

Chronic hepatitis C virus (HCV) infection is a major global public health problem. Infection often leads to severe liver injury that may progress to cirrhosis, hepatocellular carcinoma, and death. HCV coopts cellular machineries for propagation and triggers pathological processes in the liver. We previously identified a pivotal role of IKK-α in regulating cellular lipid metabolism and HCV assembly. In this study, we characterized NIK as acting upstream of IKK-α and characterized how HCV exploits this innate pathway to its advantage. Through extensive mechanistic studies, we demonstrated that NIK is a direct target of miR-122, which is regulated at the transcription level by HNF4A, a hepatocyte-specific transcription factor. We show in HCV infection that NIK expression is increased while both HNF4A and miR-122 levels are decreased. NIK represents an important host dependency that links HCV assembly, hepatic lipogenesis, and miRNA biology.
KEYWORDS: cell signaling, hepatic inflammation, lipid synthesis, microRNA, viral pathogenesis
ABSTRACT
Hepatitis C virus (HCV) harnesses host dependencies to infect human hepatocytes. We previously identified a pivotal role of IκB kinase α (IKK-α) in regulating cellular lipogenesis and HCV assembly. In this study, we defined and characterized NF-κB-inducing kinase (NIK) as an IKK-α upstream serine/threonine kinase in IKK-α-mediated proviral effects and the mechanism whereby HCV exploits this innate pathway to its advantage. We manipulated NIK expression in Huh7.5.1 cells through loss- and gain-of-function approaches and examined the effects on IKK-α activation, cellular lipid metabolism, and viral assembly. We demonstrated that NIK interacts with IKK-α to form a kinase complex in association with the stress granules, in which IKK-α is phosphorylated upon HCV infection. Depletion of NIK significantly diminished cytosolic lipid droplet content and impaired HCV particle production. NIK overexpression enhanced HCV assembly, and this process was abrogated in cells deprived of IKK-α, suggesting that NIK acts upstream of IKK-α. NIK abundance was increased in HCV-infected hepatocytes, liver tissues from Alb-uPA/Scid mice engrafted with human hepatocytes, and chronic hepatitis C patients. NIK mRNA contains an miR-122 seed sequence binding site in the 3′ untranslated region (UTR). miR-122 mimic and hairpin inhibitor directly affected NIK levels. In our hepatic models, miR-122 levels were significantly reduced by HCV infection. We demonstrated that HNF4A, a known transcriptional regulator of pri-miR-122, was downregulated by HCV infection. NIK represents a bona fide target of miR-122 whose transcription is downregulated by HCV through reduced HNF4A expression. This effect, together with the sequestering of miR-122 by HCV replication, results in “derepression” of NIK expression to deregulate lipid metabolism.
INTRODUCTION
Hepatitis C virus (HCV), a member of the genus Hepacivirus in the Flaviviridae family, is a positive-sense, single-stranded RNA virus that infects humans and other higher primates and has a selective tropism to the liver (1). Following exposure, HCV is able to evade the host’s immune system and establishes a chronic, often asymptomatic infection that may lead to liver failure, hepatocellular carcinoma (HCC), and death (2). The virus is estimated to infect 2.8% of the world’s population and constitutes a major public health burden worldwide (3). Over the past decade, great success has been achieved in discovering effective antivirals for HCV infection (4). Nevertheless, severe hepatic and extrahepatic disorders associated with chronic hepatitis C (CHC) are still common and difficult to prevent and reverse. In addition, fundamental insights into HCV virology, virus-host interactions, and the pathophysiology of HCV-mediated liver disease remain to be unveiled.
HCV extensively depends on host factors for propagation in hepatocytes and to induce various pathological processes in the liver (5). Interrogating these viral host dependencies is instrumental in elucidating HCV-related disease mechanisms and uncovering novel therapeutic strategies. Recently, applying integrative functional genomics and systems biology approaches, we globally identified host dependencies associated with the complete life cycle of HCV (6–8). Our genome-wide small interfering RNA (siRNA) screen demonstrated that IκB kinase α (IKK-α) is a crucial host factor for HCV. In a follow-up study, we uncovered a novel nuclear factor κB (NF-κB)-independent and kinase-mediated nuclear function of IKK-α in enhancing HCV assembly (9). HCV interacts with DDX3X, a putative pattern recognition receptor (PRR), through its 3′ untranslated region (UTR) RNA, which is a viral pathogen-associated molecular pattern (PAMP). This leads to the activation of IKK-α and a downstream signal cascade including the transcription factor CBP/p300, to mediate a lipogenic transcriptional program involving sterol regulatory element-binding proteins (SREBPs) to promote hepatocellular lipid droplet (LD) biogenesis and hence HCV assembly (9). Furthermore, we showed through systematic imaging and biochemical and virologic analyses that dynamic interactions occurred through the viral life cycle among IKK-α, DDX3X, and subcellular compartments, including stress granules (SGs), LDs, and HCV elements (10). These interactions provide key insights into productive HCV infection and virus-mediated liver pathogenesis.
IKK-α is the major IκB kinase in the noncanonical NF-κB activation pathway. The noncanonical pathway is initiated mainly through members of the tumor necrosis factor (TNF) receptor superfamily, such as the lymphotoxin-β receptor, resulting in activation of IKK-α by the NF-κB-inducing kinase (NIK), also known as mitogen-activated protein kinase kinase kinase 14 (MAP3K14) (11). NIK tightly associates with IKK-α to form a specific kinase complex in which NIK both activates IKK-α and functions as an adaptor that recruits IKK-α to NF-κB2 (p100) (12). NIK was previously identified as a host proviral factor (HPF) in our genome-wide RNA interference (RNAi) screen (6, 7). The aim of this study is to explore the functions and underlying mechanisms of NIK in inducing IKK-α activation, hepatic lipogenesis, and HCV assembly.
RESULTS
NIK expression is requisite for productive HCV infection.
To confirm the intrinsic function of NIK in supporting HCV propagation in hepatocytes, we knocked down its expression and examined the effect on productive HCV infection. NIK knockdown by small interfering RNA (siRNA) treatment led to about 65% repression in the endogenous NIK mRNA level in Huh7.5.1 cells (Fig. 1A) and thus reduced extracellular HCV RNA by 70%, whereas the intracellular HCV RNA level was decreased by only 32% compared to that of nontargeting control siRNA (siNT)-treated cells (Fig. 1B). The more profound reduction in extracellular HCV RNA upon NIK depletion, as well as the previously described function of its downstream substrate IKK-α (9), indicates that this gene is necessary for the assembly of HCV.
FIG 1.

Depletion of NIK disrupts productive HCV infection. (A and B) Huh7.5.1 cells were transfected with nontargeting control siRNA (siNT) or siRNA targeting NIK (siNIK) and then infected with the HCV JFH-1 strain. After 48 h, NIK mRNA level (A) and intracellular (IC) and extracellular (EC) HCV RNA levels (B) were determined by qPCR. (C) Infectivity of medium from siNT- or siNIK-treated cells was measured by performing TCID50 assay. (D and E) Representative HCV core immunostaining images (D) and quantification (E) from part-one and part-two HCV life cycle assays. The part-one assay recapitulates an impact on the early-stage HCV infection, including viral entry and viral RNA translation and replication, while the part-two assay detects an effect on the later stages of the HCV life cycle, such as virion assembly and release. (D) Green, HCV core; blue, cell nuclei. Bar, 200 μm. (E) The bar graph depicts percentages of HCV core-positive cells under each siRNA treatment condition. (A, B, and E) Values are normalized as relative values to siNT (set as 1), and error bars represent the standard error of the mean (SEM) (n = 3). *, P < 0.05; **, P < 0.01, determined by Student’s t test. Triplicates were performed for each experimental condition in all cell culture experiments. All results are representative of three separate experiments.
To further characterize this potential NIK-mediated function, we performed the 50% tissue culture infective dose (TCID50) assay to assess the infectivity of the culture supernatant in HCV-infected cells treated with either NIK or nontargeting control siRNA. NIK knockdown drastically diminished HCV infectivity assessed by the TCID50 assay (Fig. 1C), supporting a role for NIK in facilitating HCV assembly and/or secretion. In addition, in a two-part cell culture-generated HCV (HCVcc) infection assay that examines the core protein expression through immunostaining (6), siRNA-mediated silencing of NIK had little or no effect on part-one core staining that represents the early stages of the viral life cycle—from entry to viral RNA translation and replication—while it exerted a profound inhibitory effect on the part-two assay, which assesses the late stages of the HCV life cycle—assembly and/or secretion (Fig. 1D and E).
To explore the putative interactions between NIK and particular HCV life cycle stages, we surveyed the entire viral life cycle by performing multiple virological assays. First, we demonstrated that NIK knockdown had no effect on HCV entry. NIK siRNA treatment in Huh7.5.1 cells did not impede the infection of HCV pseudoparticle (HCVpp) or the control pseudovirus VSV-Gpp that bears the vesicular stomatitis virus (VSV) glycoprotein (see Fig. S1A in the supplemental material). Similarly, silencing of NIK did not influence HCV internal ribosomal entry site (IRES)-mediated translation or viral genome replication in HCV subgenomic replicon assays (Fig. S1B). These data suggest that NIK expression is dispensable for the early steps of HCV infection. In support of this notion, NIK knockdown in hepatocytes did not impact the infection of HCVsc, an HCV single-cycle infection system that captures viral entry and RNA translation and replication but not virion assembly or secretion (Fig. S1C).
Effects of NIK knockdown on HCV life cycle. Download FIG S1, DOCX file, 1.3 MB (1.3MB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
NIK enhances HCV propagation by activating IKK-α.
We next conducted gain-of-function assays to examine whether overexpression of NIK would confer a phenotype of enhanced viral assembly in hepatocytes. Cells transfected with a NIK plasmid before HCV infection exhibited a more-than-2-fold increase in viral RNA secreted into the culture supernatant over that from cells transfected with a control plasmid, despite similar intracellular viral RNA levels (Fig. 2A). NIK plasmid transfection significantly increased NIK mRNA and protein levels in Huh7.5.1 cells (Fig. 2B and C), leading to the observed upregulation of HCV RNA production and secretion described above.
FIG 2.

NIK enhances HCV RNA production and secretion that are mediated by IKK-α. (A) Effect of NIK overexpression on intracellular and extracellular HCV RNA levels. (B and C) Transfection efficiency of Flag-tagged NIK plasmid in Huh7.5.1 cells, determined by measuring NIK mRNA levels through qPCR (B) or NIK protein levels by Western blotting (C). β-Tubulin was used as a loading control. (D and E) The proviral function of NIK acts upstream of the IKK-α signaling pathway. (D) Huh7.5.1 cells were treated with siNT or siIKK-α and then transfected with control (pCtrl-Flag) or NIK (pNIK-Flag) plasmid. The cells were subsequently infected with HCV. At 48 h postinfection, extracellular HCV RNA levels were measured by qPCR (left panel). Western blots (right panel) showed overexpression and knockdown efficiency of various plasmids or siRNAs used in the left panel. (E) Cells pretreated with NT or IKK-α siRNA were transfected with control (pCtrl-HA) or IKK-α (pIKK-α-HA) plasmid and then infected with HCV. After 48 h, extracellular HCV RNA levels were measured by qPCR (left panel). The right panel shows IKK-α overexpression efficiency by Western blotting. The middle of the same blot containing other lanes was removed, as indicated by a dashed line. (A, B, D, and E) Values are normalized as relative values to the negative-control siRNA and/or plasmid (as 1), and error bars represent the standard error of the mean (SEM) (n = 3). **, P < 0.01, determined by Student’s t test. NS, not significant. Triplicates were performed for each experimental condition in all cell culture experiments. All results are representative of three separate experiments.
Since NIK mainly phosphorylates and activates IKK-α in the noncanonical NF-κβ activation pathway (13), we hypothesized that the proviral activity of NIK is mediated through IKK-α. Huh7.5.1 cells were treated with siNT or IKK-α siRNA and then transfected with a control or pNIK plasmid, followed by infection with HCV. In siNT-treated cells, overexpression of NIK elevated the extracellular HCV RNA level more than 2-fold (Fig. 2D). Nevertheless, this proviral effect of NIK was completely abrogated in cells deprived of IKK-α (Fig. 2D), indicating a direct role of IKK-α in mediating NIK’s proviral function. When IKK-α was overexpressed in Huh7.5.1 cells while NIK was depleted, the extent of enhancement of HCV RNA production and release by overexpressing IKK-α was comparable to that of NIK-intact cells (Fig. 2E), further supporting the upstream role of NIK in mediating IKK-α’s function.
NIK interacts with and activates IKK-α.
To further elucidate the interaction between NIK and IKK-α, we characterized three amino acid sequence motifs inside the NIK kinase domain that have been reported to be involved in its intrinsic kinase function (14–16). Four putative kinase-defective NIK mutants were generated and transfected into Huh7.5.1 cells (Fig. 3A). While wild-type (WT) NIK colocalized extensively with IKK-α in both cytoplasm and the nucleus, these kinase-negative mutant forms of NIK (NIK KN) caused significantly diminished nuclear localization of both NIK and IKK-α (Fig. 3B and C). The direct interaction of NIK and IKK-α was also observed through coimmunoprecipitation (co-IP) assay in Huh7.5.1 cells (Fig. 3D). Moreover, by immunofluorescence and confocal microscopy, NIK colocalized with G3BP1, a bona fide marker of cellular stress granules (SGs) (Fig. S2). Thus, we speculate that NIK constitutes a part of the protein complex shown previously to include DDX3X and IKK-α that translocates to the SGs and activates IKK-α, thereby initiating a multifaceted, dynamic cellular program that is exploited by HCV for productive infection (10).
FIG 3.

NIK complexes and phosphorylates IKK-α, leading to its nuclear localization. (A) Schematic diagram of NIK wild-type (WT) and multiple reported kinase-defective mutants (KN). (B) Immunofluorescence and confocal microscopic analyses of IKK-α and NIK subcellular localization and nuclear translocation in Huh7.5.1 cells upon transfection of HA-IKK-α with Flag-NIK WT or various KN mutants. Two days later, cells were then stained with antibodies to HA and Flag. Quantification of nuclear colocalization of IKK-α and NIK was determined by dividing the number of cells with double-positive nuclei over 100 cells with either NIK or IKK-α signal and is shown below as percentage of colocalization in the transfected cells. (C) Huh7.5.1 cells were transfected with WT or P565R mutant NIK construct and WT or mutant IKK-α KM that contains a kinase-defective mutation (K44M) and behaves as a dominant negative mutant. The cells were then immunostained as described above and imaged by immunofluorescence microscopy. (B and C) Red, Flag-NIK; green, HA-IKK-α; blue, nuclei. (D) Huh7.5.1 cells were transfected with Ctrl-HA or IKK-α-HA plasmid together with Flag-NIK WT or KN plasmid and then subjected to immunoprecipitation (IP) with anti-Flag antibody, followed by Western blotting (WB) with anti-HA antibody. Whole-cell lysates were analyzed by Western blotting using an anti-IKK-α antibody. (E) Huh7.5.1 cells were transfected with various indicated Ctrl or NIK WT or KN mutant plasmids and then subjected to immunoprecipitation with anti-IKK-α antibody, followed by Western blotting with anti-phosphorylated IKK-α antibody or anti-IKK-α antibody. Whole-cell lysates were analyzed by Western blotting using an anti-IKK-α antibody. NIK WT but not NIK KN mutant overexpression mediates IKK-α phosphorylation. Triplicates were performed for each experimental condition in all cell culture experiments. All results are representative of three separate experiments.
NIK localizes to the stress granules (SGs) upon HCV infection. Download FIG S2, DOCX file, 0.9 MB (986.2KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
We also tested whether overexpression of NIK could indeed cause an increase in phosphorylated IKK-α, the active form of IKK-α that translocates to the nucleus to activate downstream gene transcription (9). Huh7.5.1 cells were transfected with NIK wild type (WT) or P565R mutant (as a representative dominant negative [DN] form), and a co-IP assay was performed to detect the presence of phosphorylated IKK-α. Compared to the control cells, cells transfected with the NIK WT plasmid exhibited significantly elevated levels of pIKK-α (Fig. 3E), suggesting increased NIK activity and IKK-α phosphorylation. In contrast, the NIK P565R mutant could still bind to IKK-α but did not phosphorylate IKK-α (Fig. 3E).
NIK promotes HCV-induced lipogenesis and lipid droplet formation.
To investigate whether NIK, like IKK-α, mediates cellular lipogenesis and lipid droplet (LD) formation in HCV infection, we first silenced various components of the NF-κB activation pathway by siRNA treatment in Huh7.5.1 cells, followed by HCV 3′ UTR RNA transfection. HCV 3′ UTR RNA acts as an HCV pathogen-associated molecular pattern (PAMP) and has previously been shown to stimulate the expression of SREBP-1, a master regulator of cellular fatty acid synthesis, lipid metabolism, and LD formation (9). Depletion of NIK, similarly to depletion of IKK-α, significantly diminished the upregulation of SREBP-1 and LD formation by the HCV 3′ UTR RNA, while depletion of other NF-κB signaling components did not (Fig. 4A and B).
FIG 4.

NIK regulates HCV-induced hepatocellular lipogenesis and lipid droplet (LD) formation. (A and B) NIK depletion in cells abrogated HCV 3′ UTR-mediated induction of SREBP-1 mRNA expression (A) and LD biogenesis (B). (A) Huh7.5.1 cells were transfected with Ctrl or HCV 3′ UTR RNA and various siRNAs targeting components of the NF-κB pathway as indicated. mRNA levels of SREBP-1, a master regulator of cellular lipogenesis and lipid metabolism, were measured by qPCR. (B) Huh7.5.1 cells were transfected with siNT or siNIK and then treated with HCV 3′ UTR RNA. Cellular LD contents were analyzed by BODIPY staining (green). (C) Huh7.5.1 cells were treated with siNT or siNIK for 72 h before infection with HCV. At 48 h postinfection, viral core protein (red) and LDs (green) were immunostained and observed through immunofluorescence and confocal microscopy. NIK knockdown reduced HCV-mediated LD formation and core-LD association for virion assembly. (D) NIK silencing by siRNA suppressed the mRNA levels of various lipogenic enzymes in Huh7.5.1 cells. (E) NIK plasmid transfection upregulated the hepatocellular expression of these lipogenic genes in Huh7.5.1 cells. (F) Huh7 cells were transfected with either pCtrl-Flag or pNIK-Flag and immunostained after 24 h for the nucleus (blue) and LDs (green). (A, D, and E) Values are normalized as relative values to the siNT or Ctrl plasmid (set as 1), and error bars represent the standard error of the mean (SEM), n = 3. *, P < 0.05; **, P < 0.01, determined by Student’s t test. NS, not significant. Triplicates were performed for each experimental condition in all cell culture experiments. All results are representative of three separate experiments.
To further explore whether NIK is involved in hepatocellular LD biogenesis and HCV assembly, we silenced its expression in HCV-infected Huh7.5.1 cells. NIK knockdown decreased the basal level of cytosolic LDs, HCV-induced LD formation, and HCV core-LD association (Fig. 4C). In addition, transcriptomic analyses revealed that NIK knockdown significantly reduced the levels of several key lipogenic enzymes, including SREBP-1, PPARA, FASN, and ACSL3 in hepatocytes, whereas NIK overexpression exerted the opposite effects (Fig. 4D and E). NIK plasmid transfection also enhanced lipid droplet formation in Huh7 cells (Fig. 4F). When cells were transfected with various mutant forms of NIK, there were significantly fewer lipid droplet formations than that of WT NIK-transfected cells (Fig. S3). These data collectively suggested that HCV or HCV 3′ UTR induces lipogenesis and LD formation in an NIK-dependent manner.
Mutated NIK on lipid droplet accumulation. Download FIG S3, DOCX file, 1.5 MB (1.6MB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
HCV infection induces hepatic NIK expression.
HCV induces hepatocellular proviral gene expression as a strategy to achieve viral propagation and persistence in the host (17, 18). HCV also activates certain host proviral machinery or cell signaling, such as the IKK-α–lipogenic pathway, for its own benefit (9). To investigate whether HCV-induced activation of IKK-α is mediated by a manipulation of expression of its upstream kinase NIK, we infected both Huh7.5.1 and Huh7 cells with the HCV JFH-1 strain and quantified intracellular NIK transcript levels by qPCR at various time points postinfection. NIK mRNA expression was significantly induced at all time points (8 to 72 h) postinfection in Huh7.5.1 cells (Fig. 5A) and after day 2 postinfection in Huh7 cells (Fig. 5B).
FIG 5.

HCV infection increases hepatic expression of NIK. (A and B) Huh7.5.1 (A) or Huh7 (B) cells were infected with the HCV JFH-1 strain for the indicated time points. NIK mRNA levels in these cells were then quantified by qPCR. (C) HCV infection of Alb-uPA/SCID mice for 10 days or 8 weeks. Hepatic HCV RNA levels in the mouse livers were quantified by qPCR. (D) NIK expression levels are increased in livers of Alb-uPA/SCID mice infected with HCV for 10 days or 8 weeks. (E) Hepatic NIK expression levels are significantly higher among chronic hepatitis C (CHC) patients than in healthy controls. Total RNA was extracted from liver biopsy specimens of HCV-infected patients (n = 69) or healthy donors (n = 19). Relative expression of NIK mRNA was measured by qPCR and standardized to 18S RNA level. Each dot represents a single assessed individual. (F) Effect of HCV on NIK promoter activity. Huh7.5.1 cells were transfected with either NIK, beta-actin (negative-control), or NF-κB (positive-control) promoter luciferase report vector and after 24 h infected with HCV for 48 h. RLU: relative luciferase unit. Values are shown as means ± SEM. *, P < 0.05; **, P < 0.01, determined by Student’s t test. Triplicates were performed for each experimental condition in all cell culture experiments. All results are representative of three separate experiments.
We next asked whether the upregulation of NIK is pathophysiologically relevant to HCV infection in vivo. In a chimeric Alb-uPA/SCID mouse model engrafted with human hepatocytes, HCV infection resulted in elevated hepatic NIK mRNA levels at both 10 days (∼1.5-fold) and 8 weeks (∼2.5-fold) after viral exposure (Fig. 5C and D), consistent with the induction of NIK expression by HCV in cell culture. We also examined the expression levels of NIK in liver samples of healthy subjects or chronic hepatitis C (CHC) patients. There were marked increases of NIK mRNA levels in livers of CHC patients over those of healthy controls (Fig. 5E).
NIK has been previously suggested to be induced by interferons and acts as an interferon-stimulated gene (ISG) in the liver (19). This scenario could account for its elevation during HCV infection. Nevertheless, when Huh7.5.1 cells were treated with various interferons, several known ISGs, such as IFIT2 and ISG15, were significantly induced but there was no effect on NIK mRNA levels (Fig. S4). These results suggest that the increased expression of NIK in hepatocytes is not regulated by interferon-related innate immunity and is most likely mediated by a direct effect of HCV infection. In addition, HCV infection did not significantly affect the NIK promoter activities (Fig. 5F), suggesting that HCV regulates NIK expression at a posttranscriptional level.
Effects of interferons on NIK expression in hepatocytes. Download FIG S4, DOCX file, 0.4 MB (391KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
miR-122 targets and regulates NIK expression.
MicroRNA (miRNA) plays a pivotal role in HCV infection, via either a direct interaction with the viral genome—such as miR-122 (20)—or an indirect interaction through mediating gene regulation that impacts various steps of the HCV life cycle (8). Through bioinformatics-based sequence alignment, we found that NIK possesses a predicted miR-122 seed sequence binding site within its 3′ UTR. To confirm whether miR-122 targets NIK, we tested a NIK 3′ UTR construct that contains a luciferase reporter gene directly upstream of the 3′ UTR (Fig. 6A). NUAK2, a previously known miR-122 target and a proviral factor for HCV (6, 21), was employed as a positive control. The 3′ UTR assays showed that miR-122 mimic transfection in hepatocytes significantly diminished the luciferase activities of NIK (MAP3K14) and NUAK2 3′ UTRs (Fig. 6B), whereas miR-122 hairpin inhibitor overexpression exerted the opposite effects (Fig. 6C). To explore a direct interaction between NIK 3′ UTR and miR-122, we performed multiple mutagenesis analyses. First, we generated a mutant form (MUT) of miR-122 with two point mutations at its seed sequence (Fig. 6D). Compared to the wild-type (WT) miR-122, the mutant form no longer upregulates HCV infection nor suppresses the NIK 3′ UTR activity when overexpressed in Huh7.5.1 cells (Fig. 6E and F). We then tested a mutant form of the NIK 3′ UTR, which contains mutations complementary to those introduced to the miR-122 seed sequence (Fig. 6G). We found that miR-122 WT transfection had no effect on the luciferase activity of the NIK 3′ UTR mutant, while the combination of mutant NIK 3′ UTR and mutant miR-122 rescued the interaction and hence the miR-122-mediated inhibitory effect on the NIK 3′ UTR (Fig. 6H). Collectively, these data demonstrate that NIK is a direct target of miR-122.
FIG 6.

Identification of NIK as a bona fide target of miR-122 in hepatocytes. (A) Luciferase reporter-containing 3′ UTR constructs of NIK and NUAK2, a previously identified miR-122 target. Each 3′ UTR encodes an miR-122 seed sequence matching site. (B and C) Effects of miR-122 mimic (B) or hairpin inhibitor (C) transfection on the luciferase activities of NIK and NUAK2 3′ UTRs in Huh7.5.1 cells. miR-122 mimic and hairpin inhibitor exerted opposite effects by down- or upregulating the 3′ UTR activities, respectively. (D) RNA sequences of the wild-type (WT) and mutant (MUT) forms of miR-122. miR-122 seed sequence is featured in blue, with a 2-nucleotide mutation that was introduced in it shown in red. (E and F) Effects of WT or MUT miR-122 overexpression on HCVcc-Luc infection (E) and NIK 3′ UTR activity (F). (G) The NIK 3′ UTR mutant carrying a 2-nucleotide mismatch to miR-122 seed sequence was constructed. (H) Rescue of NIK 3′ UTR activity by cotransfection of MUT miR-122 and the mutant form of NIK 3′ UTR. WT miR-122 had no effect on NIK 3′ UTR mutant. (I) miR-122 inhibition in Huh7.5.1 cells significantly increased NIK mRNA expression. Relative luciferase activities are shown. (B, C, E, F, H, and I) Values are normalized relative to the miRNA mimic or inhibitor Ctrl (set as 1), and error bars represent means ± SEM. n = 5 (B, C, E, F, and H) or 3 (I). *, P < 0.05; **, P < 0.01, determined by Student’s t test; NS, not significant. Triplicates were performed for each experimental condition in all cell culture experiments. All results are representative of three separate experiments.
miR-122 level is reduced in HCV-infected hepatic cells.
miR-122, a liver-specific miRNA and the most abundantly expressed miRNA in hepatocytes, plays an important role in liver pathobiology and liver disease development and progression (22). In HCV infection, miR-122 directly binds to the viral genome and enhances viral RNA replication, thus resulting in a decrease in the availability of miR-122 within the cells (20, 23). This reduction of miR-122 leads to a global derepression of its targets in HCV-infected cells (21). We reason that the increase in NIK mRNA during HCV infection could be explained by the derepression as a bona fide miR-122 target. NIK mRNA level was significantly induced in cells where miR-122 was sequestered through the hairpin inhibitor treatment (Fig. 6I).
Through miR-122 seed sequences on HCV genomic RNA, it acts as a “sponge” and decreases the amount of available miR-122 in HCV-infected cells (21). We assessed the overall level of miR-122 in HCV-infected cells. In Huh7 cells, Huh7.5.1 cells, cultured primary human hepatocytes (PHH), and CHC patients, there were significant reductions of miR-122 copy numbers upon HCV infection (Fig. 7A to C). To validate this finding in vivo, we examined liver tissues from HCV-infected Alb-uPA/Scid mouse model and humans. Since the mature miR-122 sequences are identical between mouse and human, the only means to quantify human miR-122 levels in the Alb-uPA/Scid mouse model is to assess the pri-miR-122, the primary transcript of the miR-122 gene, which is more divergent between human and mouse. As shown in Fig. 7D, HCV infection at either day 10 or week 8 considerably decreased the hepatic abundance of pri-miR-122 in the humanized mouse livers. We also quantified human NUAK2 mRNA, a well-known target of miR-122, and showed that its hepatic level, like that of NIK, indeed increased after HCV infection (Fig. 7E).
FIG 7.

HCV infection downregulates HNF4A expression and hepatic miR-122 levels. (A) Huh7 cells were infected with the HCV JFH-1 strain for various indicated time points and subsequently assessed for intracellular miR-122 levels by qPCR. (B) Huh7.5.1 cells (left) or primary human hepatocytes (right) were infected with HCVcc for 48 h. miR-122 expression from these cells was examined by qPCR. (C) Hepatic levels of miR-122 in the liver biopsy specimens of CHC patients (n = 15) compared to those of healthy controls (n = 19), measured by qPCR. Each dot represents one individual’s liver tissue. (D) HCV infection in the chimeric Alb-uPA/Scid mouse liver downregulated pri-miR-122 expression levels. The Alb-uPA/Scid mice were infected with HCV, and livers were harvested for analysis on day 10 or week 8 post-infection. (E) NUAK2 mRNA levels were measured in livers of Alb-uPA/Scid mice infected with HCV. (F) Effect of HCV infection on HNF4A mRNA expression in the livers of Alb-uPA/Scid mice. (G) Effect of HCV infection in Huh7.5.1 on HNF4A protein level. Cells were infected with HCV for 3 days and harvested for analysis by Western blotting. Beta-actin served as a loading control. (H) Effect of HCV on HNF4A promoter activity. Huh7.5.1 cells were transfected with either HNF4A or beta-actin promoter luciferase report vector and 24 h later infected with HCV. 48 h later, cells were harvested for luciferase measurement. (I and J) Huh7 cells were infected with the HCV JFH-1 strain for various indicated time points and harvested for assessment of cellular HNF4A mRNA (I) and pri-miR-122 (J) levels by qPCR. Triplicates were performed for each experimental condition in all cell culture experiments. All values are normalized as relative values to the mock infection or healthy donors (set as 1), and error bars represent means ± SEM, n = 3 (A, B, D-F, H-J). *, P < 0.05; **, P < 0.01, determined by Student’s t test. All results are representative of three separate experiments.
HCV downregulates HNF4A expression to reduce miR-122 transcription.
It was shown previously that transcription of miR-122 (its primary transcript, pri-miR-122) is regulated by a hepatocyte-specific transcription factor, HNF4A (24). We hypothesized that HCV infection may downregulate the expression of HNF4A, leading to a reduced miR-122 transcription. To test this hypothesis, we examined the expression of HNF4A in the HCV-infected Alb-uPA/Scid mouse livers. As shown in Fig. 7F, HNF4A mRNA was significantly downregulated at both 10 days and 8 weeks in HCV-infected hepatocytes. In Huh7.5.1 cells, HNF4A protein levels were also decreased after HCV infection (Fig. 7G). The effect of HCV on HNF4A expression was tested directly with an HNF4A promoter construct, which showed a significant decrease in luciferase activity by HCV (Fig. 7H), suggesting a transcriptional downregulation of HNF4A in HCV-infected cells. Next, we assessed the time course of HNF4A and pri-miR-122 expression in HCV-infected cells. Three days after HCV infection, HNF4A mRNA levels began to decrease (Fig. 7I) while the pri-miR-122 transcript was notably lower only after 5 days of HCV infection (Fig. 7J).
DISCUSSION
We have demonstrated a previously unrecognized role of NIK and its regulation by miR-122 in enhancing HCV assembly by regulating hepatocellular lipogenesis and lipid metabolism (Fig. 8 provides a summary). We found that NIK interacts with IKK-α to form a specific kinase complex in which NIK both phosphorylates and activates IKK-α and functions as an adaptor that recruits IKK-α in the context of stress granules. Depletion of NIK in hepatocytes significantly impaired HCV particle production, whereas overexpression of NIK had an opposite effect. The proviral effect by NIK overexpression was abrogated in cells deprived of IKK-α. In NIK-silenced cells, the content of cytosolic LDs was notably reduced. NIK knockdown also substantially reduced HCV 3′-UTR-mediated induction of SREBP-1 and LD formation. Thus, NIK, a central regulatory kinase for IKK-α activation, plays an important role in the IKK-α-mediated effects on HCV assembly and lipogenic induction. Interestingly, in a recent signalome-wide study, the canonical NF-κB pathway was found to restrict HCV infection, while silencing of IRAK2 or NIK of the noncanonical NF-κB pathway reduced viral replication (25), thus supporting a proviral role of NIK in HCV infection.
FIG 8.
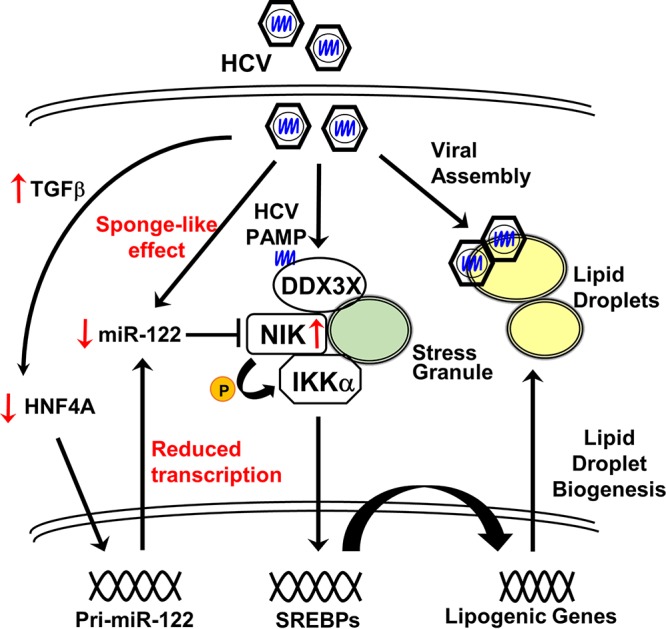
A proposed model of NIK regulation and its functions in cellular lipogenesis and thus in modulating HCV infection. See the text for discussion.
Our study demonstrates an miRNA-mediated mechanism in hepatic accumulation of cellular factors during both acute and chronic HCV infections. We showed for the first time that NIK is a previously unrecognized target of miR-122 and its expression is subject to a direct binding of miR-122 to its cognate seed sequence on the NIK 3′ UTR. miR-122 is essential for HCV genome replication (20, 23) as it binds to two sites within the viral 5′ UTR and stabilizes the viral RNA genome, thus protecting it from degradation by Xrn1 and Xrn2 (26–28). Recent studies have indicated that the cellular targets of miR-122 are functionally derepressed during HCV infection—a mechanism that is attributed to a “sponge-like” effect of the HCV genome in reducing the biologically available miR-122 to interact with its targets in hepatocytes (21). Here we report an HCV-mediated reduction of hepatic miR-122 levels in both cultured hepatocytes and livers of chronic hepatitis C patients.
Whether the overall reduced level of miR-122 may be secondary to the “sponge-like” effect leading to an increased turnover of miR-122, an independent effect of HCV on miR-122 transcription, processing, or some combination of these has not been fully addressed. We reported recently that the levels of several miRNAs involved in regulating HCV infection are also affected by the virus itself (8, 29). Transcription of the primary transcript of miR-122, pri-miR-122, has been shown to be regulated by a hepatocyte-specific transcription factor, HNF4A (24). We showed here that the HNF4A level was reduced in cells infected with HCV; this reduction has also been reported previously in HCV-infected Huh7 cells (30, 31). In support for this sequence of events, the time course experiment in Fig. 7I and J shows the reduction of HNF4A levels prior to the decrease in pri-miR-122 transcripts in HCV-infected cells. This observation explains the overall lower levels of miR-122 in HCV-infected cells. The relatively early sequestering of miR-122 by HCV RNA replication and the transcriptional decrease of miR-122 levels that occurs later in HCV-infected cells likely contribute to the derepression of miR-122 targets during HCV infection, as observed by us and others (21).
HNF4A, a hepatic cell-specific transcription factor, has been shown to be downregulated by transforming growth factor beta (TGF-β) in hepatoma cells and primary human hepatocytes (PHHs) (32, 33). TGF-β, as a bona fide inducer of epithelial-mesenchymal transition (EMT), can dedifferentiate hepatocytes, leading to a loss of hepatic cell-specific functions, such as the expression of HNF4A (34). We recently showed that TGF-β treatment of Huh7.5.1 cells resulted in the loss of the adherens junction protein E-cadherin (CDH1) and of the tight junction proteins claudin-1 (CLDN1) and occludin (OCLN) and subsequent induction of EMT (35). HCV infection diminishes CDH1 expression in hepatocytes and results in EMT, which is likely mediated by TGF-β induction in HCV-infected cells (35–37). Our data suggest that HCV-mediated downregulation of HNF4A, similarly to CDH1, is regulated by the TGF-β pathway, which triggers hepatic dedifferentiation and EMT.
We previously demonstrated the reliance of HCV-mediated alterations of lipid metabolism on IKK-α (9). The current study identifies NIK as the upstream kinase capable of activating IKK-α to induce the hepatic lipid biosynthetic pathway and lipid droplet (LD) biogenesis. LD accumulation in hepatocytes promotes viral assembly and secretion (18, 38) and meanwhile contributes to pathological processes such as the development of steatosis and metabolic syndromes (39). Hepatic steatosis also facilitates the progression of liver fibrosis in patients chronically infected with HCV (40). Therefore, we speculate that NIK functions as a critical molecule that initiates a cellular signaling cascade in induction of hepatic metabolic and inflammatory disorders. Indeed, the role of NIK in pathogenesis of hepatic steatosis has been described. NIK is abnormally activated in the liver of obese mouse models (41). In conditional knockout mice using the Cre/LoxP system, deletion of NIK in the liver protected against high-fat diet (HFD)-induced liver steatosis, glucose intolerance, and obesity (42). Mechanistically, NIK deletion suppresses liver inflammation and lipogenic programs, thus contributing to protection against hepatic steatosis (42). NIK protein levels are typically low at baseline in quiescent cells but can be increased by cytokines and oxidative stress (43), suggesting that this gene is tightly regulated to avoid untoward inflammatory and metabolic disturbance. NIK, as shown here to be an miR-122-regulated gene, may be particularly suppressed in the liver because of the overabundance of miR-122 in the liver (44).
Our study suggests that NIK partly mediates the known function of miR-122 in lipid and cholesterol metabolism (45, 46). Reduced levels of hepatic miR-122 have been reported in patients with nonalcoholic fatty liver disease (47). In addition, miR-122 has been implicated as a tumor suppressor gene in various cancers, including HCC (48). In support of a pivotal role of miR-122 in liver functions, previous studies showed that germ line or conditional deletion of miR-122 in the liver led to development of hepatic steatosis, inflammation, fibrosis, and HCC (49, 50). It is intriguing to consider that upregulation of NIK, in addition to other pathways such as peroxisome proliferator-activated receptors (51), may underlie the mechanism whereby miR-122 depletion results in dysregulated lipid metabolism, liver injury, and HCC development. Thus, NIK represents a promising target for developing unique and specific interventional strategies to prevent and cure nonalcoholic fatty liver disease, an increasingly prevalent liver disease in the world.
MATERIALS AND METHODS
Cell lines and virus.
Huh7 and its derivative cell line Huh7.5.1 (provided by F. V. Chisari of The Scripps Research Institute, La Jolla, CA) were maintained in complete growth medium (Dulbecco’s modified Eagle’s medium [DMEM]; Corning, Corning, NY) containing 10% fetal bovine serum (Corning). The cell lines were free from mycoplasma contamination and regularly tested using a mycoplasma detection kit (Thermo Scientific, Waltham, MA). Primary human hepatocytes were obtained from TRL (Morrisville, NC) and maintained in Williams medium E containing cell maintenance supplement (Invitrogen, Carlsbad, CA). HCV strain JFH-1 (genotype 2a HCVcc), provided by T. Wakita of the National Institute of Infectious Diseases, Tokyo, Japan, was propagated and infectivity was titrated as previously described (52, 53). Unless otherwise indicated, HCVcc infection was conducted at a multiplicity of infection (MOI) of 0.5, and assays were typically performed at 48 h postinfection.
Patients, liver biopsy specimens, and Alb-uPA/Scid mouse livers.
Liver biopsy specimens were obtained from healthy volunteers and chronic hepatitis C (CHC) patients (genotype 1b) with or without cirrhosis. Liver samples were harvested on day 10 and week 8 after HCV infection (genotype 1b) from Alb-uPA/Scid mice engrafted with primary human hepatocytes as described previously (54).
Immunofluorescence, lipid staining, and confocal microscopy.
Huh7.5.1 cells grown on Lab-Tek II borosilicate four-well chamber coverslips (Nunc ThermoFisher) were fixed with 4% paraformaldehyde, permeabilized in 0.3% Triton X-100, and incubated with blocking solution in phosphate-buffered saline (PBS) containing 3% bovine serum albumin (BSA) and 10% normal goat serum (Vector Laboratories, Burlingame, CA). Cells were then labeled with the appropriate primary antibodies diluted in PBS with 1% BSA followed by incubation with Alexa Fluor 488-, 568-, or 647-conjugated secondary antibodies (Invitrogen) in PBS with 1% BSA. The primary antibodies used were the following: anticore (generated from anticore 6G7 hybridoma cells, 1:500), rabbit anti-NIK polyclonal antibody (1:500) (ab19155; Abcam, Cambridge, MA), mouse anti-G3BP1 monoclonal antibody (1:300) (ab59533; Abcam), mouse anti-Flag monoclonal antibody (1:500) (F1804; Sigma, St. Louis, MO), and antihemagglutinin (anti-HA) rabbit monoclonal antibody (1:500) (C29F4; Cell Signaling, Danvers, MA). Nuclei were counterstained with Hoechst 33342 (Invitrogen) at 1:5,000 in PBS. Lipid droplets were stained with BODIPY 493/503 (Invitrogen) applied at 1 μg ml−1 for 1 h in PBS with 1% BSA. Each step was followed by two washes with PBS. Confocal laser scanning microscopy analysis was performed with an Axio Observer.Z1 microscope equipped with a Zeiss LSM 5 Live DuoScan system under an oil-immersion 1.4-numerical-aperture (NA) ×63 lens objective (Carl Zeiss, Oberkochen, Germany). Images were acquired using Zen 2010 software (Carl Zeiss). Dual- or triple-color images were acquired by consecutive scanning with only one laser line active per scan to avoid cross-excitation.
Quantitative miRNA real-time PCR assay.
Huh7.5.1 cells and PHH were transfected with various miRNA mimics for 3 days or infected with HCV for an indicated amount of time. Cell lysates were vortexed to lyse them, and biopsy specimens were lysed in a TissueLyser LT bead mill (Qiagen, Hilden, Germany). Total miRNA was isolated with an miRNeasy minikit (Qiagen) according to the manufacturer’s instructions. RNA was reverse transcribed using the TaqMan microRNA reverse transcription kit (Applied Biosystems) according to the manufacturer’s instructions. miR-122 expression level was determined by qPCR using TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA) and specific miRNA primers and probes (TaqMan MicroRNA assays; Applied Biosystems). U6 snRNA was used as an internal control.
Quantitative pri-miRNA real-time PCR assay.
Huh7.5.1 cells were mock infected or infected with HCV strain JFH-1. Total cellular RNA was prepared using the RNeasy minikit (Qiagen) per the manufacturer’s instructions. Samples were treated with the Turbo DNA-free kit (Thermo; AM1907) to remove genomic DNA. RNA quality and quantity were assessed on a NanoDrop spectrophotometer. The pri-miR-122 levels of target genes were determined by qPCR using gene-specific primers and probes (Hs03303072_pri; Thermo) and Verso 1-Step RT-qPCR kit (Thermo Scientific) on an ABI ViiA 7 real-time PCR system. Relative pri-miRNA levels were calculated using the threshold cycle (ΔΔCT) method, with 18S rRNA (Applied Biosystems) as the internal control for normalization.
Statistical analysis.
Results are presented as means ± standard deviations (SD). The two-tailed unpaired Student t test was used for statistical analysis. The level of significance is denoted in each figure (*, P < 0.05; **, P < 0.01; NS, not significant).
Ethics statement.
All patients provided written informed consent, and the protocol was approved by the Institutional Review Board of the National Institute of Diabetes and Digestive and Kidney Diseases and the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
Other materials and methods are listed in Text S1 in the supplemental material.
Supplemental materials and methods used in this study. Download Text S1, DOCX file, 0.04 MB (39.6KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
ACKNOWLEDGMENTS
We thank C. M. Rice, F. V. Chisari, T. Wakita, F.-L. Cosset, M. Niepmann, T. Suzuki, and E. Zandi for their generosity in providing various reagents. We also thank Kazuaki Chayama for providing PHH-engrafted Alb-uPA/SCID mouse samples.
Normal human liver tissues were obtained through the Liver Tissue Cell Distribution System, Minneapolis, MN, which was funded by NIH contract no. HHSN276201200017C. This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, U.S. National Institutes of Health.
Q.L. and T.J.L. conceived the project and designed the experiments; B.L., L.H., S.C., K.V., and Q.L. performed the experiments; B.L., L.H., Q.L., and T.J.L. analyzed the data and wrote the manuscript with input from all coauthors.
The authors declare no competing financial interests.
Footnotes
Citation Lowey B, Hertz L, Chiu S, Valdez K, Li Q, Liang TJ. 2019. Hepatitis C virus infection induces hepatic expression of NF-κB-inducing kinase and lipogenesis by downregulating miR-122. mBio 10:e01617-19. https://doi.org/10.1128/mBio.01617-19.
Contributor Information
Thomas Shenk, Princeton University.
Steven Weinman, University of Kansas Medical Center.
Takeshi Saito, University of Southern California.
REFERENCES
- 1.Murray CL, Rice CM. 2011. Turning hepatitis C into a real virus. Annu Rev Microbiol 65:307–327. doi: 10.1146/annurev-micro-090110-102954. [DOI] [PubMed] [Google Scholar]
- 2.Liang TJ, Rehermann B, Seeff LB, Hoofnagle JH. 2000. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann Intern Med 132:296–305. doi: 10.7326/0003-4819-132-4-200002150-00008. [DOI] [PubMed] [Google Scholar]
- 3.Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. 2013. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 57:1333–1342. doi: 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 4.Liang TJ, Ghany MG. 2013. Current and future therapies for hepatitis C virus infection. N Engl J Med 368:1907–1917. doi: 10.1056/NEJMra1213651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chisari FV. 2005. Unscrambling hepatitis C virus-host interactions. Nature 436:930–932. doi: 10.1038/nature04076. [DOI] [PubMed] [Google Scholar]
- 6.Li Q, Brass AL, Ng A, Hu Z, Xavier RJ, Liang TJ, Elledge SJ. 2009. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci U S A 106:16410–16415. doi: 10.1073/pnas.0907439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Q, Zhang YY, Chiu S, Hu Z, Lan KH, Cha H, Sodroski C, Zhang F, Hsu CS, Thomas E, Liang TJ. 2014. Integrative functional genomics of hepatitis C virus infection identifies host dependencies in complete viral replication cycle. PLoS Pathog 10:e1004163. doi: 10.1371/journal.ppat.1004163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Q, Lowey B, Sodroski C, Krishnamurthy S, Alao H, Cha H, Chiu S, El-Diwany R, Ghany MG, Liang TJ. 2017. Cellular microRNA networks regulate host dependency of hepatitis C virus infection. Nat Commun 8:1789. doi: 10.1038/s41467-017-01954-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Q, Pene V, Krishnamurthy S, Cha H, Liang TJ. 2013. Hepatitis C virus infection activates an innate pathway involving IKK-alpha in lipogenesis and viral assembly. Nat Med 19:722–729. doi: 10.1038/nm.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pene V, Li Q, Sodroski C, Hsu CS, Liang TJ. 2015. Dynamic interaction of stress granules, DDX3X, and IKK-alpha mediates multiple functions in hepatitis C virus infection. J Virol 89:5462–5477. doi: 10.1128/JVI.03197-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perkins ND. 2007. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol 8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 12.Xiao G, Fong A, Sun SC. 2004. Induction of p100 processing by NF-kappaB-inducing kinase involves docking IkappaB kinase alpha (IKKalpha) to p100 and IKKalpha-mediated phosphorylation. J Biol Chem 279:30099–30105. doi: 10.1074/jbc.M401428200. [DOI] [PubMed] [Google Scholar]
- 13.Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. 2001. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 14.Malinin NL, Boldin MP, Kovalenko AV, Wallach D. 1997. MAP3K-related kinase involved in NF-kappaB induction by TNF, CD95 and IL-1. Nature 385:540–544. doi: 10.1038/385540a0. [DOI] [PubMed] [Google Scholar]
- 15.Choudhary S, Sinha S, Zhao Y, Banerjee S, Sathyanarayana P, Shahani S, Sherman V, Tilton RG, Bajaj M. 2011. NF-kappaB-inducing kinase (NIK) mediates skeletal muscle insulin resistance: blockade by adiponectin. Endocrinology 152:3622–3627. doi: 10.1210/en.2011-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willmann KL, Klaver S, Doğu F, Santos-Valente E, Garncarz W, Bilic I, Mace E, Salzer E, Conde CD, Sic H, Májek P, Banerjee PP, Vladimer GI, Haskoloğlu S, Bolkent MG, Küpesiz A, Condino-Neto A, Colinge J, Superti-Furga G, Pickl WF, van Zelm MC, Eibel H, Orange JS, Ikincioğulları A, Boztuğ K. 2014. Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity. Nat Commun 5:5360. doi: 10.1038/ncomms6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang F, Sodroski C, Cha H, Li Q, Liang TJ. 2017. Infection of hepatocytes with HCV increases cell surface levels of heparan sulfate proteoglycans, uptake of cholesterol and lipoprotein, and virus entry by up-regulating SMAD6 and SMAD7. Gastroenterology 152:257–270.e7. doi: 10.1053/j.gastro.2016.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyer A, Park SB, de Boer Y, Li Q, Liang TJ. 2018. TM6SF2 promotes lipidation and secretion of hepatitis C virus in infected hepatocytes. Gastroenterology 155:1923. doi: 10.1053/j.gastro.2018.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309:1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 21.Luna JM, Scheel TK, Danino T, Shaw KS, Mele A, Fak JJ, Nishiuchi E, Takacs CN, Catanese MT, de Jong YP, Jacobson IM, Rice CM, Darnell RB. 2015. Hepatitis C virus RNA functionally sequesters miR-122. Cell 160:1099–1110. doi: 10.1016/j.cell.2015.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bandiera S, Pfeffer S, Baumert TF, Zeisel MB. 2015. miR-122—a key factor and therapeutic target in liver disease. J Hepatol 62:448–457. doi: 10.1016/j.jhep.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 23.Sarnow P, Sagan SM. 2016. Unraveling the mysterious interactions between hepatitis C virus RNA and liver-specific microRNA-122. Annu Rev Virol 3:309–332. doi: 10.1146/annurev-virology-110615-042409. [DOI] [PubMed] [Google Scholar]
- 24.Li ZY, Xi Y, Zhu WN, Zeng C, Zhang ZQ, Guo ZC, Hao DL, Liu G, Feng L, Chen HZ, Chen F, Lv X, Liu DP, Liang CC. 2011. Positive regulation of hepatic miR-122 expression by HNF4alpha. J Hepatol 55:602–611. doi: 10.1016/j.jhep.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, El Maadidi S, Fischer J, Grabski E, Dickhofer S, Klimosch S, Flannery SM, Filomena A, Wolz OO, Schneiderhan-Marra N, Loffler MW, Wiese M, Pichulik T, Mullhaupt B, Semela D, Dufour JF, Bochud PY, Bowie AG, Kalinke U, Berg T, Weber AN, East-German and Swiss Hepatitis C Virus Study Groups. 2015. A frequent hypofunctional IRAK2 variant is associated with reduced spontaneous hepatitis C virus clearance. Hepatology 62:1375–1387. doi: 10.1002/hep.28105. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Masaki T, Yamane D, McGivern DR, Lemon SM. 2013. Competing and noncompeting activities of miR-122 and the 5′ exonuclease Xrn1 in regulation of hepatitis C virus replication. Proc Natl Acad Sci U S A 110:1881–1886. doi: 10.1073/pnas.1213515110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Yamane D, Lemon SM. 2015. Dissecting the roles of the 5′ exoribonucleases Xrn1 and Xrn2 in restricting hepatitis C virus replication. J Virol 89:4857–4865. doi: 10.1128/JVI.03692-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sedano CD, Sarnow P. 2014. Hepatitis C virus subverts liver-specific miR-122 to protect the viral genome from exoribonuclease Xrn2. Cell Host Microbe 16:257–264. doi: 10.1016/j.chom.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sodroski C, Lowey B, Hertz L, Jake Liang T, Li Q. 2018. MicroRNA-135a modulates hepatitis C virus genome replication through downregulation of host antiviral factors. Virol Sin 34:197–210. doi: 10.1007/s12250-018-0055-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walters KA, Syder AJ, Lederer SL, Diamond DL, Paeper B, Rice CM, Katze MG. 2009. Genomic analysis reveals a potential role for cell cycle perturbation in HCV-mediated apoptosis of cultured hepatocytes. PLoS Pathog 5:e1000269. doi: 10.1371/journal.ppat.1000269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vallianou I, Dafou D, Vassilaki N, Mavromara P, Hadzopoulou-Cladaras M. 2016. Hepatitis C virus suppresses hepatocyte nuclear factor 4 alpha, a key regulator of hepatocellular carcinoma. Int J Biochem Cell Biol 78:315–326. doi: 10.1016/j.biocel.2016.07.027. [DOI] [PubMed] [Google Scholar]
- 32.Xia Y, Cheng X, Li Y, Valdez K, Chen W, Liang TJ. 2018. Hepatitis B virus deregulates the cell cycle to promote viral replication and a premalignant phenotype. J Virol 92:e00722-18. doi: 10.1128/JVI.00722-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong MH, Chou YC, Wu YC, Tsai KN, Hu CP, Jeng KS, Chen ML, Chang C. 2012. Transforming growth factor-beta1 suppresses hepatitis B virus replication by the reduction of hepatocyte nuclear factor-4alpha expression. PLoS One 7:e30360. doi: 10.1371/journal.pone.0030360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee CW, Huang WC, Huang HD, Huang YH, Ho JH, Yang MH, Yang VW, Lee OK. 2017. DNA methyltransferases modulate hepatogenic lineage plasticity of mesenchymal stromal cells. Stem Cell Reports 9:247–263. doi: 10.1016/j.stemcr.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Q, Sodroski C, Lowey B, Schweitzer CJ, Cha H, Zhang F, Liang TJ. 2016. Hepatitis C virus depends on E-cadherin as an entry factor and regulates its expression in epithelial-to-mesenchymal transition. Proc Natl Acad Sci U S A 113:7620–7625. doi: 10.1073/pnas.1602701113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Presser LD, Haskett A, Waris G. 2011. Hepatitis C virus-induced furin and thrombospondin-1 activate TGF-beta1: role of TGF-beta1 in HCV replication. Virology 412:284–296. doi: 10.1016/j.virol.2010.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin W, Tsai WL, Shao RX, Wu G, Peng LF, Barlow LL, Chung WJ, Zhang L, Zhao H, Jang JY, Chung RT. 2010. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology 138:2509–2518.e1. doi: 10.1053/j.gastro.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 39.Gluchowski NL, Becuwe M, Walther TC, Farese RV Jr.. 2017. Lipid droplets and liver disease: from basic biology to clinical implications. Nat Rev Gastroenterol Hepatol 14:343–355. doi: 10.1038/nrgastro.2017.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Negro F. 2012. HCV infection and metabolic syndrome: which is the chicken and which is the egg? Gastroenterology 142:1288–1292. doi: 10.1053/j.gastro.2011.12.063. [DOI] [PubMed] [Google Scholar]
- 41.Sheng L, Zhou Y, Chen Z, Ren D, Cho KW, Jiang L, Shen H, Sasaki Y, Rui L. 2012. NF-kappaB-inducing kinase (NIK) promotes hyperglycemia and glucose intolerance in obesity by augmenting glucagon action. Nat Med 18:943–949. doi: 10.1038/nm.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y, Sheng L, Xiong Y, Shen H, Liu Y, Rui L. 2017. Liver NF-kappaB-inducing kinase promotes liver steatosis and glucose counterregulation in male mice with obesity. Endocrinology 158:1207–1216. doi: 10.1210/en.2016-1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun SC. 2011. Non-canonical NF-kappaB signaling pathway. Cell Res 21:71–85. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. 2002. Identification of tissue-specific microRNAs from mouse. Curr Biol 12:735–739. doi: 10.1016/S0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 45.Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, Subramaniam A, Propp S, Lollo BA, Freier S, Bennett CF, Bhanot S, Monia BP. 2006. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab 3:87–98. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 46.Chai C, Rivkin M, Berkovits L, Simerzin A, Zorde-Khvalevsky E, Rosenberg N, Klein S, Yaish D, Durst R, Shpitzen S, Udi S, Tam J, Heeren J, Worthmann A, Schramm C, Kluwe J, Ravid R, Hornstein E, Giladi H, Galun E. 2017. Metabolic circuit involving free fatty acids, microRNA 122, and triglyceride synthesis in liver and muscle tissues. Gastroenterology 153:1404–1415. doi: 10.1053/j.gastro.2017.08.013. [DOI] [PubMed] [Google Scholar]
- 47.Cheung O, Puri P, Eicken C, Contos MJ, Mirshahi F, Maher JW, Kellum JM, Min H, Luketic VA, Sanyal AJ. 2008. Nonalcoholic steatohepatitis is associated with altered hepatic microRNA expression. Hepatology 48:1810–1820. doi: 10.1002/hep.22569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saito Y, Suzuki H, Matsuura M, Sato A, Kasai Y, Yamada K, Saito H, Hibi T. 2011. MicroRNAs in hepatobiliary and pancreatic cancers. Front Genet 2:66. doi: 10.3389/fgene.2011.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsu SH, Wang B, Kota J, Yu J, Costinean S, Kutay H, Yu L, Bai S, La Perle K, Chivukula RR, Mao H, Wei M, Clark KR, Mendell JR, Caligiuri MA, Jacob ST, Mendell JT, Ghoshal K. 2012. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest 122:2871–2883. doi: 10.1172/JCI63539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsai WC, Hsu SD, Hsu CS, Lai TC, Chen SJ, Shen R, Huang Y, Chen HC, Lee CH, Tsai TF, Hsu MT, Wu JC, Huang HD, Shiao MS, Hsiao M, Tsou AP. 2012. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest 122:2884–2897. doi: 10.1172/JCI63455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gatfield D, Le Martelot G, Vejnar CE, Gerlach D, Schaad O, Fleury-Olela F, Ruskeepaa AL, Oresic M, Esau CC, Zdobnov EM, Schibler U. 2009. Integration of microRNA miR-122 in hepatic circadian gene expression. Genes Dev 23:1313–1326. doi: 10.1101/gad.1781009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kato T, Matsumura T, Heller T, Saito S, Sapp RK, Murthy K, Wakita T, Liang TJ. 2007. Production of infectious hepatitis C virus of various genotypes in cell cultures. J Virol 81:4405–4411. doi: 10.1128/JVI.02334-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng X, Xia Y, Serti E, Block PD, Chung M, Chayama K, Rehermann B, Liang TJ. 2017. Hepatitis B virus evades innate immunity of hepatocytes but activates cytokine production by macrophages. Hepatology 66:1779–1793. doi: 10.1002/hep.29348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effects of NIK knockdown on HCV life cycle. Download FIG S1, DOCX file, 1.3 MB (1.3MB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
NIK localizes to the stress granules (SGs) upon HCV infection. Download FIG S2, DOCX file, 0.9 MB (986.2KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Mutated NIK on lipid droplet accumulation. Download FIG S3, DOCX file, 1.5 MB (1.6MB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Effects of interferons on NIK expression in hepatocytes. Download FIG S4, DOCX file, 0.4 MB (391KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Supplemental materials and methods used in this study. Download Text S1, DOCX file, 0.04 MB (39.6KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.