

Abstract
Excitatory amino acid transporter 2 (EAAT2) is primarily located in perisynaptic astrocytic processes (PAP) where it plays a critical role in synaptic glutamate homeostasis. Dysregulation of EAAT2 at the translational level has been implicated in a myriad of neurological diseases. We previously discovered that pyridazine analogs can activate EAAT2 translation. Here, we sought to further refine the site and mechanism of compound action. We found that in vivo, compound treatment increased EAAT2 expression only in the PAP of astrocytes where EAAT2 mRNA also was identified. Direct application of compound to isolated PAP induced de novo EAAT2 protein synthesis, indicating that compound activates translation locally in the PAP. Using a screening process, we identified a set of PAP proteins that are rapidly up-regulated following compound treatment and a subset of these PAP proteins may be locally synthesized in the PAP. Importantly, these identified proteins are associated with the structural and functional capacity of the PAP, indicating compound enhanced plasticity of the PAP. Concomitantly, we found that pyridazine analogs increase synaptic protein expression in the synapse and enhance hippocampal long-term potentiation. This was not dependent upon compound-mediated local translation in neurons. This suggests that compound enhances the structural and functional capacity of the PAP which in turn facilitates enhanced plasticity of the tripartite synapse. Overall, this provides insight into the mechanism action site of pyridazine derivatives as well as the growing appreciation of the dynamic regulation and functional aspects of the PAP at the tripartite synapse.
Keywords: glutamate transporter EAAT2, perisynaptic astrocytic processes, tripartite synapse, local translation, synaptic plasticity, pyridazine derivatives
INTRODUCTION
Excitatory amino acid transporter 2 (EAAT2) is a trimeric membrane-bound transporter primarily found in astrocytes in the brain. Individual astrocytes have 5–10 major processes from which up to 100,000 fine processes extend; these are termed peripheral astrocyte processes or perisynaptic astrocytic processes (PAPs) (Bushong et al., 2002; Halassa et al., 2007; Ventura and Harris, 1999). These PAPs wrap around pre- and post-synaptic components of the synapse and constitute the glial aspect of the tripartite synapse (Lavialle et al., 2011). At the PAP, EAAT2 forms a larger multi-protein complex to facilitate the rapid removal of glutamate from the synaptic cleft to terminate glutamatergic signaling. EAAT2 accounts for ~80–90% of glutamate uptake from the adult synaptic cleft and prevents glutamatergic excitotoxicity (Tanaka et al., 1997). In the hippocampus, for example, rapid temporal and spatial regulation of synaptic glutamate concentration by EAAT2 is integral to prevent synaptic spillover and, thus, ensures that the activity-dependent release of glutamate represents a specific, discrete signal (Asztely et al., 1997). Thus, EAAT2 plays an essential role in synaptic glutamate homeostasis under normal conditions.
To date, numerous neurological diseases have shown that EAAT2 expression is dysregulated which leads to a reduction in the functional capacity of EAAT2. This can lead to glutamate-induced excitotoxicity where prolonged, high extrasynaptic concentrations of glutamate cause synaptic bouton retraction and eventually neuronal death (Olney, 1969; Rothstein et al., 1993; Szydlowska and Tymianski, 2010). There is debate as to whether this reduced EAAT2 function is causative or epiphenomenal; however, genetic and pharmacological manipulations that increase EAAT2 expression in animal models show beneficial effect on disease phenotype in many of these diseases including: amyotrophic lateral sclerosis (Guo et al., 2003; Kong et al., 2014; Rothstein et al., 2005), Parkinson’s disease (Chotibut et al., 2014; Leung et al., 2012), Alzheimer’s disease (Takahashi et al., 2015), Huntington’s Disease (Miller et al., 2008), traumatic brain injury (Verma et al., 2010), stroke (Chu et al., 2007), pain disorders (Liaw et al., 2005; Lin et al., 2009), epilepsy (Kong et al., 2012), addictive disorders (Abulseoud et al., 2012; Kong et al., 2012), and depression (Mineur et al., 2007). This suggests that enhanced EAAT2 function may be able to modulate disease progression and outcome directly rather than having only palliative effects. Intensive study recently has been undertaken to identify compounds that can increase EAAT2 protein expression. The first drug candidates identified were the beta-lactam series of antibiotics (e.g. ceftriaxone; Rothstein et al., 2005). These antibiotics increase EAAT2 mRNA expression and, in turn, increase EAAT2 protein expression in animal models (Lee et al., 2008). However, mRNA levels are unchanged in the clinical population of many diseases with associated EAAT2 reduction (Bristol and Rothstein, 1996; Li et al., 1997; Rothstein et al., 1995). This implies that EAAT2 dysfunction is related to a post-transcriptional mechanism. There- fore, we previously implemented a high-throughput screening assay to identify compounds that increase EAAT2 by translational activation from a library of ~140,000 compounds (Colton et al., 2010). Through this screen and subsequent studies, a pyridazine-based lead series was identified and found to robustly increase the expression of EAAT2. Subsequently, we showed that LDN/OSU-0212320 exhibits profound efficacy in many neurological disease models, including amyotrophic lateral sclerosis (Kong et al., 2014), Alzheimer’s disease (Takahashi et al., 2015), and epilepsy (Kong et al., 2014). While we have shown excellent compound efficacy in these models, the compound target and detailed mechanism of action for these pyridazine derivates are still under investigation.
The current study was undertaken to further understand the mechanism and site of action for pyridazine derivatives. Previously, we observed that pyridazine analogs rapidly increase EAAT2 protein expression in vitro and in vivo as early as 1–2 h post-treatment. Since this induction is mediated through translational activation, we sought to determine if this increase is due to activation of translation directly in the PAP domain. Using biochemically isolated PAPs, we found compound treatment increases EAAT2 expression only in the PAP of intact animals and that direct application of compound ex vivo induces de novo translation of EAAT2 and other proteins in the PAP. This also demonstrated that the required signaling components for the compound mechanism of action are localized to the PAP. In parallel, we identified that compound treatment enhances synaptic plasticity in the hippocampus, but not through activation of local translation of synaptic proteins. This suggests that compound-induced changes to the PAP may facilitate enhanced synaptic plasticity.
EXPERIMENTAL PROCEDURES
Animals
In all experiments, we utilized wild-type FVB/NJ mice (Jackson; Stock #001800). Mice were housed on a 12-h light/dark cycle with access to food and water ab libitum. Mice were treated PO via oral gavage with pyridazine analog at a dose of 40 mg/kg unless otherwise noted. Mice were sacrificed post-treatment at time point indicated by isoflurane overdose and cervical dislocation. Forebrains or hippocampi of mice were collected and processed as described. All experiments were approved by the Institutional Animal Care and Use Committee of The Ohio State University and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Compounds
Compound was prepared as previously described (Kong et al., 2014) with minor modifications. Briefly, compound was dissolved in ddH20 and diluted in ~200 βl final volume of 1% polyethylene glycol 400 (Sigma–Aldrich), 0.2% Tween-80 (Sigma–Aldrich), and 10% hydroxypro pyl-β-cyclodextrin (Sigma–Aldrich) in ddH20. This was then given orally by gavage as a single bolus. Here, we used an advanced pyridazine-based compound, LDN/ OSU-0215111, that exhibits high potency, high solubility in water, and oral bioavailability. This compound is a derivative of LDN/OSU-0212320 which we previously characterized (Kong et al., 2014; Takahashi et al., 2015).
Gliosome and synaptosome preparation
The preparation was performed as described previously (Carney et al., 2014; Dunkley et al., 2008) with modifications. Briefly, brains were collected from mice and the forebrain was isolated. Forebrains were quickly cooled in sucrose-EDTA (SE) buffer (5 mM Tris, pH 7.4, 0.32 M sucrose, 1 mM EDTA) and washed 3 times. Forebrains were then suspended in 10x’s volume of homogenization buffer (SE buffer with 0.25 mM DTT and 1X protease inhibitor) and homogenized in 10 strokes using a Class B Teflon homogenizer attached to an overhead stirrer at 700 RPM. The homogenate was then centrifuged at 1,000 x g for 10 min. The supernatant (S1) was then loaded onto a Percoll (Sigma) gradient (2%, 6%, 10%, and 20% Percoll in SE buffer; 2 mL/layer). Gradients were centrifuged at 31 k x g for 260 sec in a JA 25.50 rotor. A slow stop was used to prevent the gradient from collapsing. We collected the F2 (2%−6% interphase; gliosomes) and F4 (10%–20% interphase; synaptosomes) fractions using a glass capillary pipette. The fractions were resuspended in 35 mL of SE buffer and centrifuged at 27 k x g for 30 min in a JA 25.50 rotor to remove excess Percoll. An additional centrifugation at 16 k x g for 30 min in a 1.5-mL Eppendorf tube filled with SE buffer on a table-top centrifuge was performed to condense the loose pellet that was collected from the previous step. The F2 or F4 pellet was then resuspended in either Trizol (Invitrogen) for RNA isolation procedures, 1X PBS for proteomics analysis, 1X PBS + protease inhibitor (Pierce) for Western blotting or isotonic Krebs–Henseleit (KH) Buffer (118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 25 mM NaHCO3, 11 mM glucose, 1.25 mM CaCl2,1X protease inhibitor, pH to 7.4) for use in ex vivo experiments.
Ex vivo translation
Freshly isolated gliosomes from untreated animals, resuspended in KH buffer, were held on ice until compound application. Active or inactive pyridazine derivatives were resuspended in H2O and warmed to 37 °C for 5 min before use. Gliosomes were aliquoted across multiple tubes and treated with vehicle (H2O only) or indicated amount of compound. Ex vivo samples were incubated in a water bath at 37 °C for 1 h or as indicated. Reaction was terminated by the addition of 5X SDS buffer (250 mM Tris–HCl pH 6.8, 50% Glycerol, 10% SDS, 500 mM DTT, 0.5% Bromophenol Blue, 5% β-mercaptoethanol). Experiments using the synaptosome fraction for ex vivo were carried out in the same manner. For biotinylation experiments, after the 1-h water bath incubation, samples were incubated with Ez-link Sulfo-LC-LC-Biotin (Fisher Scientific) at 1 mg/mL for 30 min to tag extracellular plasma membrane proteins. Samples were transferred to a 10-k Amicon Ultra filter (Millipore) and centrifuged at 14 k x g for 10 min. The diluted buffer was exchanged with 1X PBS. The 1X PBS was then exchanged for cold 100 mM glycine in 1X PBS to quench any remaining active biotin. The glycine buffer was then exchanged for Radioimmunoprecipitation assay (RIPA) buffer to disrupt the intact plasma membranes for the subsequent steps of binding to avidin beads. The procedure for pulldown of biotinylated proteins was performed the same as for hippocampus biotinylation.
Lipid raft microdomain isolation
Mice were given a single dose of vehicle or compound and sacrificed 24hr later. The lipid raft microdomain (LRM) of the forebrain was isolated as previously described (Butchbach et al., 2004; Tian et al., 2010). Fractions 3–6 were confirmed to contain the LRM based on the cholesterol-enriched membrane marker Flotillin-1. Functional EAAT2 expression in the forebrain was assessed by Western blot using these fractions.
Hippocampus extracellular plasma membrane biotinylation
Mice were treated for 7d with compound and sacrificed 24 h after the final treatment. The hippocampus was acutely dissected and sliced into 400-µm sections using a McIlwain tissue chopper. Sections were transferred to a 1.5-mL tube and chilled in 1X PBS. Sections were washed in 1X PBS 3 times. Sections were incubated with Ez-link Sulfo-LC-LC-Biotin (1 mg/mL) for 30 min. Sections were washed in 1X PBS 3 times and then incubated in 100 mM glycine in 1X PBS for 2 washes of 20 min each to quench biotinylation reaction. The plasma membrane of the sections was then disrupted using RIPA buffer and a total lysate aliquot was collected. 50 or 100 mg of protein was incubated with 25 µL of Neutravidin beads (Pierce) overnight at 4 °C. Beads with bound biotinylated protein were collected and washed in RIPA buffer 3 times for 5 min. Protein was dissociated from beads by resuspending beads in 40 µL of 2X SDS buffer and heated at 95 °C for 10 min. Biotinylated extracellular plasma membrane bound EAAT2 expression was assessed by Western blotting.
Plasma membrane vesicle isolation
Mice were treated for 7d with compound and sacrificed 24 h after the final treatment. The hippocampus was acutely dissected and the plasma membrane vesicle (PMV) fraction was isolated as has been previously described (Butchbach et al., 2004). The PMV fraction was resuspended in 1X PBS with protease inhibitor, sonicated, and protein was analyzed by Western blot.
Western blotting
Western blotting was performed as described previously (Tian et al., 2010). Briefly, samples were assayed for protein concentration using the DC Protein Assay (Bio-Rad). Normalized amounts of protein were resolved by SDS– PAGE (8%, 10%, or 12% polyacrylamide gel), and transferred onto nitrocellulose membranes. Membranes were probed against antibodies of interest including the following: β-Actin (Cell Signaling, AB_10950489), APPa4 (Millipore, AB_94882), Ankyrin 2 (Santa Cruz, AB_626673), EAAT1 (Abcam, AB_304334), EAAT2 (see (Guo et al., 2003)), Ezrin (Santa Cruz, AB_783303), Flotillin-1 (Cell Signaling, #18634), GAPDH (Santa Cruz, AB_10847862), GABA transporter 1 (Santa Cruz, AB_2553834), GABA transporter 3 (Santa Cruz, AB_10987703), Glutamine Synthetase (Santa Cruz, AB_2110654), HSP90ab1 (Santa Cruz, AB_675659), Phosphofructokinase Muscle (Santa Cruz, AB_2236976), ATPase Plasma Membrane Ca2 + Trans porting 1 (Santa Cruz, sc-398413), Synaptotagmin-1 (Santa Cruz, AB_10611453), Vesicular Glutamate Transporter 1 (Millipore, AB_262185), ATPase Na+/K + Tran sporting Subunit Alpha 1 (Santa Cruz, AB_626713), Glial Fibrillary Acidic Protein (Sigma, AB_477010), Synaptophysin (Cell Signaling, AB_1904154), Post-Synaptic Density 95 (Thermo Fisher, AB_2092361), Microtubule-Associated Protein 2a,b (Thermo Fisher, AB_1095983), and NMDA Receptor 2B (Millipore, AB_90772). Subsequently, goat anti-rabbit IgG or anti-mouse IgG secondary antibodies (Bio-Rad; 1:5,000) conjugated to horseradish-peroxidase were incubated with the blot. For visualization of immunoreactive bands, either WesternBright (Advansta)- or Clarity (Bio-Rad)-enhanced chemiluminescence substrates were applied according to the manufacturer’s directions. Digital images of immunoblots were captured using the ChemiDoc Imaging System (Bio-Rad) and band intensities were analyzed using Image Lab (Bio-Rad). For statistical analysis, intensities of protein bands of interest were normalized to loading controls when appropriate and fold change was determined in comparison to the vehicle condition. A one-way ANOVA with post-hoc Tukey’s test was used for statistics. Graphs are displayed as mean ± SEM.
Proteomics
Proteomic data were collected and compiled by the Proteomics Shared Resource Core at The Ohio State University. Protein from each gliosome group was extracted and digested in trypsin (Promega) with incubation at 37 °C overnight. Peptide concentration was determined by Nanodrop (A280nm). Capillary-liquid chromatography-nanospray tandem mass spectrometry (Capillary-LC/MS/MS) of protein identification was performed on an Orbitrap Fusion mass spectrometer (Thermo Scientific). Samples were separated on an Easy-Spray Nano Column (PepmapTM RSLC) using a 2D rapid separation HPLC system (Thermo Scientific). Mobile phase A was 0.1% Formic Acid in water and mobile phase B was acetonitrile (with 0.1% formic acid). To achieve high mass accuracy MS determination, the full scan sequence was performed in Fourier transformation mode. Sequence information from the MS/MS data was processed using Mascot Daemon by Matrix Science version 2.3.2 (Boston, MA) and searched against Uniprot Mouse database. A decoy database was also searched to determine the false discovery rate (FDR). The significance threshold was set at p < 0.05 and proteins with less than 1% FDR as well as a minimal of 2 significant peptides detected were considered as valid proteins. Area under the curve (AUC) quantification data was obtained using MaxQuant. AUC values were averaged across each of the 3 groups (n = 4 per group; vehicle, 4 h, and 24 h post-treatment) and data were converted to fold change in relation to the vehicle condition.
RNA array
Total RNA was isolated from the F2 fraction of the gliosome preparation under RNase-free conditions. Gliosomes were resuspended in Trizol and RNA was then collected using the RNA Concentration and Cleanup Kit (Norgen) column system. Nanodrop analysis showed all samples tested had RNA concentrations greater than 50 ng/µL, 260/230 ratios greater than 1.0, and 260/280 ratios greater than 2.0. RNA array data were collected and compiled by the Genomics Shared Resource Core at The Ohio State University utilizing the GeneChip Mouse Transcriptome Assay 1.0 (Affymetrix) kit. A total of 100 ng of total RNA was subjected to in vitro transcription, conversion to cDNA and biotinylation for hybridization to the microarray. Data were analyzed using Expression Console and Transcriptome Analysis Software (Affymetrix). Gene expression of gliosomes from vehicle-treated animals was compared with gliosomes isolated 4 h or 24 h post-treatment. Also, fluorescent scores from all samples were combined, averaged, and converted to a Z-score (from an mRNA array population of 17,856 species). A Z-score greater than 2.0 was considered significantly likely to exist in the gliosome fraction.
Reverse transcription–polymerase chain reaction
To validate a subset of the RNA array predictions of mRNAs found in gliosomes, we utilized reverse transcription-polymerase reaction (RT–PCR). RNA from S1, F2, and F4 was isolated in the same manner as for the RNA array. RNA was converted to cDNA using Super Script IV (Thermo-Fisher) reverse transcriptase using gene-specific primers. Accustart II PCR Supermix (VWR) was used for cDNA amplification. PCR cycle number and annealing temperature were optimized for each primer set. RT–PCR products were separated on 2% agarose gels, bands were stained using Radiant SmartGlow (1:20,000; AE Bios) and digital images of gels were captured using the ChemiDoc Imaging System.
Electrophysiology
Mice were treated or 7d with vehicle or compound. Brains were rapidly removed and placed in ice-cold cutting solution composed of (all in mM) 250 sucrose, 25 D-glucose, 2.5 KCl, 24 NaHCO3, 1.25 NaH2PO4, 2.0 CaCl2, 1.5 MgSO4, and 1.0 kynurenic acid (pH; 7.3– 7.4). Hippocampal coronal slices (400 µm) were prepared using a vibratome (Leica). Brain slices were transferred to a chamber containing artificial cerebral spinal fluid (aCSF; bubbled with 95% O2 and 5% CO2) composed of (all in mM) 124 NaCl, 3 KCl, 24 NaHCO3, 1.25 NaH2PO4, 2.0 CaCl2, 1.0 MgSO4, and 10 D-glucose 10, (pH; 7.3–7.4), and allowed to recover for 30 min at 37 °C followed by room temperature for at least 1 h. For recording, slices were maintained at 32 °C with gravity-fed oxygenated aCSF (2–3 ml/min). Local field excitatory synaptic postsynaptic potentials (fEPSPs) were recorded with Borosilicate glass electrodes (1.5–5 MΩ, filled with aCSF containing 100 µM Picrotoxin) from the stratum radiatum of CA1 and evoked by electrical stimulation (100 µs duration, every 20 s) of the Schaffer collateral fibers (SC-CA1) near the CA3-CA1 border. Input–output (I/O) curves were generated for each slice with stimulation intensity ascending from 0 to 1.0 mA, each step 0.1 mA. Based on I/O curves, the stimulation intensity that evoked 50% the maximum response (between 0.2 and 0.3 mA) was chosen to conduct the following experiment. Stimulation intensity and duration were constant throughout the entire experiment. Synaptic field potentials were lowpass filtered at 1 kHz and digitally sampled at 50 kHz. All data were digitized using an Axopatch 200B amplifier, Digidata 1440A, and pClamp 10.6 software (Molecular Devices). Theta-burst stimulation (TBS) was used to induce long-term potentiation (LTP). Five trains of four pulse bursts at 200 Hz separated by 200 ms, repeated six times with an intertrain interval of 10 s (5×4×6). Each LTP experiment consisted of 5 min recording for baseline before TBS and 30 min recording after TBS for LTP. The peak of the evoked fEPSPs was measured and normalized to the preconditioning 5-min averaged baseline. Data were analyzed off-line using Clampex 10.6 software. For statistical analysis, LTP recordings were binned into 5-min epochs and analyzed using a one-way ANOVA with Bonferroni’s post-hoc test in GraphPad Prism 5. Graphs are displayed as mean ± SEM.
RESULTS
Pyridazine analogs increase EAAT2 expression in perisynaptic astrocytic processes
To uncover the mechanisms of compound action, we first asked the question, ‘‘where is the compound action site – astrocyte’s process (PAP) or cell body?” If the action site is located at the cell body, we expect to detect increased EAAT2 in the cell body as well as the PAP; whereas, if induction is exclusive to the PAP, increased EAAT2 is expected only at the PAP. Nakamura et al. (1993) initially reported a procedure to isolate a subcellular faction enriched for re-sealed particles from PAPs, termed ‘‘gliosomes”. We followed a modified procedure (Dunkley et al., 2008) and prepared gliosomes from mouse forebrains, as illustrated in Fig. 1A. Harvested forebrains were homogenized and centrifuged yielding the astrocyte cell bodies and main processes in the pellet (P1) while the fine processes (PAPs) remained in the supernatant (S1). The supernatant was then layered on a discontinuous Percoll gradient. The gliosome (F2) layer between 2% and 6% was enriched for PAPs, and the synaptosome (F4) layer between 10% and 20% was enriched for nerve terminals. This was validated by Western blot analysis, which showed that gliosomes were enriched with the PAP proteins, ezrin (Derouiche and Frotscher, 2001), EAAT2, and Na+/K+ ATPase a1, while synaptosomes were enriched with the synaptic proteins, postsynaptic-density protein 95 (PSD95), vesicular glutamate transporter 1 (VGLUT1), and synaptophysin (Fig. 1B). Of note, the majority of glial fibrillary acidic protein (GFAP) was found in P1 (cell bodies and main processes) and in only small amounts in gliosomes (Fig. 1B). These results are consistent with previous reports and demonstrate that gliosomes are enriched for PAPs (Haseleu et al., 2013; Lavialle et al., 2011). This isolation procedure effectively separated the PAPs from the main processes of the astrocytes, as well as the neuronal aspect of the tripartite synapse, thus allowing us to analyze the PAP in isolation.
Fig. 1.
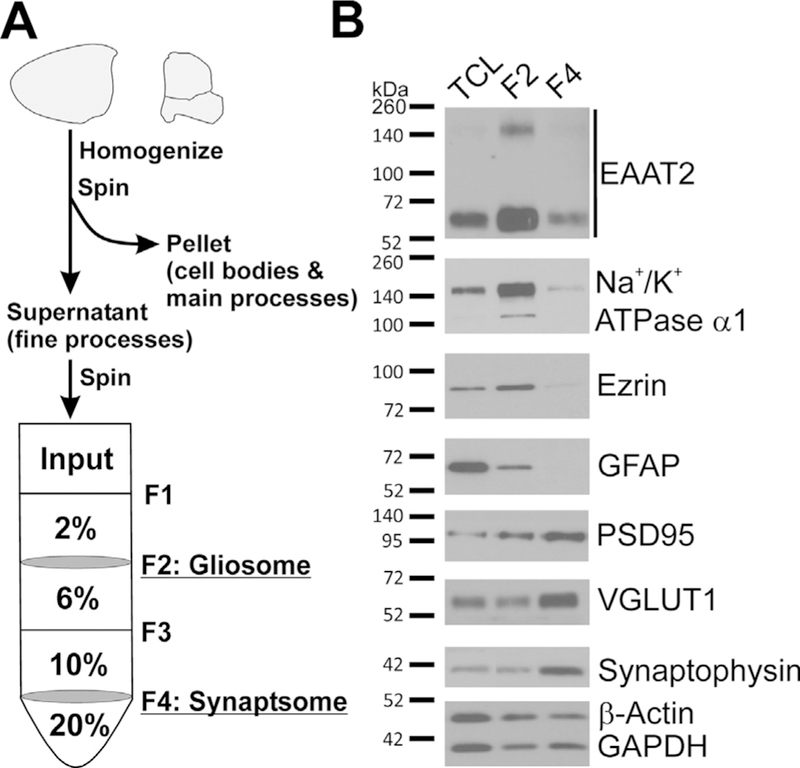
Overview and confirmation of gliosome preparation representing the PAP. (A) Schematic overview of gliosome and synaptosome preparation protocol. Once animals are euthanized, the brain is rapidly removed and the forebrain is isolated. The forebrain is homogenized and the cell bodies are removed by centrifugation. The resultant supernatant, S1, is layered on the discontinuous Percoll gradient and centrifuged to separate gliosomes (F2) from synaptosomes (F4). Excess Percoll is removed from these fractions by centrifugation and the fractions are then resuspended in an appropriate buffer for downstream applications. (B) Characterization of the protein distribution in gliosome and synaptosomes relative to total cell lysate (TCL). Gliosomes are enriched with the PAP markers EAAT2, Na+/K+ ATPase ɑ1, and ezrin, but not with the astrocyte cell body/main processes protein GFAP or the synaptic proteins PSD95, VGLUT1, and synaptophysin. Synaptosomes are also depleted of GFAP, but show inverse relationships with the above proteins as expected.
Next, we investigated where compound-induced EAAT2 is compartmentalized. Mice were treated by oral gavage with the pyridazine analog and forebrains were harvested for the gliosome preparation 24 h post-treatment. Western blot analysis showed that induction of EAAT2 was specifically limited to the gliosome (PAP) fraction and was not observed in the P1 (cell bodies and main processes) or F4 (synaptosomes) fractions (Fig. 2A). Compound treatment induced EAAT2 expression in the gliosome fraction in a dose-dependent manner (Fig. 2B). Furthermore, this induction could be seen in gliosomes as early as 4 h post-treatment (Fig. 2C). Finally, we utilized the lipid raft microdomain (LRM) isolation to confirm that compound-induced EAAT2 represents a functional enhancement of EAAT2. We have previously shown that the LRM (fractions 3–6) contains the functional population of EAAT2 at the plasma membrane (Butchbach et al., 2004; Tian et al., 2010). As seen in Fig. 2D, there is a corresponding increase in EAAT2 in the LRM that is comparable to that seen in the gliosome fraction. Therefore, the compound specifically and rapidly increases the functional expression of EAAT2 protein in the PAP compartment – the glutamate uptake action site. These results also indicate that the compound action site is located at the PAP.
Fig. 2.
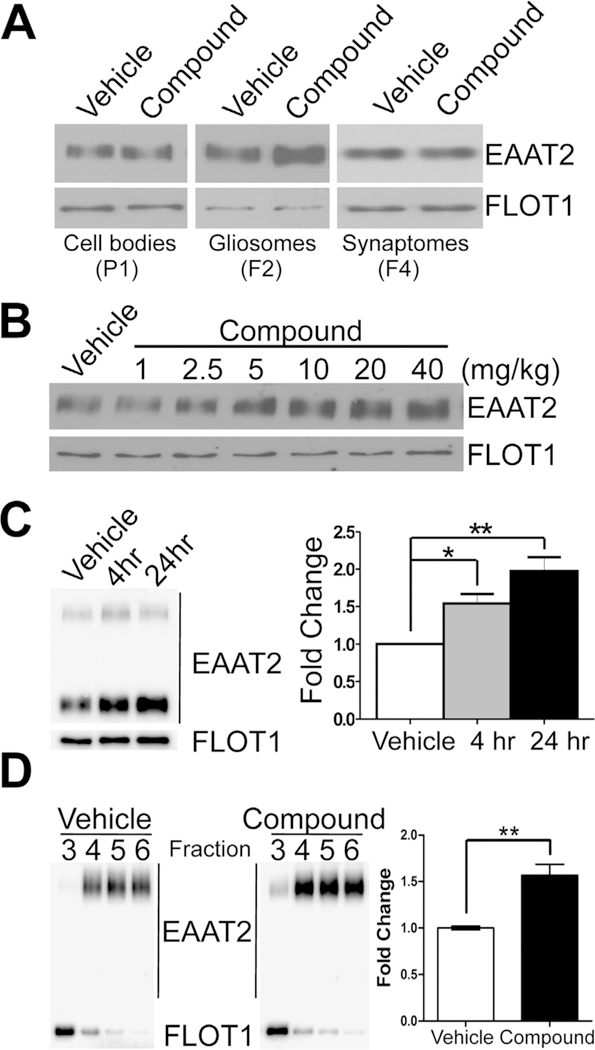
Pyridazine analogs specifically and rapidly increase EAAT2 expression only in the PAP. Mice were treated with compound at 40 mg/kg (or as depicted) and forebrains were harvested for the gliosome preparation 24 h (or as described) post-treatment. (A) Western blots show robust increases of EAAT2 protein expression were only found in gliosomes (F2) while no induction was observed in the cell bodies (P1) or in synaptosomes (F4). (B) Dose-dependent increase in EAAT2 protein expression in response to single, escalating compound dose. (C) Time-dependent fold increase in EAAT2 protein expression in response to compound treatment at 4 h (1.54 ± 0.13) and 24 h (1.98 ± 0.19) post-treatment. Quantification of EAAT2 expression time course (normalized to flotilin-1 protein; n = 4/group). (D) EAAT2 expression increased in the lipid raft micro-domain, which represents the functional EAAT2 lipid domain, of the forebrain (1.57 ± 0.12) after a single treatment. Data represented as mean ± SEM and analyzed using a one-way ANOVA with Tukey’s post-hoc test. Statistical significance denoted by *p < .05; **p < .01.
Compound activates local translation of EAAT2 at the PAP
Since EAAT2 induction occurred rapidly and was exclusive to the gliosome fraction, but not the cell body or synaptosome fraction, this suggested that compound may induce the translation of EAAT2 locally at the PAP. If this is true, the isolated gliosomes must contain all components needed for translation and may be competent in translation of EAAT2. To test this possibility, freshly isolated gliosomes from untreated animals were incubated with the compound. Western blot analysis showed that EAAT2 protein levels were increased in a dose-dependent and time-dependent manner in this ex vivo model (Fig. 3A,B). Importantly, no induction was observed when an inactive analog of the compound was applied (Fig. 3C). Furthermore, we found that the de novo-translated EAAT2 resulted in insertion into the plasma membrane of gliosomes. Pulldown of extracellular biotinylated proteins yielded increased EAAT2 plasma membrane expression after ex vivo compound treatment (Fig. 3D). To further confirm the feasibility for local protein synthesis to occur, we examined if EAAT2 mRNA and ribosomal RNA were present in the gliosome fraction. Using extracted RNA from gliosomes and RT–PCR analysis, the 18S ribosomal RNA was identified in both gliosomes and synaptosomes showing that both have translational machinery present (Fig. 3E). Full-length EAAT2 mRNA contains a ~1.7-kb coding sequence (CDS) followed by a 9.7-kb 3׳-untranslated region (3׳-UTR). We examined the CDS region as well as the proximal (last ~150 bp of CDS to first ~500 bp 3׳-UTR junction) and distal region ( 490 bp upstream of poly-A tail) of the EAAT2 3׳-UTR. Results revealed that EAAT2 mRNA, containing both the CDS and the full-length 3׳-UTR, was present in gliosomes but was absent in the synaptosome fraction (Fig. 3E). Ex vivo results indicate that compound-induced EAAT2 expression is mediated by local translation at the PAP.
Fig. 3.
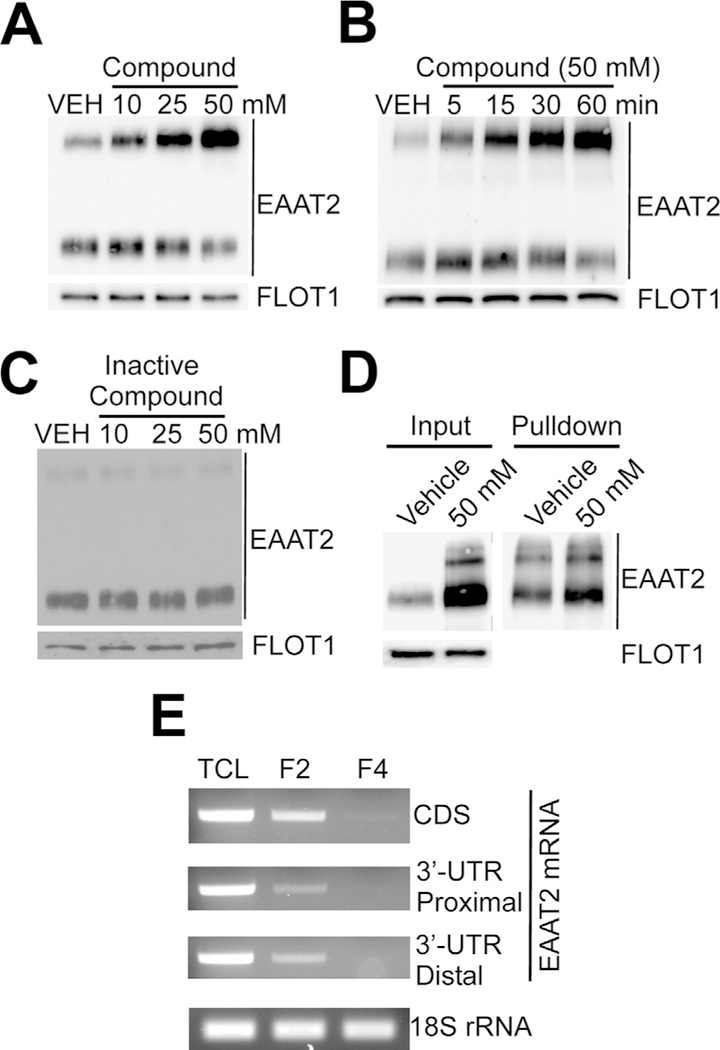
Pyridazine analogs induce EAAT2 locally translation in gliosomes. Gliosomes were isolated from untreated mice and vehicle or compound was applied directly to gliosomes ex vivo as depicted. (A, B) Western blot analysis showed that active pyridazine compounds induce de novo synthesis of EAAT2 in ex vivo gliosomes in a dose- and time-dependent manner. (C) However, inactive pyridazine analogs do not induce the local translation of EAAT2 in gliosomes. (D) Ex vivo induction of EAAT2 resulted in insertion of EAAT2 into the plasma membrane. Pulldown of biotinylated extracellular proteins after ex vivo treatment showed increased EAAT2 insertion in line with increased total expression (E) In support of the feasibility for local translation to occur, RT–PCR analysis showed expression of the full-length EAAT2 mRNA in gliosomes, but not in the synaptosome fraction, and the 18S ribosomal RNA in all fractions. This suggests that the major components necessary for translation exist in gliosomes. Each study consisted of four replicates with consistent results. Representative blots are shown.
Compound activates local translation of a subset of PAP transcripts
Next, we investigated if the expression of additional PAP proteins is also regulated by local translation, like EAAT2, following compound treatment. We conducted a proteomic analysis and an RNA array of gliosomes. Both approaches were conducted in tandem to identify proteins that are up-regulated and have mRNA present in the PAP. Gliosomes were prepared from forebrains harvested from (i) vehicle-treated mice, (ii) compound-treated mice that were euthanized 4 h post-treatment, and (iii) compound-treated mice that were euthanized 24 h post-treatment. These gliosomes (n = 4 for each group) were subjected to proteomic analysis (LC-MS/ MS). Results showed that a total of 1948 proteins were identified. Although we did not identify as many proteins, almost all the proteins that we identified overlapped with a previous mass spectrometry study of gliosomes (Carney et al., 2014). EAAT2 protein expression increased 2.2-fold and 4.6-fold at 4 h and 24 h respectively, in accordance with Western blot analysis (Fig. 2C). Of the proteins identified, 449 proteins were up-regulated 1.5-fold or greater by 4 h post-treatment. By 24 h post-treatment, a total of 790 proteins were upregulated ≥1.5 fold. All 449 proteins up-regulated at 4 h remained up-regulated at 24 h. These results indicate that compound up-regulates a subset of PAP proteins following compound treatment.
Gliosomes from the same group categories used for proteomics were prepared for the RNA array. Total RNA was extracted from gliosomes and subjected to GeneChip Mouse Transcriptome Array. Fluorescent intensity scores obtained from the array were converted to a Z-score that was used to determine which mRNAs are statistically likely to be present in gliosomes (Z-scor es ≥ 2.0). A total of 903 mRNA species and all 4 rRNA species (28S, 18S, 5.8S and 5S) reached criteria. EAAT2 mRNA yielded a standardized value of 4.288. There was no significant difference between the three groups’ gene expression results, indicating that compound treatment does not change mRNA content in PAPs.
Among the 449 proteins that showed ≥1.5 increased expression by mass spectrometry, 157 of these also had an RNA Z-score ≥2.0. The full list of proteins, which meet both the protein and mRNA expression criteria described above, is shown in Table 1. These proteins include (1) those involved in glutamate uptake and metabolism, e.g. EAAT2, Na+/K+ ATPase and glutamine synthetase; (2) glycolytic enzymes, e.g. 6-phosphofructokinase and pyruvate kinase; (3) those involved in protein folding and turnover, e.g. heat shock proteins and cullin-associated NEDD8-dissociated protein 1; (4) mitochondrial proteins, e.g. voltage-dependent anion-selective channel proteins and respiratory chain complexes; (5) signaling molecules, e.g. G-proteins and calmodulin; (6) those involved in exocytosis/endocytosis, e.g. synaptotgamin-1, synapsins and AP-2 complex subunits; (7) regulatory proteins, e.g. 14–3–3 proteins; (8) cytoskeletal proteins, e.g. tubulin and actin; (9) scaffolding proteins, e.g. ankyrin-2 and PSD-93; (10) trafficking proteins, e.g. dynamin-1 and Ras-related proteins; (11) receptors, e.g. BDNF/NT-3 growth factors receptor, GABA receptors, and metabotropic glutamate receptors; and (12) transporters, vesicular glutamate transporter 1, GABA transporter 1, and 3. This group of 157 proteins represents those most likely to be regulated by the compound-activated local translation mechanism.
Table 1.
Candidate proteins regulated via local translation in the PAP by pyridazine analogs
| Mass Spectrometry | RNA Array | ||||
|---|---|---|---|---|---|
| Primary Function | Accession | Description | 4 h Fold | 24 h Fold | Z-score |
| Antioxidant Response | Q8K4Z3 | NAD(P)H-hydrate epimerase | 2.93 | 11.42 | 2.002 |
| CO2 Regulation | P00920 | Carbonic anhydrase 2 | 2.12 | 3.13 | 2.471 |
| Cell Adhesion | Q6P9K9 | Neurexin-3 | 6.57 | 8.09 | 2.250 |
| E9QNF7 | Contactin-associated protein-like 2 | 2.57 | 4.79 | 2.184 | |
| G3XA53 | Oligodendrocyte myelin glycoprotein | 1.98 | 3.37 | 2.311 | |
| G3UZM4 | Cell adhesion molecule 2 | 3.86 | 3.30 | 2.910 | |
| Q53YX2 | Thymus cell antigen 1, theta | 1.97 | 3.28 | 2.224 | |
| D6QSS8 | A disintegrin and metallopeptidase domain 22 | 1.60 | 2.15 | 2.465 | |
| Q6PGJ3 | L1 cell adhesion molecule | 2.07 | 1.89 | 2.249 | |
| Cytoskeleton Structure | P68369 | Tubulin alpha-1A | 1.82 | 1.78 | 5.833 |
| P20357 | Microtubule-associated protein 2 | 2.23 | 5.62 | 2.833 | |
| P60710 | Actin, cytoplasmic 1 | 3.79 | 5.54 | 4.309 | |
| Q9R0P5 | Destrin | 1.62 | 5.40 | 2.159 | |
| Q9BW30 | Tubulin polymerization-promoting protein family 3 | 3.30 | 4.38 | 2.939 | |
| Q9D6F9 | Tubulin beta-4A chain | 4.76 | 4.32 | 2.796 | |
| P18760 | Cofilin-1 | 1.70 | 3.39 | 3.503 | |
| B2RQQ5 | Microtubule-associated protein 1B | 1.98 | 2.76 | 4.451 | |
| E9Q1G8 | Septin-7 | 1.67 | 2.61 | 2.758 | |
| P68368 | Tubulin alpha-4A chain | 2.10 | 2.44 | 4.786 | |
| Q3TGE1 | Actin-related protein 3 | 2.44 | 2.30 | 2.904 | |
| P99024 | Tubulin beta-5 chain | 2.64 | 2.07 | 2.105 | |
| Q7TMM9 | Tubulin beta-2A chain | 1.98 | 1.81 | 3.816 | |
| Endocytosis/Exocytosis | Q3UHD6 | Sorting nexin-27 | 5.00 | 13.99 | 2.195 |
| F7BQW7 | Synaptojanin-1 | 3.39 | 8.73 | 2.524 | |
| P18572 | Basigin | 5.50 | 8.14 | 2.404 | |
| Q80TZ3 | Putative tyrosine-protein phosphatase auxilin | 2.28 | 4.82 | 2.540 | |
| Q9JIS5 | Synaptic vesicle glycoprotein 2A | 2.29 | 3.70 | 3.657 | |
| P46096 | Synaptotagmin-1 | 2.73 | 3.17 | 4.007 | |
| Q3UHJ0 | AP2-associated protein kinase 1 | 2.01 | 3.06 | 2.195 | |
| Q9ES97 | Reticulon-3 | 1.69 | 2.95 | 3.543 | |
| O08599 | Syntaxin-binding protein 1 | 1.97 | 2.72 | 3.824 | |
| O54734 | Dolichyl-diphosphooligosaccharide–protein glycosyltransferase 48 kDa subunit | 1.54 | 2.13 | 2.306 | |
| O55100 | Synaptogyrin-1 | 19.83 | 13.87 | 2.614 | |
| Q8R191 | Synaptogyrin-3 | 2.02 | 5.06 | 2.339 | |
| P17426 | AP-2 complex subunit alpha-1 | 1.80 | 2.53 | 2.116 | |
| Q9WV55 | Vesicle-associated membrane protein-associated protein A | 1.63 | 1.51 | 2.716 | |
| Q3TXX4 | Vesicular glutamate transporter 1 | 2.06 | 11.18 | 4.170 | |
| Endoplasmic Reticulum Transporter |
Q5DTI2 | ATPase, Ca++ transporting, cardiac muscle, slow twitch 2 | 1.66 | 2.43 | 5.158 |
| Energy Metabolism | Q9CQJ8 | NADH dehydrogenase [ubiquinone] 1 beta subcomplex subunit 9 | 3.69 | 4.24 | 3.224 |
| Q3UIQ2 | NADH dehydrogenase (ubiquinone) Fe-S protein 1 | 2.47 | 2.93 | 2.303 | |
| P56391 | Cytochrome c oxidase subunit 6B1 | 1.89 | 2.56 | 3.370 | |
| Q91VA7 | Isocitrate dehydrogenase [NAD] subunit | 2.20 | 2.55 | 2.175 | |
| Q3TIC8 | Ubiquinol-cytochrome c reductase core protein 1 | 3.72 | 1.91 | 2.486 | |
| Q9DB77 | Cytochrome b-c1 complex subunit 2, mitochondrial | 1.71 | 1.83 | 2.596 | |
| Fatty Acid Metabolism | Q8BLF1 | Neutral cholesterol ester hydrolase 1 | 4.28 | 11.32 | 2.062 |
| P19096 | Fatty acid synthase | 2.74 | 3.19 | 2.122 | |
| P60202 | Myelin proteolipid protein | 4.17 | 2.20 | 5.029 | |
| Q99L43 | Phosphatidate cytidylyltransferase 2 | 1.80 | 1.68 | 4.955 | |
| Glutamate Homeostasis | P05202 | Aspartate aminotransferase, mitochondrial | 5.25 | 5.75 | 2.289 |
| P15105 | Glutamine synthetase | 2.38 | 4.94 | 3.381 | |
| P05201 | Aspartate aminotransferase, cytoplasmic | 1.75 | 3.46 | 3.073 | |
| Q8BH59 | Calcium-binding mitochondrial carrier protein Aralar1 | 2.39 | 1.70 | 2.046 | |
| Glycolysis | P47857 | ATP-dependent 6-phosphofructokinase, muscle type | 3.16 | 5.19 | 2.995 |
| P09411 | Phosphoglycerate kinase 1 | 1.83 | 2.58 | 3.403 | |
| P05063 | Fructose-bisphosphate aldolase C | 1.77 | 1.97 | 2.794 | |
| P35486 | Pyruvate dehydrogenase E1 component subunit alpha, somatic | 2.11 | 1.57 | 2.388 | |
| Glycosylation | Q9DBE8 | Alpha-1,3/1,6-mannosyltransferase ALG2 | 2.96 | 5.06 | 2.140 |
| GPCR Signaling | B2RSH2 | Guanine nucleotide-binding protein G(i) subunit a1 | 4.15 | 5.77 | 2.673 |
| Q66L47 | Guanine nucleotide binding protein, alpha stimulating, olfactory type | 4.38 | 5.54 | 2.023 | |
| O54865 | Guanylate cyclase soluble subunit beta-1 | 3.38 | 5.03 | 3.359 | |
| Hormone Production | Q543R4 | Carboxypeptidase E | 1.90 | 2.48 | 5.434 |
| Ion Channel | F7AMU5 | Sodium channel, voltage-gated, type II, alpha | 1.73 | 3.09 | 3.546 |
| Q9Z1L5 | Voltage-dependent calcium channel subunit alpha-2/delta-3 | 1.84 | 1.98 | 2.580 | |
| P63141 | Potassium voltage-gated channel subfamily A2 | 3.35 | 1.87 | 2.529 | |
| J3QMG3 | Voltage-dependent anion-selective channel protein 3 | 4.77 | 9.45 | 3.629 | |
| Ion Channel Regulator | E9QN98 | Inactive dipeptidyl peptidase 10 | 1.79 | 4.27 | 3.301 |
| E9PWX1 | Dipeptidyl aminopeptidase-like protein 6 | 1.90 | 3.04 | 2.249 | |
| Kinase/Phosphatase | P84309 | Adenylate cyclase type 5 | 3.68 | 2.13 | 2.726 |
| Signaling | B2RX66 | Serine/threonine-protein kinase TAO1 | 7.18 | 17.27 | 2.169 |
| Q9CQR6 | Serine/threonine-protein phosphatase 6 catalytic | 7.38 | 11.79 | 2.378 | |
| P52480 | Pyruvate kinase | 2.70 | 8.04 | 3.448 | |
| Q9JLM8 | Doublecortin like kinase 1 | 1.70 | 7.35 | 3.603 | |
| P63328 | Serine/threonine-protein phosphatase | 1.86 | 5.71 | 3.938 | |
| Q543F6 | Cyclin-dependent kinase 5 | 3.42 | 5.52 | 2.138 | |
| Q3TYK4 | Protein kinase, cAMP dependent regulatory, type Ia | 1.56 | 3.84 | 3.167 | |
| Q60737 | Casein kinase II subunit alpha | 2.83 | 3.19 | 2.023 | |
| Q76MZ3 | Serine/threonine-protein phosphatase 2A 65 kDa regulatory subunit A alpha isoform | 1.76 | 3.15 | 3.732 | |
| P63318 | Protein kinase C gamma | 1.95 | 2.95 | 2.994 | |
| E9QAQ5 | Glycogen synthase kinase-3 beta | 1.55 | 2.49 | 2.565 | |
| Q01065 | Calcium/calmodulin-dependent 3’,5’-cyclic nucleotide phosphodiesterase 1B | 1.61 | 2.48 | 2.152 | |
| Q924S8 | Sprouty-related, EVH1 domain-containing protein 1 | 1.59 | 2.38 | 2.195 | |
| P68404 | Protein kinase C beta | 1.78 | 2.35 | 3.530 | |
| P16054 | Protein kinase C epsilon | 1.86 | 2.35 | 2.084 | |
| P28652 | Calcium/calmodulin-dependent protein kinase type IIb | 1.63 | 2.28 | 2.481 | |
| E9Q3L2 | Phosphatidylinositol 4-kinase alpha | 1.78 | 1.62 | 3.160 | |
| Mitochondrial Transporter | Q91VR2 | ATP synthase subunit gamma | 1.68 | 3.57 | 2.145 |
| P48962 | ADP/ATP translocase 1 | 1.66 | 3.01 | 3.286 | |
| Plasma Membrane | B2BH30 | Metabotropic glutamate receptor 5 | 2.87 | 6.63 | 2.275 |
| Receptor | A2AI21 | Glutamate receptor ionotropic, NMDA 1 | 2.28 | 5.45 | 2.326 |
| Q03137 | Ephrin type-A receptor 4 | 2.95 | 5.30 | 2.723 | |
| Q9QYS2 | Metabotropic glutamate receptor 3 | 4.59 | 5.04 | 2.886 | |
| O55022 | Membrane-associated progesterone receptor component 1 | 3.27 | 4.61 | 3.220 | |
| P12023 | Amyloid beta A4 precursor protein | 3.31 | 2.94 | 4.095 | |
| P62812 | Gamma-aminobutyric acid receptor subunit alpha-1 | 3.36 | 2.84 | 3.530 | |
| P22723 | Gamma-aminobutyric acid receptor subunit gamma-2 | 2.55 | 2.45 | 2.218 | |
| B1AXV0 | DOMON domain-containing protein FRRS1L | 1.53 | 2.26 | 3.493 | |
| P15209 | BDNF/NT-3 growth factors receptor | 3.62 | 2.19 | 3.006 | |
| Q8C419 | G-protein-coupled receptor 158 | 1.73 | 2.03 | 3.192 | |
| C9K0Y3 | AMPA-selective glutamate receptor 1 flip type | 1.85 | 1.72 | 2.582 | |
| Plasma Membrane | G3X9J1 | Sodium/calcium exchanger 1 | 4.96 | 9.71 | 2.908 |
| Transporter | Q3UYK6 | Excitatory amino acid transporter 2 | 2.19 | 4.60 | 4.288 |
| P31648 | Sodium- and chloride-dependent GABA transporter 1 | 2.79 | 2.98 | 3.463 | |
| P48066 | Sodium- and chloride-dependent GABA transporter 3 | 2.20 | 4.97 | 2.666 | |
| Q3UR55 | Sodium/potassium-transporting ATPase subunit beta | 2.11 | 2.88 | 2.188 | |
| G5E829 | Plasma membrane calcium-transporting ATPase 1, PMCA1 | 1.58 | 2.52 | 3.485 | |
| F8WHB1 | Calcium-transporting ATPase, PMCA2 | 1.75 | 2.31 | 2.550 | |
| A2ALL9 | Calcium-transporting ATPase, PMCA3 | 1.90 | 2.28 | 2.155 | |
| Q3UHK1 | Proton myo-inositol cotransporter | 1.67 | 2.09 | 2.582 | |
| E9Q828 | Calcium-transporting ATPase, PMCA4 | 1.75 | 1.92 | 2.708 | |
| Protein Folding/Chaperone | Q542X7 | Chaperonin containing Tcp1, subunit 2 (beta) | 1.84 | 5.79 | 2.481 |
| P38647 | Stress-70 protein, mitochondrial | 3.52 | 5.24 | 2.345 | |
| P11499 | Heat shock protein HSP 90-beta | 2.04 | 4.23 | 4.474 | |
| P63017 | Heat shock cognate 71 kDa protein | 2.92 | 4.00 | 3.325 | |
| Q61699 | Heat shock protein 105 kDa | 5.51 | 3.92 | 2.494 | |
| Q571M2 | Heat shock protein 4 | 4.45 | 3.67 | 2.230 | |
| P17742 | Peptidyl-prolyl cis–trans isomerase A | 1.69 | 1.94 | 4.528 | |
| P14211 | Calreticulin | 2.12 | 1.70 | 2.193 | |
| Protein Turnover | P10605 | Cathepsin B | 2.78 | 5.29 | 4.259 |
| Q9QZM0 | Ubiquilin-2 | 2.44 | 4.35 | 2.216 | |
| Q6ZQ38 | Cullin-associated NEDD8-dissociated protein 1 | 3.94 | 2.90 | 2.035 | |
| Q01853 | Transitional endoplasmic reticulum ATPase | 1.91 | 2.66 | 2.655 | |
| Q7TQI3 | Ubiquitin thioesterase OTUB1 | 2.02 | 2.50 | 2.294 | |
| Q3U900 | Dolichyl-diphosphooligosaccharide–protein glycosyltransferase subunit 1 | 1.64 | 2.30 | 2.758 | |
| RNA Binding Protein | Q3U1S6 | Cold shock domain-containing protein E1 | 1.61 | 2.64 | 2.545 |
| Scaffolding | Q91XM9 | Post-synaptic density protein 93 | 2.45 | 3.06 | 2.444 |
| Q8C8R3 | Ankyrin-2 | 2.07 | 2.87 | 3.297 | |
| O54991 | Contactin-associated protein 1 | 1.63 | 1.50 | 3.241 | |
| Signal Transduction | P19157 | Glutathione S-transferase P 1 | 4.23 | 7.04 | 2.771 |
| Q9Z1B3 | 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase beta-1 | 4.31 | 6.08 | 2.042 | |
| Q3TIQ3 | Phosphatidylinositol transfer protein alpha isoform | 2.53 | 6.05 | 2.005 | |
| Q3UKW2 | Calmodulin | 2.46 | 5.91 | 4.572 | |
| Q8BVQ5 | Protein phosphatase methylesterase 1 | 4.54 | 4.61 | 2.224 | |
| Q6R891 | Neurabin-2 | 2.36 | 4.43 | 2.513 | |
| Q5SS40 | 14–3-3 protein epsilon | 3.33 | 3.95 | 3.784 | |
| Q6AXD2 | Abl-interactor 2 | 2.61 | 3.24 | 2.505 | |
| P63101 | 14–3-3 protein zeta/delta | 2.08 | 2.95 | 3.085 | |
| Q811P8 | Rho GTPase-activating protein 32 | 1.80 | 2.63 | 2.709 | |
| Q9CQV8 | 14–3-3 protein beta/alpha | 1.72 | 2.04 | 2.298 | |
| Trafficking | Q68FF6 | ARF GTPase-activating protein GIT1 | 6.92 | 11.52 | 2.182 |
| P62823 | Ras-related protein Rab-3C | 3.28 | 5.25 | 2.817 | |
| Q3TJ43 | Vacuolar protein sorting 35 | 4.26 | 4.36 | 2.690 | |
| Q3UPA3 | GDP dissociation inhibitor 2 | 2.80 | 3.93 | 3.089 | |
| P51150 | Ras-related protein Rab-7a | 2.15 | 3.60 | 2.078 | |
| Q3TD71 | Secretory carrier membrane protein 1 | 3.49 | 3.35 | 3.142 | |
| P46460 | Vesicle-fusing ATPase | 1.96 | 3.14 | 3.320 | |
| Q9JHU4 | Cytoplasmic dynein 1 heavy chain 1 | 1.73 | 2.48 | 3.818 | |
| P39053 | Dynamin-1 | 1.95 | 2.38 | 3.035 | |
| Q6PHU5 | Sortilin | 2.87 | 1.89 | 3.191 | |
| Q9D4I9 | RAB23 | 1.54 | 1.61 | 2.709 | |
| P61164 | Alpha-centractin | 2.63 | 2.66 | 2.528 | |
| Translation | O55091 | Protein IMPACT | 1.60 | 8.67 | 3.437 |
| P62274 | 40S ribosomal protein S29 | 2.63 | 3.25 | 3.500 | |
| Vacuolar Proton Pump | Q3TG21 | ATPase, H+ transporting, lysosomal V1 subunit A | 1.67 | 6.85 | 3.087 |
| P50516 | ATPase, H+ transporting, lysosomal V1 subunit A | 3.16 | 5.24 | 3.666 | |
| Q8BVE3 | ATPase, H+ transporting, lysosomal V1 subunit H | 1.98 | 3.23 | 2.134 | |
| P51863 | ATPase, H+ transporting, lysosomal V0 subunit D1 | 2.20 | 3.14 | 3.221 | |
| Q9Z1G4 | ATPase, H+ transporting, lysosomal V0 subunit A1 | 1.96 | 3.13 | 3.893 | |
Next, we validated proteomic and RNA array data by Western blotting and RT–PCR. As shown in Fig. 4A, Western blot analysis confirmed that all proteins tested and expected to show increased protein expression were up-regulated at 4 h and 24 h. As controls, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), ezrin, and glutamate transporter EAAT1 (GLAST), which showed no alteration in expression by proteomic analysis, were found to have unchanged protein expression. RT–PCR also confirmed that all mRNA species predicted to be in gliosomes were in fact present (Fig. 4B). As controls, ezrin, EAAT1, clathrin light-chain B (CLTB), and 14–3–3 protein gamma (YWHAG) mRNA species, which showed absence in gliosomes by RNA array, were not detected. Of note, not all proteins – whose mRNAs were identified in the PAP – were up-regulated following compound treatment. For example, GAPDH showed unchanged protein expression, even though its mRNA is present in gliosomes (Fig. 4A,B). This indicated that compound treatment did not induce the entire PAP transcriptome.
Fig. 4.
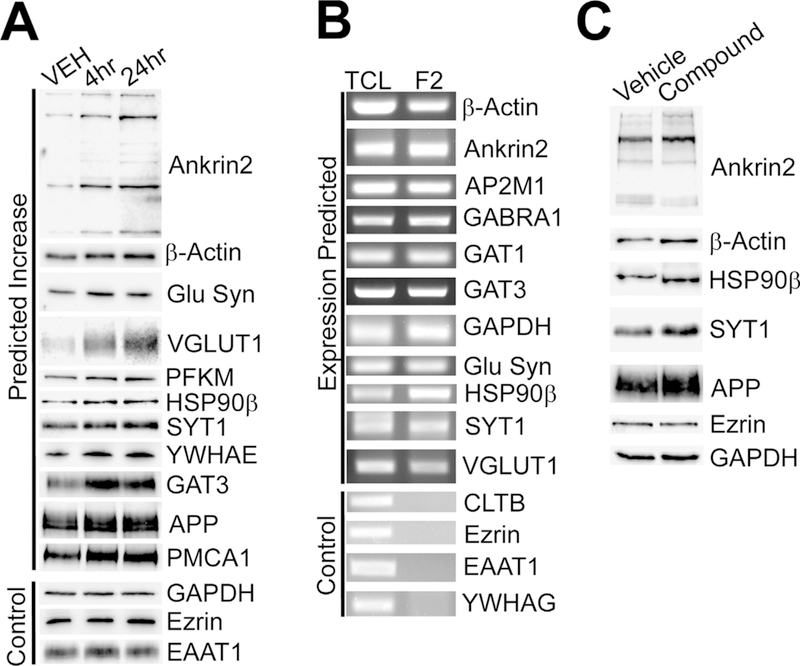
Validation of mass spectrometry and RNA array data and confirmation of more PAP proteins locally translated in gliosomes. Mice were treated with compound and gliosomes were harvested 4 h or 24 h after compound treatment. (A) Western blot validation of selected protein expression 4 and 24 h post-treatment. All proteins that were tested showed increased expression in gliosomes at both time points as predicted by mass spectrometry. The protein expression of controls was unchanged as expected based on mass spectrometry. (B) RT–PCR analysis confirmed the expression of mRNAs selected in gliosomes. As expected, control mRNAs were not expressed in gliosomes. (C) Western blot identification of additional PAP proteins that are locally translated in gliosomes of ex vivo samples. Ankyrin 2, β-actin, Hsp90β, Syt1 and APP were found to be locally translated in gliosomes as expected. The controls, ezrin and GAPDH, protein expression remained unchanged as expected. PFKM, however, did not show ex vivo translation activity suggesting that PFKM, and potentially others not tested, are trafficked to the PAP rather than locally synthesized. Each study consisted of three replicates with consistent results. Representative blots are shown.
Furthermore, we utilized the ex vivo model that we established (Fig. 3) to confirm compound induced local translation of PAP transcripts. Results showed that, in addition to EAAT2, many identified proteins, including ankyrin-2, amyloid precursor protein (APP), b-actin, heat-shock protein 90β (HSP90 β), and synaptotagmin-1 (SYT1), were up-regulated by local protein synthesis (Fig. 4C). As expected, compound did not induce a global up-regulation of the entire transcriptome as ezrin and GAPDH protein expression remained unchanged (Fig. 4C). Taken together, compound treatment results in a rapid up-regulation of a subset of PAP proteins via local translation.
Pyridazine derivatives enhance synaptic plasticity in the hippocampus
Finally, we sought to determine the functional consequences of the proteomic changes to the PAP. To answer this, wild-type mice were treated with vehicle or compound for 7 days. Acute hippocampal slices were then collected and assessed for changes in long-term potentiation (LTP). As shown in Fig. 5A, field potential recordings from CA1 of compound-treated mice (10 slices, 4 animals) showed significantly increased responses to stimulation of CA3 afferents up to 30 min after LTP induction compared to vehicle-treated animals (11 slices, 4 animals). Input/output curves showed similar initial synaptic strength before induction of LTP (Fig. 5B). LTP field potential recordings were further broken down into 5-min bins of time. At each 5-minute epoch (n = 15/epoch/group), CA1 field recordings of compound-treated slices showed significantly enhanced field potential responses relative to vehicle controls (Fig. 5C). This enhanced LTP occurred in tandem with enhanced functional EAAT2 expression. Compound treatment increased both the total and extracellular plasma membrane expression of EAAT2 in the hippocampus as assessed by the pulldown of extracellular biotinylated hippocampal proteins (Fig. 5D). These results show that compound treatment enhances LTP-related plasticity in the CA3-CA1 hippocampal circuit.
Fig. 5.
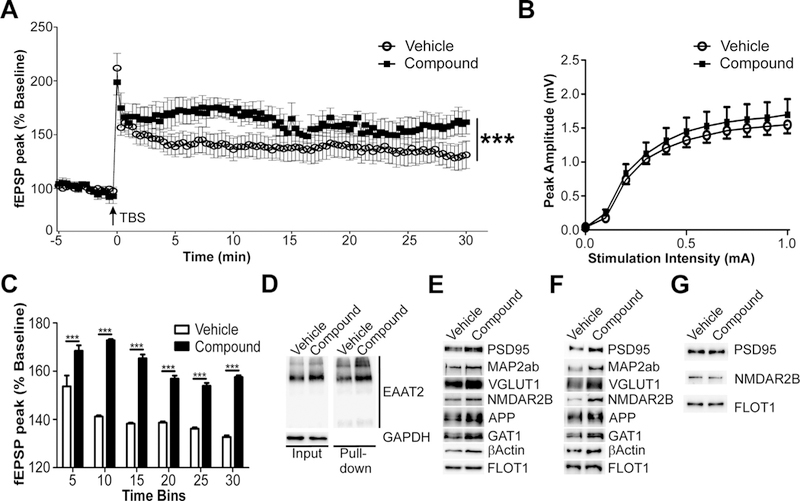
Compound treatment enhances CA3-CA1 LTP and synaptic protein expression in the hippocampus. Mice were treated with vehicle or compound for seven days and acute hippocampal sections were collected for LTP recordings. (A) Compound-treated animals (10 slices, 4 animals) exhibit increased CA1 field potential response after CA3 afferent theta-burst stimulation (TBS) compared to control animals (11 slices, 4 animals). (B) Input/output curves show similar initial synaptic strength responses between groups. (C) Hippocampal LTP recordings were broken into 5-min bins to assess the changes in LTP at different time points after TBS induction. Compound treatment significantly enhances LTP at every time point after treatment. (D) Pulldown of extracellular plasma membrane proteins of the hippocampus show that compound treatment increases functional EAAT2 expression in the hippocampus as well as total EAAT2 expression. (E) Analysis of plasma membrane vesicles from the hippocampus also revealed increased expression of the synaptic proteins PSD95, NMDAR2B, MAP2ab, and VGLUT1, while the loading control, FLOT1, remains unchanged. (F) Similarly, forebrain synaptosomes (F4) from mice treated with compound show similar increases in the same proteins. (G) Compound-treated ex vivo synaptosomes (F4) do not show up-regulation of these synaptic proteins. Therefore, compound is not directly increasing the expression of these proteins through local translation. Data represented as mean ± SEM analyzed using a one-way ANOVA with Bonferroni’s post-hoc tests. Statistical significance denoted as ***p < 0.001.
Furthermore, we examined expression levels of synaptic proteins from compound-treated mice. Results showed marked increases in the expression of post-synaptic protein PSD95 as well as other synaptic proteins including VGLUT1, N-methyl D-aspartate receptor subtype 2B (NMDAR2B), microtubule associated protein 2 a,b (MAP2a,b), APP, GABA transporter 1 (GAT1), and β-actin. Up-regulation of synaptic protein expression was observed regionally in plasma membrane vesicles of the hippocampus (Fig. 5E) and globally throughout synaptosomes of the forebrain (Fig. 5F). However, this increase was not due to compound activated local translation in the synaptosomes as compound-treated ex vivo synaptosomes did not show induction of any synaptic proteins directly (Fig. 5G). This suggests that compound-mediated PAP alterations may mediate the enhanced neuronal/synaptic response observed. Together these data show that compound treatment enhances long-term potentiation in the hippocampus by strengthening the tripartite synapse.
DISCUSSION
We provide support that gliosomes are representative of the PAP. Vesicles of glial origin, gliosomes, have been shown to express proteins found in the PAP of astrocytes, to efficiently mediate the uptake of glutamate, and can be induced to release neurotransmitters through exocytosis (Carney et al., 2014; Nakamura et al., 1993, 1994; Stigliani et al., 2006). Carney et al. (2014) identified that the PAP markers ezrin and vesicle-associated membrane protein 3 are concentrated in this compartment while neuronal markers (e.g. PSD95) are sparse. This is in line with immunostaining studies that show ezrin is concentrated while GFAP and PSD95 are deplete in PAP (Haseleu et al., 2013). Therefore, the gliosome isolation process is a way to biochemically study the PAP in isolation from both the neuronal aspect of the synapse and the main astrocyte processes/cell bodies
Previously, we determined that EAAT2 induction is mediated through translational activation and this induction appeared to be limited to the PMV fraction of the forebrain (Colton et al., 2010; Tian et al., 2007). However, the PMV fraction contains vesicles from both neurons and astrocytes. Here, we separated glial from synaptic vesicles and were able to biochemically determine that compound-mediated EAAT2 induction is limited to gliosomes and not synaptosomes (Fig. 2). This also implies EAAT2 protein expression could be increased by local synthesis in the PAP. While neuronal dendrites and axons are known to take advantage of local translation (for review see, Rangaraju et al., 2017), only recently have astrocytes been shown to locally translate proteins in distal processes (Boulay et al., 2017; Sakers et al., 2017). Sakers et al. (2017) suggested that EAAT2 may be locally translated; however, no direct protein evidence was shown. The ex vivo model provided this direct evidence as robust functional EAAT2 protein induction was observed (Fig. 3). In addition, we identified other proteins that are locally synthesized in the PAP including: HSP90b (protein folding), APP (receptor-mediated signaling), and Synaptotagmin-1 (gliotransmitter-related exocytosis) as well as β-Actin and Ank2 (PAP structure/scaffolding). In corroboration, Sakers et al, (2017) also identified EAAT2, Ank2, and a synaptotagmin (Syt7) mRNA variant in the PAP of astrocytes by TRAP-seq analysis. Using mass spectrometry, Carney et al. (2014) showed that APP and HSP90β expression is concentrated in gliosomes relative to total lysate and synaptosomes in the hip- pocampus. Of note, the concentration we used for ex vivo translation was much higher than our previous publications using cell culture (see Kong et al., 2014). The compound used in the current study was the most potent identified in this ex vivo model at the time. Recently, we have identified new derivatives of this compound series that are more potent and have activity in the low micromolar range (data not shown). However, we have found that compounds that increase EAAT2 in the ex vivo model at higher concentrations shown predict with high fidelity which compounds will function in vivo.
Pyridazine analogs increase the expression of a subset of PAP proteins. EAAT2 is part of a larger complex that mediates glutamate uptake and would, therefore, necessitate the up-regulation of other protein species to functionally alter glutamate homeostasis (Genda et al., 2011). Therefore, we surmised that more PAP proteins, especially those known to compose the EAAT2 glutamate uptake complex, must be concomitantly up-regulated as well. We indeed found that proteins involved in glutamate homeostasis were up-regulated by 4 h and have mRNA present in the PAP. Genda, et al. (2011), used immunoprecipitation to identify EAAT2-interacting proteins that compose the complex that mediates glutamate uptake into the astrocyte. They found that EAAT2 co-compartmentalizes with Na+/K+ ATPase, glycolytic enzymes, and mitochondria, providing a mechanism to spatially match energy and buffering capacity to the demands imposed by glutamate transport (Genda et al., 2011). We identified 56% (40 out of 71) of EAAT2 co-immunoprecipitated proteins listed in this study. Also, many other plasma membrane transporters, like EAAT2, were shown to be up-regulated in response to compound treatment. These include the GABA transporters as well sodium/potassium ATPases and calcium-transporting ATPases. Each of these has previously been identified as astrocytic components that mediate synaptic uptake of GABA and that maintain ion gradients necessary for neurotransmitter uptake. This suggests that compound treatment enhances the astrocytes’ functional capacity to buffer, not only glutamate through EAAT2, but also synaptic GABA and calcium.
Furthermore, we identified categories of proteins upregulated by compound that appears to be integral for PAP plasticity. (1) Proteins associated with vesicular-mediated exocytosis and calcium signaling in the PAP. Astrocytes are known to release glutamatergic vesicles under various conditions and this is believed to be important for the modulation of long-term potentiation (Sahlender et al., 2014). Compound up-regulated vesicular glutamate transporter 1 and a myriad of v-type proton pumps which facilitate the creation of ionic gradients and the loading of glutamate into vesicles. Exocytotic machinery including synaptotagmin, syntaxin binding protein, synaptogyrins and synaptojanin were also found to be up-regulated. Finally, calcium-signaling-related proteins (i.e calmodulin and calcium ATPases) were identified. Together, this suggests that calcium signaling in the PAP may be enhanced which may provide the basis for a mechanism that would facilitate enhanced astrocytic intercellular, exocytotic vesicular signaling from the PAP to the neuronal aspect of the synapse. (2) Cytoskeletal proteins in the PAP were up-regulated including β-Actin and multiple tubulins. These proteins are proposed to be involved in PAP structural plasticity which manifests as dynamic motility of the PAP in relation to synapse coverage (Heller and Rusakov, 2015; Lavialle et al., 2011; Molotkov et al., 2013). This suggests that compound modulates the structural dynamics of the PAP. Perhaps modulation of the PAP by compound is responsible for stabilizing PAP motility, enhancing synaptic bouton stability, and, thus, facilitating stabilization of the tripartite synapse. Overall, it appears that compound-treatment enhances the functional and structural plasticity potential of the PAP.
While there is limited data regarding the astrocytic role in various forms of synaptic plasticity when compared to neuronal roles, astrocytes are known to play a significant role in the maintenance, formation, and regulation of synapses (Bernardinelli et al., 2014; Chung et al., 2015; Heller and Rusakov, 2015; Perez-Alvarez et al., 2014). Hippocampal astrocytes increase EAAT2 expression within 1 h of fear-conditioning training and increased EAAT2 expression is associated with enhanced spatial memory (Choi et al., 2016; Heo et al., 2012). This rapid up-regulation of EAAT2 protein in the hippocampus mirrors what was observed in the current study suggesting that these two induction mechanisms may be one in the same. Importantly, when astrocytic PAPs were ablated in the dentate gyrus of the hippocampus, the mice were unable to retain contextual fear memories (Choi et al., 2016). Also, two separate groups have shown that the motility of the astrocyte PAP is important for stabilizing and regulating hippocampal potentiation (Bernardinelli et al., 2014; Perez-Alvarez et al., 2014). The PAP responds to synaptic activity by extending and stabilizing its coverage of individual synapses which, in turn, stabilizes the maturation of synaptic boutons (Bernardinelli et al., 2014; Perez-Alvarez et al., 2014). Together, these studies strongly suggest that the rapid up-regulation of EAAT2 and enhanced motility in the PAP are essential functions of astrocytes in the genesis of long-term potentiation and the process of learning and memory. We propose that pyridazine analogs activate an endogenous astrocytic mechanism that utilizes both trafficking and local protein synthesis to scale the astrocytic PAP and, in turn, facilitate plasticity of the neuronal aspect of the synapse.
This is the first report of a manipulation that can directly and specifically induce local translation in the PAP of astrocytes. To date, all other studies regarding local translation in the PAP have only identified that local translation occurs. Herein we have shown that pyridazine derivatives activate a mechanism that enhances, not only EAAT2 protein expression specifically in the PAP through local translation, but other PAP proteins as well. However, this is a regulated process as other mRNA species in the PAP transcriptome are not modulated by compound. Local translation of EAAT2 and additional PAP proteins identified here appears to play a significant role in modulating the functional and structural properties of the PAP, and may represent a form of astrocytic plasticity. This, in turn, may account for the enhanced LTP in hippocampal slices of compoundtreated mice. Our current data support the notion that local translation in the PAP of astrocytes represents a potential mechanism by which the PAP can respond to or induce changes in the synaptic microenvironment as well as modulate synaptic plasticity. This supports the need to further define the signaling mechanism associated with pyridazine analogs as well as to identify and understand other signaling mechanisms that activate local translation in the PAP.
ACKNOWLEDGEMENTS
We thank Ms. Liching Lai for her excellent technical support and Dr. Min Zhou for assistance with the electrophysiology study. This work was supported by grants from the BrightFocus Foundation, the Alzheimer’s Drug Discovery Foundation, the Harrington Discovery Institute, and NIA (1 U01 AG054444–01A1). Electrophysiology studies were conducted in the Ohio State University Neuroscience Center Core which is supported by NINDS Center Core Grant (P30 NS045758 & P30 NS104177).
Abbreviations:
- Ank2
ankyrin-2
- AP-2
adaptor complex 2 subunit
- APP
amyloid precursor protein
- AUC
area under the curve
- BDNF/NT-3
brain-derived neurotrophic factor and neurotrophin-3 receptor
- CDS
coding sequence
- CLTB
clathrin light-chain B
- EAAT1/GLAST
excitatory amino acid transporter 1
- EAAT2
excitatory amino acid transporter 2
- FDR
false discovery rate
- fEPSP
field excitatory post-synaptic potentials
- GABA
gamma-aminobutyric acid
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GAT1
GABA transporter 1
- GDP
guanosine diphosphate
- GFAP
glial fibrillary acidic protein
- HPLC
high-performance liquid chromatography
- HSC70
heat shock cognate 71-kDa protein
- HSP90β
heat shock protein 90-beta
- LC/MS/MS
liquid chromatography mass spectrometry/mass spectrometry
- LRM
lipid raft microdomain
- LTP
long-term potentiation
- MAP2ab
microtubule-associated protein 2
- NMDAR2B
N-methyl D-aspartate receptor subtype 2B
- PAP
perisynaptic astrocytic process
- PMV
plasma membrane vesicle
- PSD93
post-synaptic density 93
- PSD95
post-synaptic density 95
- RIPA
radioimmunoprecipitation assay
- SE
sucrose-EDTA buffer
- SYT1
synaptotagmin-1
- SYT7
synaptotagmin-7
- TBS
theta-burst stimulation
- UTR
untranslated region
- VGLUT1
vesicular glutamate transporter 1
- YWHAG
tyrosine 3/tryptophan 5-monooxygenase activation protein gamma
REFERENCES
- Abulseoud OA, Miller JD, Wu J, Choi DS, Holschneider DP (2012) Ceftriaxone upregulates the glutamate transporter in medial prefrontal cortex and blocks reinstatement of methamphetamine seeking in a condition place preference paradigm. Brain Res 1456:14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asztely F, Erdemli G, Kullmann DM (1997) Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron 18:281–293. [DOI] [PubMed] [Google Scholar]
- Bernardinelli Y, Randall J, Janett E, Nikonenko I, Konig S, Jones EV, Flores CE, Murai KK, Bochet CG, Holtmaat A, Muller D (2014) Activity-dependent structural plasticity of perisynaptic astrocytic domains promotes excitatory synapse stability. Curr Biol 24:1679–1688. [DOI] [PubMed] [Google Scholar]
- Boulay AC, Saubamea B, Adam N, Chasseigneaux S, Mazare N, Gilbert A, Bahin M, Bastianelli L, Blugeon C, Perrin S, Pouch J, Ducos B, Le Crom S, Genovesio A, Chretien F, Decleves X, Laplanche JL, Cohen-Salmon M (2017) Translation in astrocyte distal processes sets molecular heterogeneity at the gliovascular interface. Cell Discov 3:17005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristol LA, Rothstein JD (1996) Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann Neurol 39:676–679. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH (2002) Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 22:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butchbach ME, Tian G, Guo H, Lin CL (2004) Association of excitatory amino acid transporters, especially EAAT2, with cholesterol-rich lipid raft microdomains: importance for excitatory amino acid transporter localization and function. J Biol Chem 279:34388–34396. [DOI] [PubMed] [Google Scholar]
- Carney KE, Milanese M, van Nierop P, Li KW, Oliet SH, Smit AB, Bonanno G, Verheijen MH (2014) Proteomic analysis of gliosomes from mouse brain: identification and investigation of glial membrane proteins. J Proteome Res 13:5918–5927. [DOI] [PubMed] [Google Scholar]
- Choi M, Ahn S, Yang EJ, Kim H, Chong YH, Kim HS (2016) Hippocampus-based contextual memory alters the morphological characteristics of astrocytes in the dentate gyrus. Mol Brain 9:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chotibut T, Davis RW, Arnold JC, Frenchek Z, Gurwara S, Bondada V, Geddes JW, Salvatore MF (2014) Ceftriaxone increases glutamate uptake and reduces striatal tyrosine hydroxylase loss in 6-OHDA Parkinson’s model. Mol Neurobiol 49:1282–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu K, Lee ST, Sinn DI, Ko SY, Kim EH, Kim JM, Kim SJ, Park DK, Jung KH, Song EC, Lee SK, Kim M, Roh JK (2007) Pharmacological induction of ischemic tolerance by glutamate transporter-1 (EAAT2) upregulation. Stroke 38:177–182. [DOI] [PubMed] [Google Scholar]
- Chung WS, Allen NJ, Eroglu C (2015) Astrocytes control synapse formation, function, and elimination. Cold Spring Harb Perspect Biol 7 a020370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colton CK, Kong Q, Lai L, Zhu MX, Seyb KI, Cuny GD, Xian J, Glicksman MA, Lin CL (2010) Identification of translational activators of glial glutamate transporter EAAT2 through cell-based high-throughput screening: an approach to prevent excitotoxicity. J Biomol Screen 15:653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derouiche A, Frotscher M (2001) Peripheral astrocyte processes: monitoring by selective immunostaining for the actin-binding ERM proteins. Glia 36:330–341. [DOI] [PubMed] [Google Scholar]
- Dunkley PR, Jarvie PE, Robinson PJ (2008) A rapid Percoll gradient procedure for preparation of synaptosomes. Nat Protoc 3:1718–1728. [DOI] [PubMed] [Google Scholar]
- Genda EN, Jackson JG, Sheldon AL, Locke SF, Greco TM, O’Donnell JC, Spruce LA, Xiao R, Guo W, Putt M, Seeholzer S, Ischiropoulos H, Robinson MB (2011) Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J Neurosci 31:18275–18288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Lai L, Butchbach ME, Stockinger MP, Shan X, Bishop GA, Lin CL (2003) Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum Mol Genet 12:2519–2532. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG (2007) Synaptic islands defined by the territory of a single astrocyte. J Neurosci 27:6473–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseleu J, Anlauf E, Blaess S, Endl E, Derouiche A (2013) Studying subcellular detail in fixed astrocytes: dissociation of morphologically intact glial cells (DIMIGs). Front Cell Neurosci 7:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller JP, Rusakov DA (2015) Morphological plasticity of astroglia: understanding synaptic microenvironment. Glia 63:2133–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo S, Jung G, Beuk T, Hoger H, Lubec G (2012) Hippocampal glutamate transporter 1 (GLT-1) complex levels are paralleling memory training in the Multiple T-maze in C57BL/6J mice. Brain Struct Funct 217:363–378. [DOI] [PubMed] [Google Scholar]
- Kong Q, Chang LC, Takahashi K, Liu Q, Schulte DA, Lai L, Ibabao B, Lin Y, Stouffer N, Das Mukhopadhyay C, Xing X, Seyb KI, Cuny GD, Glicksman MA, Lin CL (2014) Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. J Clin Invest 124:1255–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Q, Takahashi K, Schulte D, Stouffer N, Lin Y, Lin CL (2012) Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol Dis 47:145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavialle M, Aumann G, Anlauf E, Prols F, Arpin M, Derouiche A (2011) Structural plasticity of perisynaptic astrocyte processes involves ezrin and metabotropic glutamate receptors. Proc Natl Acad Sci USA 108:12915–12919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SG, Su ZZ, Emdad L, Gupta P, Sarkar D, Borjabad A, Volsky DJ, Fisher PB (2008) Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J Biol Chem 283:13116–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung TC, Lui CN, Chen LW, Yung WH, Chan YS, Yung KK (2012) Ceftriaxone ameliorates motor deficits and protects dopaminergic neurons in 6-hydroxydopamine-lesioned rats. ACS Chem Neurosci 3:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Mallory M, Alford M, Tanaka S, Masliah E (1997) Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J Neuropathol Exp Neurol 56:901–911. [DOI] [PubMed] [Google Scholar]
- Liaw WJ, Stephens RL Jr, Binns BC, Chu Y, Sepkuty JP, Johns RA, Rothstein JD, Tao YX (2005) Spinal glutamate uptake is critical for maintaining normal sensory transmission in rat spinal cord. Pain 115:60–70. [DOI] [PubMed] [Google Scholar]
- Lin Y, Tian G, Roman K, Handy C, Travers JB, Lin CL, Stephens RL Jr (2009) Increased glial glutamate transporter EAAT2 expression reduces visceral nociceptive response in mice. Am J Physiol Gastrointest Liver Physiol 296:G129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, Dorner JL, Shou M, Sari Y, Barton SJ, Sengelaub DR, Kennedy RT, Rebec GV (2008) Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington’s disease phenotype in the R6/2 mouse. Neuroscience 153:329–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineur YS, Picciotto MR, Sanacora G (2007) Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biol Psychiatry 61:250–252. [DOI] [PubMed] [Google Scholar]
- Molotkov D, Zobova S, Arcas JM, Khiroug L (2013) Calcium-induced outgrowth of astrocytic peripheral processes requires actin binding by Profilin-1. Cell Calcium 53:338–348. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Iga K, Shibata T, Shudo M, Kataoka K (1993) Glial plasmalemmal vesicles: a subcellular fraction from rat hippocampal homogenate distinct from synaptosomes. Glia 9:48–56. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Kubo H, Kataoka K (1994) Uptake of transmitter amino acids by glial plasmalemmal vesicles from different regions of rat central nervous system. Neurochem Res 19:1145–1150. [DOI] [PubMed] [Google Scholar]
- Olney JW (1969) Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 164:719–721. [DOI] [PubMed] [Google Scholar]
- Perez-Alvarez A, Navarrete M, Covelo A, Martin ED, Araque A (2014) Structural and functional plasticity of astrocyte processes and dendritic spine interactions. J Neurosci 34:12738–12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju V, Tom Dieck S, Schuman EM (2017) Local translation in neuronal compartments: how local is local? EMBO Rep 18:693–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Jin L, Dykes-Hoberg M, Kuncl RW (1993) Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc Natl Acad Sci USA 90:6591–6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB (2005) Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433:73–77. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol 38:73–84. [DOI] [PubMed] [Google Scholar]
- Sahlender DA, Savtchouk I, Volterra A (2014) What do we know about gliotransmitter release from astrocytes? Philos T R Soc B 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakers K, Lake AM, Khazanchi R, Ouwenga R, Vasek MJ, Dani A, Dougherty JD (2017) Astrocytes locally translate transcripts in their peripheral processes. Proc Natl Acad Sci USA 114: E3830–E3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stigliani S, Zappettini S, Raiteri L, Passalacqua M, Melloni E, Venturi C, Tacchetti C, Diaspro A, Usai C, Bonanno G (2006) Glia re-sealed particles freshly prepared from adult rat brain are competent for exocytotic release of glutamate. J Neurochem 96:656–668. [DOI] [PubMed] [Google Scholar]
- Szydlowska K, Tymianski M (2010) Calcium, ischemia and excitotoxicity. Cell Calcium 47:122–129. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Kong Q, Lin Y, Stouffer N, Schulte DA, Lai L, Liu Q, Chang LC, Dominguez S, Xing X, Cuny GD, Hodgetts KJ, Glicksman MA, Lin CL (2015) Restored glial glutamate transporter EAAT2 function as a potential therapeutic approach for Alzheimer’s disease. J Exp Med 212:319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276:1699–1702. [DOI] [PubMed] [Google Scholar]
- Tian G, Kong Q, Lai L, Ray-Chaudhury A, Lin CL (2010) Increased expression of cholesterol 24S-hydroxylase results in disruption of glial glutamate transporter EAAT2 association with lipid rafts: a potential role in Alzheimer’s disease. J Neurochem 113:978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G, Lai L, Guo H, Lin Y, Butchbach ME, Chang Y, Lin CL (2007) Translational control of glial glutamate transporter EAAT2 expression. J Biol Chem 282:1727–1737. [DOI] [PubMed] [Google Scholar]
- Ventura R, Harris KM (1999) Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci 19:6897–6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Mishra V, Sasmal D, Raghubir R (2010) Pharmacological evaluation of glutamate transporter 1 (GLT-1) mediated neuroprotection following cerebral ischemia/reperfusion injury. Eur J Pharmacol 638:65–71. [DOI] [PubMed] [Google Scholar]