retention time prediction,9,10 chemical similarity,11 and biological assumptions to extend the reach of generic metabolite annotation capabilities beyond the limitations of reference data. However, to meet the emphasis placed on the biological interpretation of specific biomarkers arising from profiling studies, further advancements in de novo structure elucidation are required to accelerate the identification of unknown metabolic markers of interest, ultimately mapping the large chemical space of the human metabolome observable by analytical profiling methods.
The two widely used analytical platforms that provide information on molecular structure and elemental composition of unknown chemicals are nuclear magnetic resonance (NMR) spectroscopy12 and mass spectrometry (high-resolution MS and tandem MS/MS).13 The combination of both techniques can provide detailed chemical formula and structural information leading to comprehensive annotation. This approach is being widely employed in natural products chemistry,14,15 as well as in drug discovery,16 and in drug metabolism and metabolite discovery in the pharmaceutical industry.17 Recently, multitechnique approaches have been applied to both plant18,19 and bacterial20 metabolomics, while LC-MS has been used to assist with the structural elucidation of metabolites from NMR-based experiments.21
Traditionally in natural products chemistry, to generate the required concentration of metabolites pure enough to annotate via NMR spectroscopy preparative-scale liquid chromatography or flash chromatography systems are employed.22 However, these are often costly and are not available to analytical laboratories who routinely perform metabolic profiling studies using UHPLC-MS systems. The subsequent application of NMR spectroscopy to structurally elucidate features from LC-MS based metabolite profiling has its own challenges. NMR spectroscopic analyses do not have the sensitivity of LC-MS assays; therefore, large quantities (often in the order of milligrams) of relatively pure material isolated from biofluids, such as blood (serum and plasma) or urine, are required for structural elucidation.
Of the biofluids commonly studied in human phenotyping experiments, urine holds an advantage over blood products because of the noninvasive collection of large volumes (e.g., via routine 24 h collections which typically produce liters of biofluid per day and per person), making it an ideal starting material for purification and concentration for NMR structural elucidation. In addition, urine is a key biological matrix within metabolic profiling because it contains an extreme diversity of chemical classes, including gut microbial cometabolites and xenobiotics.23 Moreover, many metabolites present in blood are excreted and can be detected in urine23, and many metabolites circulating in blood may, therefore, be found in urine and recovered in larger amounts for further spectroscopic characterization.
At the same time, the urine matrix also poses chromatographic challenges because of its high inorganic salt content. In reversed-phase (RP) LC-MS analysis and fractionation, the presence of large salt amounts can cause various problems with both LC system and retention of the compounds, as well as with their MS detection. The salts from the urine samples can precipitate in the chromatographic column, and the amount of precipitate can increase with subsequent injections under the gradient conditions leading to a faster column degradation. Salts present in the sample or mobile phase can have also a strong influence on the solid–liquid equilibria that establish during the chromatographic run, especially in the case of ionized polar compounds.24 A high salt concentration can also impact the performance of NMR probes, decreasing the sensitivity and making it difficult to optimize experimental parameters.
To leverage the availability of human urine, its potential for use as a proxy matrix for systemic human and gut microbial metabolism, and the general availability of analytical scale instrumentation in metabolic phenotyping laboratories, we designed and implemented a novel urine-based pipeline solution that facilitates metabolite extraction, concentration, and purification using analytical scale systems coupled to a fractionation system. Prior to the primary fractionation, a urine pool underwent a desalting procedure using ion-exchange resin, which allowed for high-precision RPLC separation on an analytical scale system that demonstrated no degradation in column performance.
The resulting workflow enables purification and concentration of small molecule metabolites from urine that is easily implementable in analytical laboratories without the need for specialist purification LC systems. These purified metabolites can be subsequently decoupled from the pipeline and studied by a range of highly structural informative analytical techniques, such as such as Magnetic Resonance Mass Spectrometry (MRMS) or NMR. The efficacy and systematic deployment of the developed pipeline is demonstrated using a set of unknown metabolites of various biochemical classes and different physicochemical properties.
Materials and Methods
In-House MRC-NIHR National Phenome Centre Reversed-Phase Metabolite Profiling Method
Pooled bulk urine and all subsequent downstream fractions underwent RP profiling using the method developed in-house at the NIHR-MRC National Phenome Centre (NPC) at Imperial College London described in detail in ref (25).
To briefly summarize, RP urine profiling was completed using a Waters Acquity UPLC system coupled to a Waters Xevo G2 QToF mass spectrometer (Waters Corp., Milford, MA, USA). LC separation was conducted on a 2.1 × 150 mm HSS T3 column (Waters Corp., Milford, MA, USA) maintained at 45 °C. Mobile phase flow rate was of 0.6 mL/min. A gradient was applied consisting of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). Initial conditions of 99% A were held at isocratic conditions for 0.1 min, followed by a linear gradient elution, 99% A to 45% A, in 9.9 min, and a final rapid gradient phase, 45% A to 0% A in 0.7 min, prior to returning to initial conditions. Injection volume was 2 μL.
Mass spectrometry optimization is described in detail in ref (25). To briefly summarize, capillary voltage was set at 1.5 and 1.0 kV for positive and negative ionization, respectively. Cone voltage (20 V), source offset (80 V), StepWave 2 offset (10 V), and gas flows of 150 L/h for cone gas and 1000L/h for desolvation gas were consistent for both polarities.
Masslynx software (Waters, Manchester, U.K.) was used for data acquisition and visual inspection.
Pipeline for Systematic Biomarker Isolation and De Novo Structure Elucidation
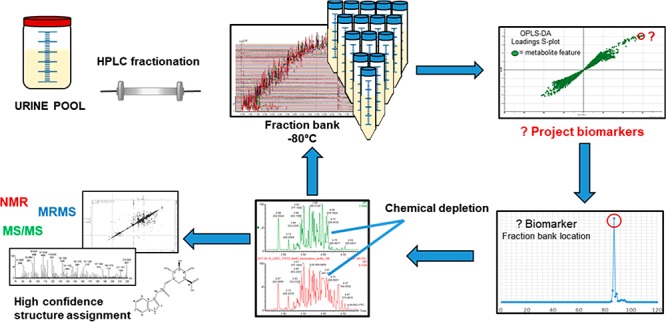
An overview of the pipeline is presented in Figure 1. First, urine from 6 volunteers was collected and pooled. The urine was split into two identical subpools. The first underwent desalting using an ion exchange protocol. The second was kept as a control. Both underwent 10-fold concentration by drying under a stream of nitrogen (Biotage Turbovap Classic), followed by primary RPLC fractionation creating both a desalted and a control fraction bank of 120 fractions. All stages underwent profiling using LC-MS so that the protocols could be compared. The desalted fraction bank, which had greater chromatographic reproducibility and long-term resolution, was utilized for downstream secondary and tertiary fractionation. The final fractions were then decoupled from the pipeline and sent to NMR spectroscopy and magnetic resonance mass spectrometry (MRMS)—traditionally known as Fourier transform ion cyclotron resonance mass spectrometry (FTICR MS), or simply Fourier transform mass spectrometry (FTMS) for annotation. In some cases, tertiary fractionation can be replaced with a computational purification by using statistical heterospectroscopy (SHY)27 in combination with NMR spectroscopy profiling.
Figure 1.

Workflow of the pipeline.
Pipeline Phase 1—Bulk Urine Preparation
Urine Collection
Individual urine was collected from a volunteer group of mixed genders (n = 6) and at multiple collection time points. No screening criteria were used to assess the health status of the donors. A total of 2 L was collected and pooled as described in ref (25). This was split into two one-liter identical aliquots. Pool one was used as an untreated control to assess the effect of the desalting protocol.
Urine Desalting
Urine pool 2 underwent a desalting process developed in-house using a two-step ion-exchange protocol. Step one used Amberlite IR120 cation exchanger (hydrogen form) (12 g per 500 mL of urine); pH readings were taken and confirmed acidic conditions resulting from the release of hydrogen ions from the ion-exchange resin. Step two consisted of the addition of 6 g of Amberlite IRN78 anion exchanger (hydroxide form) per 500 mL of urine from step 1, followed by pH reading and titration with additional Amberlite IRN78 until neutral pH 7 was reached (resulting from the release of hydroxide ions from the ion-exchange resin). The urine supernatant was removed at this point.
Urine Concentration
One liter of both control and desalted urine pools were then taken to dryness under a stream of nitrogen and subsequently resuspended in 100 mL of ultrahigh purity water (in-house Barnstead Diamond water purification system, Thermo Scientific, Waltham, MA, USA) to give a final 10-fold concentrated sample (10×). Both the control and desalted urine pools underwent metabolite profiling using the RPLC-MS method described above for comparison.
Pipeline Phase 2—Generation of the Fraction Bank
Primary Fractionation and Formation of the Fraction Bank
A bank of 120 fractions was created for each of the two concentrated urine pools using an identical analytical scale system consisting of a Waters Acquity UPLC system. RPLC-separation was achieved using a 4.6 mm × 150 mm Atlantis T3 column (Waters Corp., Milford, MA, USA). Postcolumn solvent effluent was split using a 1:20 post column splitter, with 1 part diverted to a Waters Xevo TQ-S mass spectrometer for real-time monitoring and 20 parts diverted to a Waters Fraction Collector III collection system.
Primary fractionation employed a linear gradient consisting of 0.1% formic acid in water (A) and 0.1% formic acid in methanol (B), which provided better resolution of late eluting metabolites compared to acetonitrile when using large injection volumes (1 mL). Mobile phase flow rate was 1 mL/min. Initial conditions of 99% A were held at isocratic conditions for 2.0 min, followed by a linear gradient elution of 99% A to 80% A in 13.0 min, which was followed by a linear gradient of 80% A to 50% A in 7.0 min, and then 50% A to 0% A in a further 4 min and isocratically held for a further 2 min at 0% A, prior to returning to initial conditions. To monitor the interinjection precision of the fractionation, full scan data were collected on a Waters Xevo TQ-S mass spectrometer, using single mass unit resolution. Data were collected in both positive ionization mode (100–1200 Da), and negative ionization mode (100–1200 Da). Masslynx software (Waters, Manchester, U.K.) was used for data acquisition and visual inspection.
One hundred replicates of 1 mL injection volume underwent LC separation. Individual fractions were collected for 15 s across the run creating a bank of 120 fractions. The LC-MS system was fully cleaned, and a new column was used for the fractionation of both control and desalted urine. The remaining fractions were stored at −80 °C in 50 mL Falcon tubes until use.
Fraction Bank Metabolite Mapping and Assessment of the Desalting Protocol
Subaliquots (100 μL) of each collected fraction (from both control and desalted urine) were transferred to 96-well plates and dried under a stream of nitrogen. Dried plates were then resuspended in ultra-high-purity water (in-house Barnstead Diamond water purification system, Thermo Scientific, Waltham, MA, USA) and analyzed using the RP method described above. Raw data underwent peak picking using Progenesis QI software (Nonlinear Dynamics, Newcastle upon Tyne, UK). This resulted in a “map” of metabolite locations within the fraction bank.
Pipeline Phase 3—Proof of Concept Metabolite Annotation
Proof of Concept Feature Selection
To demonstrate the applicability of the de novo annotation protocol, four target compounds were selected for annotation as proof of concept. The four features were selected from the reversed-phase profiling method to reflect a range of retention times and physicochemical properties of metabolites. Two of the features were selected from the same fraction from the fraction bank to demonstrate the requirement for secondary and tertiary isocratic fractionations. The fraction bank location matrix described above facilitates the selection of the fraction with the maximum concentration of the feature of interest. The fraction containing the highest concentration of the feature of interest was selected for downstream isocratic purification and is described in Table 1.
Table 1. Table of Four Initially Unannotated Metabolite Features Selected from RP LC-MS Urinary Metabolic Phenotyping Methods That Are Employed by the MRC-NIHR National Phenome Centre25a.
| feature | QTOF-MS observed m/z (ES+) | QTOF-MS observed m/z(ES−) | RT in RP UHPLC-MS (min) | fraction bank maximumb |
|---|---|---|---|---|
| A | 367.150 | 365.134 | 2.01 | 44 |
| B | 374.085 | 350.087 | 4.38 | 87 |
| C | NA | 283.082 | 4.29 | 87 |
| D | NA | 495.297 | 9.24 | 117 |
The metabolite features were selected because of their range of retention times in RP chromatography, suggesting different hydrophobicity, and therefore challenging the protocol. NA = no ion observed in respective polarity.
Fraction bank maximum refers to the fraction bank stored fraction (1–120) that contains the highest concentration of the feature of interest.
Secondary and Tertiary Isocratic Fractionation
Secondary and tertiary fractionation was completed using isocratic separation. To streamline the method development time, a reference set of conditions was created using a combination of different reversed-phase columns and eluents presented in Table S1.
These reference conditions were initially used to identify a developmental start point and modified accordingly to obtain maximum chromatographic resolution. This hybrid approach of using an in-house condition library combined with bespoke modification significantly reduced the overall chromatographic development time, while maintaining maximum isocratic chromatographic resolution of fractions.
The flow rate was 1 mL/min, and the column temperature was 35 °C for all instances for all chromatographic conditions. Full scan data (50–1200 Da) in both positive and negative ionization modes were collected on a Waters Xevo G2 QTOF-MS.
Following the secondary fractionation, the obtained subfractions underwent metabolite profiling using the in-house RP metabolite profiling analytical method.25 Subfractions containing the feature of interest were pooled and taken to dryness under a stream of nitrogen, before resuspension in a mobile phase (2.2 mL) that matched the isocratic conditions of tertiary fractionation. Purified features of interest were pooled for downstream decoupled analysis including NMR and MRMS, assisting de novo structural elucidation.
MS/MS Analysis of Concentrated Purified Samples
MS/MS fragmentation analysis was completed with a Waters Acquity UPLC system coupled to a Waters Xevo G2 QToF mass spectrometer (Waters Corp., Milford, MA, USA). The chromatographic method used the same conditions as the in-house RP profiling method25 with the same mass spectrometry source conditions. MS/MS target selection was performed using unit mass selection via the quadrupole with varying collision energy (5, 10, 20, 30, and 40 V) or a collision energy voltage ramp was employed in the collision cell to facilitate fragmentation (10–45 V).
MRMS Analysis
Magnetic resonance mass spectra (MRMS) were acquired with a Bruker solariX 2xR (Bruker Daltonics, Billerica, MA, US) using electrospray ionization (ESI) and direct infusion with syringe pump. Mass spectra were acquired with a mass resolution of 1.350.000 at m/z 200 using quadrupolar detection. Sixty-four single scans were added for the final mass spectrum (the details are presented in Supporting Information (SI)). Isotopic fine structure calculation was performed with DataAnalysis software (Bruker Daltonics, Billerica, MA, US).
NMR Analysis
The dried fractions of the purified metabolites were resuspended in 150 μL of LC-MS grade water or the mixture of D2O 99% D (Sigma-Aldrich) containing 10% H2O to avoid the disappearance of any NMR signals due to the exchange with the solvent; 144 μL of this solution was transferred into an Eppendorf tube together with 16 μL of D2O containing 1 mg/mL of trimethylsilylpropanoic acid (TSP) (Sigma-Aldrich). The solution was vortexed for 1 min and then centrifuged at 12000g for 5 min at 4 °C; 155 μL of this solution was transferred into a 3 mm NMR tube. This protocol was adapted from the publication of Dona et al.26 using D2O with TSP instead of the phosphate buffer solution as there was no need to control pH of the purified samples.
1H NMR spectra were acquired using either a 600 MHz Bruker Avance III HD spectrometer equipped with a BBI room temperature probe, z-gradients, and high-order shims or a 600 MHz Bruker Avance III spectrometer equipped with a Cryo-TCI triple resonance CryoProbe. A pulse sequence with two presaturation periods to deplete water signal was used to obtain the 1D data. Standard Bruker IVDr methods were used for fraction profiling by NMR26 of the purified metabolites or their subfractions. 1H NMR general profile was acquired with a higher number of scans to increase the signal-to-noise ratio when required due to low concentration. Other 1H NMR parameters were kept as previously described.26 A range of 2D NMR spectroscopy experiments including J-resolved, 1H,1H–COSY, 1H,1H-TOCSY, 2D 1H,1H-NOESY, 1H,13C-HSQC, and 1H,13C-HMBC were performed for structural elucidation of the purified metabolites (the details are presented in SI).
Data Processing and Analysis
Peak picking of raw LC-MS data was completed using Progenesis QI software (Nonlinear Dynamics, Newcastle upon Tyne, UK). This provided a matrix of features within each fraction and was utilized as a feature reference location map to facilitate the identification of the fraction of maximum concentration for each feature of interest.
The subfractions obtained from isocratic purification of the metabolite of interest were profiled by 1H NMR using the protocol described by Dona et al. for human urine samples.26 Fourier transform, phasing, baseline correction, and calibration were done after acquisition using TopSpin 3.5 (Bruker BioSpin GmbH, Rheinstetten, Germany) and then transferred to MATLAB R2016a (Mathworks) as a matrix of intensities for each part per million value of the spectra. All peaks detected in the UHPLC-MS profiles of the subfractions of the metabolites of interest were integrated using TargetLynx software (Waters, Manchester, U.K.) and transferred as a matrix of peak areas to MATLAB R2016a (Mathworks). These two matrices were combined to perform statistical spectroscopic analysis (SHY).27 The MS vector of a peak area was correlated to the full-scan NMR spectra using Spearman correlation to infer the linkage of the MS signals and the NMR peaks of a metabolite.
Principal component analysis (PCA) was performed in SIMCA 14.1 (Umetrics, Sweden).
Results and Discussion
Pipeline Phase 1—Bulk Urine Preparation
The pipeline uses an initial one-liter volume of urine to create the fraction bank. The need for a large volume is a potential limitation of the pipeline as samples from clinical studies are often volume-limited. However, many clinically relevant metabolites (e.g., disease diagnostics or prognostics) are also typically present in the biofluids of healthy individuals. Therefore, the generation of the fraction bank can be performed with urine collected from volunteer populations or purchased from commercial suppliers. In this regard urine is an ideal biofluid for the pipeline due to the possibility for the noninvasive collection of large volumes (e.g., via routine 24h collections which typically produce liters of biofluid per day and per person). Where key biomarker metabolites are disease or subpopulation specific, the pipeline can be applied to urine obtained from those individuals or subgroups.
Assessment of Desalting Urine
Urine is characterized by the high content of inorganic salts, and their increased amount in a preconcentrated urine pool in reversed-phase (RP) LC-MS analysis can cause various problems with both the LC system and retention of the compounds as well as with their MS and NMR detections. Prior to fractionation a urine pool underwent a desalting procedure using ion-exchange resin (Figure 1).
The effect the desalting step caused on the urine profile by MS is displayed in Figure S1 as heat map plots of each fraction (number 1–120) against metabolite features ordered by RP retention time. Panel A shows the untreated control urine where fractions 1–25 (green circled) appear as a wide band suggesting poor chromatographic resolution and greater breakthrough of metabolites through lack of retention. The authors believe this to be as a result of increased salt content having a detrimental effect on the chromatographic column. Metabolite features are also visibly distributed across more fractions in control urine (represented by wider bands spanning more fractions in the heat map). On the other hand, the desalted urine heat map (Figure S1a) shows a more linear distribution, while the control urine heat map has a sigmoidal shape, demonstrating a more even distribution of metabolites in the fractionation of the desalted urine.
When operating at analytical scale the number of repeat injections has to be increased to obtain the concentrations required, therefore precision of replication is critical. To further assess the fractionation quality and stability we used principal component analysis (PCA) that should reflect in the scores plot similarities in unit mass resolution profiles acquired for each 1 mL injection (Figure S2). The scores for all 100 profiles of desalted 10× urine are tightly clustered together in comparison to the scores of 100 profiles of untreated 10× urine broadly spread across the PC2. This highlights how fractions coming from desalted concentrated urine present more stable retention times for metabolites, increasing replicate precision and allowing higher column loading, resulting in a more efficient purification process. The presented desalting approach is opposite to a traditional use of ion-exchange columns for the extraction of charged molecules of polar metabolites from biological samples with their subsequent removal from the resin using acid or base solution and analysis.25 Therefore, it is unquestionable that along with the salts the protocol decreases the concentration of some of the most polar charged metabolites altering the overall metabolic profile of the desalted urine when compared to the original one. However, the overall benefit to increasing chromatographic precision was felt by the authors to outweigh the loss of some metabolite material. To date, no feature of interest that has been through the protocol for annotation has been lost by the desalting protocol.
Pipeline Phase 2—Generation of the Fraction Bank
Primary RP (RP) fractionation of desalted urine resulted in the collection of 120 fractions which formed a fraction bank repository stored at −80 °C. Each fraction was profiled using the in-house RP phenotyping method in both positive and negative ionization modes. A feature map was created for each fraction in the bank enabling identification of those fractions containing features of interest. When these were found to reside in multiple fractions, the mapping enabled selection of the fraction with their greatest concentration for downstream isocratic analysis.
To streamline the collection and initial mapping process, all eluent was recollected for subsequent isocratic fractionations. Downstream fractions that contained the feature of interest were collected and progressed to further purification and analysis, while the remaining “waste” eluent was dried, resuspended in water, and returned to the −80 °C fraction bank (Figure S3).
While drying and transfer loss inevitably occurs, the general metabolic profile is kept across the chromatogram. This novel fraction bank approach extends the lifetime of each urine pool and streamlines the overall pipeline enabling faster annotation without the need of a new urine collection for subsequent feature assignments.
Selection of Biomarkers for the Proof-of-Concept
To demonstrate the protocol and as proof of concept, four metabolite features were selected for de novo annotation. Selected features represented a variation of retention times from the in-house RP metabolic phenotyping assay (Table 1). MS/MS fragmentation data suggested some structural properties of the metabolites.
Feature A eluted at 2.01 min on the phenotyping assay and was selected because it is a relatively polar species. MS/MS fragmentation had suggested that the feature was not a phase II metabolite (e.g., a glucuronide or sulfate), and therefore would produce a different challenge to the protocol than B, C, and D.
MS/MS fragmentation indicated that Feature B (retention time of 4.38 min) and D (retention time of 9.24 min) were both glucuronides; however, a retention time separation of 4.86 min, suggested different structural families. Feature B was selected as it eluted closely to two isomers and would therefore be an appropriate challenge for the protocol.
Feature C eluted in the same fraction as B, and it was selected to demonstrate the requirement of secondary and tertiary fractionations in relation to Feature B, and the beneficial implementation of statistical heterospectroscopy in the protocol.
Secondary and tertiary conditions were selected for each of the metabolite features of interest based on the generation of the in-house isocratic reference conditions (Table S1). Two complementary sets of separation conditions were chosen that best isolated each of the features of interest. This work flow was developed to streamline the pipeline and create a “walk-up” design, which greatly reduces the time required for each feature purification as time-consuming customizable method development is bypassed.
Structure Elucidation of Urinary Biomarkers—Feature A
Feature A (Table 1) was the most polar of the selected metabolites eluting at 2.01 min in the RP chromatography metabolic profiling method described. Its purification using analytical scale RP chromatography required the highest percentage of the aqueous phase A, employing 90% in the secondary fractionation and 99% in the tertiary fractionation (Table S1).
Initial HRMS data collected by QTOF-MS generated the molecular formula C17H22N2O7 (Waters i-FIT elemental composition calculator). The search of candidate structures in Pubchem28 returned 605 theoretical candidates while HMDB yielded two theoretical metabolites tetrahydropentoxyline and semilepidinoside B. QTOF MS/MS analysis performed on starting urine material (Figure S4) gave the main fragment ion at m/z 245.094 indicating loss of 120 Da characteristic of C-glycosides.29 Tetrahydropentoxyline is a C-glycoside while semilepidinoside is an O-glycoside therefore tetrahydropentoxyline became the most likely candidate. Additionally, tetrahydropentoxyline has been previously reported as a metabolite excreted in urine of normal subjects.30
Feature A was found to be at a maximum concentration in fraction bank 44. This was used for secondary and tertiary purification using the conditions in Table S1. Following this, a final purified collection of feature A was sent for downstream NMR and MRMS analysis.
MRMS (Figure S5) confirmed the elemental composition C17H22N2O7, while complete structural elucidation of feature A was then performed by NMR spectroscopy. The result of which was consistent with previously reported data.30
The full assignment of the structure of teterahydropentoxyline purified in this work (Figure 2) is presented in the Table S2 together with the 2D NMR data Figures S6 and S7.
Figure 2.

Structures of the UHPLC-MS features A–D purified from urine using the proposed pipeline and characterized by MS, NMR, and MRMS spectroscopic analyses.
Structure Elucidation of Urinary Biomarkers—Feature B
Feature B was observed in RP metabolite profiling in both positive and negative polarities (ES+ and ES−) eluting at 4.37 min. In ES+, the sodium adduct ion was dominant and the deprotonated ion was dominant in ES– 374.085 m/z and 350.088 m/z, respectively. The tube location in the fraction bank that contained the highest amount of feature B was identified as 87; this was used for secondary and tertiary isocratic purification.
Feature B was selected for a proof of concept as it presented a number of significant challenges, including chromatographic elution with nearby peaks of an identical m/z, and chromatographic coelution with a peak of high intensity and concentration.
Feature B Challenge 1: Chromatographic Coelution
The first challenge that Feature B presented to the pipeline was that it existed in a complicated space in the chromatographic run (Figure S8). Feature B directly coeluted with a feature of high intensity (m/z 187.007), depicted with a dashed line on the chromatogram in Figure S8. The coelutant was known to be p-cresol sulfate, and is a major urinary metabolite of gut microbial origin in metabolic profiling known to exist at high concentrations. The coelutant was successfully removed in the secondary fractionation. This demonstrates the successful application of multiple rounds of purification in a complex mixture such as urine, enabling the physical purification of coeluting features of interest, which can be difficult to purify in a single step.
Feature B also presented chromatographic elution alongside nearby peaks of an identical m/z. This can be observed from the ES– chromatogram presented in the Figure S8.
First, it had to be determined if feature B was an individual metabolite feature or if it was a split peak on the chromatographic profile. This was achieved using QTOF-MS/MS collision energy dependent analysis (Figure S9a and S9b). By repeating QTOF-MS/MS fragmentation with 5 V step intervals of fixed collision energy relative intensity of each fragment was plotted (Figure S9c). Differences in the relative fragment intensity at varying collision energies suggested each chromatographic peak was represented by compounds with a different structural arrangement and therefore differing bond energies and hence fragment ion ratios. Therefore, feature B was an ideal candidate to examine if the pipeline could be used to purify and annotate features that had closely eluting isomers that would share similarities in chemical structure and therefore behave similarly chromatographically.
Initially, QTOF-MS was used to generate a high resolution accurate mass and therefore predict the elemental composition of the feature (C16H17NO8). QTOF-MS/MS had ES– fragment ions indicative of the presence of a glucuronide moiety (fragment ions 175.024 m/z and 113.024 m/z). Through database searching (Pubchem,28 HMDB,2 Metlin,1 and ChEBI31) yielded a favored candidate, indole-3-acetic-acid-O-glucuronide.
Following a successful two-step isocratic fractionation, a purified sample of Fraction B was achieved and was then decoupled from the pipeline and progressed to structural annotation via 1H NMR spectroscopy and MRMS (Figure S10).
Feature B Challenge 2: Stability during NMR Analysis
Initially, the NMR analysis of the extracted features was completed by resuspending the purified Feature B into a phosphate buffer solution used for the NMR profiling of urine samples.26 However, it was observed that the metabolite was unstable in the buffer solution during the 2D NMR acquisition experiments.
The 1H NMR spectra confirmed that the feature of interest was transforming over time to the isomeric species i and ii (Figure S11). This irreversible transformation was also confirmed by UHPLC-MS analysis of the NMR solution of the metabolite of interest (Figure S11).
The ability of acyl glucuronides to undergo spontaneous hydrolysis and intramolecular rearrangements is well documented.32,33 It has been previously reported that the glycosidic ester bond can migrate from position 1 to positions 2, 3, and 4 of glucuronic acid, and the rearrangement takes place in phosphate buffer solution at pH 8.0.33 The first step of the transformation from position 1 to position 2 is irreversible followed by further reversible steps.33 In addition, the double UHPLC peaks for the isomers is thought to result from the simultaneous presence of both α and β anomers of glucuronic acid, which can also explain more complex shape of the 1H NMR peaks of the isomers compared to the NMR signals of the pure metabolite of interest (Figure S11).
To avoid the instability of the metabolite of interest in solution, the feature was repurified and the NMR spectroscopic analyses were run in D2O containing only TSP as a reference signal. In this way, the feature was stable in solution for the whole duration of the 2D NMR characterization including 1H–1H COSY and 1H–1H TOCSY experiments to detect intercorrelations between the protons, and 1H–13C HSQC and 1H–13C HMBC experiments to detect the intercorrelations between protons and carbons in the molecule. The metabolite of interest, Feature B, was annotated as indole-3-acetic-acid-O-alpha-glucuronide. The full assignment is shown in the Table S3, and 2D NMR spectra for the metabolite of interest are presented in Figures S12 and S13.
Structure Elucidation of Urinary Biomarkers—Feature C
To annotate feature B, a two-step physical purification was required, however the pipeline was also tested to see if a computation purification step through statistical heterospectroscopy (SHY)27 could be implemented in the annotation of some molecular species. Feature C was an unknown metabolite at the time of purification with an m/z of 283.082 that was also present in fraction bank 87 along with feature B.
Initially, the elemental composition of feature C was predicted using high resolution QTOF-MS data on the primary negative ion (283.082 m/z) which was identified as C13H16O7. MS/MS analysis (ES−) of feature C was consistent with the presence of glucuronide moiety—the loss of 176 Da and the detection of characteristic fragments of glucuronic acid (Figure S14a). The feature also shared similar fragment ions with p-cresol sulfate (107.05 m/z) that suggested p-cresol within the structure. Database searching (Pubchem,28 HMDB,2 Metlin,1 and ChEBI31) suggested p-cresol glucuronide as a potential candidate.
A lack of purity meant it was not possible to immediately annotate the unknown from NMR spectra. To overcome this, several subfractions were collected with a distribution of the feature over five subfractions. These were analyzed by UHPLC-QTOF-MS and 1H NMR spectroscopy. The integrals of the UHPLC-QTOF-MS signals of the unknown in all subfractions were obtained from the profiles acquired in negative ionization mode (m/z 283.082) and were correlated to the full scan 1H NMR spectra using SHY (see Methods). This enabled the identification of the NMR spectral peaks that were specifically associated with Feature C (Figure S14b).
The resultant 1H NMR spectrum was annotated and found to have similarities with the NMR pattern of p-cresol sulfate: two doublets in aromatic region at 7.06 (d) and 7.23 (d) ppm (Figure 2—signals 3,5 and signals 2,6, respectively) indicating p-substitution in aromatic ring (Figure S14c.i); the intense singlet at 2.30 (s) ppm for the methyl group of cresol (Figures S14c.ii and 2, signal 7), and the signals in the sugar region 3.4–4 ppm and a doublet at 5.07 ppm correspond to glucuronic moiety (Figures S14c.iii and 2, signals 1′–5′). Signals in the 1H NMR were integrated to confirm the number of hydrogens in each of the chemical groups. These MS/MS and NMR data together confirm the annotation of feature C as p-cresol glucuronide (Figure 2) that has been previously annotated in animal urine.34,35
This example together with p-cresol sulfate show that the pipeline is capable to readily extract urinary metabolites of gut-microbial origin for which the authentic standards can be rarely available. The combination of high-resolution MS and NMR spectroscopic analyses together with statistical spectroscopic methods provide a fast way for annotating these metabolites.
Structure Elucidation of Urinary Biomarkers—Feature D
Feature D was selected to test the pipeline as it was a late eluting feature in RP chromatography. The feature was readily purified under the RP chromatographic conditions that were rapidly generated from the in-house isocratic database described in the methods (Table S1) because of its high retention time in the initial RP UHPLC-MS profiling (9.23 min), and therefore, higher hydrophobicity compared to the other three features reported in this study. Both HRMS QTOF-MS (Figure S15) and MRMS (Figure S16) analyses generated the molecular formula as C27H44O8.
Following database searching (Pubchem,28 HMDB,2 Metlin,1 and ChEBI31), a potential candidate was suggested as pregnanediol-3-glucuronide. The presence of glucuronide moiety was confirmed by QTOF MS/MS analysis (Figure S15) through the loss of 176 Da and detection of characteristic fragment of glucuronic acid in negative polarity. In this instance, the standard of pregnanediol-3-glucuronide was available for purchase, and it underwent analysis by UHPLC-MS/MS and 1D 1H NMR spectroscopy along with the purified metabolite, which confirmed the identity of feature D (Figure 2). The NMR spectroscopic characterization of pregnanediol-3-glucuronide and the full assignment was performed. The spectra and the data are shown in SI (Figures S17 and S18 and Table S4). Although pregnanediol-3-glucuronide has previously been reported as a biomarker in NMR metabolomics,36 this is the first time that the full NMR assignment of pregnanediol-3-glucuronide has been detailed in the literature.
Conclusions
Here, we present a systematic pipeline designed to structurally elucidate and increase the annotation confidence of unknowns in urine metabolic profiling. These biomarkers are features of interest in clinical/epidemiological studies and their structural elucidation is key for the biological interpretation of the results. By implementation of a novel ion exchange approach to desalt urine, high precision multiple repeat fractionation can be implemented using analytical scale chromatography instrumentation typical of a metabolite profiling laboratory, without purchasing specialist preparative scale equipment. Minimal column degradation was observed after multiple repeat injections of one milliliter of concentrated urine, providing a significant benefit to nondesalted urine.
The systematic pipeline results in the initial formation of a reusable fraction bank that can become a laboratory resource and revisited without time-consuming bulk urine preparation. Downstream purification can be done either through physical secondary and tertiary chromatographic fractionation, or by using computational purification, via statistical heterospectroscopy in combination with NMR. To streamline the secondary and tertiary chromatographic steps, an in-house reference library of reversed-phase conditions was used to select a development start point that enabled rapid optimization to obtain maximum chromatographic resolution.
The proposed fraction bank workflow is advantageous as purified metabolites can be decoupled from the pipeline at each stage and can be sent to a combination of downstream analytical technologies, including MRMS and NMR spectroscopy for increased confidence in structural elucidation. The pipeline is particularly relevant where analytical standards cannot be purchased, for example with biotransformed metabolites, such as glucuronides, and an increase in confidence is required.
A limitation of the pipeline is the length of time required for complete purification and structural elucidation of unknowns. Therefore, to help reduce this time window, a library of isocratic conditions was created to help enable the rapid selection of secondary and tertiary separation conditions. However, despite this, overall pipeline time scales remain constrained by the complexity of the NMR spectroscopic data and the time taken for interpretation. For instance, in the present work the structural elucidation of the feature C (p-cresol glucuronide) took just several days while the full structure assignment by NMR of the Feature D (pregnanediol-3-glucuronide), which was not previously reported in the literature and online databases, required several weeks due to the highly complex NMR spectra. Therefore, the pipeline is most suitable for scenarios where structural elucidation is critical for the understanding and interpretation of the biology behind key statistically significant metabolites that would otherwise remain unannotated and unknown.
Acknowledgments
The MRC-NIHR National Phenome Centre is supported by the UK Medical Research Council [in association with National Institute for Health Research (England)] Grant MC_PC_12025. The Clinical Phenotyping Centre (CPC) was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre (BRC) based at Imperial College Healthcare NHS Trust and Imperial College London. The UK Dementia Research Institute (DRI) is an initiative funded by the Medical Council, Alzheimer’s Society and Alzheimer’s Research UK. The authors would like to thank Dr Jose Ivan Serrano Contreras for providing advice and assistance in the acquisition of some NMR data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.9b00241.
Secondary and tertiary isocratic fractionation, MRMS analysis, and NMR analysis; chromatographic conditions used for the secondary and tertiary isocratic separations of each features of interest; NMR signal assignment of features A, B, and D; photographs and metabolite feature heat maps of desalted urine and nondesalted control urine; principal component analysis scores plot comparing LC-MS profiles of repeat injections of desalted and untreated urine; fraction bank protocol cycle; MS/MS spectra of feature A; ES+ MRMS with an isotopic fine structure confirmation of elemental composition (C17H23N2O7) of purified feature A; 1H–1H COSY NMR spectrum of feature A; 1H–13C HMBC NMR spectrum of feature A; extracted ion chromatograms of the m/z 350.088 and the coeluting feature m/z 187.007; extracted ion chromatograms and MS/MS spectra of features with m/z 350.088 (ES−); ES– MRMS with an isotopic fine structure confirmation of elemental composition (C16H16NO8) of purified feature B; evidence of the degradation of purified feature B when stored in phosphate buffer; 1H–1H COSY NMR spectrum of feature B; the 1H–13C HMBC NMR spectrum of feature B; negative MS/MS spectrum and statistical heterospectroscopy analysis of feature C; EIC chromatogram (ES−) comparing urine pool with analytical standard of pregnanediol-3-glucuronide; ES– MRMS with an isotopic fine structure confirmation of elemental composition (C27H43O8) of purified feature C; 1H–1H COSY NMR spectrum of feature D; and 1H–13C HMBC NMR spectrum of feature D (PDF)
Author Present Address
⊥ J.N.: Australian National Phenome Centre, Murdoch University, Harry Perkins Building, Perth, Western Australia 6150, Australia.
Author Contributions
# L.W. and E.C. contributed equally.
The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
Human samples (used in generation of the urine pool long-term reference matrix) used in this research project were obtained from the Imperial College Healthcare Tissue Bank (ICHTB). ICHTB is supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Imperial College Healthcare NHS Trust and Imperial College London. ICHTB is approved by NRES to release human material for research (12/WA/0196), and the samples for this project (R13053) were issued from subcollection reference number IRD-ML-13-030.
The authors declare no competing financial interest.
Supplementary Material
References
- Guijas C.; Montenegro-Burke J. R.; Domingo-Almenara X.; Palermo A.; Warth B.; Hermann G.; Koellensperger G.; Huan T.; Uritboonthai W.; Aisporna A. E.; Wolan D. W.; Spilker M. E.; Benton H. P.; Siuzdak G. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90 (5), 3156–3164. 10.1021/acs.analchem.7b04424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Feunang Y. D.; Marcu A.; Guo A. C.; Liang K.; Vázquez-Fresno R.; Sajed T.; Johnson D.; Li C.; Karu N.; Sayeeda Z.; Lo E.; Assempour N.; Berjanskii M.; Singhal S.; Arndt D.; Liang Y.; Badran H.; Grant J.; Serra-Cayuela A.; Liu Y.; Mandal R.; Neveu V.; Pon A.; Knox C.; Wilson M.; Manach C.; Scalbert A. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 2018, 46 (D1), D608–D617. 10.1093/nar/gkx1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horai H.; Arita M.; Kanaya S.; Nihei Y.; Ikeda T.; Suwa K.; Ojima Y.; Tanaka K.; Tanaka S.; Aoshima K.; Oda Y.; Kakazu Y.; Kusano M.; Tohge T.; Matsuda F.; Sawada Y.; Hirai M. Y.; Nakanishi H.; Ikeda K.; Akimoto N.; Maoka T.; Takahashi H.; Ara T.; Sakurai N.; Suzuki H.; Shibata D.; Neumann S.; Iida T.; Tanaka K.; Funatsu K.; Matsuura F.; Soga T.; Taguchi R.; Saito K.; Nishioka T. MassBank: a public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45 (7), 703–714. 10.1002/jms.1777. [DOI] [PubMed] [Google Scholar]
- Domingo-Almenara X.; Montenegro-Burke J. R.; Benton H. P.; Siuzdak G. Annotation: A Computational Solution for Streamlining Metabolomics Analysis. Anal. Chem. 2018, 90 (1), 480–489. 10.1021/acs.analchem.7b03929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruttkies C.; Schymanski E. L.; Wolf S.; Hollender J.; Neumann S. MetFrag relaunched: incorporating strategies beyond in silico fragmentation. J. Cheminf. 2016, 8 (1), 3. 10.1186/s13321-016-0115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böcker S. Searching molecular structure databases using tandem MS data: are we there yet?. Curr. Opin. Chem. Biol. 2017, 36, 1–6. 10.1016/j.cbpa.2016.12.010. [DOI] [PubMed] [Google Scholar]
- Allen F.; Pon A.; Wilson M.; Greiner R.; Wishart D. CFM-ID: a web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014, 42 (W1), W94–W99. 10.1093/nar/gku436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dührkop K.; Shen H.; Meusel M.; Rousu J.; Böcker S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (41), 12580–12585. 10.1073/pnas.1509788112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliszan R. QSRR: Quantitative Structure-(Chromatographic) Retention Relationships. Chem. Rev. 2007, 107 (7), 3212–3246. 10.1021/cr068412z. [DOI] [PubMed] [Google Scholar]
- Miller T. H.; Musenga A.; Cowan D. A.; Barron L. P. Prediction of Chromatographic Retention Time in High-Resolution Anti-Doping Screening Data Using Artificial Neural Networks. Anal. Chem. 2013, 85 (21), 10330–10337. 10.1021/ac4024878. [DOI] [PubMed] [Google Scholar]
- van der Hooft J. J. J.; Wandy J.; Barrett M. P.; Burgess K. E. V.; Rogers S. Topic modeling for untargeted substructure exploration in metabolomics. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (48), 13738–13743. 10.1073/pnas.1608041113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dona A. C.; Kyriakides M.; Scott F.; Shephard E. A.; Varshavi D.; Veselkov K.; Everett J. R. A guide to the identification of metabolites in NMR-based metabonomics/metabolomics experiments. Comput. Struct. Biotechnol. J. 2016, 14, 135–153. 10.1016/j.csbj.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn W. B.; Erban A.; Weber R. J. M.; Creek D. J.; Brown M.; Breitling R.; Hankemeier T.; Goodacre R.; Neumann S.; Kopka J.; Viant M. R. Mass appeal: metabolite identification in mass spectrometry-focused untargeted metabolomics. Metabolomics 2013, 9 (1), 44–66. 10.1007/s11306-012-0434-4. [DOI] [Google Scholar]
- Zani C. L.; Carroll A. R. Database for Rapid Dereplication of Known Natural Products Using Data from MS and Fast NMR Experiments. J. Nat. Prod. 2017, 80 (6), 1758–1766. 10.1021/acs.jnatprod.6b01093. [DOI] [PubMed] [Google Scholar]
- Wolfender J.-L.; Nuzillard J.-M.; van der Hooft J. J. J.; Renault J.-H.; Bertrand S. Accelerating Metabolite Identification in Natural Product Research: Toward an Ideal Combination of Liquid Chromatography–High-Resolution Tandem Mass Spectrometry and NMR Profiling, in Silico Databases, and Chemometrics. Anal. Chem. 2019, 91 (1), 704–742. 10.1021/acs.analchem.8b05112. [DOI] [PubMed] [Google Scholar]
- Koehn F. E.; Carter G. T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discovery 2005, 4, 206. 10.1038/nrd1657. [DOI] [PubMed] [Google Scholar]
- Dear G. J.; Ayrton J.; Plumb R.; Fraser I. J. The rapid identification of drug metabolites using capillary liquid chromatography coupled to an ion trap mass spectrometer. Rapid Commun. Mass Spectrom. 1999, 13 (5), 456–463. . [DOI] [PubMed] [Google Scholar]
- Boiteau M. R.; Hoyt W. D.; Nicora D. C.; Kinmonth-Schultz A. H.; Ward K. J.; Bingol K. Structure Elucidation of Unknown Metabolites in Metabolomics by Combined NMR and MS/MS Prediction. Metabolites 2018, 8, 8. 10.3390/metabo8010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Z.; Jing L.; Qiu F.; Zhang H.; Huhman D.; Zhou Z.; Sumner L. W. Construction of an Ultrahigh Pressure Liquid Chromatography-Tandem Mass Spectral Library of Plant Natural Products and Comparative Spectral Analyses. Anal. Chem. 2015, 87 (14), 7373–7381. 10.1021/acs.analchem.5b01559. [DOI] [PubMed] [Google Scholar]
- Thomas M.; Stuani L.; Darii E.; Lechaplais C.; Pateau E.; Tabet J.-C.; Salanoubat M.; Saaidi P.-L.; Perret A. De novo structure determination of 3-((3-aminopropyl)amino)-4-hydroxybenzoic acid, a novel and abundant metabolite in Acinetobacter baylyi ADP1. Metabolomics 2019, 15 (3), 45. 10.1007/s11306-019-1508-3. [DOI] [PubMed] [Google Scholar]
- Posma J. M.; Garcia-Perez I.; Heaton J. C.; Burdisso P.; Mathers J. C.; Draper J.; Lewis M.; Lindon J. C.; Frost G.; Holmes E.; Nicholson J. K. Integrated Analytical and Statistical Two-Dimensional Spectroscopy Strategy for Metabolite Identification: Application to Dietary Biomarkers. Anal. Chem. 2017, 89 (6), 3300–3309. 10.1021/acs.analchem.6b03324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli G. F.; Chen S.-N.; Friesen J. B.; McAlpine J. B.; Jaki B. U. Analysis and Purification of Bioactive Natural Products: The AnaPurNa Study. J. Nat. Prod. 2012, 75 (6), 1243–1255. 10.1021/np300066q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouatra S.; Aziat F.; Mandal R.; Guo A. C.; Wilson M. R.; Knox C.; Bjorndahl T. C.; Krishnamurthy R.; Saleem F.; Liu P.; Dame Z. T.; Poelzer J.; Huynh J.; Yallou F. S.; Psychogios N.; Dong E.; Bogumil R.; Roehring C.; Wishart D. S. The Human Urine Metabolome. PLoS One 2013, 8 (9), e73076. 10.1371/journal.pone.0073076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritti F.; Guiochon G. Effect of the ionic strength of salts on retention and overloading behavior of ionizable compounds in reversed-phase liquid chromatography: I. XTerra-C18. J. Chromatogr. A 2004, 1033 (1), 43–55. 10.1016/j.chroma.2004.01.027. [DOI] [PubMed] [Google Scholar]
- Lewis M. R.; Pearce J. T. M.; Spagou K.; Green M.; Dona A. C.; Yuen A. H. Y.; David M.; Berry D. J.; Chappell K.; Horneffer-van der Sluis V.; Shaw R.; Lovestone S.; Elliott P.; Shockcor J.; Lindon J. C.; Cloarec O.; Takats Z.; Holmes E.; Nicholson J. K. Development and application of ultra-performance liquid chromatography-TOF MS for precision large scale urinary metabolic phenotyping. Anal. Chem. 2016, 88 (18), 9004–9013. 10.1021/acs.analchem.6b01481. [DOI] [PubMed] [Google Scholar]
- Dona A. C.; Jiménez B.; Schäfer H.; Humpfer E.; Spraul M.; Lewis M. R.; Pearce J. T. M.; Holmes E.; Lindon J. C.; Nicholson J. K. Precision High-Throughput Proton NMR Spectroscopy of Human Urine, Serum, and Plasma for Large-Scale Metabolic Phenotyping. Anal. Chem. 2014, 86 (19), 9887–9894. 10.1021/ac5025039. [DOI] [PubMed] [Google Scholar]
- Crockford D. J.; Holmes E.; Lindon J. C.; Plumb R. S.; Zirah S.; Bruce S. J.; Rainville P.; Stumpf C. L.; Nicholson J. K. Statistical Heterospectroscopy, an Approach to the Integrated Analysis of NMR and UPLC-MS Data Sets: Application in Metabonomic Toxicology Studies. Anal. Chem. 2006, 78 (2), 363–371. 10.1021/ac051444m. [DOI] [PubMed] [Google Scholar]
- Kim S.; Thiessen P. A.; Bolton E. E.; Chen J.; Fu G.; Gindulyte A.; Han L.; He J.; He S.; Shoemaker B. A.; Wang J.; Yu B.; Zhang J.; Bryant S. H. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44 (D1), D1202–D1213. 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabryelski W.; Froese K. L. Rapid and sensitive differentiation of anomers, linkage, and position isomers of disaccharides using High-Field Asymmetric Waveform Ion Mobility Spectrometry (FAIMS). J. Am. Soc. Mass Spectrom. 2003, 14 (3), 265–277. 10.1016/S1044-0305(03)00002-3. [DOI] [PubMed] [Google Scholar]
- Horiuchi K.; Yonekawa O.; Iwahara K.; Kanno T.; Kurihara T.; Fujise Y. A hydrophilic tetrahydro-beta-carboline in human urine. J. Biochem. 1994, 115 (2), 362–366. 10.1093/oxfordjournals.jbchem.a124343. [DOI] [PubMed] [Google Scholar]
- Hastings J.; Owen G.; Dekker A.; Ennis M.; Kale N.; Muthukrishnan V.; Turner S.; Swainston N.; Mendes P.; Steinbeck C. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 2016, 44 (D1), D1214–1219. 10.1093/nar/gkv1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan S. L.; Maggs J. L.; Hammond T. G.; Lambert C.; Williams D. P.; Park B. K. Acyl glucuronides: the good, the bad and the ugly. Biopharm. Drug Dispos. 2010, 31 (7), 367–395. 10.1002/bdd.720. [DOI] [PubMed] [Google Scholar]
- Bradow G.; Kan L. S.; Fenselau C. Studies of intramolecular rearrangements of acyl-linked glucuronides using salicyclic acid, flufenamic acid, and (S)- and (R)-benoxaprofrn and confirmation of isomerization in acyl-linked.DELTA.9–11-carboxytetrahydrocannabinol glucuronide. Chem. Res. Toxicol. 1989, 2 (5), 316–324. 10.1021/tx00011a009. [DOI] [PubMed] [Google Scholar]
- Garcia-Perez I.; Couto Alves A.; Angulo S.; Li J. V.; Utzinger J.; Ebbels T. M. D.; Legido-Quigley C.; Nicholson J. K.; Holmes E.; Barbas C. Bidirectional Correlation of NMR and Capillary Electrophoresis Fingerprints: A New Approach to Investigating Schistosoma mansoni Infection in a Mouse Model. Anal. Chem. 2010, 82 (1), 203–210. 10.1021/ac901728w. [DOI] [PubMed] [Google Scholar]
- Merrifield C. A.; Lewis M.; Claus S. P.; Beckonert O. P.; Dumas M.-E.; Duncker S.; Kochhar S.; Rezzi S.; Lindon J. C.; Bailey M.; Holmes E.; Nicholson J. K. A metabolic system-wide characterisation of the pig: a model for human physiology. Mol. BioSyst. 2011, 7 (9), 2577–2588. 10.1039/c1mb05023k. [DOI] [PubMed] [Google Scholar]
- Maitre L.; Villanueva C. M.; Lewis M. R.; Ibarluzea J.; Santa-Marina L.; Vrijheid M.; Sunyer J.; Coen M.; Toledano M. B. Maternal urinary metabolic signatures of fetal growth and associated clinical and environmental factors in the INMA study. BMC Med. 2016, 14, 177. 10.1186/s12916-016-0706-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.