

Abstract
Background
It has been suggested that antidepressant benefits are smaller for mild than severe depression. Because antidepressants are also used for anxiety disorders, obsessive‐compulsive disorder (OCD), and posttraumatic stress disorder (PTSD), we examined the influence of severity for these disorders.
Methods
We used individual patient data of eight trials (3,430 participants) for generalized anxiety disorder (GAD); four trials (1,195 participants) for social anxiety disorder (SAD); four trials (1,132 participants) for OCD; three trials (1,071 participants) for PTSD; and 10 trials (2,151 participants) for panic disorder (PD). Mixed‐effects models were used to investigate an interaction between severity and treatment group.
Results
For GAD and PD, severity moderated antidepressant efficacy. The antidepressant–placebo difference was 1.4 (95% CI: 0.4–2.5; SMD: 0.21) Hamilton Anxiety Rating Scale (HAM‐A) points for participants with mild GAD (baseline HAM‐A = 10), increasing to 4.0 (3.4–4.6; SMD: 0.45) or greater for severely ill participants (HAM‐A ≥ 30). For PD, the difference was 0.4 (0.3–0.6) panic attacks/2 weeks for participants with 10 panic attacks/2 weeks at baseline, increasing to 4.7 (3.0–6.4) for participants with 40. For SAD, OCD, and PTSD, no interaction was found. Across severity levels, the differences were 16.1 (12.9–19.3; SMD: 0.59) Liebowitz Social Anxiety Scale points, 3.4 (2.5–4.4, SMD: 0.39) Yale–Brown Obsessive‐Compulsive Scale points, and 10.3 (6.9–13.6; SMD: 0.41) Clinician‐Administered PTSD Scale points.
Conclusions
Antidepressants are equally effective across severity levels for SAD, OCD, and PTSD. For GAD and PD, however, benefits are small at low severity, and the benefit–risk ratio may be unfavorable for these patients.
Keywords: antidepressive agents, anxiety disorders, meta‐analysis, obsessive‐compulsive disorder, panic disorder, posttraumatic stress disorder
1. INTRODUCTION
Antidepressants are considered effective treatments for major depressive disorder (MDD) (Qaseem, Barry, & Kansagara, 2016), anxiety disorders (Bandelow et al., 2015), obsessive‐compulsive disorder (OCD) (Soomro, Altman, Rajagopal, & Oakley Browne, 2008), and posttraumatic stress disorder (PTSD) (Stein, Ipser, & Seedat, 2006). However, research in MDD has suggested that antidepressant efficacy may depend upon initial symptom severity. Both trial‐level (Khan, Leventhal, Khan, & Brown, 2002; Kirsch et al., 2008) and individual patient data (IPD) meta‐analyses (Fournier et al., 2010; Khan, Bhat, Faucett, Kolts, & Brown, 2011; Khan, Brodhead, Kolts, & Brown, 2005) have found that antidepressants are more effective for patients with high initial severity, with some of these analyses suggesting that efficacy is minimal for patients with mild depression (Fournier et al., 2010; Kirsch et al., 2008). Consequently, many guidelines no longer recommend antidepressants for mild depression (National Institute for Health and Care Excellence, 2009; Spijker et al., 2013). Recently, however, two substantially larger IPD meta‐analyses did not find an association between severity and antidepressant efficacy for MDD (Gibbons, Hur, Brown, Davis, & Mann, 2012; Rabinowitz et al., 2016), indicating that this question is not yet settled.
Antidepressants are also commonly used for anxiety disorders, OCD, and PTSD (Wong et al., 2016), but comparatively little evidence regarding the influence of severity is available for these disorders. Trial‐level meta‐analyses have found no evidence that antidepressant efficacy increases with increasing severity (Ackerman & Greenland, 2002; Curtiss, Andrews, Davis, Smits, & Hofmann, 2017; Davis, Smits, & Hofmann, 2014; de Vries, de Jonge, van den Heuvel, Turner, & Roest, 2016; Sugarman, Loree, Baltes, Grekin, & Kirsch, 2014). However, such trial‐level analyses may be prone to the ecological fallacy (Thompson & Higgins, 2002), in which a trial‐level relationship can be found that does not exist at the participant level, or vice versa. They can also be underpowered and suffer from a restriction of range. Hence, IPD is needed to provide better insight into whether initial severity is associated with antidepressant efficacy for anxiety disorders.
However, few studies have used IPD and most of these studies had significant limitations. Two studies examined efficacy in subgroups of less and more severely anxious patients without actually testing for differences (Pae et al., 2015; Stein, Kasper, Andersen, Nil, & Lader, 2004). Two other studies tested the association between GAD severity and dichotomized outcomes, with one analysis reporting a significant association only for remission (Montgomery, Sheehan, Meoni, Haudiquet, & Hackett, 2002) and the other only for response (Pollack, Meoni, Otto, Simon, & Hackett, 2003). Two patient‐level analyses for SAD found contradictory results, with one reporting greater efficacy in more severely anxious participants than in less severely anxious participants (Montgomery, 1998), while the other reported similar efficacy (Stein, Stein, Goodwin, Kumar, & Hunter, 2001). Finally, a post hoc analysis of a trial for PTSD found no evidence for moderation by initial severity, but this was a negative trial, which may have made it impossible to detect such moderation. To our knowledge, there are no patient‐level analyses for OCD or PD. Given the limitations of the available evidence (including dichotomization of outcomes and predictors, which leads to a significant loss of power (Altman & Royston, 2006)) and the conflicting results, the question of whether initial severity moderates antidepressant efficacy for anxiety disorders, OCD, and PTSD remains unanswered. In the current study, we therefore examined this question using IPD from 29 trials enrolling 8,979 participants.
2. MATERIALS AND METHODS
2.1. Data source
We requested IPD from Clinical Study Data Request (CSDR, https://clinicalstudydatarequest.com). We first identified all selective serotonin reuptake inhibitors (SSRIs) and serotonin–norepinephrine reuptake inhibitors (SNRIs) developed by participating sponsors. These were paroxetine (GlaxoSmithKline), fluoxetine (Lilly), and duloxetine (Lilly). Although other SSRIs and SNRIs are also used for anxiety disorders, these were not available through CSDR. We then identified all double‐blind, placebo‐controlled, and short‐term (≤16 weeks) randomized controlled trials (RCTs) of these antidepressants for an anxiety disorder, OCD, or PTSD in adults that were mentioned in Food and Drug Administration (FDA) drug approval packages (Roest et al., 2015) or the GlaxoSmithKline (https://www.gsk-clinicalstudyregister.com) and Lilly (https://www.lilly.com/clinical-study-report-csr-synopses) trial registries.
2.2. Primary outcomes
As our primary outcome, we chose the outcome usually considered primary for that disorder. For GAD, this was the Hamilton Rating Scale for Anxiety (HAM‐A); for SAD, the Liebowitz Social Anxiety Scale (LSAS); for OCD, the Yale–Brown Obsessive‐Compulsive Scale (Y‐BOCS); and for PTSD, the Clinician‐Administered PTSD Scale (CAPS). For PD, most trials used response (defined as having 0 full panic attacks) as an outcome, but we selected the number of full panic attacks per 2 weeks, since dichotomization leads to a significant loss of information (Altman & Royston, 2006).
2.3. Patient population
We included patients with a valid baseline score and at least one valid follow‐up score on the primary outcome (modified intention‐to‐treat population). Patients assigned to placebo, the investigative antidepressant, or a comparator SSRI or SNRI were included. We excluded patients assigned to other active comparators.
2.4. Statistical analysis
We conducted separate analyses for each disorder. For GAD, SAD, OCD, and PTSD, we applied linear mixed models, using the nlme package (version 3.1‐127) for R (3.3.0). The effect measure of interest was the change from baseline on the primary outcome. The initial model included all fixed effects, regardless of significance. These were initial severity, treatment group, linear and quadratic terms for time (in days since baseline), and their two‐ and three‐way interactions. Baseline and change scores were grand‐mean centered and standardized, while time was centered at trial endpoint and standardized. Using this first model, we modeled the covariance structure of the nested data. We considered random intercepts at trial and participant level and random effects for linear and quadratic time. For these random effects, we examined compound symmetry, diagonal and unstructured covariance matrices, and autocorrelation terms for the residuals. We used restricted maximum likelihood (REML) for estimation and the Akaike Information Criterion (AIC) to select the best‐fitting covariance structure.
Subsequently, we refitted the model using maximum likelihood (ML) and removed nonsignificant fixed effects by backward selection. If an interaction or quadratic effect was significant, we retained all component main or linear effects regardless of significance. We used the AIC to select the best‐fitting model. However, for clarity we further simplified models containing nonsignificant terms even if the more complex model had a marginally lower AIC. In these cases, both the Bayesian Information Criterion (BIC) and the likelihood ratio test also favored the simpler models. A standardized mean difference (SMD) was calculated by dividing the drug‐placebo difference in change scores by the pooled standard deviation of the change score at endpoint (imputed where necessary and stratified by baseline severity in case of a significant interaction between severity and treatment group).
For PD, we applied a negative binomial mixed model, using the glmer.nb command from the lme4 package (version 1.1‐12). The effect measure of interest was the number of panic attacks/2 weeks. Because this measure was highly skewed, we replaced values ≥100 (45 (0.4%) of 11,785 observations) by a new value between 70 and 100 (randomly drawn from a uniform distribution) to improve model convergence. The initial model included the same fixed effects as for the other disorders. For the covariance structure, we considered only random intercepts at trial and participant level and a random effect for linear time, as models including a random effect for quadratic time failed to converge. Since lme4 does not easily allow for either autocorrelation or different covariance structures, we only modeled an unstructured covariance matrix. We subsequently selected the best‐fitting model using backward selection of the fixed effects, as done for the other disorders. Because of the nonnormal distribution of panic attacks, we did not include a standardized difference for PD.
For all disorders, we also analyzed (prespecified) models that included age and gender as covariates, which yielded similar results and hence are not described further. We conducted several other post hoc sensitivity analyses. First, we excluded dosages below the FDA‐approved dose range (e.g., 10 mg paroxetine for PD). Secondly, we excluded participants with very low baseline scores, because some trials included a small number of participants with even, in some cases, scores within the range generally thought to correspond to “absence of illness” (although all participants met diagnostic criteria for the disorder under study). Finally, we analyzed an alternative outcome for PD, the Panic Disorder Severity Scale (PDSS) (Shear et al., 1997), a more comprehensive measure that was only available in two out of 10 PD trials. For this analysis, we followed the approach also taken for GAD, SAD, OCD, and PTSD.
3. RESULTS
3.1. Trials and participants
We identified 34 trials, but we excluded one trial of paroxetine for PD (29060/108 (Oehrberg et al., 1995)) a priori, as it did not distinguish between full and limited‐symptom panic attacks. We were denied access to four other trials: electronic data was not available for a trial of fluoxetine for OCD (E079 (Montgomery et al., 1993), completed in 1991), while GlaxoSmithKline considered the translation costs for three (unpublished) Japanese trials of paroxetine for GAD or SAD (29060/661, 29060/856, and PIR104776) to be prohibitive. One of these trials was positive (PIR104776), i.e., had statistically significant results for the primary outcome, while the other three were negative.
We received access to 29 trials with 3,656 placebo‐treated and 5,323 antidepressant‐treated participants. Two trials (of duloxetine for GAD) included an active comparator (venlafaxine extended release). For GAD, we had access to eight trials (six positive) with 1,342 placebo‐treated and 2,088 antidepressant‐treated participants; for SAD, four trials (all positive) with 514 placebo‐treated and 681 antidepressant‐treated participants; for OCD, four trials (three positive) with 350 placebo‐treated and 782 antidepressant‐treated participants; for PTSD, three trials (two positive) with 459 placebo‐treated and 612 antidepressant‐treated participants; and for PD, 10 trials (five positive) with 991 placebo‐treated and 1,160 antidepressant‐treated participants (see flow chart in Supporting Information Figure 1 for trial selection, Table 1 for baseline characteristics, and Supporting Information Tables 1 and 2 for individual trial information).
Table 1.
Baseline characteristics for each disorder
| Gender (% Female) | Mean Age (SD) | Mean Baseline Score (SD) | Baseline Range | |
|---|---|---|---|---|
| GAD | 62.3 | 42.0 (13.4) | 25.1 (5.9) | 2–50 |
| SAD | 46.8 | 37.3 (11.0) | 80.2 (24.0) | 7–139 |
| OCD | 44.4 | 38.7 (12.4) | 25.0 (5.3) | 10–40 |
| PTSD | 62.6 | 41.1 (11.7) | 75.2 (16.6) | 30–132 |
| Gender (% Female) | Mean Age (SD) | Median Baseline Score (IQR) | Baseline Range | |
|---|---|---|---|---|
| Panic disorder | 61.4 | 37.3 (10.5) | 5 (3–11) | 0–99 |
Notes. The baseline score is determined as the score at baseline on the Hamilton Anxiety Rating Scale (HAM‐A) for GAD, the Liebowitz Social Anxiety Scale (LSAS) for SAD, the Yale–Brown Obsessive‐Compulsive Scale (Y‐BOCS) for OCD, the Clinician‐Administered PTSD Scale (CAPS) for PTSD, and the number of panic attacks/2 weeks for panic disorder.
3.2. GAD, SAD, OCD, and PTSD
For all four disorders, a model with an unstructured covariance matrix (including all random effects) and autocorrelated errors fit best. For GAD, the best‐fitting model included the two‐way interaction between baseline score and treatment group, but not the three‐way interactions between baseline score, treatment group, and time (Figure 1a). For SAD, OCD, and PTSD, the best‐fitting model did not include any of the interactions between baseline score and treatment group (Figure 1b–d). Model specifications are provided in Supporting Information Table 3. For all disorders, there was a significant main effect of baseline score, resulting in a greater change from baseline with increasing severity in both treatment groups, consistent with a regression to the mean effect.
Figure 1.
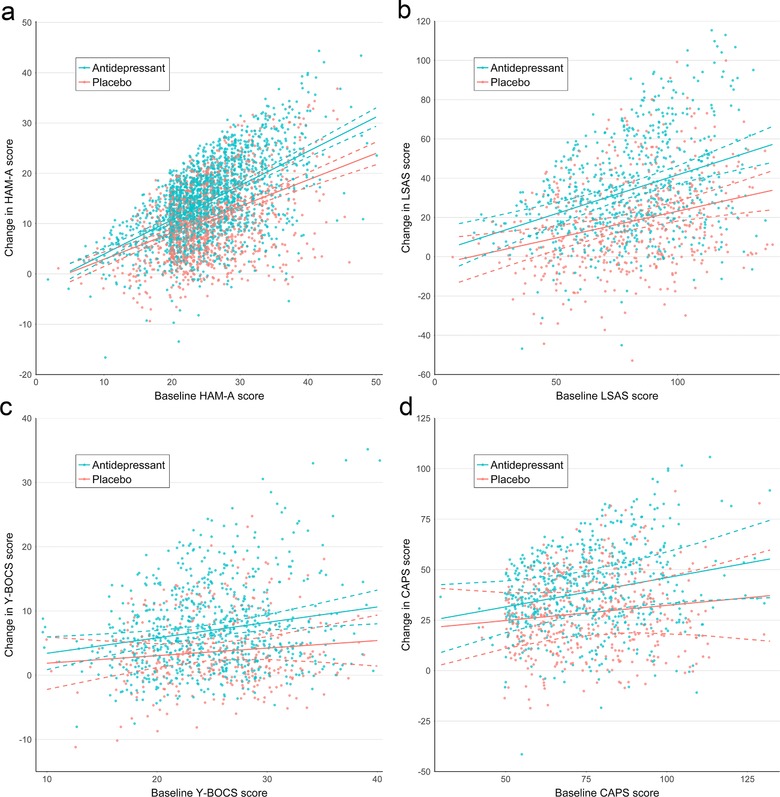
Predicted change from baseline for antidepressant‐ and placebo‐treated participants with (a) generalized anxiety disorder, (b) social anxiety disorder, (c) obsessive‐compulsive disorder, and (d) PTSD. Predictions are derived from the full model, including nonsignificant interaction terms. Data points were jittered to reduce over‐plotting
For GAD, the estimated benefit of antidepressants (compared to placebo) at trial endpoint (8 weeks) was 1.4 (95% CI: 0.4–2.5, SMD: 0.21) points on the HAM‐A for participants with a baseline score of 10, increasing to 4.0 (95% CI: 3.4–4.6, SMD: 0.45) for participants with a baseline score of 30 (Table 2). Because there was no significant relationship between initial severity and antidepressant efficacy for SAD, OCD, and PTSD, the best‐fitting model estimated the same antidepressant–placebo difference across the severity range. For SAD, it was 16.1 (95% CI: 12.9–19.3, SMD: 0.59) LSAS points at week 12; for OCD, 3.4 (95% CI: 2.5–4.4, SMD: 0.39) Y‐BOCS points at week 12; and for PTSD, 10.3 (95% CI: 6.9–13.6, SMD: 0.41) CAPS points at week 12.
Table 2.
Predicted change on the Hamilton Anxiety Rating Scale (HAM‐A) and antidepressant–placebo difference after 8 weeks of treatment for GAD
| Predicted Change (95% CI) | |||||
|---|---|---|---|---|---|
| Baseline Score | Sample Size1 | Placebo | Antidepressant | Drug‐Placebo Difference (95% CI) | SMD |
| 10 | 79 | 2.7 (1.7–3.7) | 4.1 (3.2–5.0) | 1.4 (0.4–2.5) | 0.21 |
| 15 | 259 | 5.4 (4.6–6.2) | 7.4 (6.8–8.1) | 2.1 (1.3–2.9) | 0.29 |
| 20 | 1388 | 8.1 (7.5–8.7) | 10.8 (10.3–11.3) | 2.7 (2.1–3.3) | 0.35 |
| 25 | 998 | 10.8 (10.3–11.3) | 14.2 (13.7–14.6) | 3.3 (2.8–3.9) | 0.39 |
| 30 | 442 | 13.5 (12.9–14.1) | 17.5 (17.0–18.0) | 4.0 (3.4–4.6) | 0.45 |
| 35 | 187 | 16.2 (15.5–17.0) | 20.9 (20.2–21.5) | 4.6 (3.8–5.4) | 0.43 |
| 40 | 48 | 18.9 (17.9–20.0) | 24.2 (23.3–25.1) | 5.3 (4.2–6.3) | 0.43 |
| 45 | 10 | 21.7 (20.4–22.9) | 27.6 (26.5–28.7) | 5.9 (4.6–7.2) | 0.48 |
Notes. 1Sample size indicates the number of participants with a baseline score in between the indicated score and the subsequent score (e.g., 10 includes participants with baseline scores between 10 and 14 (inclusive)).
3.3. Panic disorder
For PD, the model with the lowest AIC (38,267.2) contained the two‐way interaction between baseline severity and group, but because this term was not significant (p = 0.11), we preferred a more parsimonious model without the interaction and with an only marginally larger AIC (38,268.6 after removing two nonsignificant terms, see Supporting Information Table 3). This model indicated that the drug‐placebo difference was constant on the log scale of the negative binomial model and hence that the ratio of the endpoint number of panic attacks/2 weeks in the placebo group compared to the drug group was constant (2.46) on the original scale. Consequently, the absolute difference between drug and placebo groups actually increased with increasing severity (Figure 2). For participants experiencing two panic attacks/2 weeks at baseline, the estimated drug–placebo difference was 0.2 (95% CI: 0.2–0.3) (in favor of antidepressants) at week 10. This increased to 0.4 (95% CI: 0.3–0.6) for participants experiencing 10 panic attacks/2 weeks at baseline, 0.9 (95% CI: 0.7–1.2) for participants experiencing 20, and 4.7 (95% CI: 3.0–6.4) for participants experiencing 40 (Table 3).
Figure 2.
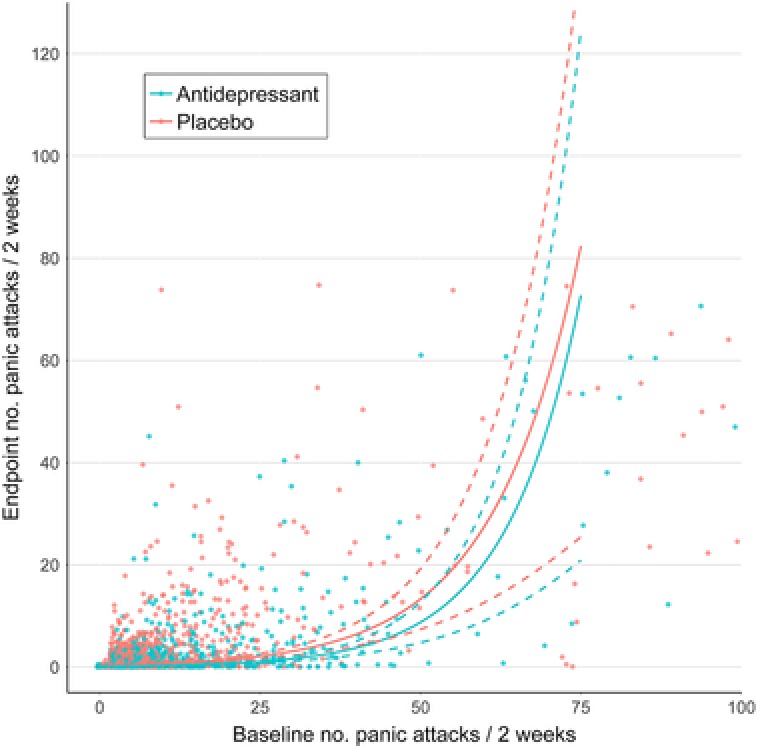
Predicted numbers of panic attacks/2 weeks at endpoint for antidepressant‐ and placebo‐treated participants with panic disorder. Predictions are derived from the full model, including nonsignificant interaction terms. Data points were jittered to reduce over‐plotting
Table 3.
Predicted endpoint number of panic attacks/2 weeks and antidepressant–placebo difference after 10 weeks of treatment for panic disorder
| Predicted No. Panic Attacks / 2 Weeks (95% CI) | ||||
|---|---|---|---|---|
| Baseline Score | Sample Size1 | Placebo | Antidepressant | Drug–Placebo Difference |
| 2 | 636 | 0.38 (0.31–0.46) | 0.15 (0.13–0.19) | 0.23 (0.16–0.29) |
| 4 | 396 | 0.44 (0.37–0.54) | 0.18 (0.15–0.22) | 0.26 (0.19–0.34) |
| 6 | 258 | 0.52 (0.43–0.63) | 0.21 (0.17–0.26) | 0.31 (0.22–0.40) |
| 8 | 175 | 0.61 (0.51–0.74) | 0.25 (0.21–0.30) | 0.36 (0.26–0.47) |
| 10 | 240 | 0.72 (0.60–0.86) | 0.29 (0.24–0.35) | 0.43 (0.30–0.55) |
| 15 | 123 | 1.07 (0.89–1.29) | 0.43 (0.36–0.52) | 0.64 (0.45–0.82) |
| 20 | 133 | 1.59 (1.31–1.93) | 0.65 (0.53–0.78) | 0.95 (0.67–1.23) |
| 30 | 58 | 3.54 (2.82–4.45) | 1.44 (1.14–1.80) | 2.10 (1.43–2.78) |
| 40 | 44 | 7.86 (5.93–10.41) | 3.19 (2.41–4.21) | 4.67 (2.98–6.37) |
| 60 | 37 | 38.76 (25.78–58.28) | 15.72 (10.50–23.55) | 23.04 (12.23–33.83) |
Notes. 1Sample size indicates the number of participants with a baseline score in between the indicated score and the subsequent score (e.g., 2 includes participants with baseline scores of 2 or 3) or the maximum score (99).
3.4. Post hoc analyses
The results of our post hoc analyses excluding unapproved dosages or participants with very low baseline scores were similar to our main analyses (Supporting Information Tables 4–6). In the post hoc analysis of the PDSS for PD, the best‐fitting model included both the two‐way interaction between baseline severity and treatment group and the three‐way interaction between severity, treatment group, and time (Supporting Information Tables 7 and 8). At endpoint (week 12), the antidepressant–placebo difference was 0.0 (95% CI: –1.9 to 2.0; SMD: 0.00) for participants with moderate illness (baseline PDSS of 12), 3.9 (95% CI: 2.7–5.2; SMD: 0.59) for participants with severe illness (baseline PDSS of 18), and 5.9 (95% CI: 4.1–7.7; SMD: 1.00) for participants with very severe illness (baseline PDSS of 21).
4. DISCUSSION
4.1. Principal findings
To our knowledge, this is the first individual patient data meta‐analysis examining the relationship between baseline severity and antidepressant efficacy for anxiety disorders, OCD, and PTSD. We showed that initial severity moderates antidepressant efficacy for GAD, but not for SAD, OCD, and PTSD. For PD, the ratio between the number of panic attacks in the placebo group compared to the drug group was constant, but the absolute antidepressant‐placebo difference was small for patients experiencing few panic attacks at baseline and increased with increasing severity. For all disorders, a regression to the mean or law of initial value effect occurred, but this cannot explain the interaction between baseline severity and treatment in GAD.
Our findings are partially in agreement with our earlier trial‐level meta‐analysis, confirming that initial severity does not influence antidepressant efficacy for SAD, OCD, and PTSD (de Vries et al., 2016). However, our finding that initial severity does affect antidepressant efficacy for GAD and PD diverges from our earlier study, in which no effect of initial severity was apparent for these disorders either. These differences are likely because of the much larger sample size and the use of IPD in this study, which allows for the detection of smaller interaction effects. Furthermore, for PD we used a different outcome in this study (number of panic attacks/2 weeks instead of remission). The SMDs for SAD and PTSD were also larger than those found earlier (Roest et al., 2015). This is probably at least in part because the paroxetine trials had higher effect sizes than trials of other drugs for these disorders, which we were unable to include in this study. The larger SMDs may also be due, in part, to different analytical techniques, since our previous meta‐analysis used last‐observation‐carried‐forward (LOCF) to handle missing data, while the current study employed mixed models.
It is unclear why we found a relationship between initial severity and antidepressant efficacy for GAD and PD, but not for the other disorders. GAD is often considered to be more closely related to MDD than the other anxiety disorders (Krueger, 1999) and the HAM‐A also overlaps with the Hamilton Depression Rating Scale (HAM‐D). On the other hand, since the association between initial severity and antidepressant efficacy in MDD has been called into question (Gibbons et al., 2012; Rabinowitz et al., 2016), a greater similarity between MDD and GAD might not explain our findings. HAM‐A items also tend to be relatively nonspecific, covering common symptoms like insomnia, tension, and worries, while the LSAS and Y‐BOCS specifically examine distress associated with respectively social situations and obsessions/compulsions, and the CAPS examines both general distress and specific trauma‐related distress. Such general distress symptoms, particularly when mild, may be more responsive to placebo or more likely to improve spontaneously. However, this would not explain the significant relationship between severity and antidepressant efficacy for PD, since both the number of panic attacks and the PDSS examine panic‐specific symptoms.
It is also important to note that we had the largest sample size for GAD, although our sensitivity analyses for PD included relatively few participants and also yielded a significant interaction effect. Since we had more than 1,000 participants for each disorder, we should have been able to detect a substantial interaction effect, but it is possible that smaller interaction effects for the other disorders were missed. However, these are less likely to be of clinical significance.
4.2. Strengths and limitations
The main strength of this study is that we used IPD and had a large sample size for each disorder. Furthermore, we used disorder‐specific primary outcomes and made full use of the longitudinal data by employing mixed models.
Our study is limited by the limitations of the included trials. In particular, minimum severity criteria restricted the number of participants at the low end of the severity range. Half of the GAD trials specified a minimum HAM‐A score of 20, for instance, even though most primary care patients with GAD have scores below 20 (Rollman et al., 2005). Patients with comorbid disorders such as MDD were also frequently excluded, even though these disorders commonly occur together (Kessler, Chiu, Demler, & Walters, 2005).
Furthermore, our findings for PD are difficult to compare to the other disorders. The best‐fitting model showed that the ratio of the number of panic attacks at endpoint in the placebo group compared to the drug group remained constant, but this means that the drug–placebo difference increased with increasing severity. We have emphasized the latter, because this measure is most comparable to the other disorders, but other choices could be made. Additionally, while the number of panic attacks is a clinically relevant outcome, it does not include other important facets of PD (e.g., agoraphobia). However, our sensitivity analyses examining the PDSS also indicated that antidepressant efficacy increased with increasing baseline severity.
We also did not receive data for four trials. Since three of these trials were negative, we may have overestimated antidepressant efficacy for GAD, SAD, and OCD, but it seems unlikely that this would have affected our findings regarding initial severity. Negative trials will probably show little evidence for differential efficacy, so it is not likely that we would have found a significant interaction effect for SAD and OCD if we had been able to include these trials. For GAD, the evidence for an interaction was sufficiently strong that it probably would have remained even if we had added an additional trial in which differential efficacy was not apparent.
Finally, because we obtained our data through CSDR, we could only include manufacturer‐sponsored trials of duloxetine, paroxetine, and fluoxetine. We used CSDR because it allowed us to obtain a nearly complete set of manufacturer‐sponsored trials for these drugs. Other approaches (e.g., a comprehensive literature search followed by requesting IPD from authors) would almost certainly have introduced much more significant biases into our trial selection, because of reporting bias (Roest et al., 2015) and refusal or inability to share data. For example, a study that took this approach for MDD trials only received data for six of 23 trials (Fournier et al., 2010). Since other SSRIs and SNRIs have similar pharmacological effects, it seems unlikely that the relationship between initial severity and antidepressant efficacy would be different. However, future research should examine other antidepressants and non‐industry‐sponsored trials.
4.3. Clinical implications
To understand the implications of these findings, it should be noted that the clinical relevance of a treatment effect is context‐specific, depending on such factors as the expected sequelae of the disease, the costs and drawbacks of the treatment, and the efficacy of alternative treatments (Kraemer et al., 2003). Without an agreed‐upon cut‐off point for a clinically relevant effect, it is difficult to establish a threshold below which antidepressant efficacy for GAD and PD is not clinically meaningful. For GAD, the SMDs do suggest that the antidepressant–placebo difference is relatively small for patients with a baseline severity score of 15 or less. Even without a definite cutoff point, though, it is clear that the risk‐benefit ratio for GAD and PD becomes less favorable as initial severity decreases. It is therefore imperative that clinicians transparently discuss the expected benefits of antidepressants with patients with mild to moderate symptoms, who constitute the majority of patients in primary care (Rollman et al., 2005).
There was no evidence for a relationship between initial severity and antidepressant efficacy for SAD, OCD, and PTSD. Nevertheless, other factors, such as anticipated course, patient preferences, and the acceptability and efficacy of alternative treatments, could still lead to different prescribing decisions for mild versus severe disorders, even in the absence of differential efficacy.
5. CONCLUSIONS
We found that antidepressants are equally effective across the severity range generally included in clinical trials for SAD, OCD, and PTSD. For GAD and PD, however, the benefits of antidepressants over and above placebo are small at low severity, and the trade‐off between benefits and risks may therefore be unfavorable for these patients.
CONFLICT OF INTEREST
All authors have declared that they have no conflict of interest.
Supporting information
Supporting Information
de Vries YA, Roest AM, Burgerhof JGM, de Jonge P. Initial severity and antidepressant efficacy for anxiety disorders, obsessive‐compulsive disorder, and posttraumatic stress disorder: an individual patient data meta‐analysis. Depress Anxiety. 2018;35:515–522. 10.1002/da.22737
Ethics statement: This study did not require ethical approval.
Registration: NCT02476136
REFERENCES
- Ackerman, D. L. , & Greenland, S. (2002). Multivariate meta‐analysis of controlled drug studies for obsessive‐compulsive disorder. Journal of Clinical Psychopharmacology, 22(3), 309–317. [DOI] [PubMed] [Google Scholar]
- Altman, D. G. , & Royston, P. (2006). The cost of dichotomising continuous variables. British Medical Journal, 332(7549), 1080–1080. 10.1136/bmj.332.7549.1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelow, B. , Reitt, M. , Röver, C. , Michaelis, S. , Görlich, Y. , & Wedekind, D. (2015). Efficacy of treatments for anxiety disorders: A meta‐analysis. International Clinical Psychopharmacology, 30(4), 183–192. 10.1097/YIC.0000000000000078 [DOI] [PubMed] [Google Scholar]
- Curtiss, J. , Andrews, L. , Davis, M. , Smits, J. , & Hofmann, S. G. (2017). A meta‐analysis of pharmacotherapy for social anxiety disorder: An examination of efficacy, moderators, and mediators. Expert Opinion on Pharmacotherapy, 18(3), 243–251. 10.1080/14656566.2017.1285907 [DOI] [PubMed] [Google Scholar]
- Davis, M. L. , Smits, J. A. , & Hofmann, S. G. (2014). Update on the efficacy of pharmacotherapy for social anxiety disorder: A meta‐analysis. Expert Opinion on Pharmacotherapy, 15(16), 2281–2291. 10.1517/14656566.2014.955472 [DOI] [PubMed] [Google Scholar]
- de Vries, Y. A. , de Jonge, P. , van den Heuvel, E. , Turner, E. H. , & Roest, A. M. (2016). Influence of baseline severity on antidepressant efficacy for anxiety disorders: Meta‐analysis and meta‐regression. British Journal of Psychiatry, 208(6), 515–521. 10.1192/bjp.bp.115.173450 [DOI] [PubMed] [Google Scholar]
- Fournier, J. C. , Derubeis, R. J. , Hollon, S. D. , Dimidjian, S. , Amsterdam, J. D. , Shelton, R. C. , & Fawcett, J. (2010). Antidepressant drug effects and depression severity. Journal of the American Medical Association, 303(1), 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons, R. D. , Hur, K. , Brown, C. H. , Davis, J. M. , & Mann, J. J. (2012). Benefits from antidepressants: Synthesis of 6‐week patient‐level outcomes from double‐blind placebo‐controlled randomized trials of fluoxetine and venlafaxine. Archives of General Psychiatry, 69(6), 572–579. 10.1001/archgenpsychiatry.2011.2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler, R. C. , Chiu, W. T. , Demler, O. , & Walters, E. E. (2005). Prevalence, severity, and comorbidity of 12‐month DSM‐IV disorders in the national comorbidity survey replication. Archives of General Psychiatry, 62, 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, A. , Bhat, A. , Faucett, J. , Kolts, R. L. , & Brown, W. A. (2011). Antidepressant‐placebo differences in 16 clinical trials over 10 years at a single site: Role of baseline severity. Psychopharmacology, 214(4), 961–965. 10.1007/s00213-010-2107-1 [DOI] [PubMed] [Google Scholar]
- Khan, A. , Brodhead, A. E. , Kolts, R. L. , & Brown, W. A. (2005). Severity of depressive symptoms and response to antidepressants and placebo in antidepressant trials. Journal of Psychiatric Research, 39(2), 145–150. 10.1016/j.jpsychires.2004.06.005 [DOI] [PubMed] [Google Scholar]
- Khan, A. , Leventhal, R. M. , Khan, S. R. F. , & Brown, W. A. (2002). Severity of depression and response to antidepressants and placebo: An analysis of the Food and Drug Administration database. Journal of Clinical Psychopharmacology, 22(1), 40–45. [DOI] [PubMed] [Google Scholar]
- Kirsch, I. , Deacon, B. J. , Huedo‐Medina, T. B. , Scoboria, A. , Moore, T. J. , & Johnson, B. T. (2008). Initial severity and antidepressant benefits: A meta‐analysis of data submitted to the food and drug administration. PLoS Medicine, 5(2), e45 10.1371/journal.pmed.0050045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer, H. , Morgan, G. A. , Leech, N. , Gliner, J. A. , Vaske, J. J. , & Harmon, R. J. (2003). Measures of clinical significance. The American Academy of Child and Adolescent Psychiatry, 42(12), 1524–1529. 10.1097/01.chi.0000091507.46853.d1 [DOI] [PubMed] [Google Scholar]
- Krueger, R. F. (1999). The structure of common mental disorders. Archives of General Psychiatry, 56(10), 921–926. 10.1001/archpsyc.56.10.921 [DOI] [PubMed] [Google Scholar]
- Montgomery, S. A. (1998). Implications of the severity of social phobia. Journal of Affective Disorders, 50 (Suppl 1), S17–S22. [DOI] [PubMed] [Google Scholar]
- Montgomery, S. A. , McIntyre, A. , Osterheider, M. , Sarteschi, P. , Zitterl, W. , Zohar, J. , … Wood, A. J. (1993). A double‐blind, placebo‐controlled study of fluoxetine in patients with DSM‐III‐R obsessive‐compulsive disorder. The Lilly European OCD Study Group. European Neuropsychopharmacology, 3(2), 143–152. [DOI] [PubMed] [Google Scholar]
- Montgomery, S. A. , Sheehan, D. V. , Meoni, P. , Haudiquet, V. , & Hackett, D. (2002). Characterization of the longitudinal course of improvement in generalized anxiety disorder during long‐term treatment with venlafaxine XR. Journal of Psychiatric Research, 36(4), 209–217. Retrieved from https://doi.org/S0022395602000055 [DOI] [PubMed] [Google Scholar]
- National Institute for Health and Care Excellence . (2009). Depression in adults: The treatment and management of depression in adults (CG90), London: Author. [PubMed] [Google Scholar]
- Oehrberg, S. , Christiansen, P. E. , Behnke, K. , Borup, A. L. , Severin, B. , Soegaard, J. , … Manniche, P. M. (1995). Paroxetine in the treatment of panic disorder. A randomised, double‐blind, placebo‐controlled study. British Journal of Psychiatry, 167, 374–379. 10.1192/bjp.167.3.374 [DOI] [PubMed] [Google Scholar]
- Pae, C.‐U. , Wang, S.‐M. , Han, C. , Lee, S.‐J. , Patkar, A. A. , Masand, P. S. , & Serretti, A. (2015). Vortioxetine, a multimodal antidepressant for generalized anxiety disorder: A systematic review and meta‐analysis. Journal of Psychiatric Research, 64, 88–98. 10.1016/j.jpsychires.2015.02.017 [DOI] [PubMed] [Google Scholar]
- Pollack, M. H. , Meoni, P. , Otto, M. W. , Simon, N. , & Hackett, D. (2003). Predictors of outcome following venlafaxine extended‐release treatment of DSM‐IV generalized anxiety disorder: A pooled analysis of short‐ and long‐term studies. Journal of Clinical Psychopharmacology, 23(3), 250–259. 10.1097/01.jcp.0000084025.22282.84 [DOI] [PubMed] [Google Scholar]
- Qaseem, A. , Barry, M. J. , & Kansagara, D. (2016). Nonpharmacologic versus pharmacologic treatment of adult patients with major depressive disorder: A clinical practice guideline from the American College of Physicians. Annals of Internal Medicine, 164, 350–359. 10.7326/M15-2570 [DOI] [PubMed] [Google Scholar]
- Rabinowitz, J. , Werbeloff, N. , Mandel, F. S. , Menard, F. , Marangell, L. , & Kapur, S. (2016). Initial depression severity and response to antidepressants v. placebo: Patient‐level data analysis from 34 randomised controlled trials. The British Journal of Psychiatry, 209(5), 427–428. 10.1192/bjp.bp.115.173906 [DOI] [PubMed] [Google Scholar]
- Roest, A. M. , de Jonge, P. , Williams, C. D. , de Vries, Y. A. , Schoevers, R. A. , & Turner, E. H. (2015). Reporting bias in clinical trials investigating the efficacy of second‐generation antidepressants in the treatment of anxiety disorders: A report of 2 meta‐analyses. JAMA Psychiatry, 72(5), 500–510. 10.1001/jamapsychiatry.2015.15 [DOI] [PubMed] [Google Scholar]
- Rollman, B. L. , Belnap, B. H. , Mazumdar, S. , Zhu, F. , Kroenke, K. , Schulberg, H. C. , & Katherine Shear, M. (2005). Symptomatic severity of PRIME‐MD diagnosed episodes of panic and generalized anxiety disorder in primary care. Journal of General Internal Medicine, 20(7), 623–628. 10.1111/j.1525-1497.2005.0120.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shear, M. K. , Brown, T. A. , Barlow, D. H. , Money, R. , Sholomskas, D. E. , Woods, S. W. , … Papp, L. A. (1997). Multicenter collaborative panic disorder severity scale. American Journal of Psychiatry, 154, 1571–1575. [DOI] [PubMed] [Google Scholar]
- Soomro, G. M. , Altman, D. G. , Rajagopal, S. , & Oakley Browne, M. (2008). Selective serotonin re‐uptake inhibitors (SSRIs) versus placebo for obsessive compulsive disorder (OCD). Cochrane Database of Systematic Reviews, 1, CD001765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spijker, J. , Bockting, C. L. H. , Meeuwissen, J. A. C. , van Vliet, I. M. , Emmelkamp, P. M. G. , Hermens, M. L. M. , & van Balkom, A. J. L. M. (2013). Multidisciplinaire richtlijn depressie (3e revisie), Utrecht: Trimbos‐instituut. [Google Scholar]
- Stein, D. J. , Ipser, J. C. , & Seedat, S. (2006). Pharmacotherapy for post traumatic stress disorder (PTSD). Cochrane Database of Systematic Reviews, 1, CD002795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, D. J. , Kasper, S. , Andersen, E. W. , Nil, R. , & Lader, M. (2004). Escitalopram in the treatment of social anxiety disorder: Analysis of efficacy for different clinical subgroups and symptom dimensions. Depression and Anxiety, 20(4), 175–181. 10.1002/da.20043 [DOI] [PubMed] [Google Scholar]
- Stein, D. J. , Stein, M. B. , Goodwin, W. , Kumar, R. , & Hunter, B. (2001). The selective serotonin reuptake inhibitor paroxetine is effective in more generalized and in less generalized social anxiety disorder. Psychopharmacology, 158(3), 267–272. 10.1007/s002130100880 [DOI] [PubMed] [Google Scholar]
- Sugarman, M. A. , Loree, A. M. , Baltes, B. B. , Grekin, E. R. , & Kirsch, I. (2014). The efficacy of paroxetine and placebo in treating anxiety and depression: A meta‐analysis of change on the Hamilton rating scales. PLoS One, 9(8), e106337 10.1371/journal.pone.0106337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, S. G. , & Higgins, J. P. T. (2002). How should meta‐regression analyses be undertaken and interpreted? Statistics in Medicine, 21(11), 1559–1573. 10.1002/sim.1187 [DOI] [PubMed] [Google Scholar]
- Wong, J. , Motulsky, A. , Eguale, T. , Buckeridge, D. L. , Abrahamowicz, M. , & Tamblyn, R. (2016). Treatment indications for antidepressants prescribed in primary care in Quebec, Canada, 2006–2015. Journal of the American Medical Association, 315(20), 2230–2231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information