

Abstract
Mapping enhancers to genes is a fundamental goal of modern biology. We have developed an innovative strategy that maps enhancers to genes in a principled manner. We illustrate its power by applying it to Myrf. Despite being a master regulator of oligodendrocytes, oligodendrocyte enhancers governing Myrf expression remain elusive. Since chromatin conformation capture studies have shown that a gene and its enhancer tend to be found in the same topologically associating domain (TAD), we started with the delineation of the Myrf TAD. A genome-wide map of putative oligodendrocyte enhancers uncovered 6 putative oligodendrocyte enhancers in the Myrf TAD, narrowing down the search space for Myrf enhancers from the entire genome to 6 loci in a principled manner. Epigenome editing experiments revealed that two of them govern Myrf expression for oligodendrocyte development. Our new method is simple, principled, and powerful, providing a systematic way to find enhancers that regulate the expression of a gene of interest. Since it can be applied to most cell types, it would greatly facilitate our effort to unravel transcriptional regulatory networks of diverse cell types.
Subject terms: Gene expression, Gene regulation, Epigenomics
Introduction
Enhancers are short segments of DNA that orchestrate cell type-specific gene expression by serving as transcription factor binding platforms1,2. A fascinating yet perplexing feature of enhancers is that they are often far away from target genes. For example, the ZRS enhancer is almost 1 Mb away from its target gene Shh3. This has made it difficult to annotate enhancers to genes. For this reason, the traditional approach to finding enhancers for a gene is to find conserved sequence segments in its vicinity and to test whether they work as enhancers in cell culture and/or transgenic animals4–9. If they do, it is assumed that they would regulate the endogenous gene in the genomic context. Although it has been extensively used to successfully characterize putative enhancers for genes of interest, this traditional approach has a couple of shortcomings. First, one has to make an arbitrary decision about where and how far to look in the genome for conserved sequence segments. Is upstream 100 Kb enough? Or do we have to look both upstream and downstream for as far as 1 Mb? Second, the traditional approach just assumes a regulatory relationship between a gene and an enhancer based on a distance criterion. If they are close to each other, which is again an arbitrary decision, it assumes that the enhancer would regulate the gene.
To tackle this fundamental issue, we have developed a novel strategy that maps enhancers to genes in a principled manner. This paper illustrates its power by applying it to the gene Myrf encoding a master regulator of oligodendrocytes (OLs)10–12. A unique aspect of Myrf, compared to other crucial OL genes, is that it is highly expressed in differentiating OLs, but not in OL precursor cells (OPCs), indicating that Myrf expression marks the onset of OL differentiation. Consistently, gene expression analysis of multiple sclerosis lesions indicated that OLs stalled in their differentiation are those that fail to upregulate Myrf expression. Hence, elucidating how Myrf expression is activated in OLs holds a great promise for revealing the molecular events underlying OL differentiation and developing novel remyelination therapies. The identity of enhancers governing Myrf expression in OLs remains elusive, and this is why Myrf was chosen as the first target for our new method. The only information available about Myrf expression regulation is that there is an enhancer called ECR9 in the first intron of Myrf5, which was active in OLs and some other cell types when tested in transgenic mice. ECR9 has been assumed, but not proved, to regulate Myrf expression in OLs. Our new method shows that ECR9 and a novel OL enhancer jointly control Myrf expression for OL development, demonstrating its effectiveness. Importantly, our new strategy can be applied to other genes and cell types and is expected to greatly accelerate our effort to unravel transcriptional regulatory networks of diverse cell types.
Results
Overview: A principled strategy for mapping enhancers to genes
Our strategy consists of three steps (Fig. 1A), and we will illustrate them by using Myrf as an exemplary case. First, a paradigm-shift discovery from chromatin conformation capture studies is that a gene and its enhancer tend to be found in the same topologically associating domain (TAD), a fundamental unit of genome organization and function13–17. With the TAD knowledge, one does not have to make an arbitrary decision about where and how far to look in the genome for enhancer candidates. The TAD information allows one to narrow down the enhancer search space in a principled manner. There are two features of a TAD that need to be distinguished – internal detail and boundaries18. The internal detail of a TAD reflects cell type-specific interactions among genes and enhancers, and it differs between cell types. In contrast, the boundaries of a TAD tend to be conserved between cell types and species13. Thus, even if there is no chromatin interaction data for OLs, we can still delineate the Myrf TAD by analyzing public chromatin interaction data for other cell types, as shown below. Second, we identify putative OL enhancers in the Myrf TAD, which are Myrf enhancer candidates because they are in the same TAD as Myrf. By comparing the Myrf TAD with a genome-wide map of putative OL enhancers (see below), we were able to identify 6 Myrf enhancer candidates. Third, we interrogate the Myrf enhancer candidates with CRISPRi19–22, a cutting-edge epigenome editing technique, to determine whether they govern Myrf expression in OLs. A definitive way of proving an enhancer-target gene relationship is to demonstrate that the inactivation of the enhancer downregulates the target gene. Unfortunately, this seemingly simple experiment used to be almost impossible due to the lack of tools that manipulate enhancer activity in the genomic context. This is why the traditional approach just assumes a regulatory relationship between a gene and an enhancer based on a distance criterion. With the advent of CRISPRi, we can inactivate enhancers in the genomic context, linking enhancers to target genes on the basis of experimental proof of causality in gene expression. Below, we describe each step in detail and demonstrate how principled and powerful this new strategy is for finding enhancers that govern the expression of a given gene.
Figure 1.
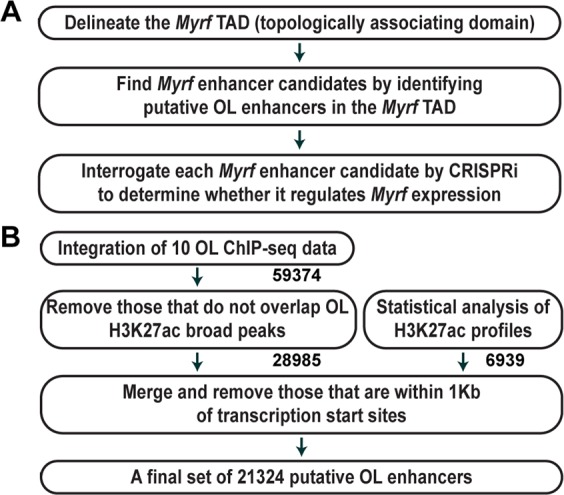
Schemes for identifying Myrf enhancers and deriving a genome-wide map of putative OL enhancers. (A) A principled approach that we propose to find Myrf enhancers. (B) An overview of computational analysis that generates a genome-wide map of 21324 putative promoter-distal OL enhancers.
TAD analysis for Myrf
In order to delineate the Myrf TAD, we analyzed publicly available kilobase-resolution Hi-C data for 7 diverse cell types from human and mouse23. The location of the MYRF/Myrf promoter is indicated by thin crossing lines in Fig. 2. The TAD organization around MYRF is well defined and conserved between different cell types (Fig. 2). The MYRF syntenic region is flipped in mouse compared to human. Strikingly, this flip is also mirrored in the Myrf TAD (CH12-LX, Fig. 2), highlighting a high degree conservation of the Myrf TAD through evolution. The Myrf TAD is about 300 Kb long (a thick blue square in CH12-LX), suggesting that critical Myrf enhancers would be found in the region spanning downstream 30 Kb and upstream 270 Kb of Myrf.
Figure 2.

The Myrf TAD is conserved between cell types and species. The publicly available 5 Kb-resolution Hi-C data for 7 diverse cell types from human and mouse23. The interaction frequency between two loci is indicated by color: white means no interaction, and red the strongest possible interaction. The Myrf promoter position is marked by thin crossing lines. The Myrf TAD is marked by a blue box for CH12-LX. The corresponding TAD for human is marked for the IMR90 data. IMR90: lung fibroblast. K562 and KBM7: chronic myelogenous leukemia cells. HeLa: cervical cancer cell. HUVEC: human umbilical vein endothelial cell. NHEK: normal human epidermal keratinocyte. CH12-LX: murine CH12 B cell lymphoma cell. This figure was generated by Juicebox52,53.
A genome-wide map of 21324 putative promoter-distal OL enhancers
Although the TAD analysis helps to narrow down the search space for Myrf enhancers, the Myrf TAD is still quite large. It is impractical to apply CRISPRi (CRISPR interference) to such a large genomic region for enhancer discovery (see below). To remedy this situation, we have derived a genome-wide map of putative OL enhancers by analyzing public OL ChIP-seq (chromatin immunoprecipitation coupled with high-throughput sequencing) data, with the idea being that the genome-wide map of putative OL enhancers would reveal putative OL enhancers in the Myrf TAD, which are Myrf enhancer candidates because they are in the same TAD as Myrf.
To predict OL enhancers on a genome-wide scale, we first identified 59374 genomic regions bound by Olig2, Sox10, Myrf, Brg1, Tcf7l2, or Chd7 (Fig. 1B)24–27. Of note, all these ChIP-seq data are from cultured rat OL lineage cells. Second, they were filtered by the H3K27ac broad peaks of rat OL lineage cells24 because H3K27ac marks active enhancers28, leaving 28985 genomic regions. Third, since ChIP-seq data are not available for all OL transcription factors, we have developed a statistical method that predicts enhancers independent of transcription factor ChIP-seq data. We took advantage of a unique feature of enhancers in H3K27ac ChIP-seq profiles. Since enhancers are bound by transcription factors, they are usually depleted of nucleosomes. Hence, although H3K27ac is known to mark active enhancers, its enrichment is found in shoulders flanking an enhancer rather than in the enhancer itself. This is why a peak-valley-peak pattern is observed for enhancers in H3K27ac ChIP-seq data (Fig. 3A). However, a reasonable null model would posit that H3K27ac ChIP-seq reads are distributed randomly (i.e., uniformly). Under the null model, the peak-valley-peak pattern of H3K27ac ChIP-seq reads for an enhancer would be unlikely, and this deviation from the null model can be quantified by the binomial cumulative distribution function (see Methods). Analysis of the public H3K27ac ChIP-seq data for OL lineage cells24 with the binomial cumulative distribution function revealed that 5804 of the 45212 H3K27ac broad peaks have at least one incidence of the peak-valley-peak pattern, leading to the prediction of 6939 OL enhancers. Interestingly, the 5804 broad peaks display a significantly higher level of H3K27ac signals than the rest (Fig. S1A). Finally, the 28985 genomic regions from the second step were merged with the 6939 putative OL enhancers from the third step (Fig. 1B). From the resulting set, we removed those within 1 Kb of transcription start sites, ending up with a final set of 21324 putative promoter-distal OL enhancers.
Figure 3.

Characterization of the genome-wide map of putative OL enhancers. (A) A typical peak-valley-peak pattern for an enhancer in H3K27ac ChIP-seq data. (B) The Sox10 spinal cord ChIP-seq data30 were aligned with the 21324 putative OL enhancers. (C) Gene ontology analysis by GREAT33. (D) The brain RNA-seq database34 was looked up to estimate the expression level of genes in in vivo OL lineage cells. A box plot shows that genes neighboring the putative OL enhancers, as defined by GREAT, are expressed at a significantly higher level than the rest. *p value ≈ 0 by the Mann–Whitney–Wilcoxon test corrected by the Bonferroni procedure.
We examined several aspects of the predicted OL enhancers to estimate its quality. First, we examined the size distribution of putative OL enhancers with the H3K27ac peak-valley-peak configuration. 3612 of the 21324 putative OL enhancers are overlaid with the H3K27ac peak-valley-peak pattern (Fig. S1B). The average size of the 3612 putative OL enhancers, as defined by the distance between the half-points of the flanking H3K27ac peaks, is about 390 base pairs (Fig. S1B). In other words, the 3612 putative OL enhancers are characterized by a 390 base pair-long nucleosome-depleted region, which is in line with a previous estimate29. Nucleosome depletion for the 3612 putative OL enhancers is likely due to the competitive DNA binding of transcription factors because transcription factor ChIP-seq peaks fall within the 390 base pair-long region (Fig. S1C,D). Second, we looked into the in vivo relevance of the 21324 putative OL enhancers. The Svaren laboratory published Sox10 ChIP-seq data for the spinal cord30. Our putative OL enhancers were derived independent of it. Sox10 is a key transcriptional regulator of OL lineage cells5,31,32, and a subset of important OL enhancers is expected to be marked by it. For Sox10 ChIP-seq peaks that are within ±4 Kb of the putative OL enhancers, about 85% of their ChIP-seq reads are mapped to the putative OL enhancers (defined as 390 base pairs long, Figs 3B and S1E). This high concentration of Sox10 ChIP-seq reads in the putative OL enhancers is not expected by chance (p value ≈ 0 by the binomial cumulative distribution function), supporting the in vivo relevance of the putative OL enhancers. Third, gene ontology (GO) analysis by GREAT33 indicates that the putative OL enhancers are significantly associated with GO terms related to OL development and central nervous system (CNS) myelination (Fig. 3C). Fourth, genes neighboring the putative OL enhancers, as defined by GREAT, tend to be expressed at a higher level in in vivo OL lineage cells than the rest34 (Fig. 3D), suggesting that our putative OL enhancers are active in the mouse brain. Overall, these results suggest a high quality for our putative OL enhancers.
Identification and CRISPRi analysis of 6 Myrf enhancer candidates
We compared our genome-wide map of putative OL enhancers with the Myrf TAD, finding 6 putative OL enhancers in the Myrf TAD. Since these 6 putative OL enhancers are in the same TAD as Myrf, they are Myrf enhancer candidates (EC1, 2, 3, 4, 5, and 6 in Fig. 4A). The 6 putative OL enhancers in the Myrf TAD were sorted by the strength of the underlying ChIP-seq data (Fig. 4B) and named accordingly. The one with the strongest evidence was named EC1, and the one with the weakest evidence EC6. The two best Myrf enhancer candidates (EC1 and EC2) are overlaid with the H3K27ac peak-valley-peak pattern. In addition, EC1 and EC2 are strongly bound by Sox10. The other four enhancer candidates are only associated with weak binding of Olig2 and Sox10. Four Myrf enhancer candidates are in the upstream region (EC2, EC4, EC5, and EC6), one in the first intron (EC1), and one in the downstream region (EC3). The one in the first intron (EC1 in Fig. 4A) is the same as ECR9 discovered by the Wegner laboratory on the basis of interspecies sequence conservation5. Bound by Sox10, ECR9 was shown to work as an enhancer in OL lineage cells of transgenic mice. Thus, ECR9 has been assumed, but not proved, to regulate Myrf expression in OLs. Our approach to identifying Myrf enhancer candidates does not rely on sequence conservation (Fig. 1B). Nonetheless, it successfully recovered ECR9, a known enhancer in the vicinity of Myrf, attesting to its good sensitivity.
Figure 4.

Six Myrf enhancer candidates. (A) Genomic locations of the 6 Myrf enhancer candidates. (B) Rat OL ChIP-seq data underlying the 6 Myrf enhancer candidates. iOL: immature OL. mOL: mature OL. OPC: oligodendrocyte precursor cells. SC: spinal cord.
When deriving the genome-wide map of putative OL enhancers, we used lenient criteria to minimize false negatives. Consequently, the six Myrf enhancer candidates may not all be OL enhancers. Even if they are OL enhancers, they may not regulate Myrf expression. Whether they are OL enhancers that govern Myrf expression needs to be determined experimentally. To this end, we resorted to CRISPRi in which dCas9-KRAB, a fusion protein between a nuclease-null Cas9 (dCas9) and a KRAB (Krüppel associated box) domain, is targeted to a specific locus by single guide RNAs (sgRNAs)19–22. When targeted to a promoter, dCas9-KRAB silences it, decreasing gene expression. When targeted to an enhancer, dCas9-KRAB inactivates it, which in turn downregulates target genes. To avoid the danger of false positive and false negative, each Myrf enhancer candidate was tested with at least 5 independent sgRNAs. In our CRISPRi experiment, dCas9-KRAB and sgRNA plasmids were transfected into primary mouse OPCs purified by immunopanning35,36, and transfected OPCs were cultured in a differentiation condition for 3 days to induce their differentiation into OLs and Myrf expression35. To easily monitor the expression level of Myrf, Rffl (an OL enhancer in the Rffl locus [rn4 chr10:71034166–71034749], which is a well-characterized Myrf luciferase reporter25,37,38) was co-transfected. Since co-transfection efficiency is high and luciferase assay extraordinarily sensitive, co-transfection of Rffl allowed us to detect changes in Myrf expression in the few transfected cells by a simple luciferase assay without selecting them. Of note, it remains unknown whether Rffl governs the expression of Rffl, which does not concern the current study.
Before interrogating the six Myrf enhancer candidates with CRISPRi, we validated CRISPRi for the Myrf locus. We designed six sgRNAs for the Myrf promoter and tested them in Oli-neu cells, a widely used OL cell line39 (G1-G6 in Fig. 5A). Scr1 and Scr2 are non-targeting negative control sgRNAs. When dCas9-KRAB was targeted by Scr2, there was no change in Myrf expression compared to Scr1, as expected (Fig. S2). Since Scr1 and Scr2 allow us to estimate non-specific effects associated with the expression of the CRISPRi components, we normalized all our data to the average of Scr1 and Scr2 for robust statistical analysis. When dCas9-KRAB was delivered to the Myrf promoter by the 6 sgRNAs, Myrf expression went down by 65–85% compared to the average of Scr1 and Scr2 (Fig. 5A), demonstrating that dCas9-KRAB works well for Myrf. Of the 6 sgRNAs, G5 was the most potent, and we used it throughout our study as a positive control. Having validated CRISPRi for Myrf, we tiled each Myrf enhancer candidate with 5 independent sgRNAs and used them to determine whether it regulates Myrf expression in primary mouse OLs. For a true Myrf enhancer, most of the 5 sgRNAs would lead to a significant drop in Myrf expression. For other genomic regions, few of the 5 sgRNAs would result in such change. Targeting dCas9-KRAB to EC1 or EC2 by the 5 sgRNAs significantly downregulated Myrf expression (Fig. 5B). In contrast, delivery of dCas9-KRAB to the other four Myrf enhancer candidates did not affect Myrf expression. Thus, the luciferase assay-based epigenome editing analysis indicates that EC1 and EC2 govern Myrf expression in primary mouse OLs. We also got the same results with Oli-neu cells (Fig. S2).
Figure 5.

Interrogation of the 6 Myrf enhancer candidates by CRISPRi. (A) CRISPRi validation for the Myrf locus. The expression level of Myrf was estimated by the reporter activity of Rffl, a highly specific and sensitive Myrf luciferase reporter25,37. The Rffl activity for each sgRNA was divided by the average Rffl activity for Scr1 and Scr2 to get the relative Rffl activity. For each sgRNA, the mean and standard error are shown. *p value < 1.4 × 10−4 by two-sided one sample Student’s t test corrected by the Bonferroni procedure (n = 4). (B) For each of the 6 Myrf enhancer candidates, 5 independent sgRNAs were used. For each sgRNA, the mean and standard error of the relative Rffl activity are shown. *p value < 3.3 × 10−2 by two-sided one sample Student’s t test corrected by the Bonferroni procedure (n = 9). (C) The signal from each fluorescence channel was quantified for individual cells by CellProfiler40. The number of cells analyzed is as follows: Scr1 (91), Pro (79), EC1 (42), EC2 (46), EC1&2 (82). Scale bar, 20 µm. AU: arbitrary unit. Targeting dCas9-KRAB to the Myrf promoter (Pro, by G5 in panel A), EC1 (by 5 in panel B), EC2 (by 4 in panel B), or EC1&2 (by 5 and 4 in panel B, respectively) led to a significant drop in Myrf expression. *p value < 4.6 × 10−10 by two-sided unpaired Student’s t test corrected by the Bonferroni procedure (comparison with Scr1). (D) Epigenome editing analysis was repeated for two negative control regions, NC1 and NC2. The mean and standard error for 8 and 7 sgRNAs that tile NC1 and NC2, respectively, are shown. *p value < 2.4 × 10−3 by two-sided one sample Student’s t test corrected by the Bonferroni procedure (n = 8).
To corroborate the above result, we performed a quantitative immunofluorescence experiment. A plasmid expressing dCas9-KRAB and tdTomato was transfected into primary mouse OPCs, together with sgRNA plasmids. Transfected OPCs were cultured in the differentiation condition for 3 days to induce their differentiation into OLs. They were then stained for Myrf and tdTomato (identifying transfected cells). The Myrf antibody used for this experiment has been previously validated for immunofluorescence5,12. We further confirmed it by comparing its immunofluorescence with that of a well-established Flag antibody for Flag-tagged Myrf constructs in Oli-neu cells (Fig. S3). Co-expression of dCas9-KRAB with Scr1 did not interfere with Myrf expression (Fig. 5C). However, when dCas9-KRAB was targeted to the Myrf promoter (Pro, Fig. 5C), Myrf expression was significantly downregulated. For an objective image analysis, the signal from each fluorescence channel (Hoechst, Myrf, and tdTomato) was quantified for individual OLs by CellProfiler40. This revealed that Hoechst and tdTomato signals were comparable across the samples. In contrast, Myrf signals were much lower when dCas9-KRAB was targeted to the Myrf promoter, EC1, EC2, or EC1&2 (Fig. 5C). These observations reinforce our epigenome editing analysis that EC1 and EC2 are required for the expression of Myrf.
Confirming the epigenome editing analysis
EC1 and EC2 happen to be the closest ones to Myrf. EC1 is in the first intron of Myrf, and EC2 is located 2 Kb upstream of Myrf. Creating repressive chromatin in an intron may hinder transcriptional elongation. Similarly, repressive epigenetic modifications induced by dCas9-KRAB for EC2 may spread to the Myrf promoter. Hence, it is possible that EC1 and EC2 came out positive in our epigenome editing analysis due to the non-specific effect of dCas9-KRAB. An exhaustive analysis for the GATA1 and MYC loci by dCas9-KRAB has shown that dCas9-KRAB is highly specific21, not displaying non-specific effects in promoter upstream regions and gene bodies (Fig. S4). This known specificity of dCas9-KRAB-mediated epigenome editing makes it unlikely that EC1 and EC2 are false positives. To experimentally confirm this conjecture, we repeated epigenome editing analysis for two negative control regions, NC1 and NC2 (Fig. 5D). NC1 is located between EC1 and the Myrf promoter. NC2 is found between EC2 and Myrf. No significant peak is observed for them in the rat OL ChIP-seq data (data not shown) and the public single-cell ATAC-seq data (see below). These features make NC1 and NC2 suitable negative controls. Of the 8 sgRNAs tested for NC1, only one came out positive (Fig. 5D). For NC2, only one came out positive from the 7 tested sgRNAs. These results are consistent with the known specificity of dCas9-KRAB and indicate that the positive epigenome editing results for EC1 and EC2 cannot be explained by non-specific effects of dCas9-KRAB.
To gain further support for this conclusion, we analyzed public single-cell ATAC-seq data. By using a single-cell ATAC-seq method, Shendure and co-workers determined chromatin accessibility for 13 different mouse tissues at a single cell resolution41. The resulting data were clustered into 27 broadly defined cell types. Notably, of the first intron of Myrf, EC1 is uniquely accessible (Fig. 6A). Likewise, EC2 is a specific peak in the upstream region of Myrf. These observations underscore the remarkable specificity of EC1 and EC2 despite their proximity to the Myrf promoter. We also analyzed the single-cell ATAC-seq data for the other four negative Myrf enhancer candidates (EC3, EC4, EC5, and EC6). In agreement with our epigenome editing analysis, they were not accessible in OLs (Fig. S5). Overall, these analyses reinforce our epigenome editing analysis that EC1 and EC2 are the only positive ones from the 6 Myrf enhancer candidates.
Figure 6.

Specificity of EC1 and EC2. (A) Single-cell ATAC-seq data for 13 mouse tissues41, which were clustered into 27 broadly defined cell types. (B) RT-qPCR data showing that CRISPRi silencing of EC1 and EC2 downregulates Myrf expression but does not impact the expression of nearby genes. Shown are the mean and standard error after normalization by Scr2. *p value < 1.3 × 10−5 by two-sided unpaired Student’s t test corrected by the Bonferroni procedure (n = 4).
Having confirmed that EC1 and EC2 govern Myrf expression, we wondered whether EC1 and EC2 are specific to Myrf. In other words, do EC1 and EC2 also regulate the expression of other genes in the vicinity of Myrf? To address this issue, we generated four Oli-neu cell lines where both EC1 and EC2 can be inducibly silenced by CRISPRi. The first and second cell lines express Scr1 and Scr2, respectively. The third cell line expresses two sgRNAs that deliver dCas9-KRAB to EC1 and EC2 (4 of Fig. 5B for EC1 and 3 of Fig. 5B for EC2, called Mix1 in Fig. 6B). To corroborate the results for this cell line, the fourth cell line expresses two other sgRNAs that deliver dCas9-KRAB to EC1 and EC2 (3 of Fig. 5B for EC1 and 5 of Fig. 5B for EC2, called Mix2 in Fig. 6B). These four Oli-neu cell lines express dCas9-KRAB in a doxycycline-dependent manner. To execute CRISPRi, doxycycline was added to the culture media for 2 days before RNA harvest. The expression level of Myrf and nearby genes that are expressed in Oli-neu cells was quantified by RT-qPCR where Gapdh was used as a control. When dCas9-KRAB was targeted to EC1 and EC2 by Mix1 or Mix2, Myrf expression went down by more than 65% (*p < 1.3 × 10−5 by unpaired two-sided Student’s t test corrected by the Bonferroni procedure, Fig. 6B), consistent with the above epigenome editing analyses. We analyzed the same RNA samples for other genes, finding no significant change in their expression levels (Fig. 6B). Collectively, these results demonstrate that EC1 and EC2 are highly specific to Myrf.
EC1 and EC2 are OL-specific enhancers
EC1, which is the same as ECR9, was previously shown to be activated by Sox10 and work as an enhancer in OLs5. Thus, there is strong evidence supporting its enhancer identity, although it remains unknown whether it is an OL-specific enhancer and whether it is also active in the human CNS. Regarding EC2, virtually nothing is known. In order to address these issues, we first tested the enhancer activity of EC1 and EC2 in primary mouse OLs. EC1 and EC2 were cloned into pGL3-promoter, and they were transfected into primary mouse OPCs. Transfected OPCs were cultured in the differentiation condition for 3 days to induce their differentiation. The SV40 promoter of pGL3-promoter had a very low basal activity in differentiating OLs (Vec in Fig. 7A). As a control, a genomic segment around Myrf that is not thought to work as an OL enhancer based on the OL ChIP-seq data (mm9 chr19:10318801–10319600) was cloned into pGL3-promoter (NC in Fig. 7A). NC failed to activate the SV40 promoter. In contrast, Rffl significantly activated the SV40 promoter. EC1 and EC2 were as powerful as Rffl (Fig. 7A), indicating that they work as enhancers in OLs. The same results were obtained when we replaced the SV40 promoter in pGL3-promoter with a minimal Myrf promoter (mm9 chr19:10315178–10315547)5. The minimal Myrf promoter had a very low basal activity in OLs (Myrf pro in Fig. 7B). However, its activity was significantly upregulated when EC1 or EC2 was placed upstream of it (EC1-Myrf pro and EC2-Myrf pro in Fig. 7B).
Figure 7.

EC1 and EC2 are OL-specific enhancers. (A) EC1 and EC2 are as good at activating the SV40 promoter of pGL3-promoter as Rffl, a known OL enhancer. The mean and standard error of the luciferase activity are shown for each construct. *p value < 4.3 × 10−8 by two-sided unpaired Student’s t test corrected by the Bonferroni procedure (comparison with Vec, n = 8). (B) EC1 and EC2 activate the Myrf promoter. The SV40 promoter of pGL3-promoter was replaced by the Myrf promoter, and the luciferase assay repeated. Shown are the mean and standard error relative to Vec (pGL3-promoter). *p value < 3.4 × 10−3 by two-sided unpaired Student’s t test corrected by the Bonferroni procedure (comparison with the Myrf promoter alone “Myrf pro”, n = 4). (C) The H3K27ac ChIP-seq data for EC1 and EC2 from the NIH Roadmap Epigenomics Project42. A complete dataset for EC1 and EC2 that encompasses more diverse tissues and cell types is available in Fig. S6. SM: smooth muscle.
Having confirmed the OL enhancer activity of EC1 and EC2, we analyzed the single-cell ATAC-seq data to determine whether they are OL-specific enhancers. Of the 27 broadly defined cell types, EC1 and EC2 are accessible only in OLs (Fig. 6A), indicating that they work as OL-specific enhancers in mouse. We also looked up the H3K27ac ChIP-seq data from the NIH Roadmap Epigenomics Project42 to determine the tissue specificity and conservation of EC1 and EC2 in human. Consistent with the mouse single-cell ATAC-seq data, EC1 and EC2 exhibit the peak-valley-peak pattern only in the brain tissue (Fig. 7C; see also Fig. S6). EC1 and EC2 are also overlaid with the H3K4me1 peak-valley-peak pattern (Fig. S7; H3K4me1 marking enhancers28). EC1 and EC2 are not marked by H3K9me3 and H3K27me3 in the human brain (Figs S8,S9; both being repressive histone marks). Taken together, we conclude that EC1 and EC2 are OL-specific enhancers that are conserved between human and mouse.
EC1 and EC2 are required for OL differentiation
Since Myrf is indispensable for OL development10 and EC1 and EC2 are essential for the expression of Myrf in OLs, we hypothesized that EC1 and EC2 would be required for OL differentiation. To test this hypothesis, primary mouse OPCs were transfected with two plasmids: one co-expressing dCas9-KRAB and tdTomato, and the other expressing sgRNAs. Transfected OPCs were cultured in the differentiation condition for 3 days and stained for tdTomato and myelin basic protein (MBP, a mature OL marker). The signal from each fluorescence channel was quantified for individual cells by CellProfiler. The quantitative image analysis revealed that targeting dCas9-KRAB to the Myrf promoter, EC1, EC2, or EC1&2 significantly decreases Mbp expression in primary mouse OLs (Fig. 8A), supporting our hypothesis that EC1 and EC2 are critical to OL differentiation in vitro. A similar experiment with Plp1, another OL maker, corroborated this conclusion (Fig. S10). However, under the condition where the expression of Plp1 was significantly downregulated, Myrf knockdown via CRISPRi silencing of the Myrf promoter, EC1, and EC2 did not consistently affect the morphological complexity of differentiating OLs (Fig. S10).
Figure 8.

EC1 and EC2 are required for OL differentiation. (A) Targeting dCas9-KRAB to the Myrf promoter (Pro, by G5 in Fig. 5A), EC1 (by 5 in Fig. 5B), EC2 (by 4 in Fig. 5B), or EC1&2 (by 5 and 4 in Fig. 5B, respectively) decreased Mbp expression in primary mouse OLs. Scale bar, 20 µm. The signal from each fluorescence channel was quantified for individual cells by CellProfiler. AU: arbitrary unit. *p value < 1.6 × 10−2 by two-sided unpaired Student’s t test corrected by the Bonferroni procedure (comparison with Scr1). The number of cells analyzed is as follows: Scr1 (165), Pro (142), EC1 (177), EC2 (46), and EC1&2 (99). (B) Three plasmids electroporated into SVZ neural stem cells of P1 CnpCre/+ mice. ITR: piggyBac inverted terminal repeat. hypBase: hyperactive piggyBac transposase45. IRES: internal ribosome entry site. (C) Targeting dCas9-KRAB to the Myrf promoter (Pro, by G5 in Fig. 5A), EC1 (by 5 in Fig. 5B), EC2 (by 4 in Fig. 5B), or EC1&2 (by 5 and 4 in Fig. 5B, respectively) downregulated the expression of Gst-π in OL lineage cells in the mouse brain. Scale bar, 20 µm. *p value < 3.6 × 10−30 by the cumulative binomial distribution function corrected by the Bonferroni procedure. See Fig. S11B for the uncropped brain section images.
To test the importance of EC1 and EC2 for OL differentiation in vivo, we silenced EC1 and EC2 in the mouse brain by CRISPRi and examined its effect on OL differentiation by Gst-π and CC1 (mature OL markers allowing for easy cell counts12). Three plasmids were co-electroporated into subventricular zone (SVZ) neural stem cells (NSCs) of CnpCre/+ mice at P1 (postnatal day 1)43,44 (Fig. 8B). The first plasmid is a piggyBac-based one that expresses dCas9-KRAB and nuclear-targeted mCherry in a Cre-dependent manner. The second is a piggyBac-based plasmid that constitutively expresses sgRNAs. The third plasmid encodes hypBase, a hyperactive piggyBac transposase that integrates the first and second plasmids into the genome of SVZ NSCs45. Our electroporation protocol primarily targets SVZ NSCs on the striatum side44 (Fig. S11A). Since the expression of dCas9-KRAB and mCherry depends on the Cre recombinase, epigenome editing occurs mainly in Cnp-positive progenies of P1 SVZ NSCs, which are OPCs46. Electroporated brains were harvested at P28 for immunohistochemistry with mCherry and Gst-π. By blind cell count, we determined the fraction of mCherry-positive cells that are also positive for Gst-π (a late-stage OL marker). When dCas9-KRAB was targeted by Scr1, more than 87% of mCherry-positive cells were positive for Gst-π (Fig. 8C). It demonstrates that epigenome editing was specifically targeted to OL lineage cells, as we intended with the Cnp1Cre/+ system. When dCas9-KRAB was targeted to the Myrf promoter, EC1, EC2, or EC1&2, less than 12% of mCherry-positive cells were positive for Gst-π. The same results were obtained for CC1 (marking an earlier stage than Gst-π12; Fig. S11C,D). We conclude that EC1 and EC2 are crucial for OL differentiation in vivo.
Discussion
Mapping enhancers to genes is a fundamental goal of modern biology. By combining recent advances in diverse fields into a coherent analysis pipeline, we have developed a simple yet powerful strategy that links enhancers to genes. This study illustrates its power by applying it to Myrf, a key OL gene that is indispensable for CNS myelination10–12. Three innovative features of our method enable a streamlined enhancer mapping for a gene of interest. First, it takes advantage of the transformative discovery from chromatin conformation capture studies that a gene and its enhancer tend to be found in the same TAD13–15. This provides strong spatial constraints on the possible locations of enhancers. Without TAD information, it is not clear where and how far to look in the genome for enhancers. TAD boundaries tend to be conserved between cell types and species13. Therefore, even if there is no Hi-C data for a particular cell type under study, one can still delineate the TAD by analyzing public Hi-C data23, as we did for Myrf. Second, we have derived a genome-wide map of 21324 putative OL enhancers. Without such map, the reduction of the enhancer search space by TAD analysis would not be really helpful because TADs are usually several hundred kilobases long. By comparing the genome-wide map of putative OL enhancers with the Myrf TAD, we uncovered 6 putative OL enhancers in the Myrf TAD, which are Myrf enhancer candidates because they are in the same TAD as Myrf. It is remarkable that the TAD information, together with our genome-wide map of putative OL enhancers, drastically reduces the search space for Myrf enhancers from the entire genome to just 6 loci, greatly facilitating the downstream CRISPRi analysis. Third, we have successfully used CRISPRi19–22, a cutting-edge epigenome editing technique, to interrogate enhancer candidates, finding two Myrf enhancers that are essential for OL development (EC1 and EC2). By inactivating enhancers in the genomic context, CRISPRi allows one to determine whether an enhancer candidate governs the expression of a given gene in the genomic context. As shown by this study and others, CRISPRi is exquisitely specific, even in promoter upstream regions and gene bodies, enabling the discovery of promoter-distal enhancers as well as those in promoter upstream regions and gene bodies. In sum, the combination of TAD, a cell type-specific genome-wide map of putative enhancers, and CRISPRi would significantly accelerate mapping enhancers to target genes. For cell type-specific genome-wide maps of putative enhancers, the single-cell ATAC-seq data and the NIH Roadmap Epigenomics Project data that we analyzed are good enough to cover most cell types. Of course, one can add publicly available cell type-specific genomic data to further strengthen such maps, as we did for OL lineage cells.
Encouraged by the positive results for Myrf, we are currently applying our new method to other OL genes and other cell types. For example, our ongoing study on Olig2, a gene essential for oligodendrogenesis, found that the Olig2 TAD is well defined and conserved between cell types and species, leading to a small number of Olig2 enhancer candidates for CRISPRi analysis. The same was also true for key myelin components (Cnp, Mbp, Plp1), critical OL transcriptional regulators (Olig1, Zeb2, Sox10, Nkx2.2, Sox2, Tcf7l2), and OL differentiation inhibitors that are potential therapeutic targets for remyelination therapies (Gpr17 and Gpr37).
If an enhancer is fully redundant with another, it may elude discovery because its inactivation would not show any effect. It is not clear how such redundancy would affect CRISPRi. In this regard, a previous analysis of HS2, an enhancer in the β-globin locus control region, by dCas9-KRAB is informative. Genetic deletion of HS2 in mice had only a mild effect on the expression of β-globin genes despite it being a strong enhancer47. In contrast, epigenome editing analysis of HS2 in human cells revealed that its inactivation by dCas9-KRAB significantly downregulates the expression of several β-globin genes20. Even though it is not possible to directly compare the two studies because of several differences (e.g., one study in mice while the other in a human cell line), the human cell line study suggests that CRISPRi may be able to overcome the redundancy issue. On the other hand, CRISPRi can easily be multiplexed, and one potential way to overcome enhancer redundancy is to silence multiple enhancer candidates simultaneously by co-expressing different sgRNAs, as we did for EC1 and EC2. Alternatively, one may supplement CRISPRi with CRISPRa, an epigenome editing technique that activates enhancers and upregulates target genes in the genomic context22,48, which may suffer less from enhancer redundancy than CRISPRi.
Methods
Reagents
A mouse Myrf cDNA that encodes the 1139 amino acid-long isoform was kindly provided by Dr. Ben Emery. The Myrf cDNA was cloned into pcDNA3. To generate epigenome editing constructs, we amplified dCas9-KRAB from pHAGE EF1α dCas9-KRAB (Addgene #50919) by PCR and replaced the Cre portion of pCAG-Cre (Addgene #13775) with it and an IRES (internal ribosome entry site)-tdTomato cassette. For SVZ electroporation, the piggyBac ITRs (inverted terminal repeat) were inserted, and tdTomato replaced with nuclear-targeted mCherry (Addgene #20972). To generate sgRNA expression vectors, the EF-1α promoter of pSBbi-RN (Addgene #60519) was replaced by the sgRNA scaffold taken from lentiCRISPR v2 (Addgene #52961). For the SVZ electroporation of sgRNAs, the content of PB-CA (Addgene #20960) was replaced by the sgRNA scaffold. Rffl, a Myrf luciferase reporter, was generated by cloning a rat genomic fragment (rn4 chr10:71034166–71034749) into pGL3-promoter (Promega). The sequence information of all constructs was verified by Sanger sequencing, and protein expression was confirmed by Western blot. The Myrf antibody was generously provided by Michael Wegner5. The sources of the commercial antibodies used for the study are as follows: FLAG (Sigma F1804) RFP (Rockland 600-901-379), MBP (Millipore MAB386), CC1 (Millipore OP80), Gst-π (MBL 311), donkey anti-rat IgG, Alexa Fluor 594 (Invitrogen A21209), donkey anti-rabbit IgG, Alexa Fluor 488 (Invitrogen A21206), donkey anti-mouse IgG, Alexa Fluor 488 (Invitrogen A21202), and donkey anti-chicken IgY, Alexa Fluor 594 (Jackson 703-585-155).
Animal procedures, tissue harvest, and cell culture
The current study was conducted in strict accordance with the protocol (approved protocol number #NA-Park2) approved by the Institutional Animal Care and Use Committee of SUNY Buffalo, which is licensed by the National Institutes of Health Office of Laboratory Animal Welfare (animal welfare assurance number: D16-00231). OPCs were purified from mouse pups of P7 ~ P9 by immunopanning36. The original immunopanning protocol for mouse OPCs35 did not work well in our hands. Instead, we found that the immunopanning protocol for rat OPCs works well for mouse OPCs, and this is why we used it to purify mouse OPCs. Primary mouse OPCs and Oli-neu cells39 were kept in a proliferative condition by supplementing the Sato media36 with PDGF (10 μg/mL), NT3 (1 μg/mL), CNTF (10 μg/mL), and NeuroCult™ SM1 Neuronal Supplement. They were maintained in a humidified 8% CO2 incubator at 37 °C. Transient transfection was performed using Lipofectamine 2000 as per the manufacturer’s instructions.
Genome-wide map of putative OL enhancers
The OL ChIP-seq data were downloaded from the Sequence Read Archive49. All the OL ChIP-seq data are from cultured rat OL lineage cells, and their accession numbers are as follows: Brg1 (GSM1040154, GSM1040155), Chd7 (GSM1869162), H3K27ac (GSM1040159, GSM1040160, GSM1040161), H3K4me3 (GSM1040162, GSM1040163, GSM1040164), Olig2 (GSM1040156, GSM1040157, GSM1040158), Sox10 (GSM1869163, GSM1577133, GSM1577134), Tcf7l2 (GSM1587566, GSM1587567, GSM1587568). The Myrf ChIP-seq data were downloaded from the journal website (https://journals.plos.org/plosbiology/article?id = 10.1371/journal.pbio.1001625). ChIP-seq reads were mapped to rn4 by Bowtie50, and peaks called by MACS251. The genome-wide map of 21324 putative OL enhancers were derived in rn4 and mapped to mm9 by liftOver using default options. For comparison with the Roadmap Epigenomics Project data, the six Myrf enhancer candidates were mapped to hg19 by liftOver using default options.
For the statistical analysis of OL H3K27ac profiles, the three OL H3K27ac ChIP-seq data (GSM1040159, GSM1040160, and GSM1040161) were merged, and the merged H3K27ac profile analyzed by the cumulative binomial distribution function. For a hypothetical valley (V) flanked by the left and right shoulders (LS and RS), we count the number of reads mapped to each region. VR: # of reads for V, LSR: # of reads for LS, and RSR: # of reads for RS. Also we define their sizes in base pairs (bps). VS: size of V, LSS: size of LS, and RSS: size of RS. Under the null model, H3K27ac ChIP-seq reads would be distributed randomly (i.e., uniformly). We quantify the deviation of the observed read distribution around the valley from the null model by the cumulative binomial distribution function as follows. For the left shoulder, compute the cumulative binomial probability of observing VR or less reads for V given the total number of reads being VR + LSR under the probability of VS/(VS + LSS). Since the sizes of valleys and shoulders are not pre-defined, we allow the valley to take any value from 250, 300, 350, and 400 bps. Shoulders are allowed to take any value from 300, 400, and 500 bps. From these 12 combinations, we take the lowest p value as a score for the valley. We repeat the same calculation for the right shoulder. Then we take the greater of the two p values as the final score for the valley. Once this computation was finished for the entire genome, the valleys were ranked by their scores. We leniently picked the top 6939 valleys as putative OL enhancers by cross-examining the valleys’ scores with their distributions of H3K27ac ChIP-seq reads.
ATAC-seq and the NIH roadmap epigenomics project data
The ATAC-seq data were downloaded from the Shendure laboratory website (http://atlas.gs.washington.edu/mouse-atac). The NIH Roadmap Epigenomics Project data were visualized by the WASHU Epigenome Browswer.
Luciferase assay-based epigenome editing analysis
Single guide RNAs (sgRNAs) were designed by using a web service from the Zhang laboratory (zlab.bio/guide-design-resources). For epigenome editing, dCas9-KRAB, gRNAs, Rffl, and pRL-TK (an internal control for ratiometric luciferase analysis, Promega) were transfected into primary mouse OPCs, which were then cultured for 3 days in the differentiation condition. The reporter activity of Rffl relative to that of pRL-TK was determined by the Promega dual luciferase reporter assay kit, as per the manufacturer’s instructions.
RT-qPCR
Total RNA was purified using Trizol (Thermo Fisher Scientific #15596026), and cDNA synthesized by the SuperScript First-Strand kit (Invitrogen #11904-018). Quantitative PCR was performed by C1000 Touch thermal cycler with the CFX384 optical reaction module (Bio-rad). The expression level of a gene was normalized to that of Gapdh. Each PCR reaction contained 2 µL of cDNA, 5 µL of the iTaq Universal SYBR Green Supermix (Bio-rad #1725124), and 500 nM of forward and reverse primers. The primer sequences are shown in Table 1.
Table 1.
Primer sequences for RT-qPCR.
| Myrf | Forward | CATTGTGCGGGCCTCTAACCC |
| Reverse | CCTCATCTGGCCGGTCGG | |
| Dagla | Forward | GCCGCACCTTCGTCAAGC |
| Reverse | GACCAGCTGGTGGCCTGAC | |
| Syt7 | Forward | CCACTGGTGTCAGCGCAAACTG |
| Reverse | GCTTTCTTCTCACCGCGCCC | |
| Sdhaf2 | Forward | CCTTGATCCCGACGCTGGC |
| Reverse | GAGTCTGTTGGGCTGTCACCTCTG | |
| Cpsf7 | Forward | CCCAAGAGGGGGAATACCTCCAC |
| Reverse | GGGCTTATCCACACGAGCAGATGAG | |
| Tmem258 | Forward | CCTGGTTCTTCGTTTACGAGGTCAC |
| Reverse | GGAGGAAGAGGACTCCAAAGCC | |
| Fads1 | Forward | CCCCTCTTCTTCGCCCTG |
| Reverse | GGGGTCCGATGAGGAAGAAGTAC | |
| Gapdh | Forward | GGTGAAGGTCGGTGTGAACGG |
| Reverse | CTGGAACATGTAGACCATGTAGTTGAGG |
In vitro differentiation assay of primary OPCs
DNA plasmids that express dCas9-KRAB and sgRNAs were transfected into primary mouse OPCs using Lipofectamine 2000. Transfected OPCs were kept in the differentiation condition for 3 days. Cells were fixed with 4% formaldehyde and permeabilized with 0.1% Triton X-100. Upon blocking with 1% BSA, they were incubated with primary antibodies diluted in the blocking buffer at 4 °C overnight, followed by incubation with fluorochrome-conjugated secondary antibodies. Nuclei were stained with Hoechst 33342 (Invitrogen). Fluorescence was visualized with a Leica DMi8 microscope with an ORCA-Flash4.0 sCMOS camera. Images were taken in a blind manner. The signal from each fluorescence channel (Hoechst, GFP, and RFP) was quantified by CellProfiler for an objective quantitative analysis.
SVZ electroporation
P1 CnpCre/+ pups were anesthetized by hypothermia. Five µl of DNA solution, including dCas9-KRAB, gRNA, the hyperactive piggyBac transposase, and 0.05% of Fast Green Dye for visualization, was injected into brain ventricles with a fine-tipped micropipette. After injection, electroporation paddles (wet with saline) were placed on the sides of the pup head, and five 40 V, 50 ms square pulses at a frequency of 60 Hz, were delivered. Our electroporation protocol mainly targets SVZ NSCs on the striatum side. The pup was allowed to recover on a heating pad. Electroporated pups were euthanized at P28. The brain tissue was fixed in 4% paraformaldehyde in PBS for 2 days, which was followed by an incubation in 30% sucrose for 1~2 days. The brain tissue was cut in 10 µm thickness. Brain sections were washed in PBS, permeabilized in 0.3% Trition-X100, and blocked in 5% FBS, 0.3~0.6% Triton-X100 in PBS. The sections were incubated with primary antibodies, washed in PBS, incubated with secondary antibodies, and washed in PBS. They were mounted in a mounting medium (VectaShield H-1000). Fluorescence was visualized with the Leica DMi8 microscope in a blind manner. Cells were counted in a blind manner by two independent researchers for robust statistical analysis.
Supplementary information
Acknowledgements
We thank Joshua Breunig and Hannah Park for teaching us SVZ electroporation and providing piggyBac plasmids; Dr. Klaus Nave for permission to use CnpCre/+ mice; Dr. Bogdan Beirowski for providing CnpCre/+ mice; Dr. Fraser Sim and Dr. Ben Emery for their critical comments on the manuscript; Dr. Laura Feltri for the advice during the course of this project. This work was supported by grants from the National Institutes of Health (R01NS094181 and R21NS102558).
Author Contributions
Y.P. and D.K. conceived the study. Y.P. directed it, performed computational analysis, and drafted the manuscript. D.K., H.A., R.S.S., M.S., C.F. and J.C. performed molecular cloning. D.K., R.S.S., M.S. and J.C. perform luciferase assays. D.K. performed quantitative immunofluorescence. H.A. performed S.V.Z. electroporation and immunohistochemistry. D.K., H.A. and R.S. performed blind cell counting. All the authors discussed the data and read and approved the manuscript.
Data Availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-47521-w.
References
- 1.Buecker C, Wysocka J. Enhancers as information integration hubs in development: lessons from genomics. Trends Genet. 2012;28:276–284. doi: 10.1016/j.tig.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nature Reviews Genetics. 2014;15:272–286. doi: 10.1038/nrg3682. [DOI] [PubMed] [Google Scholar]
- 3.Symmons O, et al. The Shh topological domain facilitates the action of remote enhancers by reducing the effects of genomic distances. Developmental Cell. 2016;39:529–543. doi: 10.1016/j.devcel.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Werner T, Hammer A, Wahlbuhl M, Bösl MR, Wegner M. Multiple conserved regulatory elements with overlapping functions determine Sox10 expression in mouse embryogenesis. Nucleic Acids Research. 2007;35:6526–6538. doi: 10.1093/nar/gkm727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hornig J, et al. The transcription factors Sox10 and Myrf define an essential regulatory network module in differentiating oligodendrocytes. Plos Genet. 2013;9:e1003907. doi: 10.1371/journal.pgen.1003907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weider M, et al. Elevated in vivo levels of a single transcription factor directly convert satellite glia into oligodendrocyte-like cells. Plos Genet. 2015;11:e1005008. doi: 10.1371/journal.pgen.1005008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weider M, et al. Nfat/calcineurin signaling promotes oligodendrocyte differentiation and myelination by transcription factor network tuning. Nature Communications. 2018;9:899. doi: 10.1038/s41467-018-03336-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forghani R, et al. A distal upstream enhancer from the Myelin Basic Protein gene regulates expression in myelin-forming Schwann cells. The Journal of Neuroscience. 2001;21:3780–3787. doi: 10.1523/JNEUROSCI.21-11-03780.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farhadi HF, et al. A combinatorial network of evolutionarily conserved Myelin Basic Protein regulatory sequences confers distinct glial-specific phenotypes. The Journal of Neuroscience. 2003;23:10214–10223. doi: 10.1523/JNEUROSCI.23-32-10214.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emery B, et al. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell. 2009;138:172–185. doi: 10.1016/j.cell.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koenning M, et al. Myelin gene regulatory factor is required for maintenance of myelin and mature oligodendrocyte identity in the adult CNS. The Journal of Neuroscience. 2012;32:12528–12542. doi: 10.1523/JNEUROSCI.1069-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duncan GJ, et al. Myelin regulatory factor drives remyelination in multiple sclerosis. Acta Neuropathologica. 2017;134:403–422. doi: 10.1007/s00401-017-1741-7. [DOI] [PubMed] [Google Scholar]
- 13.Dixon JR, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin F, et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503:290–294. doi: 10.1038/nature12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nature Reviews Genetics. 2013;14:390–403. doi: 10.1038/nrg3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nora EP, Dekker J, Heard E. Segmental folding of chromosomes: A basis for structural and regulatory chromosomal neighborhoods? BioEssays. 2013;35:818–828. doi: 10.1002/bies.201300040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pope BD, et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014;515:402–405. doi: 10.1038/nature13986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dixon JR, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–336. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kearns NA, et al. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods. 2015;12:401–403. doi: 10.1038/nmeth.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thakore PI, et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods. 2015;12:1143–1149. doi: 10.1038/nmeth.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fulco CP, et al. Systematic mapping of functional enhancer–promoter connections with CRISPR interference. Science. 2016;354:769–773. doi: 10.1126/science.aag2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klann TS, et al. CRISPR–Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat Biotechnol. 2017;35:561–568. doi: 10.1038/nbt.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rao SS, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu Y, et al. Olig2 targets chromatin remodelers to enhancers to initiate oligodendrocyte differentiation. Cell. 2013;152:248–261. doi: 10.1016/j.cell.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bujalka H, et al. MYRF is a membrane-associated transcription factor that autoproteolytically cleaves to directly activate myelin genes. Plos Biol. 2013;11:e1001625. doi: 10.1371/journal.pbio.1001625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He D, et al. Chd7 cooperates with Sox10 and regulates the onset of CNS myelination and remyelination. Nat Neurosci. 2016;19:678–689. doi: 10.1038/nn.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao C, et al. Dual regulatory switch through interactions of Tcf7l2/Tcf4 with stage-specific partners propels oligodendroglial maturation. Nature Communications. 2016;7:10883. doi: 10.1038/ncomms10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grossman SR, et al. Positional specificity of different transcription factor classes within enhancers. Proceedings of the National Academy of Sciences. 2018;115:E7222–E7230. doi: 10.1073/pnas.1804663115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez-Anido C, et al. Differential Sox10 genomic occupancy in myelinating glia. Glia. 2015;63:1897–1914. doi: 10.1002/glia.22855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stolt CC, et al. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes and Development. 2002;16:165–170. doi: 10.1101/gad.215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takada N, Kucenas S, Appel B. Sox10 Is Necessary for Oligodendrocyte Survival Following Axon Wrapping. Glia. 2010;58:996–1006. doi: 10.1002/glia.20981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLean CY, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. The Journal of Neuroscience. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emery B, Dugas JC. Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harbor Protocols. 2013;2013:854–868. doi: 10.1101/pdb.prot073973. [DOI] [PubMed] [Google Scholar]
- 36.Dugas JC, Emery B. Purification of oligodendrocyte precursor cells from rat cortices by immunopanning. Cold Spring Harbor Protocols. 2013;2013:745–758. doi: 10.1101/pdb.prot070862. [DOI] [PubMed] [Google Scholar]
- 37.Kim D, et al. Homo-trimerization is essential for the transcription factor function of Myrf for oligodendrocyte differentiation. Nucleic Acids Research. 2017;45:5112–5125. doi: 10.1093/nar/gkx080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi J-o, et al. Elucidating the transactivation domain of the pleiotropic transcription factor Myrf. Scientific Reports. 2018;8:13075. doi: 10.1038/s41598-018-31477-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jung M, et al. Lines of murine oligodendroglial precursor cells immortalized by an activated neu tyrosine kinase show distinct degrees of interaction with axons in vitro and in vivo. European Journal of Neuroscience. 1995;7:1245–1265. doi: 10.1111/j.1460-9568.1995.tb01115.x. [DOI] [PubMed] [Google Scholar]
- 40.Carpenter AE, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cusanovich DA, et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell. 2018;174:1309–1324. doi: 10.1016/j.cell.2018.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.The Roadmap Epigenomics Consortium Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lappe-Siefke C, et al. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nature Genetics. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- 44.Breunig JJ, et al. Ets factors regulate neural stem cell depletion and gliogenesis in Ras pathway glioma. Cell Reports. 2015;12:258–271. doi: 10.1016/j.celrep.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 45.Yusa K, Zhou L, Li MA, Bradley A, Craig NL. A hyperactive piggyBac transposase for mammalian applications. Proceedings of the National Academy of Sciences. 2011;108:1531–1536. doi: 10.1073/pnas.1008322108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kessaris N, et al. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. 2006;9:173–179. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fiering S, et al. Targeted deletion of 5′HS2 of the murine beta-globin LCR reveals that it is not essential for proper regulation of the beta-globin locus. Gene Dev. 1995;9:2203–2213. doi: 10.1101/gad.9.18.2203. [DOI] [PubMed] [Google Scholar]
- 48.Hilton IB, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33:510–517. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.International Nucleotide Sequence Database Collaboration The Sequence Read Archive: explosive growth of sequencing data. Nucleic Acids Research. 2012;40:D54–56. doi: 10.1093/nar/gkr854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Durand NC, et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Systems. 2016;3:99–101. doi: 10.1016/j.cels.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson JT, et al. Juicebox.js provides a cloud-based visualization system for Hi-C data. Cell Systems. 2018;6:256–258.e251. doi: 10.1016/j.cels.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.