

Abstract
Objective
Reelin (RELN) is a large glycoprotein involved in synapse maturation and neuronal organization throughout development. Deficits in RELN signaling contribute to multiple psychological disorders, such as autism spectrum disorder, schizophrenia, and bipolar disorder. Nutritional stress alters RELN expression in brain regions associated with these disorders; however, the involvement of RELN in the neural circuits involved in energy metabolism is unknown. The RELN receptors apolipoprotein E receptor 2 (ApoER2) and very low-density lipoprotein receptor (VLDLR) are involved in lipid metabolism and expressed in the hypothalamus. Here we explored the involvement of RELN in hypothalamic signaling and the impact of diet-induced obesity (DIO) on this system.
Methods
Adult male mice were fed a chow diet or maintained on a high-fat diet (HFD) for 12–16 weeks. HFD-fed DIO mice exhibited decreased ApoER2 and VLDLR expression and increased RELN protein in the hypothalamus. Electrophysiology was used to determine the mechanism by which the central fragment of RELN (CF-RELN) acts on arcuate nucleus (ARH) satiety-promoting proopiomelanocortin (POMC) neurons and the impact of DIO on this circuitry.
Results
CF-RELN exhibited heterogeneous presynaptic actions on inhibitory inputs onto ARH-POMC-EGFP neurons and consistent postsynaptic actions. Additionally, central administration of CF-RELN caused a significant increase in ARH c-Fos expression and an acute decrease in food intake and body weight.
Conclusions
We conclude that RELN signaling is modulated by diet, that RELN is involved in synaptic signaling onto ARH-POMC neurons, and that altering central CF-RELN levels can impact food intake and body weight.
Keywords: Reelin, Proopiomelanocortin, Arcuate nucleus, Obesity
Highlights
-
•
Diet-induced obesity alters reelin protein levels and expression of ApoER2 and VLDLR.
-
•
Reelin has direct, but divergent actions on GABAergic inputs onto POMC neurons.
-
•
Central administration of reelin protein decreases food intake and body weight.
1. Introduction
Reelin (RELN) is a large extracellular glycoprotein that is implicated in synaptic formation, remodeling, neuronal migration, and development [1]. Disruption of RELN signaling leads to abnormal architectural patterning in cortical, cerebellar, and hippocampal neurons [2], [3], [4]. RELN is also linked to cognitive defects associated with psychiatric and developmental disorders, such as autism spectrum disorder (ASD) [5], [6]. Nutritional stress from consumption of a high-fat diet (HFD) decreases RELN expression in brain regions associated with developmental disorders, including the hippocampus and medial prefrontal cortex (mPFC) [4], [7]. However, the synaptic mechanisms underlying hypothalamic RELN signaling and their involvement with energy metabolism are unknown.
Full-length (FL) RELN is comprised of 6 unique RELN repeat domains and is cleaved by ADAMTS-4 and ADAMTS-5 into three primary fragments, the N-terminus (NT; N-R2, 180 kDa), central fragment (CF; R3-6, 164 kDa), and the C-terminus fragment (CT; R7-C, 80 kDa) [8], [9]. There are two known receptors for RELN, the very low-density lipoprotein receptor (VLDLR) and the apolipoprotein E receptor 2 (ApoER2), both of which have a high affinity for FL-RELN and CF-RELN, but not for NT-RELN or CT-RELN [8], [10]. CF-RELN alone can activate ApoER2 and VLDLR, leading to Disabled-1 (Dab1) phosphorylation and downstream signaling cascades that result in neuronal maturation [11]. VLDLR is primarily found in the periphery and is implicated in lipid metabolism, adipose tissue inflammation, and adipocyte-macrophage interactions, while ApoER2 is primarily found in the brain and contributes to lipoprotein metabolism, neuronal migration, and long-term potentiation [12], [13], [14], [15].
The arcuate nucleus of the hypothalamus (ARH) plays a major role in energy balance and food intake [16]. Two ARH neuronal cell types that are important in the central regulation of energy homeostasis are neurons that co-express neuropeptide Y (NPY), agouti-related peptide (AgRP), and GABA, and those that express proopiomelanocortin (POMC) [17], [18], [19]. ARH-POMC neurons send projections throughout the brain and are involved in glucose homeostasis, thermogenesis, satiety pathways, and hedonic drive [20], [21], [22]. They are directly activated by anorexigenic signals, including insulin, high glucose, and leptin [23], [24], [25], [26]. Attenuated hypothalamic POMC expression is the earliest parameter that distinguishes obesity-prone from obesity-resistant mice before challenge with a HFD [27]. ARH-POMC neurons receive direct inhibitory GABAergic inputs from ARH-NPY neurons [28], [29]. Many GABAergic interneurons in the adult cortex co-express RELN; however, the role of RELN in inhibitory GABAergic inputs onto ARH-POMC neurons and how its signaling is altered in different metabolic states is unknown [30], [31].
Here we determined the impact of DIO on RELN, ApoER2, and VLDLR expression in the hypothalamus. Next, we identified a synaptic mechanism by which CF-RELN acts on ARH-POMC neurons and the impact of DIO on this system. Lastly, we investigated the in vivo effects of central CF-RELN administration.
2. Materials and methods
2.1. Animals
All animal procedures and experiments were approved by the Oregon National Primate Research Center Animal Institutional Care and Use Committee (IACUC) and the Novo Nordisk Ethical Review Committee in accordance with the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals (PHS Policy) and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Guide). Male POMC-EGFP transgenic mice on a C57BL/6 background were bred in-house (JAX stock #009593, The Jackson Laboratory). All animals were group-housed at 25 °C, under a 12/12-hr light/dark cycle, with food and water available ad libitum. For induction of DIO, animals were placed on a 60% high-fat diet (HFD) for 12–16 weeks (D12492, Research Diets). POMC-EGFP mice on a C57BL/6 background at ages 12–24 weeks were used for these studies [28] (JAX stock #009593, The Jackson Laboratory).
2.2. Surgery
Animals were anesthetized for surgery using 3% isoflurane in oxygen delivered by nose cone. Intracerebroventricular (ICV) implantation of guide cannula (C315GS-4/SPC, cut 2 mm below the pedestal, PlasticsOne) was conducted under sterile conditions and cannula were placed in the lateral ventricle using a stereotax (David Kopf Instruments, Tujunga, CA) at the following coordinates relative to bregma: AP = −0.7 mm, ML = 1.2 mm. Mice were administered carprofen at 5 mg/kg/day for 2 additional days and allowed to recover for 7 days before initiating experimental tests. For verification of cannula placement, angiotensin II (2.5 μg/mouse, 5-μL injection volume, Sigma–Aldrich) was administered via i.c.v. injection and water intake was recorded after one hour. Mice that consumed <1 mL of water were not included in the study.
2.3. Euthanasia
Animals were anesthetized by intraperitoneal injection of a ketamine/xylazine cocktail (120 mg/kg) and then transcardially perfused with 0.9% ice-cold saline solution followed by a 10% neutral-buffered formalin solution (NBF; Fisher Scientific). Brains were post-fixed in the same fixatives overnight and transferred to 25% sucrose buffer solution. For electrophysiology, qPCR, and western blot studies, animals were anesthetized in a chamber with isoflurane before sacrifice by decapitation.
2.4. In situ hybridization
PCR primers to generate riboprobes for ApoER2, VLDLR, and RELN gene expression were designed using NIH BLAST. PCR products were ligated into a pGEM-T vector and sequenced to verify their identity. Plasmids containing the probes were linearized using the appropriate restriction enzymes and 33P-UTP labeled probes generated. In situ hybridization was performed as previously described [32]. Adult male mouse brains were sectioned at 20 μm on a microtome, brain sections were fixed in 4% paraformaldehyde and then run through a series of washes for dehydration, delipidation, and rehydration. Sections were exposed to 33P-UTP-labeled probes overnight in a moist chamber at 55 °C. After hybridization, the slides were washed in 4x saline sodium citrate (SSC), then incubated in buffer containing RNase A at 37 °C, followed by washes in 0.1xSSC at 60 °C. Slides were then dehydrated through a graded ethanol series and dried. Sections were exposed to film overnight for visualization of the probe (Biomax MR, Kodak).
2.5. Quantitative PCR
Hypothalami were collected from control and DIO mice. Tissue was homogenized in Trizol and RNA was isolated using the RNeasy micro kit with on-column deoxyribonuclease I treatment (Qiagen). Quality and integrity of RNA was determined using a ND-1000 Nanodrop spectrophotometer. Reverse-transcription reactions were prepared using 2 μg of RNA and a cDNA Synthesis Kit (Promega). Quantitative real-time PCR was performed using TaqMan probes (Applied Biosystems) for RELN (Mm00465200_m1), ApoER2 (Mm00474030_m1), and VLDLR (Mm00443298_m1). The level of 18s rRNA (Hs03003631_g1) was used as an endogenous control for normalization. PCRs were performed in a 10-μl volume using 0.5 μl TaqMan probe, 20 ng cDNA template, 5 μl TaqMan Gene Expression Master Mix II with UNG (Applied Biosystems), and 2.5 μl DNase/RNase molecular-grade water (Qiagen). Real-time PCR was run using a 7900HT Fast Real-Time PCR system with initial denaturation at 50 °C for 2 min and 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s, and annealing at 60 °C for 1 min.
2.6. Western blot
Hypothalamic protein samples from control and DIO mice were heat-denatured at 95 °C for 5 min in denaturing loading buffer and separated on 8–16% Tris-glycine pre-cast gels (Invitrogen) using a mini-gel electrophoresis system (Bio-Rad Laboratories) at 35 mA per gel for 80 min. Protein was transferred to nitrocellulose membranes using iBlot (Life Technologies). Equal protein loading was assessed using Ponceau S staining (Sigma–Aldrich). Following transfer, membranes were blocked for 2 h with Tris-buffered saline/0.1% Tween-20 (TBST) containing 5% bovine serum albumin (BSA) or 5% non-fat milk protein (Mid-America Farms). Nitrocellulose membranes were incubated with a mouse monoclonal anti-RELN (Abcam, ab78540, 1:500) or rabbit ApoER2 (Abcam, ab215274, 1:500) primary antibody overnight at 4 °C in 1% BSA prepared in TBST. To confirm equal protein loading, membranes were later stripped using Re-Blot solution (Chemicon International) and incubated with anti-β-actin antibody (Santa Cruz Biotechnology; Santa Cruz, CA; 1:1000) in TBST containing 5% non-fat milk protein. Following primary antibody incubation, membranes were washed in TBST prior to incubation with an HRP-conjugated donkey anti-mouse or goat anti-rabbit (Invitrogen, 1:10,000) secondary antibody (Invitrogen; 1:10,000) in TBST containing 5% nonfat milk protein (Mid-America Farms) for 1 h at room temperature. Signals were detected using an enhanced chemiluminescence detection system (ECL; Pierce) and detected on photosensitive film (Hyperfilm-ECL; Kodak). Protein band intensity was determined using scanning densitometry and normalized to β-actin for analysis.
2.7. Protein verification and ApoER2 binding assay
Enzyme-linked immunosorbent assays (ELISA) were performed to assess the binding of various preparations of human CF-RELN stored under different conditions to recombinant human ApoER2 (R&D Systems Cat. No. 3520-AR). The format and description of the assay is as follows. E. coli-expressed CF-RELN protein (AA 1220-2660 based on GenBank: AAC51105.1) supplied by both Novo Nordisk and R&D Systems (Cat. No. 8546-MR-050) was coated onto 96-well microtiter plates (Nunc Polystyrene MaxiSorp ThermoScientific Cat. No. 442404) using 100 μL/well at 1 μg/mL overnight at 4 °C. After a wash step using 400 μL/well x 5 cycles (BioTek 405 Select Washer), plates were blocked using 200 μL/well of SuperBlock T20 in PBS (ThermoScientific/Pierce Cat. No. 37516) for 1 h at ambient room temperature (ART) with shaking (IKA MTS 2/4 set at 600 rpm). After another wash step, biotinylated ApoER2 was titrated into SuperBlock to concentrations ranging from 1000 to 15.6 ng/mL. Biotinylation of recombinant human ApoER2 was performed in-house. The titrations were then loaded in duplicate wells per condition tested using 100 μL/well and incubated at ART for 1 h with shaking. After another wash step, streptavidin-conjugated horseradish peroxidase (Pierce Cat. No. 21130) diluted into SuperBlock to 0.5 μg/mL was added to wells using 100 μL/well and incubated at ART for 30 min with shaking. Following a final wash step, plates were developed to sufficient color for approximately 3 min using 100 μL/well tetramethylbenzidine (SurModics TMB One Component Cat. No. TMBW-1000-01) and the reaction stopped with 1 N H2SO4 (SurModics TMB Stop Solution Cat. No. LSTP-1000-01). Plates were read on a microplate reader (Molecular Devices SpectraMax M5) for absorbance at 450 nm. SDS PAGE analysis of CF-RELN employed a 4–12% gradient gel run in MOPS buffer followed by Coomassie staining. Chromatography experiments were run using an Agilent 1290 UHPLC system (America, Agilent Corp.), on a SEC BEH 200A column (Waters Corporation) at 0.3 mL/min in PBS mobile phase controlled by a workstation. Data visualization was compiled using OpenLab Software (Version 2.2, Agilent Corp.).
2.8. Brain slice electrophysiology
The forebrain was removed and placed for 1 min in cold (0–4 °C) artificial cerebrospinal fluid (aCSF) sucrose solution composed of (in mM): 2 KCl, 1 MgCl2, 1.25 NaH2PO4, 10 HEPES, 26 NaHCO3, 1CaCl2, 2 MgSO4, 10 glucose, 208 sucrose, 0.7 ascorbic acid, and bubbled using 95% 02/5% C02. Final sucrose concentration was altered to adjust the osmolarity to between 310 and 314 mOsm. The forebrain region containing the ARH was blocked (rostral-caudal) and mounted in a vibrating microtome (Leica VT-1000S). Brains were sectioned with a sapphire knife (Delaware Diamond Knives) yielding roughly three slices (250-μm) per mouse. Slices were transferred and maintained at room temperature in a six-well dish containing a recording aCSF solution composed of (mM): 124 NaCl, 5 KCl, 2.6 NaH2PO4, 10 HEPES, 26 NaHCO3, 2 CaCl2, MgSO4, 5 dextrose, and bubbled using 95% 02/5% C02. For recordings, brain slices were transferred to a perfusion chamber containing aCSF maintained at 34–37 °C. Neurons were visualized using an upright microscope (Zeiss Axoskop 2). Recording electrodes were back-filled with experiment-specific internal solutions as follows (mM): Current-clamp; 125 K-gluconate, 2 KCl, 5 HEPES, 10 EGTA, 5 MgATP, 0.25 NaGTP. Voltage-clamp IPSC; 140 CsCl, 5 MgCl2, 1 BAPTA, 10 HEPES, 5 MgATP, 0.25 NaGTP. All internal solutions were brought to pH 7.3 using KOH (voltage-clamp) or CsOH (IPSC) at 301–304 mOsm. Neurons were recorded from the lateral ARH, away from the median eminence and third ventricle. Patch electrodes with a resistance of 3–5MΩ were guided to neurons using differential interference contrast (DIC) optics (Hammamatsu). Patch-clamp recordings were made with Axopatch 700B (Molecular Devices), a Digidata 1322A digitizer (Molecular Devices), and Clampex 10 recording software. Only neurons with holding currents not exceeding 100 pA at VH = −60mV for the 10-min control period (input resistance > 150 MΩ) were studied further. Series resistance was monitored throughout the recording, and neurons were not considered for further analysis if it exceeded 24 MΩ or drifted >25%. Series resistance did not differ between control (aCSF) and treatment. Current-clamp recordings were made at resting membrane potentials, and current injections were not used to hold the membrane at set potentials. All membrane potentials reported were corrected for junction potential (13 mV). All compounds were obtained from Tocris Cookson or Sigma Aldrich. CF-RELN protein was produced and provided by Novo Nordisk.
2.9. Immunohistochemistry
ICV-cannulated wild-type C57/BL mice were injected with vehicle (phosphate-buffered saline (PBS)) or CF-RELN (0.32 nmol/mouse, 5 μl/mouse). Ninety minutes later, mice were anaesthetized with ketamine/xylazine, then transcardially perfused with PBS followed by 4% paraformaldehyde in PBS. Brains were post-fixed in the same fixative overnight and transferred to 25% sucrose buffer solution. Forebrains were then frozen and sectioned at 25 μm on a microtome into six (6) serial sets. Tissue sections were washed in 50 mM potassium phosphate-buffered saline (KPBS) several times and pre-incubated in blocking buffer (KPBS + 0.4% triton X + 2% normal donkey serum) for 30 min before incubating in rabbit anti-c-Fos, 1:10,000 (sc-52, Santa Cruz Biotechnology, Santa Cruz, CA) containing blocking buffer for 24 h at 4 °C. Following 3 10-min washes in KPBS, tissue sections were incubated for 1 h in biotinylated donkey anti-rabbit antibody (1:600, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), then subsequently washed and incubated in avidin-biotin solution (Vectastain elite ABC, Vector laboratories, Burlingame, CA) for 30 min. The signals were then labeled using a nickel DAB peroxidase substrate kit (NiDAB) and incubating tissue for 1 min (SK-1000, Vector Laboratories, Burlingame, CA). Tissue was then washed 3 × 10 min in KPBS, incubated in a DAPI nuclear stain, followed by 3 10-min washes in KPBS (D1306, ThermoFisher Scientific). Tissue was then mounted and imaged using an upright Olympus BX6IVS microscope with VS-ASW imaging software (Olympus). Cell counts of c-Fos positive neurons from each mouse (n = 3–4 sections/mouse) were performed manually using ImageJ software. Cell counts were performed blind before the final analysis.
2.10. Food intake
For food intake and body weight studies, animals were single-housed and measurements were taken by weighing food hoppers and animals on a scale 1.5 h before the dark cycle. CF-RELN endotoxin levels measured at <0.05 EU per 1 mg of protein. Animals were randomized into groups according to food intake on day −1 of the first injection. Injections were given ICV after food intake and body weight measurements and received either 2 μl of vehicle (PBS), or CF-RELN at 0.15 nmol, 0.30 nmol, or 0.63 nmol. Baseline values for change in food intake and body weight were made by averaging day −3 to day 0 data points to normalize average starting food intake and body weight.
2.11. Statistical analysis
For electrophysiology experiments, statistical comparison of drug effect between groups was made using one-way ANOVA with Tukey's or Bonferroni post hoc analysis as noted (see results). In repeated-measures designed experiments that were missing data sets for the wash period, we used a mixed-model design. For experiments that classified effects as bidirectional, the Kolmogorov–Smirnov (KS) test was used to determine the significance of drug effect on mIPSC frequency and action potential firing frequency within individual neurons (Mini Analysis, Synaptosoft). To classify quiescent neurons in current-clamp studies, we used a cut off of a 0.5 mV change, which is greater than the mean of our vehicle-treated neurons. For all experiments, error bars are presented as mean ± standard error of the mean (SEM) and statistics were calculated using Prism7 software (Graphpad).
3. Results
3.1. Hypothalamic distribution of RELN, ApoER2, and VLDLR expression
Areas of the hypothalamus with the highest levels of RELN, VLDLR, and ApoER2 mRNA expression were identified by in situ hybridization (ISH) of hypothalamic sections. Photomicrographs in Figure 1A show that RELN mRNA expression was detected in multiple hypothalamic nuclei, including the ARH, paraventricular nucleus (PVH), and dorsal medial nucleus (DMH). VLDLR mRNA expression was detectable primarily in the PVH, ARH, ventral medial hypothalamus (VMH), and suprachiasmatic nucleus (SCh; Figure 1B). Additionally, within the hypothalamus, ApoER2 mRNA expression was very abundant, with the highest levels detected in the PVH, DMH, ARH, ventral medial nucleus (VMH), and SCh (Figure 1C). RELN, ApoER2, and VLDLR were all also expressed in the cortex and mPFC (data not shown). We also noted high levels of RELN and ApoER2 expression in the medial amygdala (MeA), which is implicated in both social and repetitive behaviors (Figure 1A,C) [33]. These results show that RELN, ApoER2, and VLDLR are expressed throughout the hypothalamus, including areas responsible for food intake and energy metabolism.
Figure 1.

RELN, VLDLR, and ApoER2 gene expression in the hypothalamus.A,In situ hybridization showing expression of RELN (left), B,VLDLR (center), and C,ApoER2 (right) mRNA in the ARH (arcuate nucleus), DMH (dorsal medial hypothalamus), MeA (medial amygdala), PVH (paraventricular nucleus of the hypothalamus), SCh (suprachiasmatic nucleus), and VMH (ventral medial nucleus of the hypothalamus).
3.2. DIO alters hypothalamic levels of RELN and its receptors VLDLR and ApoER2
RELN mRNA expression is decreased in the dorsal hippocampus of rats maintained on a high-fat, high-sugar diet and in the mPFC of mice maintained on a HFD [4], [7]. However, it is unknown whether DIO alters RELN, VLDLR, or ApoER2 expression in brain areas involved in energy metabolism, such as the hypothalamus. We used qPCR on hypothalamic blocks to quantify hypothalamic RELN, ApoER2, and VLDLR gene expression in lean control and DIO mice. Unlike previous reports demonstrating decreased RELN gene expression in the mPFC and hippocampus of rats, hypothalamic RELN gene expression was not altered in DIO mice when normalized to 18s rRNA (Quantity/18s) (Control: 0.13 ± 0.02, DIO: 0.09 ± 0.01; t (13) = 1.72, p = 0.11; Figure 2A) [4], [7]. However, both ApoER2 (Control: 1.34 ± 0.33, DIO: 0.59 ± 0.09; Students t-test, t (13) = 2.3, p = 0.04) and VLDLR (Control: 0.99 ± 0.13, DIO: 0.36 ± 0.05; t (13) = 4.7, p < 0.001) gene expression were decreased in the hypothalamus of DIO mice (Figure 2B,C).
Figure 2.

DIO alters hypothalamic VLDLR and ApoER2 mRNA and RELN protein levels.A, Relative hypothalamic expression of RELN in control (CTRL; n = 7) and DIO (n = 8) mice. B, Relative hypothalamic expression of ApoER2 in CTRL (n = 7) and DIO (n = 8) mice. C, Relative hypothalamic expression of VLDLR in CTRL (n = 7) and DIO (n = 8) mice. Individual data points from qPCR analyses are shown for each mouse. D, Western blot of FL-RELN, NT-RELN protein fragments, ApoER2, and β-actin in CTRL (n = 6) and DIO (n = 6) mice E, Relative optical density of Nt-RELN (F), FL-RELN and (G) ApoER2 protein levels in CTRL and DIO mice normalized to β-actin. (Error bars indicate ±SEM; student's t-test *p < 0.05).
To further examine whether DIO impacts RELN expression in the hypothalamus, western blot analysis was used to determine whether N-terminus (NT) RELN or full-length (FL) RELN protein levels in the hypothalamus were altered in DIO mice. The relative optical density (R.O.D.), normalized to β-actin, of FL-RELN protein levels were increased by 2.4-fold (Control: 156.5 ± 39.8 R.O.D., DIO: 372.2 ± 30.7; student's t-test, t (10) = 4.29, p = 0.002; Figure 2D,E). However, the NT-RELN (Control: 388.6 ± 29.8 R.O.D., DIO: 407.6 ± 18.8 R.O.D.; t (10) = 0.54. p = 0.60) was not altered in DIO mice (Figure 1D,F). Next, we looked at the protein levels of ApoER2, as this is the receptor primarily found in the brain and found that similar to its expression profile, ApoER2 protein levels were decreased in DIO mice Control: 635.9.6 ± 29.8 R.O.D., DIO: 516.9 ± 18.8 R.O.D.; t (10) = 2.25. p = 0.048; Figure 2D,G). These data show that the RELN system within the hypothalamus is altered in DIO mice, including increased FL-RELN protein, decreased ApoER2 protein, and decreased VLDLR and ApoER2 gene expression, all of which may impact RELN signaling in the hypothalamus.
3.3. Recombinant CF-RELN binding affinity to ApoER2 in vitro
RELN protein is made up of 8 repeats (R1-8) and is cleaved into three fragments in vivo, the N terminal (N-t), central (CF), and C terminal (C-t) fragments (Figure 3A). Previous studies demonstrated that the ApoER2 and VLDLR binding site is located in the R5-R6 region of the central fragment [34]. For our studies, we produced recombinant human CF-RELN (R3-R6 fragment), which shares 95% homology with mouse, to determine the central effects ex vivo and in vivo. High performance liquid chromatography (HPLC) with CF-RELN (24 mg/mL) either immediately after stock production or subjected to three freeze thaw cycles demonstrated no impact on the monomer:dimer ratio. An overnight incubation resulted in some aggregation, with a slight shift towards CF-RELN present as high-molecular-weight dimers or aggregates (data not shown). A Coomassie stain was performed to confirm the molecular weight of recombinant CF-RELN, which was present as a monomer and dimer in the absence of reducing agent (NR) and as a monomer in the presence of reducing agent (R) (Figure 3B). To determine the binding affinity of CF-RELN onto ApoER2, biotinylated ApoER2 was treated with 6 nM of synthesized CF-RELN and compared to CF-RELN obtained from R&D Systems. The binding affinity of synthesized CF-RELN was similar to CF-RELN obtained from R&D Systems (Figure 3C). Lastly, we performed a competition assay with non-labeled (cold) and biotinylated ApoER2 at increasing molar ratios, demonstrating that observed binding is due to a specific interaction between CF-RELN and ApoER2 (Figure 3D). These experiments demonstrate that CF-RELN is stable within our experimental parameters and physically bind to ApoER2 in vitro. However, it cannot be excluded that there are differences in their ability to functionally activate the receptor, or differences in its binding characteristics ex vivo or in vivo.
Figure 3.

Recombinant CF-RELN stability and in vitro ApoER2 binding affinity. A, Schematic of the full-length RELN protein (N: N terminus, SP: signal peptide, F-SL: F-spondin like domain, U: Unique domain, A/B: subdomains of the eight RELN repeats, C+: positively charged C-terminus) illustrating the three fragments and ApoER2/VLDLR binding site. B, Coomassie stain of CF-RELN without (NR) and with (R) the presence of reducing agent (M: molecular weight marker). C, Absorption as measured by optical density (OD) for binding of in-house and R&D Systems CF-RELN to biotinylated ApoER2 D, ApoER2 competitive binding assay for CF-RELN (R&D Systems) expressed as molar ratio of non-biotinylated (cold) ApoER2 to biotinylated ApoER2. (Error bars expressed as ± SEM * p < 0.05).
3.4. CF-RELN has distinct effects on action potential firing rate and membrane potential in ARH-POMC-EGFP neurons
To determine whether CF-RELN alters the activity of POMC neurons that participate in body weight regulation, whole-cell patch-clamp recordings were performed on ARH-POMC-EGFP neurons in hypothalamic brain slices of mice to measure action potential firing rates and the resting membrane potential in these neurons. ARH-POMC-EGFP neurons were easily identified and visualized in the coronal brain slice preparation for electrophysiology recordings. In 23 out of 38 recorded ARH-POMC-EGFP neurons, bath application of CF-RELN (100 nM) depolarized membrane potential (aCSF: −51.8 ± 2.0 mV, CF-RELN: −45.9 ± 1.6 mV, Wash: −43.6 ± 1.5 mV; Mixed-effects model, Tukey's post hoc, F (2, 37)) = 8.94, p < 0.001; Figure 4A,C,D) and increased the frequency of action potential firing in non-quiescent cells (aCSF: 1.4 ± 0.6 Hz, CF-RELN: 1.6 ± 0.5 Hz; 3.8 ± 0.9-fold; t (11) = 3.12, p < 0.001; Figure 4A,F). In 12 out of 38 recorded ARH-POMC-EGFP neurons, a 10-min bath application of CF-RELN (100 nM) induced a within-cell hyperpolarization of membrane potential without a significant combined effect (aCSF: −52.8 ± 1.5 mV, CF-RELN: −56.43 ± 1.8 mV, Wash: −55.8 ± 1.8 mV; Mixed-effects model, Tukey's post hoc, F (2, 18) = 2.89, p = 0.08; Figure 4B–D) and decreased the frequency of action potential firing in non-quiescent cells (aCSF: 0.8 ± 0.3 Hz, CF-RELN: 0.2 ± 0.1 Hz; 0.4 ± 0.1-fold; t (5) = 7.61, p = 0.001; Figure 4B,F). The combined effect of CF-RELN on action potential firing among all cells was a 2.5 ± 0.7-fold increase (t (17) = 2.23, p = 0.04; Figure 4F). The RELN effect on membrane potential was not dependent on resting membrane potential (Pearson R correlation, R2 = 0.008; p = 0.64; data not shown) and there was no significant difference between baseline action potential firing frequency of CF-RELN excited and inhibited neurons (Excited: 0.6 ± 0.3 Hz, Inhibited: 0.8 ± 0.4 Hz; t (24) = 0.28, p = 0.78; Figure 4E). The effects of CF-RELN did not disappear after 20 min of an aCSF wash perfusion, and three out of 38 cells did not respond to CF-RELN (Figure 4C,G).
Figure 4.

CF-RELN has two distinct effects on action potential firing and membrane potential in ARH-POMC-EGFP neurons. A, B, Representative traces showing the firing rate and membrane potential before and after bath application of CF-RELN (100 nM) in a depolarized (A) and hyperpolarized (B) POMC-EGFP neuron. C, Mean membrane potential in CF-RELN excited (left; n = 23) and inhibited (right; n = 12) neurons (one-way ANOVA, Tukey's post-hoc) D, Change in membrane potential in all recorded neurons. E, Baseline action potential firing frequency of CF-RELN-excited (E) and inhibited (I) neurons. F, Normalized frequency of action potential firing from individual POMC-EGFP neurons before and after CF-RELN treatment (2.5 ± 0.7-fold; n = 20; One-way ANOVA with pairwise analysis; Bold line represents cumulative mean change; aCSF error bar represents pooled between-cell variance from all neurons). G, Ratio of CF-RELN in inhibited, excited, and non-responsive (n = 3) neurons as determined by Kolmogorov–Smirnov (KS) test. (Error bars indicate ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001).
3.5. CF-RELN increases mIPSC frequency in ARH-POMC-EGFP neurons
To establish whether CF-RELN acts on ARH-POMC-EGFP neurons through a pre- or post-synaptic mechanism, we examined miniature inhibitory post-synaptic currents (mIPSCs). To isolate synaptic inhibitory currents and inputs, recordings were performed in the presence of TTX (1 μM), the AMPA receptor antagonist CNQX (10 μM) and the NMDA receptor antagonist APV (50 μM). In 9 out of 14 neurons, bath application of CF-RELN (100 nM) reduced mIPSC frequency from 2.4 ± 0.3 Hz to 1.9 ± 0.3 Hz (0.8 ± 0.1-fold; Figure 5A–C). In 5 out of 14 neurons, bath application of CF-RELN (100 nM) enhanced mIPSC frequency from 2.3 ± 0.6 Hz to 2.5 ± 0.7 Hz (1.1 ± 0.04-fold; Figure 5A–C). There was no significant difference in baseline mIPSC frequency between neurons in which CF-RELN reduced (2.4 ± 0.3 Hz) or enhanced (2.3 ± 0.6 Hz) mIPSC frequency (t (12) = 0.28, p = 0.78; Figure 5D). Bath application of CF-RELN (100nM) decreased mIPSC amplitude in both the reduced and enhanced groups (mIPSC control: 52.1 ± 6.4 pA, CF-RELN: 43.4 ± 5.5 pA; t (13) = 2.26, p = 0.04; Figure 5E). CF-RELN did not have a significant effect on holding current (t (13) = 0.46, p = 0.65; data not shown). These data suggest that CF-RELN has both pre- and post-synaptic actions on ARH-POMC-EGFP neurons.
Figure 5.

CF-RELN alters pre- and post-synaptic inputs onto ARH-POMC-EGFP neurons. A, Representative traces from a voltage-clamp experiment showing both reduced and enhanced mIPSC frequency after bath application of CF-RELN (100 nM) in POMC-EGFP neurons. B, Normalized mIPSC frequency (n = 14) before and after treatment of CF-RELN (0.9 ± 0.1-fold; Bold line, cumulative mean change; one-way ANOVA with pairwise analysis) C, Fold change of mIPSC frequency in CF-RELN reduced (0.8 ± 0.1-fold; left) and enhanced (1.1 ± 0.04-fold; right) as determined by KS-test. D, Baseline mIPSC frequency of neurons which CF-RELN reduced (R; n = 5) or enhanced (E; n = 9) mIPSC frequency E, Mean amplitude of mIPSCs before and after CF-RELN application (n = 14). Between-cell analysis performed using a paired t-test or mixed-effects model with Tukey's post-hoc analysis. (Error bars indicate ± SEM *p < 0.05).
3.6. CF-RELN has multiple modes of action on ARH-POMC-EGFP neurons
To investigate whether the actions of CF-RELN on ARH-POMC-EGFP neurons are dependent on the modulation of GABA mediated inhibitory inputs, we switched to the current-clamp configuration and recorded CF-RELN effects on membrane potential in the presence of multiple synaptic blocker combinations. Since we did not observe a washout of CF-RELN in most neurons, we wanted to confirm that our observed effect of CF-RELN was not an artifact due to a drifting membrane potential over time. We recorded a small sample of neurons with bath application of vehicle only and did not observe any drift in these recordings (aCSF: −52.6 ± 4.4 mV, Vehicle: −52.1 ± 4.5 mV; t (3) = 1.67, p = 0.20; Figure 6A,B,K).
Figure 6.

CF-RELN has actions on ARH-POMC-EGFP neurons independent of GABAergic and glutamatergic inputs. Representative trace and mean membrane potential before and after bath application of (A-B) vehicle (1:600 dilution) (C-D) CF-RELN in the presence of TTX (n = 11) (E-F) CF-RELN in the presence of TTX + BIC (n = 8), (G-H) TTX + APV (50 μM) + CNQX (10 μM; n = 9) (I-J), and TTX + APV + CNQX + BIC (n = 7). K, Mean change in membrane potential for CF-RELN excited and inhibited neurons, all neurons combined, and experiments in panels A-J. Statistical differences were determined using a paired t-test or mixed-effects model with Tukey's post-hoc (Error bars indicate ± SEM; *p < 0.05, **p < 0.01).
Pretreatment with the sodium channel blocker TTX (1 μM) did not block CF-RELN effects on membrane potential, suggesting that CF-RELN acts directly at the synapse (TTX: −56.1 ± 1.9 mV, TTX + CF-RELN: −54.3 ± 1.8 mV, TTX wash: −56.7 ± 1.5 mV; n = 11; Mixed-effects model, Tukey's post-hoc, F (2, 17) = 8.89, p = 0.002; Figure 6C,D,K). Interestingly, in the presence of TTX no neurons were hyperpolarized by CF-RELN. Next, we determined whether GABAergic inputs were required for the effects of CF-RELN on membrane potential using the GABAAR antagonist bicuculline (BIC) in combination with TTX. Similar to TTX, in this preparation CF-RELN significantly depolarized ARH-POMC-EGFP neurons and no recorded neurons hyperpolarized in response to CF-RELN (TTX + BIC: −52.9 ± 2.8 mV, CF-RELN: −50.6 ± 2.6 mV, Wash: −48.4 ± 2.7 mV; Mixed-effects model, Tukey's post hoc, F (2, 11) = 16.40, p < 0.001; Figure 6E,F,K). Similar to CF-RELN effects on mIPSC frequency, in the presence of TTX + CNQX + APV CF-RELN did not have a net effect on membrane potential, but approached significance (TTX + CNQX + APV: −56.9 ± 2.0 mV, CF-RELN: −55.8 ± 3.5 mV, Wash: −53.6 ± 1.8 mV; Mixed-effects model, Tukey's post hoc, F (2, 13) = 4.66, p = 0.055; Figure 6G,H,K). Lastly, we tested CF-RELN in the presence of all blockers combined, which independently depolarized ARH-POMC-EGFP neurons, and the addition of CF-RELN significantly hyperpolarized neurons independent of GABA and glutamate signaling (aCSF: −57.1 ± 1.2 mV, TTX + BIC + CNQX + APV: −47.2 ± 2.0 mV, CF-RELN: −49.2 ± 2.0; One-way ANOVA, Tukey's post-hoc, F (6, 12) = 4.42, p = 0.01; Figure 6I,J,K). Taken together, these data demonstrate that CF-RELN has complex actions on ARH-POMC-EGFP neurons, some of which are indirect through influencing presynaptic inputs onto ARH-POMC-EGFP neurons, and direct inhibitory effects that are independent of GABAergic and glutamatergic signaling.
3.7. DIO blunts post-synaptic actions of CF-RELN
Given that DIO decreases ApoER2 and VLDLR gene expression (Figure 1B,C), we wanted to determine the impact of DIO on the synaptic actions of CF-RELN on ARH-POMC-EGFP neurons. To determine whether DIO affects the pre- or postsynaptic effect of CF-RELN, recordings of inhibitory currents were made from ARH-POMC-EGFP neurons in DIO mice. Baseline mIPSC frequency was significantly lower in DIO mice when compared to control (Chow: 2.4 ± 0.3 Hz, DIO: 1.3 ± 0.2 Hz; unpaired t-test, F13,14 = 2.1, p = 0.002; Figure 7B). However, similar to control animals, there were two populations of ARH-POMC-EGFP neurons in DIO mice. CF-RELN reduced mIPSC frequency in 7 out of 15 neurons (Control: 1.3 ± 0.2 Hz, CF-RELN: 1.0 ± 0.1 Hz; 0.8 ± 0.0 fold; t (6) = 3.72, p = 0.0098) and enhanced mIPSC frequency in 8 out of 15 neurons (Control: 1.4 ± 0.3 Hz, CF-RELN: 1.5 ± 0.3 Hz; 1.1 ± 0.1 fold; t (7) = 4.21, p = 0.004; Figure 7C). Interestingly, the effect of CF-RELN on mIPSC amplitude was lost in DIO mice (Control: −60.5 ± 5.5 pA, CF-RELN: −58.4 ± 4.9 pA; Figure 7D). There was no significant difference in mIPSC amplitude between control and DIO mice. Next, we switched to the current-clamp configuration and determined that CF-RELN was still able to hyperpolarize ARH-POMC-EGFP neurons in DIO mice (aCSF: −54.33 ± 4.2 mV, CF-RELN: −50.0 ± 4.3 mV, −49.9 ± 5.8 mV; Mixed-effects model, Tukey's post hoc, F (2, 11) = 5.79, p = 0.02; Figure 7E,F,G). These data suggest that DIO mice have a reduced presynaptic inhibitory tone, though the presynaptic effects of CF-RELN on inhibitory inputs, along with the depolarizing effects of CF-RELN, are not altered. In contrast, the post-synaptic actions of CF-RELN on ARH-POMC-EGFP neurons were blunted by DIO.
Figure 7.
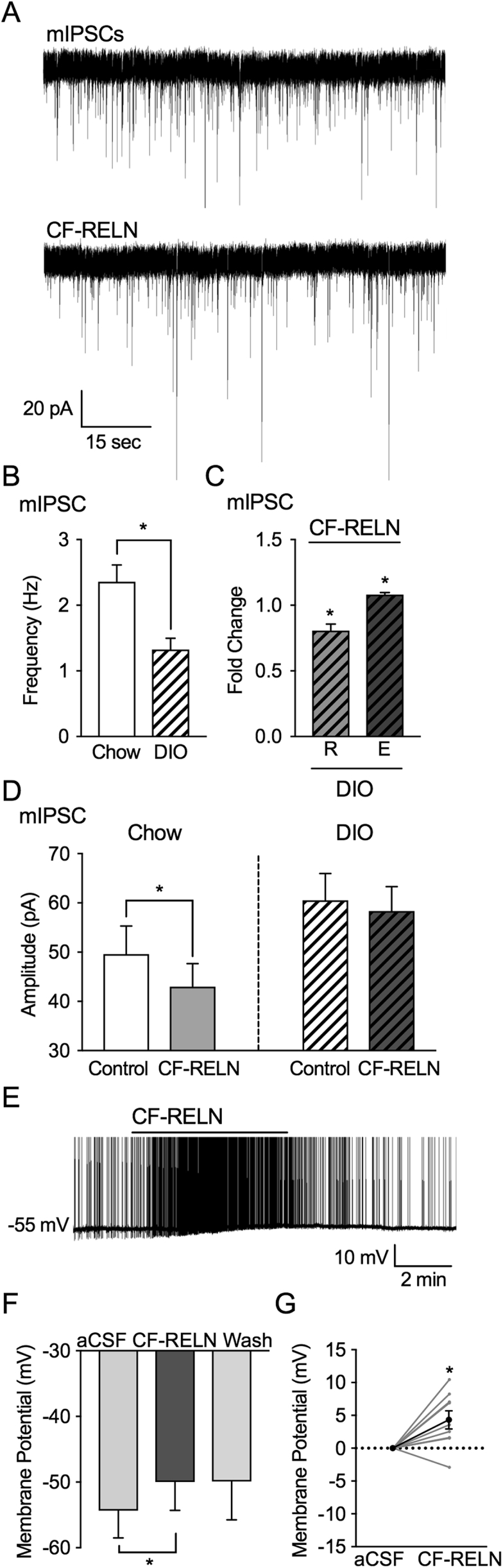
DIO alters actions of CF-RELN on mIPSC amplitude in ARH-POMC-EGFP neurons, but not membrane potential. A, Representative trace from a voltage-clamp experiment showing reduced mIPSC frequency after bath application of RELN (100 nM) in a POMC-EGFP neuron from DIO mice. B, Basal mIPSC frequency in POMC-EGFP neurons from chow fed (n = 14) and DIO mice (n = 15) C, Fold change of mIPSC frequency in neurons which CF-RELN reduced (R; 0.8 ± 0.0-fold; n = 7) or enhanced (E; 1.1 ± 0.1-fold; n = 8) mIPSC frequency as determined by KS-test D, Mean amplitude of mIPSCs in POMC-EGFP neurons from chow fed (n = 14) and DIO mice (n = 15) before and after RELN application. E, Representative current-clamp trace of showing membrane depolarization of a POMC-EGFP neuron in a DIO mouse after bath application of CF-RELN (100 nM). (F) Mean and change in (G) membrane potential before and after application of CF-RELN in DIO mice (n = 10). (Between-cell analysis using a Mixed-effects model with Tukey's post-hoc analysis *p < 0.05).
3.8. Central administration of CF-RELN increases ARH c-Fos expression and results in a reduction in food intake and body weight
To determine whether CF-RELN can activate hypothalamic neurons in vivo, c-Fos activation was quantified and visualized using NiDAB staining, following ICV injections of vehicle (n = 6) or CF-RELN (0.3 nmol; n = 5) into the lateral ventricle of wild-type mice (Figure 8A). When compared to vehicle (3.71 ± 0.87 c-Fos + neurons/section), CF-RELN significantly increased c-Fos activation in the ARH (32.53 ± 13.53 c-Fos + neurons/section; student's t-test, t (9) = 2.35, p = 0.04; Figure 8A,B). These data demonstrate that central administration of CF-RELN activates hypothalamic neurons, including in the ARH. Given that CF-RELN alters the activity of ARH-POMC-EGFP neurons ex vivo and increases cFos expression in vivo, we next determined the effects of CF-RELN on food intake and body weight. Lean mice on a control diet received daily lateral cerebral ventricle injections of vehicle, or one of three concentrations of CF-RELN (0.15 nmol, 0.30 nmol, or 0.63 nmol). CF-RELN had a significant effect on food intake 24 h after the first injection in mice receiving 0.15 nmol (p = 0.003) or 0.63 nmol (p = 0.015) CF-RELN; however, additional daily injections did not result in further decreases in food intake (two-way ANOVA, F3, 99 = 2.40 p = 0.05, Dunnet's posthoc; Figure 8C,D). Overall body weight trended down in CF-RELN-treated mice and this effect was only significant in mice treated with 0.15 nmol (p = 0.023) CF-RELN when measured as delta body weight (two-way ANOVA, F3, 100 = 4.53 p = 0.005, Dunnet's posthoc; Figure 8E,F). These data show that, in mice fed a regular chow diet, CF-RELN can induce a temporary reduction in food intake and body weight.
Figure 8.

Effect of central CF-RELN administration on c-Fos expression, food intake, and body weight. A, Representative immunohistochemistry images of c-Fos expression in the ARH of vehicle (n = 6) and CF-RELN (n = 5) treated mice. B, Average number of c-Fos + neurons in the ARH per section (unpaired t-test). C, Daily and delta (D) food intake after daily lateral ventricle ICV administration of vehicle (n = 6), or CF-RELN at 0.15 nmol/day (n = 6; p = 0.001), 0.30 nmol/day (n = 5), or 0.625 nmol/day (n = 6; p = 0.02; two-way ANOVA Dunnett's post-hoc) E, Daily and delta (F) body weight measured in animals injected with vehicle (n = 6), or CF-RELN at 0.15 nmol/day (n = 6; p = 0.02), 0.30 nmol/day (n = 5), or 0.625 nmol/day (n = 6; two-way ANOVA Dunnett's post-hoc; Error bars indicate ± SEM; *p < 0.05, **p < 0.01).
4. Discussion
Obesity is a risk factor for multiple neurological disorders associated with RELN, and an HFD disrupts RELN expression in brain areas associated with these disorders [4], [6], [7], [35]. The current studies demonstrate for the first time that the RELN system in the hypothalamus is altered by DIO, and that RELN has direct actions on ARH-POMC neurons, a key population of neurons regulating appetite and body weight homeostasis. Here we report four key findings: first, hypothalamic protein levels of RELN and the expression of ApoER2 and VLDLR are altered by DIO; second, CF-RELN activates ARH neurons in vivo; third, CF-RELN acts pre- and post-synaptically on ARH-POMC neurons; lastly, the post-synaptic effects of CF-RELN are blunted by DIO.
RELN acts through the lipoprotein receptors ApoER2 and VLDLR, which together play an important role in development, synaptic maturation, and cholesterol and fatty acid metabolism [15]. Notably, VLDLR deficiency protects against DIO; however, the synaptic mechanisms underlying RELN, ApoER2, and VLDLR signaling in hypothalamic nuclei and the impact of DIO on this system have not been explored [36]. Here we show that VLDLR and ApoER2 gene expression is decreased in the hypothalamus of DIO mice. Obesity is known to increase plasma triglycerides, cholesterol levels, and blood glucose, and VLDLR-deficient mice exhibit improved glucose clearance, reduced adipose tissue size, protection against DIO, and decreased levels of inflammatory markers [14], [37], [38]. This suggests that the decreased expression of VLDLR and ApoER2 may be a protective mechanism or compensatory response to elevated free fatty acids and circulating lipoproteins. Interestingly, obesity upregulates VLDLR expression in human adipose tissue, suggesting that the impact of obesity on VLDLR expression is likely tissue-specific [39].
Although hypothalamic RELN expression was unchanged, protein levels of RELN were increased in the hypothalamus of DIO mice. This could either be due to changes in the translation of RELN mRNA to RELN protein or decreased degradation processes. For example, expression of ADAMTS-4 and ADAMTS-5, the proteases responsible for the degradation of RELN, are regulated by leptin, a hormone involved in both metabolism and mood disorders, whose circulating levels are increased in DIO animals [8], [40], [41], [42], [43].
The majority of studies characterizing hypothalamic RELN expression have focused on embryonic and postnatal developmental periods, reporting strong RELN expression during development and a slow decline in expression into adulthood [11], [44]. However, the function of RELN in adulthood, particularly in the hypothalamus, remains largely unknown. Similar to previous findings, we observed the highest hypothalamic RELN gene expression in the PVH, which receives direct connections from ARH-POMC neurons and is involved in both mood disorders and satiety pathways [11], [20], [45]. However, neither RELN nor its receptors have been studied in regard to hypothalamic metabolic processes. VLDLR plays a major role in cholesterol and fatty acid metabolism; however, the few studies looking at the role of VLDLR in the hypothalamus have primarily focused on reproductive endocrine signals [15], [46]. The function of RELN receptors and the expression of RELN, ApoER2, and VLDLR in hypothalamic nuclei such as the ARH, DMH, PVH, and VMH suggest interplay between RELN and metabolic circuitry.
Further evidence that RELN could be an endogenous regulator of metabolic circuitry in the hypothalamus comes from the current study's findings that CF-RELN alters the electrophysiological properties ARH-POMC neurons. Here we report two distinct responses of RELN on ARH-POMC neurons. Depending on the neuron, CF-RELN was able to either excite or inhibit the activity of ARH-POMC neurons. It is well established that responses from ARH-POMC neurons are often heterogeneous, with previously documented heterogeneous responses to leptin, insulin and glucose [26], [47], [48], [49]. Additionally, ARH-POMC neurons can be either glutamatergic or GABAergic and also produce opposing peptides, such as the orexigenic peptide β-endorphin and the anorexigenic peptide α-MSH, both of which are also involved in affective disorders [50], [51], [52], [53], [54], [55]. This finding indicates that RELN may have several different modes of action to alter firing of the ARH-POMC neuron population which could influence the activity of downstream targets involved in metabolic regulation, such as the PVH [11], [20], [45].
To explore potential mechanisms by which RELN inhibits a subpopulation of ARH-POMC neurons, the current study investigated RELN modulation of inhibitory input to this population. ARH-POMC neurons can self-regulate via inhibitory inputs and also receive inhibitory inputs from ARH-NPY neurons [26], [28], [29]. In other brain areas such as the cortex, RELN is co-expressed with both NPY neurons and GABAergic neurons [30]. We observed that, similar to CF-RELN actions on ARH-POMC neuron excitability, CF-RELN has differential effects on inhibitory inputs onto ARH-POMC neurons, either reducing or enhancing the presynaptic release of GABA onto these neurons. Our results show that CF-RELN alters both mIPSC frequency, which is associated with presynaptic actions, and amplitude, which is associated with a postsynaptic effect [56], [57].
Interestingly, when we isolated the synapse and measured membrane potential, ARH-POMC neurons depolarized homogenously. This also occurred when we blocked the GABAAR. One possible mechanism for this finding is that CF-RELN actions on ARH-POMC neurons also involve excitatory neurotransmitters such as glutamate. RELN is involved in synapse formation and in the hippocampus RELN enhances NMDA and AMPA receptor activity through different mechanisms [58], [59]. When blocking NMDA and AMPA receptor activity, multiple neurons did depolarize; however, CF-RELN did not have a net effect on membrane potential, suggesting the involvement of glutamatergic inputs in CF-RELN actions on ARH-POMC neurons. Lastly, we tested whether CF-RELN had direct actions on membrane potential independent of both glutamatergic and GABAergic signaling. When we blocked GABAA, NMDA, and AMPA receptors, CF-RELN consistently inhibited ARH-POMC neurons. Together these data demonstrate that CF-RELN has complex actions on ARH-POMC neurons, and while excitatory and inhibitory presynaptic inputs are involved in the actions of CF-RELN on ARH-POMC neurons, CF-RELN has independent postsynaptic actions on these neurons. It is possible that presynaptic actions of CF-RELN are variable due to the number, strength, and types of inputs on each individual ARH-POMC neuron. How all these components work together, and the significance of each component in the system as a whole, is still unclear and requires future studies.
Consistent with RELN's ability to alter ARH-POMC neurons through differential pre- and post-synaptic mechanisms, there is evidence that both VLDLR and ApoER2 are located pre- and post-synaptically in other neuronal phenotypes [60], [61], [62]. Here we show that DIO attenuates CF-RELN's actions on inhibitory postsynaptic signals in ARH-POMC neurons, which is consistent with our data showing a decrease in VLDLR and ApoER2 expression. However, in DIO CF-RELN still has an excitatory action on ARH-POMC neurons. This suggests that, although the RELN system is altered in DIO, the functional role of RELN in metabolic circuitry is still intact. Whether CF-RELN actions in DIO are mediated by ApoER2 and VLDLR or if an additional unknown receptor is involved is yet to be determined.
Lastly, we investigated the effects of CF-RELN in vivo. Our results indicate that exogenous CF-RELN can activate ARH neurons and has a modest effect on food intake and body weight. Similar to known orexigenic hormones, CF-RELN treatment does not have long-lasting effects in lean animals. RELN might be altering hypothalamic systems that influence long-term synaptic organization and play a role in non-metabolic processes. The more long-term influence of RELN on metabolic circuitry is supported by our initial results that show hypothalamic ApoER2 and VLDLR expression are decreased, and RELN protein levels are increased, in DIO mice. It remains unclear whether CF-RELN treatment would be effective in DIO and if one or both of the known RELN receptors mediate the postsynaptic response in ARH-POMC neurons.
5. Conclusions
While the current study focuses on the role of RELN in the hypothalamus and in metabolic circuitry, it does raise the question of whether RELN is an intermediary link between obesity and other disorders. The hypothalamic-pituitary-adrenal axis is highly involved in metabolic processes and is associated with mood and developmental disorders. Additionally, the hypothalamus is smaller in patients with ASD [63], [64]. Given that maternal obesity is a risk factor for ASD, RELN is associated with ASD, and RELN signaling is altered in the hypothalamus of DIO animals, it is reasonable to question whether RELN is the common denominator [35], [65], [66], [67]. Further studies are needed to investigate the exact role of RELN in metabolic circuitry, whether maternal obesity impacts RELN signaling in developing offspring, and how these systems are related.
Taken together, these studies demonstrate a possible mechanism by which RELN could influence energy homeostasis, potentially via actions on ARH-POMC neurons. In addition, consumption of an HFD blunts VLDLR and ApoER2 expression in the hypothalamus and disrupts at least one mechanism by which RELN acts on ARH-POMC neurons. While future work is required to determine the exact role of RELN in energy homeostasis, these studies provide an interesting direction for future work exploring the relationship between development, metabolic circuitry, and their associated pathologies.
Grants
This work was supported in part by research funding from Novo Nordisk to PK, CTR, MWS, and MAC and NIH grant P51 OD 01192 for operation of the Oregon National Primate Research Center.
Conflict of interest
BLR, BJB, JMC, MAK, SRL, KGT and CAT declare no competing financial interests. CTR, MWS, MAC and PK receive research funding from Novo Nordisk. MAC is a consultant for Novo Nordisk, iNova and Valeant. CMB, LSD, CH, WH, KMH, AW, ACW, MTC and KLG are employees of Novo Nordisk.
References
- 1.Stranahan A.M., Erion J.R., Wosiski-Kuhn M. Reelin signaling in development, maintenance, and plasticity of neural networks. Ageing Research Reviews. 2013;12(3):815–822. doi: 10.1016/j.arr.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D'Arcangelo G., Miao G.G., Chen S.C., Soares H.D., Morgan J.I., Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374(6524):719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 3.Tissir F., Goffinet A.M. Reelin and brain development. Nature Reviews Neuroscience. 2003;4(6):496–505. doi: 10.1038/nrn1113. [DOI] [PubMed] [Google Scholar]
- 4.Reichelt A.C., Maniam J., Westbrook R.F., Morris M.J. Dietary-induced obesity disrupts trace fear conditioning and decreases hippocampal reelin expression. Brain, Behavior, and Immunity. 2015;43:68–75. doi: 10.1016/j.bbi.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Caruncho H.J., Brymer K., Romay-Tallon R., Mitchell M.A., Rivera-Baltanas T., Botterill J. Reelin-related disturbances in depression: implications for translational studies. Frontiers in Cellular Neuroscience. 2016;10:48. doi: 10.3389/fncel.2016.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lammert D.B., Howell B.W. RELN mutations in autism spectrum disorder. Frontiers in Cellular Neuroscience. 2016;10:84. doi: 10.3389/fncel.2016.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Labouesse M.A., Lassalle O., Richetto J., Iafrati J., Weber-Stadlbauer U., Notter T. Hypervulnerability of the adolescent prefrontal cortex to nutritional stress via reelin deficiency. Molecular Psychiatry. 2017;22(7):961–971. doi: 10.1038/mp.2016.193. [DOI] [PubMed] [Google Scholar]
- 8.Krstic D., Rodriguez M., Knuesel I. Regulated proteolytic processing of Reelin through interplay of tissue plasminogen activator (tPA), ADAMTS-4, ADAMTS-5, and their modulators. PLoS One. 2012;7(10):e47793. doi: 10.1371/journal.pone.0047793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kohno S., Kohno T., Nakano Y., Suzuki K., Ishii M., Tagami H. Mechanism and significance of specific proteolytic cleavage of Reelin. Biochemical and Biophysical Research Communications. 2009;380(1):93–97. doi: 10.1016/j.bbrc.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 10.Lee G.H., Chhangawala Z., von Daake S., Savas J.N., Yates J.R., 3rd, Comoletti D. Reelin induces Erk1/2 signaling in cortical neurons through a non-canonical pathway. Journal of Biological Chemistry. 2014;289(29):20307–20317. doi: 10.1074/jbc.M114.576249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jossin Y., Ignatova N., Hiesberger T., Herz J., Lambert de Rouvroit C., Goffinet A.M. The central fragment of Reelin, generated by proteolytic processing in vivo, is critical to its function during cortical plate development. Journal of Neuroscience. 2004;24(2):514–521. doi: 10.1523/JNEUROSCI.3408-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Myant N.B. Reelin and apolipoprotein E receptor 2 in the embryonic and mature brain: effects of an evolutionary change in the apoER2 gene. Proceedings of the Royal Society B: Biological Sciences. 2010;277(1680):345–351. doi: 10.1098/rspb.2009.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim D.H., Iijima H., Goto K., Sakai J., Ishii H., Kim H.J. Human apolipoprotein E receptor 2. A novel lipoprotein receptor of the low density lipoprotein receptor family predominantly expressed in brain. Journal of Biological Chemistry. 1996;271(14):8373–8380. doi: 10.1074/jbc.271.14.8373. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen A., Tao H., Metrione M., Hajri T. Very low density lipoprotein receptor (VLDLR) expression is a determinant factor in adipose tissue inflammation and adipocyte-macrophage interaction. Journal of Biological Chemistry. 2014;289(3):1688–1703. doi: 10.1074/jbc.M113.515320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tacken P.J., Hofker M.H., Havekes L.M., van Dijk K.W. Living up to a name: the role of the VLDL receptor in lipid metabolism. Current Opinion in Lipidology. 2001;12(3):275–279. doi: 10.1097/00041433-200106000-00006. [DOI] [PubMed] [Google Scholar]
- 16.Smith P.M., Ferguson A.V. Neurophysiology of hunger and satiety. Developmental Disabilities Research Reviews. 2008;14(2):96–104. doi: 10.1002/ddrr.13. [DOI] [PubMed] [Google Scholar]
- 17.Myers M.G., Jr., Olson D.P. Central nervous system control of metabolism. Nature. 2012;491(7424):357–363. doi: 10.1038/nature11705. [DOI] [PubMed] [Google Scholar]
- 18.Morton G.J., Meek T.H., Schwartz M.W. Neurobiology of food intake in health and disease. Nature Reviews Neuroscience. 2014;15(6):367–378. doi: 10.1038/nrn3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gautron L., Elmquist J.K., Williams K.W. Neural control of energy balance: translating circuits to therapies. Cell. 2015;161(1):133–145. doi: 10.1016/j.cell.2015.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fenselau H., Campbell J.N., Verstegen A.M., Madara J.C., Xu J., Shah B.P. A rapidly acting glutamatergic ARC→PVH satiety circuit postsynaptically regulated by alpha-MSH. Nature Neuroscience. 2017;20(1):42–51. doi: 10.1038/nn.4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwartz M.W. Brain pathways controlling food intake and body weight. Experimental Biology and Medicine. 2001;226(11):978–981. doi: 10.1177/153537020122601103. Maywood. [DOI] [PubMed] [Google Scholar]
- 22.Lutter M., Nestler E.J. Homeostatic and hedonic signals interact in the regulation of food intake. Journal of Nutrition. 2009;139(3):629–632. doi: 10.3945/jn.108.097618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Batterham R.L., Cowley M.A., Small C.J., Herzog H., Cohen M.A., Dakin C.L. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature. 2002;418(6898):650–654. doi: 10.1038/nature00887. [DOI] [PubMed] [Google Scholar]
- 24.Sohn Jong-Woo, Kevin J.K.E., Williams W. Neuronal circuits that regulate feeding behavior and metabolism. Trends in Neurosciences. 2013;36(9):504–512. doi: 10.1016/j.tins.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiu J., Zhang C., Borgquist A., Nestor C.C., Smith A.W., Bosch M.A. Insulin excites anorexigenic proopiomelanocortin neurons via activation of canonical transient receptor potential channels. Cell Metabolism. 2014;19(4):682–693. doi: 10.1016/j.cmet.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee D.K., Jeong J.H., Chun S.K., Chua S., Jr., Jo Y.H. Interplay between glucose and leptin signalling determines the strength of GABAergic synapses at POMC neurons. Nature Communications. 2015;6:6618. doi: 10.1038/ncomms7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Souza G.F., Solon C., Nascimento L.F., De-Lima-Junior J.C., Nogueira G., Moura R. Defective regulation of POMC precedes hypothalamic inflammation in diet-induced obesity. Scientific Reports. 2016;6:29290. doi: 10.1038/srep29290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cowley M.A., Smart J.L., Rubinstein M., Cerdan M.G., Diano S., Horvath T.L. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411(6836):480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 29.Tong Q., Ye C.P., Jones J.E., Elmquist J.K., Lowell B.B. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nature Neuroscience. 2008;11(9):998–1000. doi: 10.1038/nn.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pohlkamp T., David C., Cauli B., Gallopin T., Bouche E., Karagiannis A. Characterization and distribution of Reelin-positive interneuron subtypes in the rat barrel cortex. Cerebral Cortex. 2014;24(11):3046–3058. doi: 10.1093/cercor/bht161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alcantara S., Ruiz M., D'Arcangelo G., Ezan F., de Lecea L., Curran T. Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. Journal of Neuroscience. 1998;18(19):7779–7799. doi: 10.1523/JNEUROSCI.18-19-07779.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Draper S., Kirigiti M., Glavas M., Grayson B., Chong C.N., Jiang B. Differential gene expression between neuropeptide Y expressing neurons of the dorsomedial nucleus of the hypothalamus and the arcuate nucleus: microarray analysis study. Brain Research. 2010;1350:139–150. doi: 10.1016/j.brainres.2010.03.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong W., Kim D.W., Anderson D.J. Antagonistic control of social versus repetitive self-grooming behaviors by separable amygdala neuronal subsets. Cell. 2014;158(6):1348–1361. doi: 10.1016/j.cell.2014.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yasui N., Nogi T., Takagi J. Structural basis for specific recognition of reelin by its receptors. Structure. 2010;18(3):320–331. doi: 10.1016/j.str.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 35.Contu L., Hawkes C.A. A Review of the impact of maternal obesity on the cognitive function and mental health of the offspring. International Journal of Molecular Sciences. 2017;18(5) doi: 10.3390/ijms18051093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goudriaan J.R., Tacken P.J., Dahlmans V.E., Gijbels M.J., van Dijk K.W., Havekes L.M. Protection from obesity in mice lacking the VLDL receptor. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21(9):1488–1493. doi: 10.1161/hq0901.095147. [DOI] [PubMed] [Google Scholar]
- 37.Franssen R., Monajemi H., Stroes E.S., Kastelein J.J. Obesity and dyslipidemia. Medical Clinics of North America. 2011;95(5):893–902. doi: 10.1016/j.mcna.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 38.Wang H., Peng D.Q. New insights into the mechanism of low high-density lipoprotein cholesterol in obesity. Lipids in Health and Disease. 2011;10:176. doi: 10.1186/1476-511X-10-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tahar Hajri B.J., Talishinskiy Toghrul, Mazpule George, Ewing Douglas, Eid Sebastian, Novack Richard., Jr. Obesity upregulates the expression of VLDL in adipose tissue. Advances in Nutrition. 2017;8(8) [Google Scholar]
- 40.Yaykasli K.O., Hatipoglu O.F., Yaykasli E., Yildirim K., Kaya E., Ozsahin M. Leptin induces ADAMTS-4, ADAMTS-5, and ADAMTS-9 genes expression by mitogen-activated protein kinases and NF-kB signaling pathways in human chondrocytes. Cell Biology International. 2015;39(1):104–112. doi: 10.1002/cbin.10336. [DOI] [PubMed] [Google Scholar]
- 41.Liu Z.J., Bian J., Liu J., Endoh A. Obesity reduced the gene expressions of leptin receptors in hypothalamus and liver. Hormone and Metabolic Research. 2007;39(7):489–494. doi: 10.1055/s-2007-981680. [DOI] [PubMed] [Google Scholar]
- 42.Considine R.V., Sinha M.K., Heiman M.L., Kriauciunas A., Stephens T.W., Nyce M.R. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. New England Journal of Medicine. 1996;334(5):292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 43.Maffei M., Halaas J., Ravussin E., Pratley R.E., Lee G.H., Zhang Y. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nature Medicine. 1995;1(11):1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 44.Bernier B., Bar I., D'Arcangelo G., Curran T., Goffinet A.M. Reelin mRNA expression during embryonic brain development in the chick. Journal of Comparative Neurology. 2000;422(3):448–463. [PubMed] [Google Scholar]
- 45.Purba J.S., Hoogendijk W.J., Hofman M.A., Swaab D.F. Increased number of vasopressin- and oxytocin-expressing neurons in the paraventricular nucleus of the hypothalamus in depression. Archives of General Psychiatry. 1996;53(2):137–143. doi: 10.1001/archpsyc.1996.01830020055007. [DOI] [PubMed] [Google Scholar]
- 46.Cariboni A., Rakic S., Liapi A., Maggi R., Goffinet A., Parnavelas J.G. Reelin provides an inhibitory signal in the migration of gonadotropin-releasing hormone neurons. Development. 2005;132(21):4709–4718. doi: 10.1242/dev.02033. [DOI] [PubMed] [Google Scholar]
- 47.Williams K.W., Margatho L.O., Lee C.E., Choi M., Lee S., Scott M.M. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. Journal of Neuroscience. 2010;30(7):2472–2479. doi: 10.1523/JNEUROSCI.3118-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu J., Jiang L., Low M.J., Rui L. Glucose rapidly induces different forms of excitatory synaptic plasticity in hypothalamic POMC neurons. PLoS One. 2014;9(8) doi: 10.1371/journal.pone.0105080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dodd G.T., Michael N.J., Lee-Young R.S., Mangiafico S.P., Pryor J.T., Munder A.C. Insulin regulates POMC neuronal plasticity to control glucose metabolism. Elife. 2018;7 doi: 10.7554/eLife.38704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jarvie B.C., Hentges S.T. Expression of GABAergic and glutamatergic phenotypic markers in hypothalamic proopiomelanocortin neurons. The Journal of Comparative Neurology. 2012;520(17):3863–3876. doi: 10.1002/cne.23127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wittmann G., Hrabovszky E., Lechan R.M. Distinct glutamatergic and GABAergic subsets of hypothalamic pro-opiomelanocortin neurons revealed by in situ hybridization in male rats and mice. The Journal of Comparative Neurology. 2013;521(14):3287–3302. doi: 10.1002/cne.23350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bodnar R.J. Endogenous opioids and feeding behavior: a 30-year historical perspective. Peptides. 2004;25(4):697–725. doi: 10.1016/j.peptides.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 53.Kalra S.P., Horvath T.L. Neuroendocrine interactions between galanin, opioids, and neuropeptide Y in the control of reproduction and appetite. Annals of the New York Academy of Sciences. 1998;863:236–240. doi: 10.1111/j.1749-6632.1998.tb10698.x. [DOI] [PubMed] [Google Scholar]
- 54.Kokare D.M., Dandekar M.P., Singru P.S., Gupta G.L., Subhedar N.K. Involvement of alpha-MSH in the social isolation induced anxiety- and depression-like behaviors in rat. Neuropharmacology. 2010;58(7):1009–1018. doi: 10.1016/j.neuropharm.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 55.Lutz P.E., Kieffer B.L. Opioid receptors: distinct roles in mood disorders. Trends in Neurosciences. 2013;36(3):195–206. doi: 10.1016/j.tins.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Del Castillo J., Katz B. Quantal components of the end-plate potential. Journal of Physiology. 1954;124(3):560–573. doi: 10.1113/jphysiol.1954.sp005129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fatt P., Katz B. Spontaneous subthreshold activity at motor nerve endings. The Journal of Physiology. 1952;117(1):109–128. [PMC free article] [PubMed] [Google Scholar]
- 58.Qiu S., Zhao L.F., Korwek K.M., Weeber E.J. Differential reelin-induced enhancement of NMDA and AMPA receptor activity in the adult hippocampus. Journal of Neuroscience. 2006;26(50):12943–12955. doi: 10.1523/JNEUROSCI.2561-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qiu S., Weeber E.J. Reelin signaling facilitates maturation of CA1 glutamatergic synapses. Journal of Neurophysiology. 2007;97(3):2312–2321. doi: 10.1152/jn.00869.2006. [DOI] [PubMed] [Google Scholar]
- 60.DiBattista A.M., Dumanis S.B., Song J.M., Bu G., Weeber E., Rebeck G.W. Very low density lipoprotein receptor regulates dendritic spine formation in a RasGRF1/CaMKII dependent manner. Biochimica et Biophysica Acta. 2015;1853(5):904–917. doi: 10.1016/j.bbamcr.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.D'Arcangelo G. Apoer2: a reelin receptor to remember. Neuron. 2005;47(4):471–473. doi: 10.1016/j.neuron.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 62.Barger S.W. Apolipoprotein E acts at pre-synaptic sites...among others. Journal of Neurochemistry. 2013;124(1):1–3. doi: 10.1111/j.1471-4159.2012.07935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zarouna S., Wozniak G., Papachristou A.I. Mood disorders: a potential link between ghrelin and leptin on human body? World Journal of Experimental Medicine. 2015;5(2):103–109. doi: 10.5493/wjem.v5.i2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Corbett B.A., Simon D. Adolescence, stress and cortisol in autism spectrum disorders. OA Autism. 2014;1(1):2. [PMC free article] [PubMed] [Google Scholar]
- 65.Kurth F., Narr K.L., Woods R.P., O'Neill J., Alger J.R., Caplan R. Diminished gray matter within the hypothalamus in autism disorder: a potential link to hormonal effects? Biological Psychiatry. 2011;70(3):278–282. doi: 10.1016/j.biopsych.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Barbosa D.A.N., de Oliveira-Souza R., Monte Santo F., de Oliveira Faria A.C., Gorgulho A.A., De Salles A.A.F. The hypothalamus at the crossroads of psychopathology and neurosurgery. Neurosurgical Focus. 2017;43(3):E15. doi: 10.3171/2017.6.FOCUS17256. [DOI] [PubMed] [Google Scholar]
- 67.Liu W., Pappas G.D., Carter C.S. Oxytocin receptors in brain cortical regions are reduced in haploinsufficient (+/-) reeler mice. Neurological Research. 2005;27(4):339–345. doi: 10.1179/016164105X35602. [DOI] [PubMed] [Google Scholar]