

Abstract
Since 2005, more than 40 new medicines for the treatment of type 2 diabetes have been introduced on the market. These consist of 15 new active substances establishing three new classes of non‐insulin products, and several new or modified insulin products and combinations. The approval of these products in Europe is regulated via the centralized procedure at the European Medicines Agency. Demonstration of benefit with regard to improved glucose control remains the principal outcome required from confirmatory studies to demonstrate efficacy. For the majority of these new medicines approved since 2005, cardiovascular outcome trials have now been completed, and have invariably supported the cardiovascular safety of these products. In some of these trials additional important benefits have been observed, for instance, a reduction in major adverse cardiovascular events and improvement of renal outcome. The existing regulatory framework and the continuous adaption of regulatory requirements to emerging developments will continue to guide the approval of new products in the future.
Keywords: antidiabetic drug, drug development, systematic review
1. INTRODUCTION
Diabetes mellitus affects about 7% of the population in the European Union (EU)1; its increase in prevalence over recent decades is largely attributable to type 2 diabetes (T2D). Recognizing the importance of this disease, the approval of human medicines containing a new active substance for the treatment of diabetes in the EU was made compulsory by European legislation in 2005 via one single, centralized application procedure at the European Medicines Agency (EMA). One of the advantages of this procedure is that it is based on a single EU‐wide assessment and the marketing authorization becomes valid throughout the EU member states. Once pharmaceutical companies submit an authorization application for their product to EMA, the Agency's Committee for Medicinal Products for Human Use (CHMP) carries out a scientific assessment of the application, typically including several review cycles which may not exceed 210 days. The CHMP then gives a recommendation to the European Commission on whether or not to grant a marketing authorization, on average about 1 year after submission of the initial application. Each outcome is described in detail in a European Public Assessment Report (EPAR), available on EMA's website (http://www.ema.europa.eu). This article provides an overview of the approvals of such medicines via this centralized procedure and highlights current and future regulatory trends in T2D.
2. APPROVALS OF GLUCOSE‐LOWERING AGENTS BEFORE 2005
Before the EMA's establishment in 1995, only insulins, biguanides, sulphonylureas and alpha‐glucosidase inhibitors were authorized as glucose‐lowering agents in member states of the EU. Between 1995 and 2005, marketing authorizations for new products for the treatment of diabetes could be obtained both via applications in EU member states or via EMA's centralized procedure. The products evaluated via EMA during that time are listed in Table S1 (see the supporting information for this article) and include insulins and insulin analogues, and, as oral glucose‐lowering agents, thiazolidinediones and meglitinides.
3. APPROVALS SINCE 2005
Since the centralized application procedure was made compulsory for medicinal products for the treatment of diabetes in 2005, more than 40 new products have been reviewed by EMA and subsequently authorized in the EU (Figure 1, Table S2). These include both monotherapies and an increasing number of fixed dose combinations, either as oral tablets or, more recently, as fixed‐ratio combinations of injectable medicines (see section 3.2 on insulins); this trend reflects the need for the combination of several agents when the disease progresses.
Figure 1.
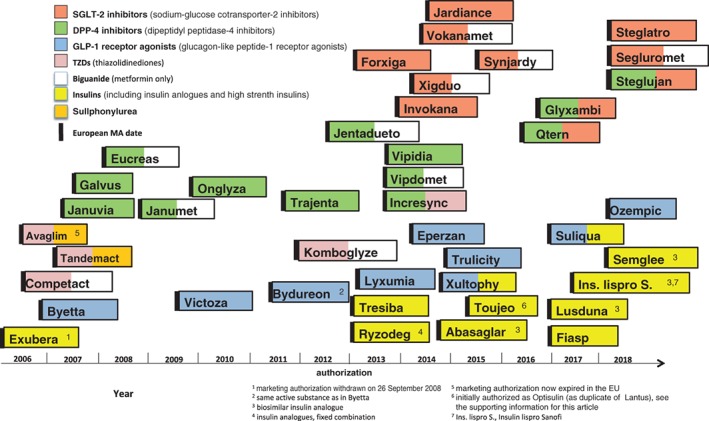
Approval of new products for the treatment of type 2 diabetes since 2005
3.1. Non‐insulin glucose‐lowering agents
Three new classes have been introduced since 2005, the glucagon‐like peptide‐1 (GLP‐1) receptor agonists, the dipeptidyl peptidase‐4 (DPP‐4) inhibitors, and the sodium‐glucose cotransporter‐2 (SGLT‐2) inhibitors. These three new classes comprise 15 new active substances approved in the EU (Figure 2).
Figure 2.
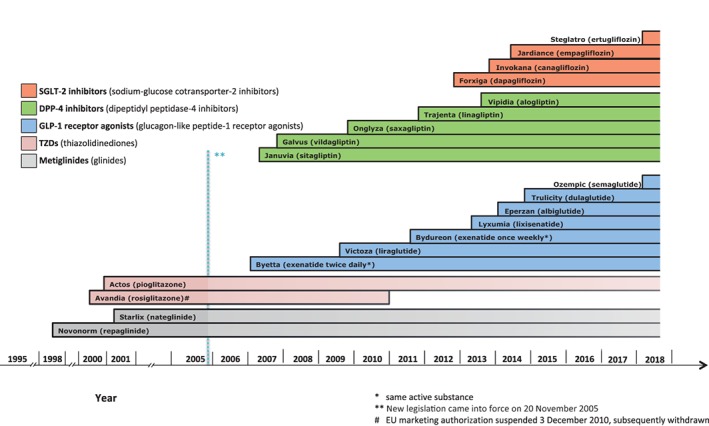
Approval of new active substances (non‐insulin only) for the treatment of type 2 diabetes
The first such product approved in the EU was exenatide in 2006, a GLP‐1 receptor agonist developed as a twice‐daily subcutaneous formulation. Thereafter, five more products of that class have been authorized, most recently semaglutide in 2018. In addition to lowering glucose, these products showed reductions in body weight, which is important in the treatment of patients with T2D. One of the products, liraglutide, approved for treatment of T2D with a maximal dose of 1.8 mg/d, has been further developed for body weight management in patients without T2D with a maintenance dose of 3 mg/d.
DPP‐4 inhibitors were the next new class to become available in the EU, with the approval of five products between 2007 and 2013. Development of a new DPP‐4 inhibitor intended for once‐weekly use and tested in phase 3 trials—omarigliptin—was discontinued in 2016. Even though this class of drugs in general appears to have a lower glucose‐lowering capacity compared to the class of GLP‐1 receptor agonists, they are useful in some patients due to the low risk of hypoglycaemia.
The most recent new class consists of the SGLT‐2 inhibitors, four of which have been approved between 2012 and 2018. This class also has a body weight‐lowering capacity in addition to lowering glucose.
All non‐insulin products are currently approved only for the treatment of T2D. However, studies of SGLT‐1/SGLT‐2 inhibitors2 and SGLT‐2 inhibitors, investigating their use as add‐ons to insulin in the treatment of type 1 diabetes (T1D), have recently been completed.
The emergence of these new products significantly expanded the spectrum of available treatment options for T2D. This is reflected in the inclusion of these three new classes in the current treatment algorithms for T2D in a joint position paper by EASD and ADA,3 in contrast to the publication of an earlier paper in 2006.4 Cardiovascular (CV) outcomes observed with these products are covered later in section 5; comparative reviews of these new classes are beyond the scope of this paper, but are available elsewhere (e.g.5, 6, 7).
3.2. Insulins
Most of the insulins and insulin analogues currently authorized in the EU were already available before 2005. Fast‐acting, mealtime insulin analogues were first authorized in 1996 (insulin lispro), and long‐acting, basal insulin analogues in 2000 (insulin glargine) (Table S1). All insulin products in the EU are authorized for use in both T1D and T2D. Recently, a new group of injectable, fixed‐dose combinations of long‐acting insulins with GLP‐1 receptor agonists has been assessed by EMA and subsequently authorized (Figure 1); these new combination products offer a lower body weight gain and a beneficial profile regarding hypoglycaemic events compared to high doses of insulin on its own.
Another recent development is the approval of higher strength pre‐filled pen insulins, exceeding the 100 U/mL strength that was previously in general the only one available in the EU. Both higher strength insulins and fixed‐dose combinations containing insulin may present added challenges in terms of potential risks of medication errors, the mitigation of which was addressed in a recent guideline dedicated to this matter.8
The choice of insulin analogues has been broadened by the EU approval of four biosimilar insulin analogues since 2014 (Figure 1). The EMA has issued specific guidance for the development of biosimilar insulins and insulin analogues.9 While an inhalable insulin was approved in 2006 (Exubera), the product was withdrawn shortly after licensing for commercial reasons (Figure 1, Table S2), and therefore no insulin with non‐parenteral delivery is currently authorized in the EU. Further development of new administration routes, new insulin analogues, and non‐insulin peptide agonists of the insulin receptor, is ongoing.10
4. GENERAL REGULATORY ASPECTS OF APPROVALS FOR MEDICINES FOR DIABETES
Since 2005, the average time from filing an application to receiving the opinion given by EMA's CHMP was 377 days, which is close to the average for other medicines. For three out of four diabetes product applications since 2005, the applicants obtained prior specific EMA Scientific Advice on at least one aspect of the development programme. Since 2005, only one application, for a biosimilar insulin, was refused a marketing authorization (Table S2).
In 2002, the EMA published a guideline for the development of medicines for diabetes. The guideline11 has been revised previously and is currently being revised again to reflect recent developments.12, 13 Guidance by both the EMA and the US Food and Drug Administration (US FDA) stipulates HbA1c (as a measure of glucose control) as the main primary outcome parameter for pivotal trials for the approval of new medicines for the treatment of diabetes.11, 14 This is a reflection of the expected long‐term benefit of glucose control on vascular outcomes as the main component of the disease burden in diabetes. While there is general agreement that improved glucose control is linked to a reduction in microvascular events, the long‐term benefit with regard to macrovascular events remains uncertain. However, recently completed cardiovascular outcome trials (CVOTs) are starting to provide a better understanding of that relationship (see later).
Beyond HbA1c as a primary outcome, other outcomes are also of importance to patients, and these may in the future help to further refine the profile of diabetes medicines addressing the needs of patients, for instance, quality of life captured as patient‐reported outcomes, or periods of hypoglycaemia, captured by continuous glucose‐monitoring .15
5. CARDIOVASCULAR OUTCOME TRIALS IN TYPE 2 DIABETES
General adaptation to emerging knowledge over time, but in particular concerns over the CV safety profile of rosiglitazone, which had its marketing authorization suspended16 and was subsequently withdrawn in the EU, led to a revision of guidance relating to the safety data required for regulatory purposes.17, 18 Current EU guidance requires comprehensive safety data, including a comprehensive assessment of the CV safety of a new medicine developed for the treatment of diabetes.19 The guidance explains that CVOTs are not mandatory for such products in the EU but that they can be required. Nevertheless, for most of the new active substances for T2D reviewed since 2005, CVOTs have been completed or are ongoing, due to applicants' globally defined development programmes based on requirements stipulated by the US FDA.
Several of these CVOTs, having been designed to exclude any detrimental effect on CV safety, demonstrated non‐inferiority compared to placebo with respect to the incidence of major adverse CV events (MACE, a composite of non‐fatal myocardial infarction, non‐fatal stroke, and CV death): this was the case for all three of the five authorized DPP‐4 inhibitors, for which such data are already available.20, 21, 22 These trials also provided new data regarding known and (in some cases) new safety concerns (e.g. the occurrence of heart failure in the case of certain DPP‐4 inhibitors). Beyond their stated primary goal, i.e. to expand the knowledge of the CV safety profiles of these medicines, data from such trials have, to some extent, contributed to gaining further information on their safety profiles in general (e.g. on pancreatic safety), confirming regulatory data obtained earlier.23
For four of the authorized GLP‐1 receptor agonists, CVOTs have been completed,24, 25, 26, 27 and in two cases (liraglutide and semaglutide) these showed a reduction of at least some components of MACE in selected populations with T2D and a high CV risk compared to placebo.26, 27 Similarly, for two of the SGLT‐2 inhibitors (empagliflozin and canagliflozin), CVOTs have been completed recently, also showing a reduction of at least some components of MACE.28, 29 The data from the CVOTs of empagliflozin and liraglutide were the first of their respective classes to be assessed by CHMP, and their product information has been updated accordingly.
Beyond macrovascular outcomes, these CVOTs have provided additional observations of outcomes for organ damage associated with T2D, such as kidney (renal insufficiency), heart (heart failure), and eye (diabetic retinopathy). Beneficial effects have been shown in terms of delayed progression of albuminuria, and lower rates of new and worsening nephropathy have been shown in CVOTs in examples from both GLP‐1 agonist and SGLT‐2 inhibitor classes.26, 27, 30 As potential benefits for those organs are not necessarily mediated exclusively by glucose‐lowering effects, and as mechanisms of action differ between the various classes, further studies are needed to unravel these mechanisms and justify more specific claims. Also, unexpected effects in terms of rates of retinopathy complications have been observed,27 also requiring further studies.
6. MODIFICATION OF USE OF APPROVED MEDICINES FOR TYPE 2 DIABETES
All medicines for T2D approved in the EU are constantly monitored with regard to safety. Since 2012, EMA has a committee dedicated to the safety of medicines for human use, the Pharmacovigilance Risk Assessment Committee (PRAC), where all major safety issues are discussed. As an example, this has led to the inclusion of the occurrence of keto‐acidosis as a side effect of SGLT‐2 inhibitors. All suspected side effects that are reported by patients and healthcare professionals must be entered into EudraVigilance, the EU web‐based information system operated by EMA, that collects, manages and analyzes reports of suspected side effects of medicines.
New information can also lead to expansion of use. In a recent important case of regulatory action on metformin, new clinical data, epidemiological studies, clinical guidelines from medical bodies and scientific literature were considered, and led the EMA's scientific committee CHMP to conclude that metformin's permitted use in T2D should be expanded to patients with moderately reduced renal function (GFR [glomerular filtration rate] = 30‐59 mL/min).31 As an outcome of this referral procedure, the product information of all metformin‐containing products for the treatment of T2D in the EU was revised, including clear dosing recommendations and advice on the monitoring of these patients.
7. FUTURE PERSPECTIVES
Over the last 10 years a large number of new medicines and major new product classes for diabetes have been approved in the EU, significantly widening the therapeutic options now available for the treatment of T2D. Increased safety monitoring has led to new insights. Continuing innovation of these existing product classes hopefully complements preventive measures and non‐medical interventions in the short term, further improving the outcome of patients with T2D. Also, better insights into the mechanisms of action obtained by information from additional trials with existing drugs may provide further guidance in their optimal use.
While a direct treatment of the root cause of this complex disease is not yet possible, the potential of some molecules to modify fuel metabolism as well as centrally controlling satiety and hunger signals can be increasingly exploited, and the continuously emerging knowledge of the role of gut hormones may give rise to additional new therapeutic options for T2D. The existing regulatory framework and continually adapted EU guidance for clinical development of those products will hopefully continue to help bring to fruition those new therapies in the future.
Supporting information
Table S1 New products for the treatment of diabetes approved between 1995 and 2005 (via EMA's centralised procedure only*).
Table S2 Approval of new products for the treatment of diabetes in the European Union since 2005*.
ACKNOWLEDGMENTS
The authors wish to thank Caroline Snacke and Anthony O'Shea for assembling data, and Mia van Petegem and Benjamin Pelle for valuable comments.
Conflict of interest
The authors declare no competing interests.
1. Disclaimer
The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of the European Medicines Agency or one of its committees or working parties.
Blind E, Janssen H, Dunder K, de Graeff PA. The European Medicines Agency's approval of new medicines for type 2 diabetes. Diabetes Obes Metab. 2018;20:2059–2063. 10.1111/dom.13349
REFERENCES
- 1. OECD/EU . Health at a Glance: Europe 2016 – State of Health in the EU Cycle. Paris: OECD Publishing; 2016. [Google Scholar]
- 2. Garg SK, Henry RR, Banks P, et al. Effects of Sotagliflozin added to insulin in patients with type 1 diabetes. N Engl J Med. 2017;377:2337‐2348. [DOI] [PubMed] [Google Scholar]
- 3. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycaemia in type 2 diabetes, 2015: a patient‐centred approach. Update to a position statement of the American Diabetes Association and the European Association for the Study of diabetes. Diabetologia. 2015;58:429‐442. [DOI] [PubMed] [Google Scholar]
- 4. Nathan DM, Buse JB, Davidson MB, et al. Management of hyperglycaemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy. A consensus statement from the American Diabetes Association and the European Association for the Study of diabetes. Diabetologia. 2006;49:1711‐1721. [DOI] [PubMed] [Google Scholar]
- 5. Deacon CF, Lebovitz HE. Comparative review of dipeptidyl peptidase‐4 inhibitors and sulphonylureas. Diabetes Obes Metab. 2016;18:333‐347. [DOI] [PubMed] [Google Scholar]
- 6. Nauck M. Incretin therapies: highlighting common features and differences in the modes of action of glucagon‐like peptide‐1 receptor agonists and dipeptidyl peptidase‐4 inhibitors. Diabetes Obes Metab. 2016;18:203‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zaccardi F, Webb DR, Htike ZZ, Youssef D, Khunti K, Davies MJ. Efficacy and safety of sodium‐glucose co‐transporter‐2 inhibitors in type 2 diabetes mellitus: systematic review and network meta‐analysis. Diabetes Obes Metab. 2016;18:783‐794. [DOI] [PubMed] [Google Scholar]
- 8. EMA . Risk minimisation strategy for high‐strength and fixed‐combination insulin products; 2015. http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2015/11/WC500196980.pdf. Accessed March 31, 2018.
- 9. EMA . Guideline on non‐clinical and clinical development of similar biological medicinal products containing recombinant human insulin and insulin analogues; 2015. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/03/WC500184161.pdf. Accessed March 31, 2018.
- 10. Zaykov AN, Mayer JP, DiMarchi RD. Pursuit of a perfect insulin. Nat Rev Drug Discov. 2016;15:425‐439. [DOI] [PubMed] [Google Scholar]
- 11. EMA . Guideline on clinical investigation of medicinal products in the treatment or prevention of diabetes mellitus; 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500129256.pdf. Accessed March 31, 2018.
- 12. EMA . Draft concept paper on the need for revision of the guideline on clinical investigation of medicinal products in the treatment or prevention of diabetes mellitus; 2016. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/07/WC500211234.pdf. Accessed March 31, 2018.
- 13. EMA . Draft Guideline on clinical investigation of medicinal products in the treatment or prevention of diabetes mellitus; 2018. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2018/02/WC500243464.pdf. Accessed March 31, 2018.
- 14. US FDA . Guidance for industry. Diabetes mellitus: developing drugs and therapeutic biologics for treatment and prevention ‐ draft; 2008. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071624.pdf. Accessed March 31, 2018.
- 15. Kingwell K. FDA eyes new diabetes end points. Nat Rev Drug Discov. 2016;15:666‐667. [DOI] [PubMed] [Google Scholar]
- 16. Blind E, Dunder K, de Graeff PA, Abadie E. Rosiglitazone: a European regulatory perspective. Diabetologia. 2011;54:213‐218. [DOI] [PubMed] [Google Scholar]
- 17. EMA . Concept paper on the need for revision of the note for guidance on clinical investigation of medicinal products in the treatment of diabetes mellitus; 2008. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003182.pdf. Accessed March 31, 2018.
- 18. US FDA . Guidance for industry. Diabetes mellitus — evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 Diabetes; 2008. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071627.pdf. Accessed March 31, 2018.
- 19. EMA . Reflection paper on assessment of cardiovascular safety profile of medicinal products; 2016. http://www.ema.europa/docs/en_GB/document_library/Scientific_guideline/2016/03/WC500203804.pdf. Accessed March 31, 2018.
- 20. Green JB, Bethel MA, Armstrong PW, et al. Effect of Sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373:232‐242. [DOI] [PubMed] [Google Scholar]
- 21. Scirica BM, Bhatt DL, Braunwald E, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317‐1326. [DOI] [PubMed] [Google Scholar]
- 22. White WB, Cannon CP, Heller SR, et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327‐1335. [DOI] [PubMed] [Google Scholar]
- 23. Egan AG, Blind E, Dunder K, et al. Pancreatic safety of incretin‐based drugs‐‐FDA and EMA assessment. N Engl J Med. 2014;370:794‐797. [DOI] [PubMed] [Google Scholar]
- 24. Holman RR, Bethel MA, Mentz RJ, et al. Effects of once‐weekly Exenatide on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2017;377:1228‐1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pfeffer MA, Claggett B, Diaz R, et al. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med. 2015;373:2247‐2257. [DOI] [PubMed] [Google Scholar]
- 26. Marso SP, Daniels GH, Brown‐Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marso SP, Bain SC, Consoli A, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834‐1844. [DOI] [PubMed] [Google Scholar]
- 28. Neal B, Perkovic V, Mahaffey KW, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644‐657. [DOI] [PubMed] [Google Scholar]
- 29. Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117‐2128. [DOI] [PubMed] [Google Scholar]
- 30. Wanner C, Inzucchi SE, Lachin JM, et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med. 2016;375:323‐334. [DOI] [PubMed] [Google Scholar]
- 31. EMA . Assessment report. Referral under Article 31 of Directive 2001/83/EC ‐ Metformin‐containing medicinal products; 2016. http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/Metformin_31/WC500218638.pdf. Accessed March 31, 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 New products for the treatment of diabetes approved between 1995 and 2005 (via EMA's centralised procedure only*).
Table S2 Approval of new products for the treatment of diabetes in the European Union since 2005*.