

SUMMARY
Ectopic expression of transcription factors (TFs) can reprogram cell state. However, because of the large combinatorial space of possible TF cocktails, it remains difficult to identify TFs that reprogram specific cell types. Here, we develop Reprogram-Seq to experimentally screen thousands of TF cocktails for reprogramming performance. Reprogram-Seq leverages organ-specific cell-atlas data with single-cell perturbation and computational analysis to predict, evaluate, and optimize TF combinations that reprogram a cell type of interest. Focusing on the cardiac system, we perform Reprogram-Seq on MEFs using an undirected library of 48 cardiac factors and, separately, a directed library of 10 epicardial-related TFs. We identify a combination of three TFs, which efficiently reprogram MEFs to epicardial-like cells that are transcriptionally, molecularly, morphologically, and functionally similar to primary epicardial cells. Reprogram-Seq holds promise to accelerate the generation of specific cell types for regenerative medicine.
Graphical Abstract
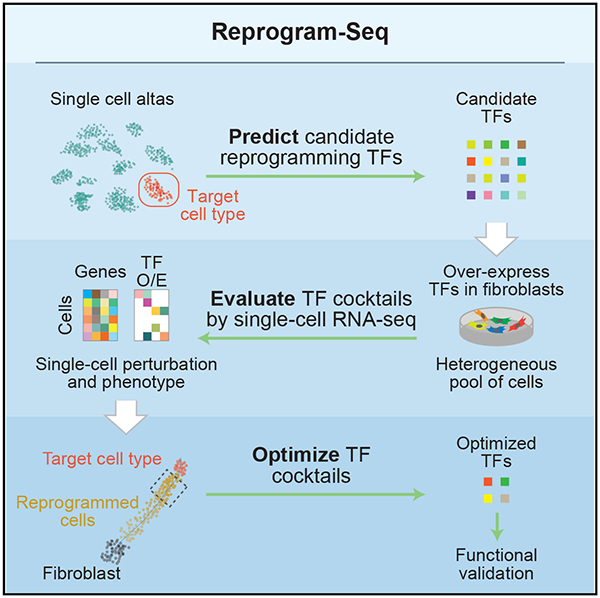
In Brief
Direct reprogramming of a cellular state holds promise for regenerative medicine. Duan et al. present Reprogram-Seq to identify, evaluate, and optimize transcription factor cocktails that drive direct reprogramming of a cell state. They apply Reprogram-Seq to generate epicardial-like cells and show how the approach can be leveraged for rational cellular reprogramming.
INTRODUCTION
Ectopic expression of transcription factors (TFs) can reprogram cellular states. For example, forced expression of MyoD1 alone reprograms fibroblasts to myoblasts (Tapscott et al., 1988). In addition, transduction of Oct4, Sox2, Klf4, and Myc converts mouse embryonic fibroblasts (MEFs) to an induced pluripotent stem (iPS) cell state (Takahashi and Yamanaka, 2006). This seminal study required three time- and labor- intensive features: (1) an extensive prior knowledge base to inform selection of the original 24 TF pool, (2) generation of a knockin mouse to report on pluripotency (Fbx15-bGeo), and (3) laborious “N-1” TF evaluation that required multiple iterative rounds of screening. Subsequent studies have used similar strategies to optimize iPS reprogramming efficiency or identify TF cocktails to reprogram other cell states. For example, Gata4, Hand2, Mef2c, and Tbx5 reprogram fibroblasts into cardiomyocyte-like cells (Ieda et al., 2010; Song et al., 2012b). However, efficiently generating a multitude of cell types by direct reprogramming will require new approaches for systematic TF identification, reprogramming evaluation, and cell-type assessment. To address this problem, several computational approaches have recently been described (Cahan et al., 2014; D’Alessio et al., 2015; Morris et al., 2014; Rackham et al., 2016). However, empiric experimental evaluation is not a component of those methods. As the rules governing cellular reprogramming are poorly defined, integrating both computational and experimental observations to identify TF cocktails capable of reprogramming specific cell types could be a viable alternative strategy.
Here, we describe Reprogram-Seq, an approach that leverages organ-specific cell-atlas data with single-cell perturbation and computational analysis to predict, evaluate, and optimize TF combinations that reprogram a cell type of interest. Focusing on the cardiac system, we demonstrate two orthogonal approaches for reprogramming epicardial-like cells. First, we apply Reprogram-Seq to screen random combinations of a large 48-factor (48F) library for reprogramming potential. Second, in a more focused approach, we establish a single-cell atlas of the P0 mouse heart to subsequently identify 10 candidate TFs (10F) for epicardial reprogramming. Reprogram-Seq analysis on these TFs identified a three-TF combination (3F) that more efficiently generated cells resembling an epicardial cell state. Importantly, we show that 3F-reprogrammed MEFs resemble genuine epicardial cells morphologically and functionally. Unlike recent approaches relying purely on computational prediction, Reprogram-Seq empirically tests and evaluates thousands of TF cocktails by direct experimental measurement. Direct reprogramming has emerged as a promising approach for cellular therapy (Srivastava and DeWitt, 2016), but the generation of specific cell types on demand remains challenging. Thus, Reprogram-Seq can be applied to reprogram cell types defined by single-cell genomics with implications for regenerative medicine.
RESULTS
Single-Cell Combinatorial Reprogramming with Reprogram-Seq
For a given in vivo cell type defined by single-cell RNA sequencing (scRNA-Seq), we hypothesize that overexpression of a cocktail of cell-type specific TFs can drive cellular reprogramming toward these cells. To enumerate, test, and identify combinations of TFs that can efficiently reprogram fibroblasts to this cell type, we have developed an approach called Reprogram-Seq (Figure 1), which measures one key phenotype of reprogramming: the transcriptome. Briefly, we infect MEFs with a retroviral library of candidate TFs. At high infectivity, each cell expresses multiple exogenous TFs that drive transcriptional reprogramming. This results in a library of perturbed cells, in which different cells can express various combinations of exogenous TFs. To characterize these cells phenotypically, we perform scRNA-Seq. For each cell sequenced, we measure its full transcriptome, which we use to identify groups of cells in which exogenous TFs have driven transcriptional reprogramming. Importantly, detection of exogenous TFs does not rely on distal barcoding, thus avoiding complications with barcode recombination (Hill et al., 2018; Sack et al., 2016; Xie et al., 2018). By scaling the reprogramming experiments to the single-cell level, Reprogram-Seq allows a parallel search for reprogramming factors. Furthermore, profiling thousands of individual cells using scRNA-Seq allows many distinct combinations of TF cocktails to be tested simultaneously in a single, well-controlled experiment.
Figure 1. Combinatorial Cellular Reprogramming with Reprogram-Seq.

Analysis of single-cell transcriptomes or expert knowledge identifies a set of candidate TFs for direct reprogramming. TFs are cloned into retroviral expression vectors and induced in MEFs. In Reprogram-Seq, we performed scRNA-Seq on thousands of individual cells to identify successfully reprogrammed cells and the exogenous factors expressed in those cells. The process can then be repeated with those factors to optimize TF cocktails for direct cellular reprogramming.
Unbiased Reprogramming with 48 Factors
To test the feasibility of Reprogram-Seq on the cardiac system (Figure 2A), we first defined the transcriptional state of P0 mouse hearts. Our analysis of 15,684 primary, single-cell transcriptomes identified known cardiac cell types and their molecular markers, including cardiomyocytes (Tnnt2+), cardiac fibroblasts (Col1a1+), epicardial cells (Wt1+), and macrophages (Lys6c+) (Figures 2B, S1A, and S1B; Table S1).
Figure 2. Application of Reprogram-Seq for Unbiased Reprogramming with 48 Factors.

(A) Experimental design. We performed scRNA-Seq on MEFs reprogrammed with 48F, uninfected MEFs, and primary P0 mouse heart cells.
(B) The 25,776 cells were clustered and plotted using t-distributed stochastic neighbor embedding (t-SNE; MEF-derived, red; primary cells, non-red).
(C) Heatmap of cells by Myod1 expression. MEF-derived cells and Cluster 20 are indicated.
(D) Expression of Myod1 in MEF-derived cells.
(E) Differential-expression analysis of 48F in MEF-derived clusters, as compared with all other reprogrammed cells. Each dot represents a gene (colored by fold change and sized by p value).
(F) Gene ontology analysis for genes differentially expressed in Cluster 20.
(G) Heatmap of cells by Wt1 expression. Indicated is Cluster 12, which consists of MEF-derived cells and primary cells.
(H) Expression of Wt1 and Bnc1 in MEF-derived cells.
Next, we tested whether Reprogram-Seq could perform combinatorial TF reprogramming of many TF cocktails at the same time. We created a complex library of 48 TFs and genes (48F) expressed in cardiac cells based on literature curation and bulk transcriptome analysis (Table S2). Some of those genes have known roles in cardiac biology, including members of the Gata, Hand, Mef2, and T-box families. As a positive control, we also included MyoD1, which alone can reprogram fibro-blasts to skeletal muscle cells. We pack aged a pooled retroviral library, infected MEFs, and profiled their transcriptomes by scRNA-Seq. The vast majority of infected MEFs clustered together with uninfected MEFs (Figures 2B and S1C), suggesting that most TFs do not dramatically alter the MEF transcriptome when compared with in vivo cell types. However, there were two notable exceptions.
Almost all of the cells in Cluster 20 were MEF-derived and transcriptionally distinct from the other MEF-derived cells (Figure 2B). This cluster had the highest levels of MyoD1 expression compared with other MEF-derived cell clusters (Figures 2C–2E), suggesting that these cells are derived from MEFs infected with the retrovirus driving MyoD1 expression. Supporting that possibility, the genes induced in Cluster 20 were significantly enriched for gene ontology annotations related to muscle development (Figure 2F), and the cells in Cluster 20 exhibited endogenous MyoD1 gene expression (Figure S2A). In addition, MEFs separately infected with MyoD1 retrovirus correlated more with Cluster 20 than they did with any other cluster (Figure S2B). Taken together, these results indicate that MyoD1 successfully outcompetes 47 other exogenous factors in a subpopulation of cells to push cell state toward a skeletal muscle cell fate and highlights the fidelity of combinatorial reprogramming achieved by Reprogram-Seq.
Next, we wondered whether Reprogram-Seq could identify MEFs reprogrammed to a cardiac-like cell fate. Interestingly, Cluster 12 contains a mixture of in vivo (63.1%) and MEF-derived(36.9%) cells (Figure 2G). All the in vivo epicardial cells were found in this cluster, suggesting that the MEF-derived cells may have activated an epicardial program. Consistent with that possibility, the MEF-derived cells in Cluster 12 expressed known markers of epicardial cells, including Wt1 and Bnc1, which are not in the 48F (Figure 2H). To exclude the possibility that these reprogrammed epicardial-like cells were derived from the proliferation of epicardial progenitor cells in MEFs, we examined the expression of cell proliferation gene Cenpp (Figure S1B). Cenpp expression was depleted in primary epicardial cells as well as in MEF-derived epicardial-like cells, suggesting that these cells were derived from reprogramming rather than from proliferation. To identify the exogenous TFs induced in these cells, we examined the expression of 48F. Strikingly, 78.6% of MEF-derived cells in Cluster 12 expressed Gata6, compared with only 22.1% in other MEF-derived cells (Figures 2E and S2C) (p = 4.4E−62; binomial). Similarly, Hand2 expression was highly enriched (48.8% Cluster 12; 9.6% other; p = 5.2E−43). These results suggest that Hand2 and Gata6 may transcriptionally reprogram MEFs toward an epicardial-like state. Consistent with these findings, mouse genetic studies have previously shown that Gata6 and Hand2 function in epicardial development and maintenance (Barnes et al., 2011; Ko-lander et al., 2014).
Together, these results indicate that Reprogram-Seq can efficiently search a large combinatorial space to simultaneously identify multiple TF cocktails (MyoD1 and Gata6/Hand2) that reprogram MEFs to distinct cell states.
Rational Reprogramming of Epicardial Cells
Our single-cell analysis of in vivo heart cells identified several cell types. Although many TF cocktails can drive MEFs toward a cardiomyocyte cell fate, TF cocktails for other cardiac cell types remain limited. Next, we applied Reprogram-Seq to engineer epicardial cells rationally.
To identify a set of candidate TFs for epicardial reprogramming, we performed differential gene-expression analysis of in vivo epicardial cells (the destination state of reprogramming) compared with MEFs (the origin state). This analysis identified 10 transcription factors (10F) (Figure 3A). We were encouraged by the presence of known markers of epicardial cells, including Tbx18 (Cai et al., 2008; Witty et al., 2014; Wu et al., 2013), Tcf21 (Acharya et al., 2012; Tandon et al., 2013), and Wt1 (von Gise et al., 2011; Guadix et al., 2011), as well as TFs identified in our previous, unbiased reprogramming (Gata6 and Hand2).
Figure 3. Application of Reprogram-Seq for Epicardial Reprogramming.

(A) We identified 10 TFs differentially expressed in primary epicardial cells compared with uninfected MEFs. Shown is the expression of 10F in MEFs and P0 mouse heart cells.
(B) Experimental design. We performed scRNA-Seq on MEFs reprogrammed with 10F, uninfected MEFs, and primary P0 mouse hearts.
(C) t-SNE plots consisting of all sequenced cells in (B). The 10F-reprogrammed MEFs (top) and uninfected MEFs (bottom) are colored orange. Primary epicardial cells are indicated in blue. Cluster 10 is outlined in black.
(D) Enrichment of reprogrammed MEFs compared with uninfected MEFs in different cell clusters.
(E) Expression of epicardial marker genes Gata5 and Upk1b in primary and MEF-derived cells.
(F) We performed pseudotime analysis on all MEF-derived cells and primary epicardial cells (blue). Uninfected MEFs (left), Cluster 10 reprogrammed cells (right), and other reprogrammed cells (middle) are indicated in orange.
(G) Bar chart indicating the percentage of cells in each group belonging to the terminal pseudotime branch (F).
(H) Heatmap of cells by (left) Gpm6a and (right) Upk1b expression.
To test the ability of 10F to convert MEFs to an epicardial-like fate, we performed Reprogram-Seq with 10F after 7 days of retroviral induction (Figure 3B). As a control, we also sequenced uninfected MEFs. Notably, we found that the vast majority of cells in Cluster 10 (90%) were derived from 10F-reprogrammed MEFs, rather than from control MEFs (10%) (Figures 3C and 3D). Consistently, the cells in Cluster 10 were highly enriched for exogenously expressed TFs (Figure S3A).
To assess whether Cluster 10 cells share molecular similarities with in vivo epicardial cells, we examined the expression of known epicardial marker genes Anxa8, Gata5, Gpm6a, Krt8, Krt19, and Upk1b (Witty et al., 2014), none of which are in 10F. We observed significant induction of those epicardial markers in Cluster 10 cells compared with uninfected MEFs and other MEF-derived clusters (Figures 3E, S3B, and S3C).
Several lines of evidence suggest that cells in Cluster 10 are derived from reprogramming, rather than from proliferation of MEFs. First, consistent with the idea that reprogrammed cells retain a memory of their origin (Kim et al., 2010), Cluster 10 cells retain residual expression of fibroblast marker genes, including Spp1 (Figure S3B). Those fibroblast markers are more highly expressed in other MEF-derived clusters and are not expressed in primary epicardial cells, suggesting that Cluster 10 cells are undergoing reprogramming. Second, we observed cell proliferation marker genes, including Cenpp, were depleted in both primary epicardial cells and Cluster 10 epicardial-like cells (Figure S3B).
To more precisely quantify the efficiency of epicardial reprogramming across the transcriptome, we performed pseudotime analysis across reprogrammed MEFs, control MEFs (origin cell), and primary epicardial cells (target cell). Pseudotime analysis reveals that the starting population of uninfected MEFs was somewhat heterogeneous but largely distinct from the destination population of primary epicardial cells (Figure 3F, left). A critical juncture in that pseudotime was the branch between uninfected MEFs and primary cells, which we denoted as the terminal branch. Surprisingly, we observed that the vast majority of reprogrammed cells in Cluster 10 belong to the terminal branch (91.6%) and were particularly enriched in the subset of cells in pseudotime space closest to primary epicardial cells (Figures 3F and 3G). In comparison, only 13.57% of other 10F-reprogrammed MEFs occupied the terminal branch. Consistent with these data, known epicardial markers were highly expressed in the terminal branch cells (Figure 3H).
Optimized Epicardial Reprogramming with 3F
The above results suggest that 10F-reprogrammed MEFs have undergone transcriptional reprogramming toward an epicardial-like state. However, it is possible that only a subset of genes in 10F drives that outcome. To identify subsets of key factors, previous studies have relied on labor-intensive, “N-1” approaches, whereby distinct TF cocktails are iteratively tested for reprogramming performance. To accelerate that process, we devised an alternative strategy relying on the single-cell measurements of Reprogram-Seq (see STAR Methods). We observed dramatic enrichment of Atf3, Gata6, and Hand2 (3F) expression in the reprogrammed cells of Cluster 10, with more than half of the cells expressing those factors (Figure S3A). Collectively, 3F is expressed >2.6-fold more in Cluster 10 than it is in other reprogrammed cells. In addition, we confirmed that cells in Cluster 10 exhibited significant exogenous expression of 3F (Figures 4A and S4A). These results suggest that 3F may be an optimized TF cocktail for epicardial reprogramming.
Figure 4. Optimized Epicardial Reprogramming with Atf3, Gata6, and Hand2.

(A) Estimates of exogenous and endogenous expression for 10F in Cluster 10 reprogrammed cells.
(B) Experimental design. We performed scRNA-Seq on uninfected MEFs and MEFs reprogrammed with 10F for 7 days, 10F for 14 days, and 3F for 7 days.
(C–F) We performed pseudotime analysis on the MEF-derived cells above and primary epicardial cells (blue). Indicated in orange are uninfected MEFs (C) and MEFs reprogrammed with 10F for 7 days (D), 10F for 14 days (E), and 3F for 7 days (F). States A–H of pseudotime are indicated in (C).
(G) Bar plot indicating the fraction of cells in each experiment that belong to the initial State A (left) or terminal State H (right) of pseudotime.
(H) Heatmap of cells by the expression of the epicardial marker gene Gpm6a (left) and Upk1b (right).
To test whether exogenous expression of 3F was sufficient to transcriptionally reprogram MEFs to epicardial-like cells, we performed Reprogram-Seq with 3F for 7 days. To assess performance, we compared 10F reprogramming for 7 days and 14 days (Figure 4B), as increased duration of reprogramming has been observed to improve efficacy (Ieda et al., 2010; Song et al., 2012a). Combined pseudotime analysis of all cells indicated a trajectory mirroring the previous analysis. Uninfected MEFs were predominantly enriched at the initial pseudotime state A, whereas all the primary epicardial cells clustered in terminal pseudotime state H (Figure 4C). Reprogrammed cells occupied states between those two extremes (Figures 4D–4F). Interestingly, a proportion of uninfected MEFs resides in pseudotime state I. Those cells are transcriptionally distinct from uninfected cells in state A, indicating that heterogeneity already exists in the original MEF population (Figure S4E; Table S3). In addition, we observed that reprogrammed epicardial-like cells transcriptionally resemble cells in state A (Figure S4F), suggesting that epicardial-like reprogramming proceeds from state A to state H, rather than from state I.
As a measure of reprogramming efficiency, we compared the fraction of cells in terminal state H. Among 10F cells, we observed a dramatic 3.6-fold (p < 2.2E 16) increase in cells attaining the terminal state H after 14 days (26.0%) of reprogramming compared with 7 days (7.3%), which was accompanied by a corresponding decrease in the initial state A (Figures 4G and S4B). These results confirm that reprogramming efficiency increases with time. Next, we assessed the efficiency of 3F reprogramming. Surprisingly, we found that the performance of 3F for 7 days (31.4% of cells reaching the epicardial state H) exceeded that of 10F reprogramming for both 7 days and 14 days. We also observed similar performance of 3F reprogramming after 14 days (Figure S4C). 3F reprogramming was efficient: only11.2% of cells remained in the initial state A. In addition, 42.3% of cells were on the pseudotime trajectory toward epicardial cells (states BDFH), as indicated by increased expression of the epicardial markers in this path (Figures 4H and S4D). These observations suggest that 3F is an optimized cocktail for transcriptional reprogramming of MEFs toward epicardial-like cells.
3F Activates the Endogenous Epicardial Gene Regulatory Network
To understand the molecular basis of epicardial-like reprogramming, we examined 3F reprogramming along the main pseudo-time trajectory consisting of the MEF state A, intermediate states BDF, and epicardial state H (Figure 5A). As pseudotime increased along the main trajectory, the expression of 3F increased (Figures 5B and S5A). This indicates that attaining high expression of exogenous 3F is a key step in epicardial reprogramming, which agrees with previous studies on cellular reprogramming (Papapetrou et al., 2009; Wang et al., 2015; Xu et al., 2015). In contrast to the gradual increase of 3F, it is not until terminal state H that the epicardial marker genes Myrf, Wt1, and Bnc1 become endogenously activated (Figures 5A and S5C).
Figure 5. Atf3, Gata6, and Hand2 Activate the Epicardial Transcriptional Network.

(A) (left) The main trajectory of pseudotime is highlighted in orange. (middle-right) For MEFs reprogrammed with 3F for 7 days, we plot the expression of epicardial marker genes Wt1, Bnc1, and Myrf.
(B) Expression of exogenous genes (Atf3, Hand2, Gata6) and endogenous genes (Myrf, Wt1, Bnc1) across pseudotime states in MEFs reprogrammed with 3F for 7 days.
(C) We identified 2,040 genes differentially expressed between MEFs and primary epicardial cells. The genes cluster into six gene expression signatures during pseudotime. Expression is log10 transformed and Z normalized.
(D) Heatmap of the six gene-expression signatures. Columns indicate cells ordered by pseudotime (with primary epicardial cells at the far right); rows indicate genes. For visualization purposes, groups of 20 cells are binned.
(E) Gene ontology analysis of genes with expression Signature 6.
To further assess how the MEF gene regulatory network becomes rewired during 3F reprogramming, we clustered genes during pseudotime to identify six distinct expression signatures (Figures 5C and S5F). We observed that 3F significantly reconfigured the expression of ~2,000 genes toward the epicardial state: MEF-specific genes were repressed (Signatures 1–3) and epicardial-specific genes become activated (Signatures 4–6) (Figures 5D and S5G). Consistent with their intermediate position in pseudotime, the BDF state represents a transitional state of gene expression between states A and H. A few epicardial markers, such as Sox6, are induced early upon commitment to the intermediate BDF branch. Most epicardial markers exhibit the strongest induction late in pseudotime in state H. Indeed, key epicardial genes in Signature 6 (Gata5, Gpm6a, Myrf, Wt1, and Bnc1) exhibited sustained induction throughout state H, reaching their maximal expression only at the end of pseudo-time. Consistent with their roles in epicardial cells, Signature 6 genes were enriched for gene ontology terms relevant to epicardial function, including cell motility (p = 9.5E−11) and cell adhesion (p = 3.4E−06) (Figure 5E) (Bochmann et al., 2010). Thus, activation of the endogenous epicardial gene regulatory network occurs late during reprogramming.
Pseudotime also identifies two states (E and G) that branch off the main trajectory. Our analysis indicates that these cells represent alternative cell fates of 3F reprogramming with altered expression of cell cycle genes and signal transduction pathways (see STAR Methods) (Figure S5).
Functional Validation of Epicardial Reprogramming
To confirm that Reprogram-Seq has identified TF cocktails driving conversion of MEFs to epicardial-like cells, we performed reprogramming experiments using traditional bulk approaches and examined several features of epicardial cells (Figure 6A). We confirmed that MEF infectivity is high, with >90% of pBabe-GFP-infected MEFs exhibiting GFP fluorescence (Figure S6A). First, we measured the expression of key marker genes by qPCR. For both 10F and 3F, we observed induction of epicardial marker genes Upk1b, Gpm6a, and Krt19 (Figure 6B; Table S4) (Bochmann et al., 2010), which are not part of either reprogramming cocktail. Induction of epicardial markers is sustained after 7 and 14 days of reprogramming. Second, microscopy confirmed that 10F- and 3F-reprogrammed MEFs adopted a cobblestone morphology, which is consistent with that of epicardial cells and which is distinct from the morphology of uninfected MEFs (Figure 6C) (Witty et al., 2014). Third, we performed immunostaining for the tight junction marker ZO-1, a molecular marker of epithelial cells, such as epicardial cells (Witty et al., 2014). Consistent with epicardial reprogramming, both 10F and 3F cells exhibited ZO-1 protein localized to the cell membrane (Figure 6D). Importantly, reprogramming toward an epicardial cell-like state is specific to 10F and 3F because GHMT (Gata4, Hand2, Mef2c, Tbx5) could only generate ɑ-actinin+ reprogrammed cells without ZO-1 cell membrane localization (Figure S6C). Strikingly, at high confluency, sheets of ZO-1+ cells were present in 10F-reprogrammed MEFs (Figure S6B), in contrast to the irregular pattern in uninfected cells. Additionally, we performed immunostaining for UPK1B, the protein product of an epicardial marker gene Upk1b (Figures S6D and S6E) (Bochmann et al., 2010). Interestingly, we observed perinuclear localization of UPK1B in 10F- and 3F-reprogrammed MEFs, which is rare in empty-vector infected MEFs and not observed in GHMT-reprogrammed MEFs. As a positive control, an epicardial-derived cell line (Russell et al., 2011) also showed consistent UPK1B perinuclear localization. Fourth, we stimulated 10F- and 3F-reprogrammed cells with transforming growth factor β1 (TGF-β1) to induce epithelial-to-mesenchymal transition (EMT) (Figure 6A), a well-known feature of epicardial cells (Russell et al., 2011; Witty et al., 2014). Consistent with EMT, we observed continuous decreases in the expression of epithelial marker genes, including Krt19 and E-Cadherin, after TGFß1 treatment (Figure 6E) (Nieto et al., 2016). Finally, as primary epicardial cells exhibit aldehyde dehydrogenase activity (Witty et al., 2014), we performed the Aldefluor assay to directly measure that activity in reprogrammed cells. Confirming an epicardial-like cell state, we observed a nearly 10-fold induction of aldehyde dehydrogenase activity in 10F and 3F cells (Figure 6F). Together, these experiments indicate that 10F and 3F can drive the conversion of MEFs toward an epicardial-like cell fate.
Figure 6. Functional Validation of Epicardial-like Reprogramming.

(A) Experimental design to functionally validate epicardial-like reprogramming.
(B) qRT-PCR analysis of epicardial markers (Upk1b, Gpm6a, Krt19) in reprogrammed MEFs (10F, 3F, and GHMT), negative-control-infected MEFs (empty vector), and whole mouse heart. Each bar shows the mean of three technical replicates. Error bars denote SEM.
(C) Phase contrast images of primary epicardial cells, uninfected MEFs, and reprogrammed MEFs. Scale bar indicates 100 microns.
(D) Immunocytochemistry with DAPI of ZO-1 in primary epicardial cells and reprogrammed MEFs. Insets: Zoomed-in views of example cells. Scale bar indicates 50 microns.
(E) qRT-PCR analysis of epithelial marker genes (Krt19 and E-Cadherin) during a time-course of EMT mediated by TGFß1 induction. Each bar shows the mean of three technical replicates. Error bars denote SEM.
(F) Aldehyde dehydrogenase activity of reprogrammed MEFs as measured by the Aldefluor assay.
Predicting TF Cocktails to Reprogram Mouse Cell Types
We anticipate that Reprogram-Seq can be applied to identify new reprogramming cocktails for cell types defined by cell-atlas efforts. The Tabula Muris Consortium et al., (2018) has generated scRNA-Seq atlases for 20 organs and tissues spanning ~60 mouse cell types. Because of the diversity of cell types in Tabula Muris, most in vivo cells do not cluster with MEFs from our cardiac-centric 48F experiment (Figure S7C). The exception is a set of cardiac muscle cells annotated by Tabula Muris, which clusters with seven cells from reprogrammed MEFs and no cells from control MEFs, although we are unable to draw strong statistical conclusions given the small number of cells.
Next, we computationally identified candidate TFs to drive cellular conversion to those cells from MEFs (Table S5). We found known reprogramming factors for hepatocytes (Hnf4a, Foxa3) (Huang et al., 2011, 2014; Sekiya and Suzuki, 2011) and renal tubular epithelial cells (Hnf1b, Hnf4a, Pax8) (Kaminski et al., 2016) (Figures 7A and S7A). We also identified potential reprogramming factors for cells such as skeletal muscle satellite cells that include TFs with known roles in those cells (MyoD1, Myf5, and Pax7) (Tapscott et al., 1988) (Figure 7A).
Figure 7. Predicting TF Cocktails to Reprogram Mouse Cell Types.

(A) Differential expression analysis of TFs in three primary mouse cell types (hepatocytes, skeletal muscle satellite cells, and neuroendocrine cells) defined by Tabula Muris, as compared with MEFs.
(B) Box plots indicating the cell-type-specific expression of TF families based on Shannon entropy.
(C) Dot plot indicating the expression of 11 TFs from the GATA family across the Tabula Muris cell types.
(D) Dot plot indicating the expression of 145 low-entropy TFs across cell types defined by the Tabula Muris.
(E) Dot plot of differentially expressed TFs for endothelial cells across four mouse organs (left) and three types of epithelial cells in mouse kidney (right).
To attain cell-type specificity, we reasoned that reprogramming cocktails likely contained cell-type-specifically expressed TFs. Using Shannon entropy (Schug et al., 2005), we observe that the GATA and T-box families were the most tissue specific (Figures 7B and 7C), and many members had known roles in cellular reprogramming (Batta et al., 2014; Ieda et al., 2010; Kubaczka et al., 2015; Song et al., 2012b). Therefore, we used Shannon entropy to define 145 cell-type-specifically expressed TFs across Tabula Muris cell types. Clustering of those cell-type-specific TFs revealed several known TFs for direct reprogramming. For example, several known cardiomyocyte reprogramming factors (Gata4, Hand2, and Tbx5) were significantly enriched in cardiac muscle cells (Figures 7D and S7B). Interestingly, Tabula Muris defines endothelial cells in four tissues, and we identified TFs specifically expressed in each one: kidney capillary (Irx3, Rarb), lung (Smad6, Tbx3), hepatic sinusoid (Maf, Gata4), and other tissue (Meox2, Nr4a2) (Figure 7E). In addition, Tabula Muris defines three types of epithelial cells in the kidney, each of which has cell-type specifically expressed TFs: proximal straight tubule (Hnf4a), collecting duct (Emx1), and loop of Henle ascending loop (Irx1). These unique TFs could have applications in cell-type-specific, direct reprogramming.
DISCUSSION
Reprogram-Seq provides a functional platform for both undirected and directed cellular engineering. For undirected reprogramming, we provide two examples that highlight the robustness of Reprogram-Seq performed on 48F, which were curated based on their role in heart development and/or function from the literature. First, Reprogram-Seq accurately identified MyoD1-reprogrammed cells as induced skeletal myoblasts. Second, Reprogram-Seq specifically identified Hand2 and Gata6 as reprogramming factors for epicardial-like cells among a milieu of 48 factors. Alternatively, Reprogram-Seq can be used for rational reprogramming when a cell atlas is available. In our study, Reprogram-Seq precisely identified 10 TFs that were subsequently refined to an optimized three-TF combination capable of generating epicardial-like cells. Remarkably, Hand2 and Gata6 were identified by both undirected and directed reprogramming, thus confirming that both approaches achieve consistent solutions for generating epicardial-like cells and underscoring the robustness of Reprogram-Seq.
Successful application of Reprogram-Seq depends on several key factors. First, choosing which transcription factors to test is critical. Here, we predicted the TFs to test based on differential gene expression analysis between initial and destination cell states. More-sophisticated methods, for example using gene regulatory networks (Cahan et al., 2014; D’Alessio et al., 2015; Morris et al., 2014; Rackham et al., 2016), may offer improvements. Second, because of noise in single-cell genomics measurements, sequencing many cells is important. Detecting multiple reprogrammed cells aids in statistical analysis and in the identification of potential TF cocktails. Third, sampling TF cocktails with high complexity requires sequencing many cells.
In our most-complex reprogramming experiment with 48F, we observed that most TFs only subtly perturb MEF cell state. Although some MEF-derived cells cluster with primary cells, those situations are rare. There are several explanations for that rare event. First, many cases of cellular reprogramming are relatively inefficient. For example, GHMT reprogramming only reaches ~1% efficiency in generating cardiomyocytes (Nam et al., 2014). Second, we have not sufficiently sampled the full combinatorial space of 48 TFs, which is still too prohibitive to exhaustively search. Third, we focused our analyses on a small number of growth conditions and sampling times. New technologies for higher throughput scRNA-Seq (Cao et al., 2017; Rosenberg et al., 2018) will enable future experiments to more effectively sample TF combinations, growth conditions, and sampling times to better optimize cellular reprogramming.
Several advantages of Reprogram-Seq distinguish it from previously described strategies for cellular engineering. First, Reprogram-Seq can be applied agnostic to literature. TF candidates for reprogramming can be derived entirely from cell-type-specific, single-cell atlases. In principle, each cell type expresses a unique profile of TFs, a subset of which is sufficient to establish the cell state. Second, as Reprogram-Seq can be applied in undirected reprogramming, it can be used for cellular engineering even if a single-cell atlas is not available for a particular cell type. Third, one Reprogram-Seq experiment can give insights into multiple TF cocktails. Thus, Reprogram-Seq is capable of providing refined TF cocktails, cell-fate trajectories, and alternative cell fates by examining different subsets of cells from the same single-cell perturbation dataset.
Finally, Reprogram-Seq establishes an accelerated timeline of TF cocktail identification, optimization, and induced cell-type characterization. To illustrate how Reprogram-Seq compares with traditional strategies for cellular engineering, we highlight induced cardiomyocyte (iCM) reprogramming as a well-studied example (Kojima and Ieda, 2017). The knowledge base for generating the original pool of candidate TFs for iCM reprogramming derived from many foundational studies using mouse genetics to understand heart development that spanned two decades (Galdos et al., 2017). Then, the candidate pool was refined to GMT through an iterative screening process (Ieda et al., 2010). Subsequent work identified additional factors to improve iCM reprogramming, identify alternative TF combinations, and further characterize the iCM phenotype (Kojima and Ieda, 2017). In addition, the trajectory of the early steps of iCM reprogramming was recently described using scRNA-Seq (Liu et al., 2017). Taken together, these studies span more than twenty years of active investigation. Our example of applying Reprogram-seq for epicardial reprogramming significantly shortens this time. Thus, Reprogram-Seq is an important step toward acquiring the capability to reprogram new cell types for translational purposes on a reasonable timescale.
With the future completion of mammalian cell atlas projects (Regev et al., 2017; The Tabula Muris Consortium et al., 2018), every cell type in human and mouse will be defined. This massive undertaking will provide a foundation for future mechanistic studies. We suggest that Reprogram-Seq can leverage this information to identify new TF cocktails for cell-type specific reprogramming. Reprogrammed human cells could then be made on demand and used for cell-based therapy, drug testing, and disease modeling to improve our understanding of human physiology.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: Gary Hon (Gary.Hon@UTSouthwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal experiments described in this study were conducted under the oversight of the University of Texas Southwestern Institutional Animal Care and Use Committee. Equal ratios of male and female mice were harvested at E12.5 for this study. Unless otherwise stated, all animals used in this study were wild-type CD-1 mice purchased from Charles River and maintained under normal husbandry conditions, including standardized light/dark cycles and normal chow. Normal health and immune status of the mice was ensured by regular visits by a veterinarian, and none of the mice underwent procedures or drug treatment prior to primary cell collection.
Primary sample collection
Pregnant dams were housed in a standard facility 12 hr light/dark cycle, 70 – 75°F, and ~50% humidity. Primary cardiac cells were harvested from neonatal mouse hearts at P0 by dissecting and mincing the hearts. The tissue was then incubated in trypsin for a total of 30 minutes in a shaking water bath at 37°C. Supernatant was strained and transferred to a flask containing cold growth media every 5 minutes-6 fractions total. Cells were spun at 500X G for 5 minutes at 4°C and resuspended in cold growth media. Incubation times in trypsin were adjusted depending on the dissected area of interest.
MEF isolation
Mouse embryonic fibroblasts (MEFs) were isolated from timed pregnant E12 CD-1 mice purchased from Charles River as described previously (Nam et al., 2014). Briefly, uterine horns containing embryos were removed from the pregnant dam and placed in cold DPBS. Subsequently, embryos were removed from the uterus and amniotic sac. Head, limbs, tail, and internal organs and viscera were removed and discarded. Remaining tissue was minced, washed with DPBS, and incubated in trypsin 0.25% for 10 minutes at 37°C. Cells were strained using 100 μm filter. Trypsin was inactivated with growth media (high glucose DMEM, 10% FBS, 1X penicillin/streptomycin). Cells were spun at 500X G for 5 minutes at 4°C and resuspended in growth media and plated at ~100 K cells per cm2. MEFs were maintained in growth media and incubated at 37°C in 5% CO2. After 24 hours, cells were frozen at −80°C in aliquots of 5 – 10 million cells suspended in 90% FBS and 10% DMSO.
Epicardial-derived cell line
An epicardial-derived cell line(Russell et al., 2011) was used as a control in ICC experiments. The cell line was grown in growth media (1 volume Media 199 + 3 volumes DMEM + 10% FBS, Sigma F6178) until confluency before being harvested for ICC.
METHOD DETAILS
Construction of retroviral vectors
Retroviral vectors were generated by subcloning individual TF coding sequences into the pBABE plasmid (Nam et al., 2014). TF coding sequences were amplified from mouse or human heart cDNA using primers containing linkers with appropriate restriction enzyme sites. Amplified PCR products were cut with restriction enzymes, gel purified, and ligated into the pBABE vector. Positive clones were verified by Sanger sequencing.
Viral packaging, infection, and single cell reprogramming
A packaging cell line Platinum-E (Cell Biolabs, San Diego, CA) was used to produce retrovirus for reprogramming (Nam et al., 2014). Packaging cells were transfected using Fugene 6 (Promega, Madison, WI) per manufacturers’ instructions with retroviral vectors (pBabe) expressing specific transcription factors. Virus was harvested, filtered (0.45 μm pore) and applied to MEFs at 24 hr and 48 hr post-transfection after the addition of polybrene 8 μg/mL (Sigma-Aldrich, St. Louis, MO). The cells were incubated in growth media (DMEM high-glucose + 10% FBS + 1% penicillin/streptomycin + 1% 100X GlutaMAX) for 7 or 14 days and subsequently washed with DPBS and trypsinized into a single cell suspension. Cells were spun at 500X G for 5 minutes at 4°C and resuspended in cold growth media.
Flow cytometry analysis of GFP+ cells
For flow cytometry, cells were trypsinized and resuspended in growth media. Cells were then spun at 500X G for 5 minutes at 4°C and washed with room temperature DPBS. Cells were spun again at 500X G for 5 minutes at 4°C and resuspended into a single-cell suspension in DPBS with 0.04% BSA. Flow cytometry was carried out on a BD FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ).
Phase contrast microscopy
For phase contrast microscopy, cells were imaged live in media on a 24-well plate with EVOS FL Auto Imaging System (Thermo Fisher Scientific, Waltham, MA). Phase contrast mode was used for imaging.
Cell staining
For ICC staining, MEFs were plated on 12 mm glass coverslips #1.5 for 24 hr then infected with retrovirus. After 7 days, cells were washed with DPBS and fixed using 4% Paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) for 15 minutes at room temperature. Cells were stored in DPBS for up to a month, or directly permeabilized using PBST (0.1% Triton X-100 in PBS) 3 times for 5 minutes. Cells were then blocked for 10 minutes at room temperature using 10x Universal Blocking Buffer diluted to 1x. Primary antibodies were diluted in 1:1 1x Universal Blocking Buffer and DPBS—ZO1(1:5) (DSHB), a-actinin (1:200) (Sigma-Aldrich, St. Louis, MO), UPK1B (1:200) (Invitrogen, Carlsbad, CA)—and incubated overnight at 4°C after application to the cells. Cells were then washed 3 times with PBST and incubated with secondary antibody for 1 hr at room temperature or for overnight at 4C. All secondary antibodies were obtained from Life Technologies’ (Carlsbad, CA) Alexa Fluor Dyes 488, 555, and 647. Cells were washed 3 times and coverslips were mounted using Vectashield with DAPI (Vector Laboratories, Burlingame, CA) and imaged using a ZEISS LSM 880 with Airyscan confocal microscope.
EMT induction of reprogrammed cells with TGF-β1 protein
For EMT induction with TGF-β1 protein, cells were incubated in growth media with 5 ng/mL TGF-β1 protein (R&D Systems, Minneapolis, MN) (reconstituted per manufacturer’s instructions). Media was changed every two days till 7 days of treatment. Cells were then harvested with Trizol (Life Technologies, Carlsbad, CA) for RNA extraction.
Aldefluor assay
MEFs were incubated for 7 days and 14 days post induction to assess aldehyde dehydrogenase activity (STEMCELL Technologies, Vancouver, Canada). Aldefluor staining was carried out according to the manufacturers’ directions using an incubation time of 50 minutes at 37°C. Flow cytometry was carried out on a BD FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ) and control signals was subtracted from each sample.
Single cell RNA sequencing library preparation
To confirm the single cell resolution of our RNA sequencing, prior to real experiments, mixed-species experiments were performed. Briefly, human cell lines and mouse primary/reprogrammed cells were mixed together and scRNA-seq libraries were constructed using the Drop-seq and 10x Genomics platforms. The resulting data of both platforms are highly organism-specific, suggesting a low cell multiplet rate and low cross-cell contamination.
Drop-seq procedure
Drop-seq was performed as described previously (Macosko et al., 2015; Xie et al., 2017) with small modifications. Briefly, droplets were generated using microfluidic devices, which encapsulated single cells and barcoded beads (ChemGenes Corporation, Wilmington, MA, catalog number Macosko201110). Beads were collected after the breakage of individual droplets using perfluorooctanol (Sigma-Aldrich, St. Louis, MO, catalog number 370533). RNA hybridized on the barcoded beads was reverse transcribed using Maxima H minus reverse transcriptase (Thermo Fisher Scientific, Waltham, MA, catalog number EP0751) and cDNA was amplified with 13 cycles (KAPA HotStart ReadyMix, Kapa Biosystems, Wilmington, MA, catalog number KK2602). In-house Tn5 transposase was used to insert sequencing adapters and fragmentate cDNA. Illumina Nextera adapters i5 and i7 were used to amplify the fragmented cDNA with 12 cycles (KAPA HiFi PCR Kits, catalog number KK2102). Agarose gel size selection was performed to recover fragments with length of 400 to 600 bp.
10x Genomics single cell RNA sequencing procedure
The concentration of single cell suspension was adjusted to 500–1000 cells/mL and was loaded on the 10x Genomics Chromium system (10x Genomics, Pleasanton, CA) with the aim of generating 6000 to 10000 transcriptomes per channel (Chromium Single Cell 3′ Library & Gel Bead Kit v2, catalog number 120237). Single cell RNA sequencing libraries were constructed following the manufacturer’s instructions (Zheng et al., 2017).
QUANTIFICATION AND STATISTICAL ANALYSIS
Sequencing, basecalling and demultiplexing
Libraries were sequenced on Illumina NextSeq 500/550 sequencing systems (Illumina, San Diego, CA). BCL files generated by Illumina sequencing systems were demultiplexed and converted to standard FASTQ files using bcl2fastq (version 2.17.1.14). For 10x Genomics libraries, the mkfastq function from Cell Ranger pipeline (version 2.1.0) was used to perform basecalling and demultiplexing for downstream analyses.
Read alignment and generation of gene expression matrix
For Drop-seq data, demultiplexed reads were processed using Drop-seq Tools (version 1.12) as described in Macosko et al. (2015) with small modifications. Briefly, low quality reads containing cell barcodes and UMIs were filtered and polyA tails (longer than 6 bases) were trimmed before mapping. Processed reads were mapped to mouse mm10 reference using STAR (version 2.5.3a) (Dobin et al., 2013). Only uniquely aligned reads were kept. Cell barcodes within one edit distance were collapsed together. UMI-based PCR duplicate removal was performed using the directional method of UMI-tools with default parameters (version0.5.3) (Smith et al., 2017). An inflection point of cumulative number of reads of cells in each library was calculated to determine the number of cells in each experiment. Gene expression matrices were generated using featureCounts (version 1.5.3) (Liao et al., 2014). The gene annotation used is based on Ensembl release 84 (GENCODE Gene Set M9).
For 10x Genomics data, the gene expression matrix was generated for each experiment using Cell Ranger pipeline (version 2.1.0) with default parameters, except the parameter of expected number of cells, which was adjusted based on each individual experiment. The genome reference and gene annotation used were the same as Drop-seq data.
Dimensionality reduction
Filtering low quality cells and uninformative genes
All Drop-seq and 10x Genomics data were combined together respectively and analyzed separately. Cells with fewer than 200 genes detected were filtered. Genes detected in fewer than 30 cells or with fewer than 60 UMIs in total across all cells were filtered. In total, 25,776 cells from Drop-seq and 34,564 cells from 10x Genomics data were kept for downstream analyses.
Normalization and batch correction
The total UMI counts for each cell were normalized toward the median UMI counts per cell by multiplying each cell with a scaling factor. The resulting matrix was natural-log-transformed with the addition of a pseudocount of 1 prior taking the log-transformation. The ComBat (Johnson et al., 2007) method from sva package (version 3.28.0) was used to correct the batch effect with the default parametric adjustment parameters (Dixit et al., 2016).
Principal component analysis (PCA)
The median-normalized natural-log-transformed batch-effect-corrected matrix was centered and scaled per gene so that the mean expression value of each gene is 0 and the standard deviation is 1 across all cells. The resulting matrix was used to perform PCA using the prcomp function from built-in R package stats (version 3.5.1). To determine the number of principal components to use for clustering and visualization, permutation tests were performed (Macosko et al., 2015). PCA was performed on 500 permuted versions of the original matrix. In each version, 10% of the total genes were randomly selected and their expression values were independently permuted. To speed up the calculation, prcomp_irlba function from irlba package (version 2.3.2) was used to perform PCA on the original and 500 permuted matrices, and only the top 40 principal component vectors were calculated. The average proportions of variance explained by each principal component of the 500 permutations were compared to the proportions of variance explained by each principal component of the original matrix.
t-distributed stochastic neighbor embedding (t-SNE) visualization
A two-dimensional non-linear embedding of the single cells was computed using Barnes-Hut implementation of t-SNE method (van der Maaten, 2014). The scores of the significant principal components estimated above (output of the prcomp function) were used as input to Rtsne package (version 0.13). The initial PCA step was disabled, and perplexity parameter was set to 30 with 3000 iterations.
Graph clustering
The Louvain-Jaccard method (Shekhar et al., 2016) was used to cluster single cells. The input is the scores of the chosen principal components. Briefly, a k-nearest neighbor (k-NN) graph was built based on euclidean distance in chosen principal component scores using nn2 function from RANN package (version 2.6, k set to 30). Jaccard overlap index of each edge in the graph was calculated and used as input to the Louvain algorithm implemented in igraph package (version 1.2.1).
Binomial test for differentially expressed genes
The method used for testing if a gene is expressed more frequently between different group of cells was described in Shekhar et al. (2016).
Identification of exogenous and endogenous expression
The plasmid sequences of Myrf Sox6 Tbx20 and Wt1 are from human. To accurately calculate their expression values, demultiplexed reads were also mapped to human GRCh38 reference. Reads uniquely mapped to mouse genomic regions of these four genes and reads uniquely mapped to the orthologs of those fours gene in human genome were retrieved. The source of each read was determined based on their genotypes.
Genomic tracks were generated using uniquely mapped non-PCR duplicate reads from selected group of cells. The distribution pattern of reads were compared among different cells groups. The gene sequences on the retroviral plasmids have no 3′ untranslated regions (3′ UTRs).
Pseudotime construction
Monocle (version 2.8.0) was used for pseudotime analysis of single cells (Qiu et al., 2017). Raw UMI counts were used as input and the distribution was set to “negbinomial.size().” The minimum expression level that constitutes true expression was set to 0.5. Differentially expressed genes between starting cells and target primary cells were used to measure cells’ progresses during reprogramming. The data dimensionality was reduced to 2 using “DDRTree” method.
Gene ontology (GO) enrichment analysis
Enrichment analysis for GO terms was performed using topGO (version 2.32.0). Fisher’s exact test was used for enrichment tests.
Transcription factor (TF) annotation
TFs were defined based on GO terms (org.Mm.eg.db, version 3.4.1; GO.db, version 3.4.1). Genes need to meet both criterias to be considered as TFs: a) have either “regulation of transcription” or “transcription factor activity” GO terms in BP sub-ontology; b) “DNA binding” or “transcription factor activity” GO terms in MF sub-ontology.
Transcription factor family categorization
A full list of curated human TFs was obtained from the “HumanTFs” website (http://humantfs.ccbr.utoronto.ca/) (Lambert et al., 2018). The identification of TF families was based on the third column (DNA-binding domain, or DBD) of the list. Next, the mouse orthologs of all curated human TFs were identified through an online tool, bioDBnet (https://biodbnet-abcc.ncifcrf.gov/db/dbOrtho.php). Human Ensembl IDs were converted to mouse gene symbols, and the generated list was overlapped with the list of all annotated mouse TFs mentioned above. Only the common TFs between the two lists were used for downstream analysis.
Additional analysis of cluster 20 cells from Figure 1
On average, cells in Cluster 20 express 61.3 times more MyoD1 as compared to other reprogrammed MEFs (Figures 1D and 1E) (Cluster 20: 254 cpm; other reprogrammed MEFs: 4.1 cpm; p < 2.2e-16; Wilcoxon test). In addition, 164 of the 165 cells in Cluster 20 are reprogrammed cells, and none belong to uninfected MEFs. Cluster 20 exhibits both exogenous as well as endogenous expression of MyoD1 (Figure S3A).
Additional analysis supporting 3F as an optimized reprogramming cocktail
First, we examined the expression of the transcription factors in the epicardial-like Cluster 10. While each of the factors in 10F exhibited some induction, we observed dramatic enrichment of Atf3, Gata6, and Hand2 expression in the reprogrammed cells of Cluster 10, with over half of the cells expressing these factors (Figures S5A and S5B). We denote this combination of TFs as 3F. Collectively, 3F is > 8.1-fold more enriched in Cluster 10 reprogrammed cells compared to uninfected MEFs, and > 2.6-fold more enriched compared with other reprogrammed MEFs. Supporting the consistency of Reprogram-Seq, Gata6 and Hand2 were also identified in our unbiased analysis of 48F above.
Next, we sought to confirm that reprogrammed cells in Cluster 10 have exogenous expression of 3F, as we reasoned that enrichment of exogenously expressed trans-genes is indicative of successful viral induction. We took advantage of our cloning strategy in that exogenous TFs do not have 3′UTRs. Therefore, reads derived from exogenous TFs will be strictly upstream of the 3′UTR while reads derived from endogenous TFs will be enriched downstream of the 3′UTR. In primary epicardial cells, we observed that 92.0% of reads for Hand2 fall into the 3′UTR, which is consistent with endogenous expression. In contrast, this number is only 2.5% for cells in Cluster 10, which is indicative of exogenous expression (Figure S5C). Control genes including Gapdh and Bnc1 are almost exclusively endogenously expressed (Figure S5C). By using this observation to estimate the exogenous and endogenous expression of 10F, we confirmed that cells in Cluster 10 exhibit significant exogenous expression of 3F (Figure 3A). We also examined whether cells with 3F expression are more likely to be in Cluster 10. Of the 1026 MEF-derived cells expressing 3F, we observe that 555 (54.1%) belong to Cluster 10 as compared to 18.4% expected by chance (p = 4.10e-197). These results suggest that 3F may be an optimized TF cocktail for epicardial reprogramming.
Additional analysis of alternative cell fates during 3F reprogramming
Pseudotime also identifies two states (E and G) that branch off the main trajectory. These cells exhibit expression of 3F (Figures S6A and S6C), indicating that they are indeed derived from MEF reprogramming. However, the expression of endogenous epicardial markers is lower in these cells compared to the terminal state H (Figure S6C). These observations suggest that states E and G could be alternative cell fates of 3F reprogramming. Next, we examine potential molecular pathways interfering with epicardial reprogramming. We observe that cells in state E exhibited higher expression of cell division genes (Figure S6D) (p = 2.4E-11) including the DNA topoisomerase Top2a, the cytokinesis regulator Prc1, and the mitotic regulator Cdca8 (Figure S6E). Consistent with previous studies (Bektik et al., 2018; Jiang et al., 2015; Liu et al., 2017; Treutlein et al., 2016), these results suggest that inhibiting the cell cycle could improve epicardial reprogramming. Finally, extending this analysis to state G, we find that this alternative cell fate exhibits failed activation of several tyrosine kinases with roles in cell signaling, including Abl2, Ptk2, and Src. Consistent with this observation, gene ontology analysis reveals that genes repressed in state G are enriched for small GTPase mediated signal transduction (p = 1.2E-09) and epidermal growth factor receptor signaling pathway (p = 9.5e-06) (Figure S6B). These results suggest that activation of specific signal transduction pathways may be an important step in epicardial reprogramming.
DATA AND SOFTWARE AVAILABILITY
The single cell RNA sequencing data reported in this paper were deposited in NCBI GeneExpression Omnibus (GEO) under the accession number GEO: GSE117795. Scripts for analyzing Reprogram-Seq data are available at https://github.com/jlduan/Reprogram-Seq.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-α-actinin antibody | Sigma-Aldrich | Cat#: A7811; RRID: AB_476766 |
| Rat anti-ZO1 antibody | DSHB | Cat#: R26.4C; RRID: AB_2205518 |
| Rabbit anti-UPK1B antibody | Thermo Fisher Scientific | Cat#: PA5–72711; RRID: AB_2718565 |
| Alexa Fluor 488 Goat Anti-Rabbit IgG | Thermo Fisher Scientific | Cat#: A-11034; RRID: AB_2576217 |
| Alexa Fluor 555 Goat Anti-Mouse IgG | Thermo Fisher Scientific | Cat#: A-21422; RRID: AB_141822 |
| Alexa Fluor 647 Goat Anti-Rabbit IgG | Thermo Fisher Scientific | Cat#: A-21245; RRID: AB_2535813 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant human TGF-beta 1 protein | R&D Systems | Cat#: 240-B |
| Critical Commercial Assays | ||
| Aldefluor Kit | STEMCELL Technologies | Cat#: #01700 |
| Deposited Data | ||
| Raw reads | This paper | GEO: GSE117795 |
| Gene expression matrices | This paper | GEO: GSE117795 |
| Oligonucleotides | ||
| gcgcATCGATATGATGCTTCAACATCCAGGCCAG | This paper | Clal_Atf3_5 |
| gcgcGTCGACTTAGCTCTGCAATGTTCCTTCTTTTATCTGTTGG | This paper | Sall_Atf3_3 |
| gcgcATCGATATGCGGCGGTCGCC | This paper | Clal_Bnc1_5 |
| gcgcGTCGACTTACTGGAGGTGGCTTGGAGATGAAG | This paper | Sall_Bnc1_3 |
| gcgcATCGATATGTCCACTGGCTCCCTCAGC | This paper | Clal_Tcf21_5 |
| gcgcGTCGACTCAGGATGCTGTAGTTCCACACAAGC | This paper | Sall_Tcf21_3 |
| gcgcATCGATATGAGTCTGGTGGGGGGC | This paper | Clal_Hand2_5 |
| gcgcGAATTCTCACTGCTTGAGCTCCAGGG | This paper | EcoRl_Hand2_3 |
| gcgcgcCAATTGATGGCCTTGACTGACGGCGGC | This paper | Mfel_Gata6_5cds |
| gcgcgcCTCGAGTCAGGCCAGGGCCAGAGCAC | This paper | Xhol_Gata6_3cds |
| qPCR primers | This paper | Table S3 |
| Software and Algorithms | ||
| bcl2fastq (version 2.17.1.14) | Illumina | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| Cell Ranger (version 2.1.0) | 10x Genomics | https://www.10xgenomics.com |
| Drop-seq Tools (version 1.12) | Macosko et al., (2015) | http://mccarrolllab.com/dropseq |
| featureCounts (version 1.5.3) | Liao et al., (2014) | http://bioinf.wehi.edu.au/featureCounts |
| Gviz (version 1.24.0) | Hahne and Ivanek, (2016) | https://bioconductor.org/packages/release/bioc/html/Gviz.html |
| irlba (version 2.3.2) | Jim Baglama, Lothar Reichel, B. W. Lewis | https://cran.r-project.org/web/packages/irlba/index.html |
| Monocle (version 2.8.0) | Qiu etal., (2017) | http://cole-trapnell-lab.github.io/monocle-release/ |
| R (version 3.5.1) | R Foundation | https://www.r-project.org |
| Rtsne (version 0.13) | van der Maaten, (2014) | https://cran.r-project.org/web/packages/Rtsne/index.html |
| SAMtools (version 1.8) | Li et al., (2009) | https://www.htslib.org |
| STAR (version 2.5.3a) | Dobin et al., (2013) | https://github.com/alexdobin/STAR |
| sva (version 3.28.0) | Johnson et al., (2007) | https://bioconductor.org/packages/release/bioc/html/sva.html |
| topGO (version 2.32.0) | Adrian Alexa, Jorg Rahnenfuhrer | https://bioconductor.org/packages/release/bioc/html/topGO.html |
| UMI-tools (version 0.5.3) | Smith etal., (2017) | https://github.com/CGATOxford/UMI-tools |
| org.Mm.eg.db (version 3.4.1) | Marc Carlson | https://bioconductor.org/packages/release/data/annotation/html/org.Mm.eg.db.html |
| GO.db (version 3.4.1) | Marc Carlson | https://bioconductor.org/packages/release/data/annotation/html/GO.db.html |
| igraph (version 1.2.1) | https://cran.r-project.org/web/packages/igraph/AUTHORS | https://igraph.org/ |
| RANN (version 2.6) | Sunil Arya and David Mount (for ANN), Samuel E. Kemp, Gregory Jefferis | https://cran.r-project.org/web/packages/RANN/index.html |
| Code for this manuscript | This paper | https://github.com/jlduan/Reprogram-Seq |
| Other | ||
| Medium 199 | GIBCO | Cat#: 11150059 |
| Fetal bovine serum | Sigma-Aldrich | Cat#: F6178/F0926 |
| GlutaMAX | GIBCO | Cat#: 35050061 |
Highlights.
Reprogram-Seq screens thousands of TF cocktails for reprogramming performance
Reprogram-Seq finds three TFs that convert MEFs to an epicardial transcriptional state
Reprogrammed cells’ morphology and function resemble primary epicardial cells
Reprogram-Seq accelerates rational cellular reprogramming
ACKNOWLEDGMENTS
We thank all the members in Hon and Munshi laboratories for insightful discussions and Ning Liu for reviewing the manuscript. This work is supported by the Cancer Prevention Research Institute of Texas (CPRIT) (RR140023 and RP190451 to G.C.H.), NIH (DP2GM128203 to G.C.H.; HL136604, HL133642, and HL133642 to N.V.M), the Department of Defense (PR172060 to G.C.H. and N.V.M.), the Welch Foundation (I-1926–20170325 to G.C.H.), the Burroughs Wellcome Fund (1009838 to N.V.M.), the March of Dimes Foundation (5-FY13–203 to N.V.M.), and the Green Center for Reproductive Biology. G.C.H. is a CPRIT Scholar in Cancer Research. We acknowledge the BioHPC computational infrastructure at UT Southwestern for providing HPC and storage resources that have contributed to the research results reported within this paper. We acknowledge UT Southwestern’s McDermott Center, Flow Cytometry Core, and the O’Brien Kidney Center: Cell Biology and Imaging Core for providing next-generation sequencing services, flow cytometry services, and confocal microscopy services for this work, respectively.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.05.079.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, Sung CY, Banfi S, Sauer MF, Olsen GS, Duffield JS, et al. (2012). The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 139, 2139–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes RM, Firulli BA, VanDusen NJ, Morikawa Y, Conway SJ, Cserjesi P, Vincentz JW, and Firulli AB (2011). Hand2 loss-of-function in Hand1-expressing cells reveals distinct roles in epicardial and coronary vessel development. Circ. Res 108, 940–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batta K, Florkowska M, Kouskoff V, and Lacaud G (2014). Direct reprogramming of murine fibroblasts to hematopoietic progenitor cells. Cell Rep 9, 1871–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bektik E, Dennis A, Pawlowski G, Zhou C, Maleski D, Takahashi S, Laurita KR, Deschênes I, and Fu J-D (2018). S-phase synchronization facilitates the early progression of induced-cardiomyocyte reprogramming through enhanced cell-cycle exit. Int. J. Mol. Sci 19, E1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochmann L, Sarathchandra P, Mori F, Lara-Pezzi E, Lazzaro D, and Rosenthal N (2010). Revealing new mouse epicardial cell markers through transcriptomics. PLoS ONE 5, e11429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahan P, Li H, Morris SA, Lummertz da Rocha E, Daley GQ, and Collins JJ (2014). CellNet: network biology applied to stem cell engineering. Cell 158, 903–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C-L, Martin JC, Sun Y, Cui L, Wang L, Ouyang K, Yang L, Bu L, Liang X, Zhang X, et al. (2008). A myocardial lineage derives from Tbx18 epicardial cells. Nature 454, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, Qiu X, Lee C, Furlan SN, Steemers FJ, et al. (2017). Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessio AC, Fan ZP, Wert KJ, Baranov P, Cohen MA, Saini JS, Cohick E, Charniga C, Dadon D, Hannett NM, et al. (2015). A Systematic Approach to Identify Candidate Transcription Factors that Control Cell Identity. Stem Cell Reports 5, 763–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. (2016). Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167, 1853–1866.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdos FX, Guo Y, Paige SL, VanDusen NJ, Wu SM, and Pu WT (2017). Cardiac regeneration: lessons from development. Circ. Res 120, 941–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guadix JA, Ruiz-Villalba A, Lettice L, Velecela V, Muñoz-Chápuli R, Hastie ND, Pérez-Pomares JM, and Martínez-Estrada OM (2011). Wt1 controls retinoic acid signalling in embryonic epicardium through transcriptional activation of Raldh2. Development 138, 1093–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, and Ivanek R (2016). Visualizing genomic data using Gviz and Bio-conductor. Methods Mol. Biol 1418, 335–351. [DOI] [PubMed] [Google Scholar]
- Hill AJ, McFaline-Figueroa JL, Starita LM, Gasperini MJ, Matreyek KA, Packer J, Jackson D, Shendure J, and Trapnell C (2018). On the design of CRISPR-based single-cell molecular screens. Nat. Methods 15, 271–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, He Z, Ji S, Sun H, Xiang D, Liu C, Hu Y, Wang X, and Hui L (2011). Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature 475, 386–389. [DOI] [PubMed] [Google Scholar]
- Huang P, Zhang L, Gao Y, He Z, Yao D, Wu Z, Cen J, Chen X, Liu C, Hu Y, et al. (2014). Direct reprogramming of human fibroblasts to functional and expandable hepatocytes. Cell Stem Cell 14, 370–384. [DOI] [PubMed] [Google Scholar]
- Ieda M, Fu J-D, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, and Srivastava D (2010). Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Xu Z, Zhong P, Ren Y, Liang G, Schilling HA, Hu Z, Zhang Y, Wang X, Chen S, et al. (2015). Cell cycle and p53 gate the direct conversion of human fibroblasts to dopaminergic neurons. Nat. Commun 6, 10100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, and Rabinovic A (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Kaminski MM, Tosic J, Kresbach C, Engel H, Klockenbusch J, Müller A-L, Pichler R, Grahammer F, Kretz O, Huber TB, et al. (2016). Direct reprogramming of fibroblasts into renal tubular epithelial cells by defined transcription factors. Nat. Cell Biol 18, 1269–1280. [DOI] [PubMed] [Google Scholar]
- Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LIR, et al. (2010). Epigenetic memory in induced pluripotent stem cells. Nature 467, 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima H, and Ieda M (2017). Discovery and progress of direct cardiac reprogramming. Cell. Mol. Life Sci 74, 2203–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolander KD, Holtz ML, Cossette SM, Duncan SA, and Misra RP (2014). Epicardial GATA factors regulate early coronary vascular plexus formation. Dev. Biol 386, 204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubaczka C, Senner CE, Cierlitza M, Araúzo-Bravo MJ, Kuckenberg P, Peitz M, Hemberger M, and Schorle H (2015). Direct induction of tropho-blast stem cells from murine fibroblasts. Cell Stem Cell 17, 557–568. [DOI] [PubMed] [Google Scholar]
- Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, and Weirauch MT (2018). The human transcription factors. Cell 172, 650–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu Z, Wang L, Welch JD, Ma H, Zhou Y, Vaseghi HR, Yu S, Wall JB, Alimohamadi S, Zheng M, et al. (2017). Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nature 551, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SA, Cahan P, Li H, Zhao AM, San Roman AK, Shivdasani RA, Collins JJ, and Daley GQ (2014). Dissecting engineered cell types and enhancing cell fate conversion via CellNet. Cell 158, 889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam Y-J, Lubczyk C, Bhakta M, Zang T, Fernandez-Perez A, McAnally J, Bassel-Duby R, Olson EN, and Munshi NV (2014). Induction of diverse cardiac cell types by reprogramming fibroblasts with cardiac transcription factors. Development 141, 4267–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Huang RY-J, Jackson RA, and Thiery JP (2016). EMT: 2016. Cell 166, 21–45. [DOI] [PubMed] [Google Scholar]
- Papapetrou EP, Tomishima MJ, Chambers SM, Mica Y, Reed E, Menon J, Tabar V, Mo Q, Studer L, and Sadelain M (2009). Stoichiometric and temporal requirements of Oct4, Sox2, Klf4, and c-Myc expression for efficient human iPSC induction and differentiation. Proc. Natl. Acad. Sci. USA 106, 12759–12764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rackham OJL, Firas J, Fang H, Oates ME, Holmes ML, Knaupp AS, Suzuki H, Nefzger CM, Daub CO, Shin JW, et al. ; FANTOM Consortium (2016). A predictive computational framework for direct reprogramming between human cell types. Nat. Genet 48, 331–335. [DOI] [PubMed] [Google Scholar]
- Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, Bodenmiller B, Campbell P, Carninci P, Clatworthy M, et al. ; Human Cell Atlas Meeting Participants (2017). The Human Cell Atlas. eLife 6, e27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, Gray L, Peeler DJ, Mukherjee S, Chen W, et al. (2018). Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JL, Goetsch SC, Gaiano NR, Hill JA, Olson EN, and Schneider JW (2011). A dynamic notch injury response activates epicardium and contributes to fibrosis repair. Circ. Res 108, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack LM, Davoli T, Xu Q, Li MZ, and Elledge SJ (2016). Sources of error in mammalian genetic screens. G3 (Bethesda) 6, 2781–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug J, Schuller W-P, Kappen C, Salbaum JM, Bucan M, and Stoeckert CJ Jr. (2005). Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol 6, R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya S, and Suzuki A (2011). Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature 475, 390–393. [DOI] [PubMed] [Google Scholar]
- Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, et al. (2016). Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell 166, 1308–1323.e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res 27, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Zhong C, Bonaguidi MA, Sun GJ, Hsu D, Gu Y, Meletis K, Huang ZJ, Ge S, Enikolopov G, et al. (2012a). Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature 489, 150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K, Nam Y-J, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, et al. (2012b). Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485, 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava D, and DeWitt N (2016). In vivo cellular reprogramming: the next generation. Cell 166, 1386–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Tandon P, Miteva YV, Kuchenbrod LM, Cristea IM, and Conlon FL (2013). Tcf21 regulates the specification and maturation of proepicardial cells. Development 140, 2409–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapscott SJ, Davis RL, Thayer MJ, Cheng PF, Weintraub H, and Lassar AB (1988). MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science 242, 405–411. [DOI] [PubMed] [Google Scholar]
- The Tabula Muris Consortium; Quake SR, Wyss-Coray T, and Darmanis S (2018). Single-cell transcriptomic characterization of 20 organs and tissues from individual mice creates a Tabula Muris. bioRxiv https://www.biorxiv.org/content/10.1101/237446v2. [Google Scholar]
- Treutlein B, Lee QY, Camp JG, Mall M, Koh W, Shariati SAM, Sim S, Neff NF, Skotheim JM, Wernig M, and Quake SR (2016). Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq. Nature 534, 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L (2014). Accelerating t-SNE using tree-based algorithms. J. Mach. Learn. Res 15, 3221–3245. [Google Scholar]
- von Gise A, Zhou B, Honor LB, Ma Q, Petryk A, and Pu WT (2011). WT1 regulates epicardial epithelial to mesenchymal transition through β-catenin and retinoic acid signaling pathways. Dev. Biol 356, 421–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Liu Z, Yin C, Asfour H, Chen O, Li Y, Bursac N, Liu J, and Qian L (2015). Stoichiometry of Gata4, Mef2c, and Tbx5 influences the efficiency and quality of induced cardiac myocyte reprogramming. Circ. Res 116, 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witty AD, Mihic A, Tam RY, Fisher SA, Mikryukov A, Shoichet MS, Li R-K, Kattman SJ, and Keller G (2014). Generation of the epicardial lineage from human pluripotent stem cells. Nat. Biotechnol 32, 1026–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S-P, Dong X-R, Regan JN, Su C, and Majesky MW (2013). Tbx18 regulates development of the epicardium and coronary vessels. Dev. Biol 383, 307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S, Duan J, Li B, Zhou P, and Hon GC (2017). Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells. Mol. Cell 66, 285–299.e5. [DOI] [PubMed] [Google Scholar]
- Xie S, Cooley A, Armendariz D, Zhou P, and Hon G (2018). Frequent sgRNA-barcode recombination in single-cell perturbation assays. PLoS One 13, e0198634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Du Y, and Deng H (2015). Direct lineage reprogramming: strategies, mechanisms, and applications. Cell Stem Cell 16, 119–134. [DOI] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The single cell RNA sequencing data reported in this paper were deposited in NCBI GeneExpression Omnibus (GEO) under the accession number GEO: GSE117795. Scripts for analyzing Reprogram-Seq data are available at https://github.com/jlduan/Reprogram-Seq.