

Abstract
Inhalational anthrax is a highly lethal infection caused by Bacillus anthracis and a serious bioterrorism threat. Protective antigen (PA) is a critical component required for the virulence of Bacillus anthracis. Obiltoxaximab, a high‐affinity monoclonal antibody that neutralizes PA, is approved in the United States for intravenous use for the treatment of inhalational anthrax in combination with appropriate antibacterial drugs and for prophylaxis of inhalational anthrax when alternative therapies are not available or appropriate. Here, we explored the safety, pharmacokinetics (PK), and immunogenicity of obiltoxaximab administered by intramuscular injection at doses of 4, 8, 16, 20, and 24 mg/kg in healthy humans. Systemic exposures were approximately dose proportional, maximum serum concentrations were observed after 6–9 days, and terminal half‐life ranged from 16 to 23 days. Average absolute intramuscular bioavailability was 64%. Obiltoxaximab was well tolerated, and local tolerability was acceptable up to 24 mg/kg intramuscularly, up to 6 injections per dose, and up to 5 mL per injection. No injection‐site abscesses or hypersensitivity reactions occurred; no subjects developed treatment‐emergent antitherapeutic antibodies over the study period of 71 days.
Keywords: anthrax, obiltoxaximab; intramuscular; pharmacokinetics; safety
Inhalational anthrax is a highly lethal infection caused by Bacillus anthracis, a gram‐positive aerobic, encapsulated, endospore‐forming, rod‐shaped bacterial pathogen1, 2 and a serious bioterrorism threat.3, 4, 5, 6 B. anthracis has been identified as a top priority biowarfare target by the Department of Defense and a category A agent (ie, agents that pose the highest risk to the public and national security because they are easily spread and cause severe illness or death) by the Centers for Disease Control and Prevention. B. anthracis spores can survive for decades in soil and can be used in aerosol form as a biological weapon.3, 4, 7
Anthrax can manifest as cutaneous, gastrointestinal, inhalational or injection‐related infections, depending on the route of exposure, with the inhalational form having a fatality rate of ∼50% even under optimal treatment conditions.3, 4, 5, 6 In humans, inhalational anthrax has a typical incubation period of 1 to 6 days; the mean time to death after onset of symptoms is 3 days in the absence of immediate antibiotic therapy.8
Obiltoxaximab (Anthim) is a novel chimeric therapeutic IgG1 monoclonal antibody (∼148 kDa), produced via cultures of stably transfected nonsecreting GS‐NS0 myeloma cells, that binds and neutralizes Bacillus anthracis protective antigen (PA), a critical component required for the virulence of B. anthracis. Obiltoxaximab was approved in the United States for the treatment of adult and pediatric patients with inhalational anthrax because of B. anthracis in combination with appropriate antibacterial drugs and for prophylaxis of inhalational anthrax when alternative therapies are not available or are not appropriate. The approved dosage and mode of administration of obiltoxaximab is as a single intravenous 16 mg/kg infusion over 90 minutes.
Obiltoxaximab was developed under the US Food and Drug Administration's Animal Rule regulation (21 CFR 601.90), which is specifically intended for agents for which the conduct of definitive human efficacy studies is not ethical or feasible, as in the case of agents for the treatment of anthrax.9 The obiltoxaximab intravenous dose of 16 mg/kg was selected and justified based on efficacy data in animals and pharmacokinetic (PK) data in animals and healthy human volunteers, as well as population modeling of PK and efficacy data.10, 11, 12
Efficacy following intramuscular administration of obiltoxaximab for pre‐ and postexposure prophylaxis of inhalational anthrax has previously been demonstrated in animals.13 In this study we investigated the safety, PK, and immunogenicity of obiltoxaximab following intramuscular administration to healthy adult subjects.
Methods
Study Design
The study was conducted at Covance Clinical Research Unit, Inc. (Dallas, Texas) between July 26, 2012, and July 3, 2014, in accordance with Good Clinical Practice, the ethical principles that have their origin in the Declaration of Helsinki, the Belmont Report, Title 21 of the Code of Federal Regulations (CFR; Parts 50, 56, and 312), Title 45 of the CFR (Part 46), the International Conference on Harmonisation (E6), and any applicable regulatory requirements. The study protocol, including all amendments, was approved by the investigational review board (Schulman Associates IRB, Inc.), and written informed consent was obtained from each subject prior to performing any screening procedures.
This was a randomized, double‐blind, placebo‐controlled single dose‐escalation study. Thirty‐six healthy adult subjects were enrolled in 5 cohorts (cohort 1, 4 subjects; cohorts 2–5, 8 subjects per cohort) and randomized on day 1 in a 3:1 ratio to receive an intramuscular dose of either 4, 8, 12, 16, or 24 mg/kg obiltoxaximab or a matching placebo.
During the conduct of the study the protocol was amended to include pretreatment with 50 mg of oral diphenhydramine 30 minutes prior to study drug administration. This was based on preliminary results from clinical trials in healthy subjects administered obiltoxaximab intravenously, in which some subjects experienced hypersensitivity reactions during or shortly after the infusion of obiltoxaximab.11
Healthy adult men and nonpregnant women of any race ≥ 18 years of age with a body weight ≤ 100 kg and a body mass index < 32 kg/m2 without clinically significant comorbidities or test results were included in this study. Other significant exclusion criteria included a history of allergic or hypersensitivity reactions to other therapeutic antibodies or immunoglobulins; prior immunization with any approved or investigational anthrax vaccine or anthrax treatment (eg, raxibacumab, anthrax immune globulin); poor muscle mass; a personal or family history of a bleeding disorder or unexplained bleeding; low platelet count; coagulation defects; use of any anticoagulant or antiplatelet drug within 3 months prior to screening; and therapeutic use of systemic steroids, immunosuppressive agents, or antiarrhythmics within 1 year prior to study drug administration. Subjects who did not meet all the inclusion criteria or who met any of the exclusion criteria at screening or on day ‐1 were not eligible for study participation. Waivers for deviations from the eligibility criteria were not granted.
Dosing and Sampling Schedules
Obiltoxaximab (600 mg/6 mL) and matching placebo were supplied as a liquid formulation in sterile single‐dose vials and stored prior to use at 2°C to 8°C. Each milliliter contained l‐histidine (6.2 mg), polysorbate 80 (0.1 mg), and sorbitol (36 mg) with a pH of 5.5. Study drug was prepared by an unblinded pharmacist not involved in the conduct of the study based on the assigned randomization, dose level, and the subject's weight. Study drug vials were removed from the refrigerator approximately 30 minutes prior to dose preparation.
Doses were administered at the clinic on day 1 in a blinded fashion by bilateral injection into the vastus lateralis muscles using a separate syringe with a 21‐gauge, 1.5‐inch needle for each injection, with the subject in a supine position. Multiple injections were required at higher doses, especially in heavier individuals. When 2 or more intramuscular injections were needed, injections were given simultaneously or in quick succession. The number of injections and injection volume increased with subject weight and increasing dose from 2 injections with a maximum volume of 2 mL for the 4 mg/kg dose group to up to 6 injections with a maximum volume of 5 mL at 1 site and maximum volume of 4 mL at the other sites for the 24 mg/kg dose group, allowing for an assessment of the safety of increasing intramuscular obiltoxaximab doses and the tolerability of a larger number of injections and injection volume.
Subjects were discharged from the clinic on day 4 and returned for additional visits on days 7, 10, 15, 29, 43, and 71. After each cohort, a decision whether to dose‐escalate was made in a blinded fashion by the investigator in conjunction with the sponsor based on available safety data. Safety data up to and including day 4 were considered before escalating to the next higher dose.
Blood samples for obiltoxaximab PK analysis were collected predose and 1.5, 4, 8, 24, 36, 48, and 72 hours after the first injection and on days 7, 10, 15, 29, 43, and 71. Blood samples for screening of antitherapeutic antibodies (ATAs) were collected predose and on days 10, 43, and 71. Serum was separated, and PK and ATA serum samples were kept frozen (‐70°C to ‐80°C) until shipped on dry ice and analyzed at Eurofins, St. Charles, Missouri.
Pharmacokinetic Assessments
Serum samples were assayed for free obiltoxaximab concentrations using a validated enzyme‐linked immunosorbent assay method in which PA83 is used as the capture reagent.11 Selectivity was demonstrated in individual lots of normal human serum in the presence of spiked obiltoxaximab at 300 and 3000 ng/mL. Selectivity was also tested in individual lots of normal human serum spiked with an irrelevant antibody; all results were below the assay lower limit of quantification (LLOQ) of 100 ng/mL. The accuracy (relative error) ranged from –2.4% to 4.7%, and the precision (coefficient of variation [CV%]) ranged from 4.4% to 15.2%. There was no interference from diphenhydramine. PK parameters were derived by noncompartmental methods using WinNonlin Professional version 6.2.1. Maximum concentration (Cmax) and time of maximum concentration (Tmax) were observed values. The terminal‐phase rate constant (k) was determined by linear regression of the terminal phase of the log concentration–time profile and half‐life (t1/2) was calculated as ln(2)/k. Area under the concentration–time curve (AUC) was calculated using the trapezoidal method to the last quantifiable concentration (AUC0–last) and AUC extrapolated to infinity (AUC0–inf) was calculated as AUC0–last + Clast/k, where Clast is the last quantifiable concentration. Apparent clearance (CL/F) was calculated as dose divided by AUC0–inf. Values below the limit of quantification were treated as 0 for calculation of descriptive summary statistics. Only subjects from the PK populations in each study were included in the analyses. Any subject who received a partial obiltoxaximab dose or for whom the dosing record was missing was excluded from the PK population.
As intramuscular and intravenous obiltoxaximab data were not available within the same clinical study, intramuscular obiltoxaximab bioavailability in humans was estimated based on a cross‐study evaluation using ratios of mean systemic CL following a 16 mg/kg intravenous dose of obiltoxaximab11 to mean CL/F values for each dose group after intramuscular administration of obiltoxaximab in the current study based on the formula:
Immunogenicity Assessments
ATA detection followed a tiered approach (screening and confirmatory assays) using a validated electrochemiluminescence method.11 Serum samples were first subjected to a minimum required dilution of 1:10. Samples were then acidified to release ATAs from obiltoxaximab complexes, followed by neutralization and capture of the ATAs with biotinylated obiltoxaximab. The biotinylated obiltoxaximab/ATA complex was subsequently bound to streptavidin‐coated plates to immobilize the complex. The samples were washed and reacidified to free captured ATAs. The solution containing the acidified ATAs was added to a MesoScale Discovery (MSD) plate, neutralized, incubated, and then washed, and the detection antibody, ruthenium‐labeled obiltoxaximab, was added. A signal was generated from ruthenylated‐obiltoxaximab antibody on the MSD plate integrated with carbon electrodes after the addition of an MSD read buffer. This qualitative method used rabbit antiobiltoxaximab polyclonal antisera positive controls spiked at low (1:10 000 dilution) and high (1:1000 dilution) concentrations in pooled human serum to monitor the assay. The negative control was nonspiked pooled human sera. Serum samples were assayed for ATAs at an initial dilution of 1:10. Samples that had detectable ATAs in the confirmatory assay at an initial dilution of 1:10 were serially diluted 1:2 and assayed until a negative result was attained. The titer of the most dilute sample yielding a measurable result was recorded as the titer for that point. Subjects were considered to have had an immune reaction if the titer of 1 or more posttreatment samples was ≥4 times higher than baseline in subjects who had detectable ATAs at baseline or ≥1:20 in subjects who had undetectable ATAs (titer < 1:10) at baseline. Postbaseline samples with a titer ≤ 1:20 were not tested further. Positive posttreatment ATA samples with a titer of 1:40 or greater were considered potentially clinically meaningful.
Safety and Tolerability Assessments
Safety assessments included adverse event (AE) monitoring (including skin evaluation for rash), vital signs, clinical laboratory tests (including hematology, serum chemistry, urinalysis, free T3, free T4, thyroid‐stimulating hormone, thyroid antibodies, and creatinine clearance), electrocardiograms, and physical examinations. Intramuscular tolerance was monitored by measurement of serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), and creatine phosphokinase (CPK) concentrations. Injection sites were evaluated before and for 72 hours after drug administration for pain, tenderness, erythema/redness, and induration/swelling using a toxicity grading scale for local tolerability.14 In addition, subjects assessed injection‐site pain by using a 100‐mm visual analog scale (VAS).
Statistics
The safety population consisted of all subjects who received obiltoxaximab or placebo, whether prematurely withdrawn from the study or not. For qualitative variables, the population size (N for sample size and n for available data) and the percentage (of available data) for each class of the variable are presented. Quantitative variables are summarized using descriptive statistics, including n, mean, standard deviation (SD), CV%, median, minimum, and maximum values. Safety data are summarized separately for each obiltoxaximab dose group, a pooled placebo group, and a pooled obiltoxaximab group.
The dose proportionality of the PK parameters, AUC0–inf, AUC0–last, and Cmax, over the administered dose range was investigated by linear regression using the following power model: log (parameter) = a + b × log (dose), where a is the intercept and b is the slope. Dose proportionality was assessed based on whether the 90% confidence interval (CI) constructed for the estimate of b was contained within the interval (0.80–1.25). The power model parameters were estimated using least‐squares regression. A minimum of 3 values per dose had to be available for a given parameter to estimate dose proportionality with the power model. Data analysis was performed using SAS version 9.3.
Results
Subject Characteristics and Disposition
A total of 36 subjects were randomized: 27 to obiltoxaximab and 9 to placebo. Of the 27 obiltoxaximab‐randomized subjects, 3 received 4 mg/kg, and 6 each received 8, 16, 20, and 24 mg/kg. All subjects completed their scheduled intramuscular injections and were included in the safety population. Most subjects in the pooled obiltoxaximab group (96.3%) and all subjects in the pooled placebo group (100%) completed the study. One subject in the 24 mg/kg obiltoxaximab group did not complete the study and was lost to follow‐up on day 44.
Baseline characteristics were similar between the obiltoxaximab and placebo groups (Table 1). Subjects in the pooled obiltoxaximab group were 24 to 77 years of age, 59.3% were male, 44.4% white, 48.1% black or African American, 3.7% Asian, and 18.5% Hispanic. Except for the first 4 randomized subjects, all subjects were premedicated with diphenhydramine before study drug injection.
Table 1.
Demographics and Baseline Characteristics
| Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Demographic | Statistic | Pooled Placebo (N = 9) | 4 mg/kg Drug (N = 3) | Placebo (N = 1) | 8 mg/kg Drug (N = 6) | Placebo (N = 2) | 16 mg/kg Drug (N = 6) | Placebo (N = 2) | 20 mg/kg Drug (N = 6) | Placebo (N = 2) | 24 mg/kg Drug (N = 6) | Placebo (N = 2) | Overall Drug (N = 27) | |
| Age (years) | n | 9 | 3 | 1 | 6 | 2 | 6 | 2 | 6 | 2 | 6 | 2 | 27 | |
| Mean (SD) | 45.8 (11.53) | 52.0 (1.00) | NC | 44.8 (16.02) | NC | 55.0 (18.26) | NC | 44.3 (11.22) | NC | 43.3 (15.10) | NC | 47.4 (14.31) | ||
| Median (range) | 47.0 (27–59) | 52.0 (51–53) | NC (54) | 43.0 (28–67) | NC (30–46) | 57.5 (24–77) | NC (47–56) | 48.5 (24–54) | NC (39–59) | 41.0 (26–63) | NC (27–54) | 49.0 (24–77) | ||
| Weight (kg) | Mean (SD) | 67.71 (13.406) | 81.20 (5.840) | NC | 73.50 (13.335) | NC | 84.33 (9.639) | NC | 74.28 (14.992) | NC | 70.77 (11.882) | NC | 76.33 (12.351) | |
| Median (range) | 68.30 (50.9–95.1) | 83.30 (74.6–85.7) | NC | 70.65 (55.8–91.7) | NC | 84.75 (70.4–99.2) | NC | 79.20 (51.7–92.5) | NC | 68.85 (57.8–88.2) | NC | 78.80 (51.7–99.2) | ||
| Height (cm) | Mean (SD) | 164.18 (8.859) | 165.53 (1.674) | NC | 172.58 (8.070) | NC | 172.00 (8.525) | NC | 171.45 (10.461) | NC | 170.73 (8.194) | NC | 171.01 (8.062) | |
| Median (range) | 165.30 (153.4–179.6) | 166.50 (163.6–166.5) | NC (167.5) | 173.75 (158.0–179.8) | NC (161.0–165.3) | 174.00 (159.0–180.6) | NC (153.4–179.6) | 171.40 (157.7–184.0) | NC (153.9–156.8) | 171.40 (159.7–179.8) | NC (166.7–173.4) | 171.60 (157.7–184.0) | ||
| Sex | Male | n (%) | 3 (33.3) | 2 (66.7) | 1 (100.0) | 3 (50.0) | 0 (0) | 5 (83.3) | 1 (50.0) | 3 (50.0) | 0 (0) | 3 (50.0) | 1 (50.0) | 16 (59.3) |
| Female | n (%) | 6 (66.7) | 1 (33.3) | 0 (0) | 3 (50.0) | 2 (100.0) | 1 (16.7) | 1 (50.0) | 3 (50.0) | 2 (100.0) | 3 (50.0) | 1 (50.0) | 11 (40.7) | |
| Race | White | n (%) | 5 (55.6) | 2 (66.7) | 1 (100.0) | 3 (50.0) | 0 (0) | 3 (50.0) | 2 (100.0) | 2 (33.3) | 1 (50.0) | 2 (33.3) | 1 (50.0) | 12 (44.4) |
| Black or African American | n (%) | 4 (44.4) | 1 (33.3) | 0 (0) | 3 (50.0) | 2 (100.0) | 2 (33.3) | 0 (0) | 4 (66.7) | 1 (50.0) | 3 (50.0) | 1 (50.0) | 13 (48.1) | |
| Asian | n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) | 0 (0) | 1 (3.7) | |
| Other | n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (3.7) | |
| Ethnicity | Hispanic or Latino | n (%) | 2 (22.2) | 2 (66.7) | 0 (0) | 1 (16.7) | 0 (0) | 0 (0) | 1 (50.0) | 1 (16.7) | 1 (50.0) | 1 (16.7) | 0 (0) | 5 (18.5) |
| Not Hispanic or Latino | n (%) | 7 (77.8) | 1 (33.3) | 1 (100.0) | 5 (83.3) | 2 (100.0) | 5 (83.3) | 1 (50.0) | 5 (83.3) | 1 (50.0) | 5 (83.3) | 2 (100.0) | 21 (77.8) | |
| Unknown | n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (3.7) | |
Pooled placebo, subjects who received placebo in each cohort; N, number of subjects randomized; n (%), number and percentage of subjects in a given category; NC, not calculated; other, American Indian.
Pharmacokinetics
Of the 27 subjects who received obiltoxaximab, 26 were included in the PK population. One subject in the 8 mg/kg group was excluded from the PK population because the dosing record was missing. Concentration data were reported for this subject, but were excluded from summary statistics and additional PK analysis. One subject in the 24 mg/kg group did not complete the study and was lost to follow‐up. Blood samples were collected from this subject through day 43, and the data were considered adequate to characterize obiltoxaximab PK in this individual.
All samples collected from subjects in the PK population from the first postdose sample through day 71 had quantifiable obiltoxaximab concentrations except for 1 subject in the 4 mg/kg dose group, in whom the first postdose sample was below the LLOQ. For the semilog plot, mean obiltoxaximab serum concentration‐versus‐time profiles were parallel at all doses, indicating that the obiltoxaximab kinetics are not dose dependent (Figure 1). Mean serum concentrations declined mono‐ or biexponentially after Tmax, with the terminal phase beginning around day 15.
Figure 1.
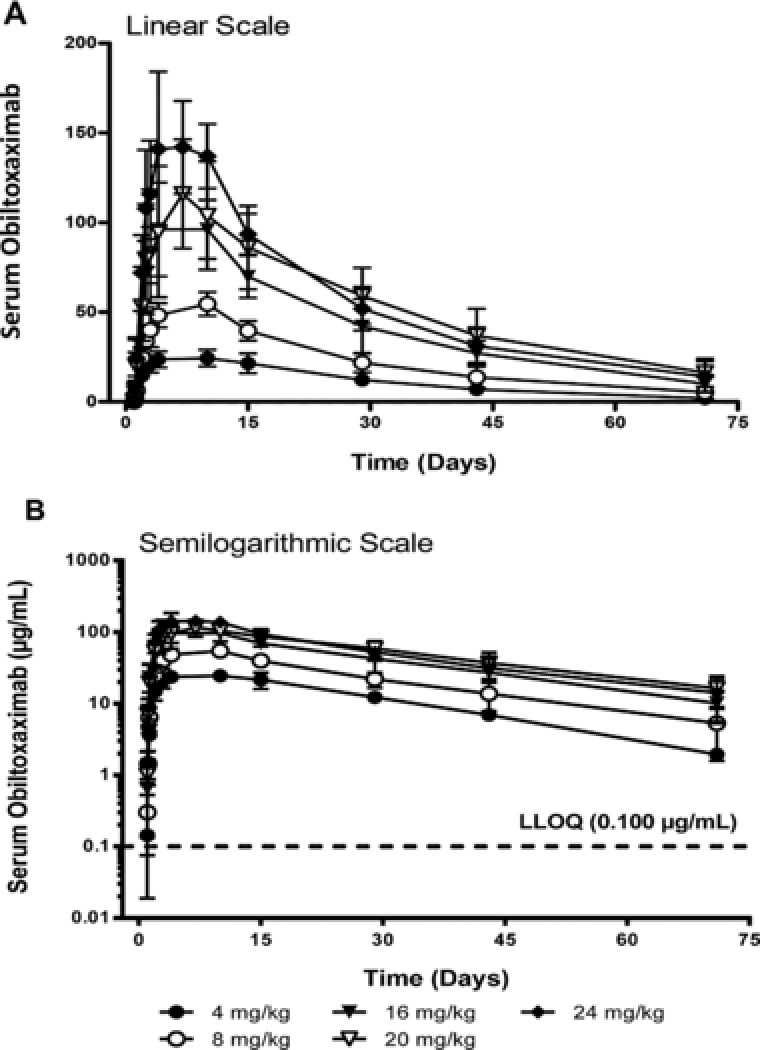
Arithmetic mean ± SD serum obiltoxaximab concentration–time profiles after single‐dose intramuscular administration.
Obiltoxaximab PK parameters are summarized by dose level in Table 2. Obiltoxaximab attained maximum serum concentrations within a range of 3 to 14 days postdose across all individuals, with similar median values (6 to 9 days postdose) across groups. The slightly higher ranges and medians in the lower‐dose groups may be related in part to a day 7 sample not being included in the profile until the dose had escalated to 20 mg/kg (adjacent samples were on day 4 and day 10 in the first 3 cohorts). Mean AUC0–inf decreased from 20 to 24 mg/kg; however, it is worth noting that AUC0–last increased from 20 to 24 mg/kg. AUC0–inf values for 1 subject at 20 mg/kg and 2 subjects at 24 mg/kg could not be used in the analysis (>20% area extrapolated).
Table 2.
Obiltoxaximab Pharmacokinetic Parameters Following Single‐Dose Intramuscular Administration to Healthy Humans
| Treatment/Statistic | Cmax (μg/mL) | Tmax (d) | AUC0–last (μg·d/mL) | AUC0–inf (μg·d/mL) | t1/2 (d) | CL/F (L/d) |
|---|---|---|---|---|---|---|
| 4 mg/kg, n | 3 | 3 | 3 | 3 | 3 | 3 |
| Mean (SD)a | 24.9 (4.70) | 9.00 (3.00‐9.00) | 774 (92.3) | 819 (85.9) | 16.0 (1.60) | 0.410 (0.0545) |
| 8 mg/kg, n | 5 | 5 | 5 | 4 | 5 | 4 |
| Mean (SD)a | 54.8 (6.45) | 9.00 (3.00‐9.00) | 1540 (328) | 1580 (441) | 20.2 (8.47) | 0.415 (0.117) |
| 16 mg/kg, n | 6 | 6 | 6 | 6 | 6 | 6 |
| Mean (SD)a | 105 (22.5) | 6.00 (3.00‐9.00) | 2890 (578) | 3200 (755) | 19.6 (4.10) | 0.445 (0.113) |
| 20 mg/kg, n | 6 | 6 | 6 | 5 | 6 | 5 |
| Mean (SD)a | 118 (28.2) | 6.00 (6.00‐14.0) | 3660 (945) | 4220 (1360) | 23.3 (6.17) | 0.382 (0.0736) |
| 24 mg/kg, n | 6 | 6 | 6 | 4 | 6 | 4 |
| Mean (SD)a | 154 (32.4) | 6.00 (3.00‐9.00) | 3740 (570) | 3960 (823) | 20.0 (7.20) | 0.478 (0.202) |
PK population n = 26.
AUC0–last, area under the concentration‐versus‐time curve from time 0 to time of last quantifiable concentration; AUC0–inf, area under the concentration‐versus‐time curve from time 0 extrapolated to infinity; CL/F, apparent clearance; Cmax, maximum concentration; n, number of subjects analyzed; SD, standard deviation; t1/2, half‐life; Tmax, time of Cmax.
Median and range are reported for Tmax.
Mean t1/2 was slightly shorter at 4 mg/kg than in the remaining dose groups, although the individual values in the 4 mg/kg group were contained within the ranges of individual values in all the remaining dose groups, except 20 mg/kg. This suggests that there were no meaningful differences in t1/2 across doses. Mean CL/F values were similar across dose groups.
Cmax and AUC values increased with dose. The similarities in mean t1/2 and CL/F values across groups from 4 to 24 mg/kg suggest that obiltoxaximab pharmacokinetics are not dose‐dependent. For Cmax, the CI criterion was strictly met (slope estimate, 0.97; 90%CI, 0.86–1.08), indicating that this parameter increased in a dose‐proportional manner from 4 to 24 mg/kg. For AUC0–last (slope estimate, 0.90; 90%CI, 0.78–1.01) and AUC0–inf (slope estimate, 0.93; 90%CI, 0.78–1.09), the 90%CIs for the slopes did not strictly meet the CI criterion; however, that the CIs fell just below the lower limit of the target interval suggests that the increases in AUC were approximately dose‐proportional.
Bioavailability estimates based on a cross‐study evaluation using ratios of mean systemic CL (0.270 L/day) following a 16 mg/kg intravenous dose of obiltoxaximab11 to mean CL/F values for each dose group after intramuscular administration of obiltoxaximab in the current study (Table 2) did not appear to be dose related over the 4 to 24 mg/kg range; overall average bioavailability was 64% (range, 57% to 71%).
Safety and Tolerability
Obiltoxaximab was well tolerated when administered intramuscularly at doses from 4 to 24 mg/kg to healthy subjects. The percentage of subjects with at least 1 AE was higher in the pooled placebo group (55.6%) than in the pooled obiltoxaximab group (33.3%); see Table 3. AEs reported by more than 1 subject in the pooled obiltoxaximab group were injection‐site pain (obiltoxaximab, 7.4%; placebo, 22.2%), dizziness (obiltoxaximab, 7.4%; placebo, no subjects), and vomiting (obiltoxaximab, 7.4%; placebo, no subjects). No hypersensitivity reactions were reported, and no AEs indicative of hypersensitivity were identified. No injection‐site abscesses occurred.
Table 3.
Frequency of Adverse Events
| Placeboa (N = 9), n (%) | Obiltoxaximabb (N = 27), n (%) | |
|---|---|---|
| Adverse event | 5 (55.6) | 9 (33.3) |
| Injection‐site pain | 2 (22.2) | 2 (7.4) |
| Dizziness | 0 | 2 (7.4) |
| Vomiting | 0 | 2 (7.4) |
| Back pain | 0 | 1 (3.7) |
| Constipation | 0 | 1 (3.7) |
| Diarrhea | 0 | 1 (3.7) |
| Excoriation | 0 | 1 (3.7) |
| Headache | 1 (11.1) | 1 (3.7) |
| Lip blister | 0 | 1 (3.7) |
| Menstruation irregular | 0 | 1 (3.7) |
| Nausea | 1 (11.1) | 1 (3.7) |
| Oropharyngeal pain | 0 | 1 (3.7) |
| Procedural dizziness | 0 | 1 (3.7) |
| Pruritus | 0 | 1 (3.7) |
| Rash | 0 | 1 (3.7) |
| Rhinorrhea | 0 | 1 (3.7) |
| Tooth infection | 0 | 1 (3.7) |
N, number of subjects randomized; n (%), number and percentage of subjects with adverse events.
Placebo is all subjects who received placebo in each dose group.
Obiltoxaximab is all subjects who received obiltoxaximab in each dose group.
All AEs were mild except for 1 female subject in the placebo group who experienced an ectopic pregnancy, which was considered moderate, and treated medically in an outpatient setting. No marked differences were observed in overall incidence or types of AEs based on sex, race, body mass index, or age. No relationship was seen for either the overall incidence of AEs or any individual AE with increasing obiltoxaximab dose, number of intramuscular injections, or injection volume. No deaths, serious AEs, or discontinuations of study drug because of AEs occurred.
Changes from baseline in laboratory, electrocardiograms, and vital signs were isolated, generally minor, not consistent across the study population, and not clinically meaningful. For each clinical laboratory parameter, mean and median values were within the normal range and similar to those in the obiltoxaximab and placebo groups at baseline and at each postdose time. In addition, no consistent pattern was observed for the percentage of subjects with values outside the normal range in either the obiltoxaximab or the placebo group. No clinically significant abnormalities were reported.
Injection site tolerability was acceptable. Erythema and induration were not observed on skin assessment in either the pooled obiltoxaximab or placebo group. Mild pain (obiltoxaximab, 11.1%; placebo, 55.6%) and mild tenderness (obiltoxaximab, 11.1%; placebo, 33.3%) during intramuscular dosing were the most frequently reported skin assessment findings. Following dosing, mild pain (obiltoxaximab range, no subjects to 11.1%; placebo range, no subjects to 55.6%) and mild tenderness (obiltoxaximab range, no subjects to 14.8%; placebo range, no subjects to 33.3%) were reported by similar percentages of subjects in the pooled obiltoxaximab and placebo groups. No relationship was seen for either pain or tenderness with increasing obiltoxaximab dose, number of intramuscular injections, or injection volume.
Mean VAS pain measurements were similar in the pooled obiltoxaximab and placebo groups throughout the study, with no evidence for an increase in pain as the obiltoxaximab dose, number of intramuscular injections, and injection volume were increased. The highest mean VAS pain measurement was in the 20 mg/kg obiltoxaximab cohort (26.7 mm) shortly after the first injection; however, this decreased to 4.2 mm within 2 hours and remained at or below 4.0 mm for the rest of the study.
Mean ALT, AST, and CPK values were within the normal range at all times and similar in the obiltoxaximab and placebo groups. There were no increases in the percentage of subjects with ALT, AST, or CPK values outside the normal range with increasing dose, number of intramuscular injections, or injection volume. In addition, no clinically significant changes for ALT, AST, or CPK were observed in either the obiltoxaximab or placebo group.
No subject met the criteria for a positive ATA response (see Methods, Immunogenicity Assessment) over the study period of 71 days.
Discussion
Intramuscular injection as an alternative to oral or intravenous administration is a common route for drug delivery and has been in use for many decades. Although intramuscular dosing is more convenient, the rate of absorption is often slower and bioavailability less than after oral or intravenous administration, especially for large water‐soluble molecules such as monoclonal antibodies that are thought to be removed from the injection site primarily via the lymphatic system.15, 16, 17, 18 In this study the PK properties of obiltoxaximab after intramuscular administration of doses from 4 to 24 mg/kg to healthy adult subjects have been examined. After a single intramuscular dose, the median time to maximum obiltoxaximab serum concentrations was 6 to 9 days postdose, which is consistent with the rate of absorption of other monoclonal antibodies following intramuscular or subcutaneous administration.16, 17 Serum concentrations declined mono‐ or biexponentially thereafter, with mean terminal t1/2 values ranging from 16 to 23 days. These terminal t1/2 estimates are comparable to those observed after administration of an intravenous dose of obiltoxaximab; mean intravenous t1/2 values ranged from approximately 17 to 23 days at doses of approximately 1.49 mg/kg and above.11 This suggests that the elimination of obiltoxaximab after intramuscular administration is not absorption rate–limited. Systemic exposures increased after intramuscular administration in an approximately dose‐proportional fashion from 4 to 24 mg/kg. The absolute mean intramuscular bioavailability of obiltoxaximab in humans was 64%, which is consistent with 50% to 100% bioavailability generally reported for other therapeutic monoclonal antibodies after intramuscular or subcutaneous administration.16, 17
Obiltoxaximab administered intramuscularly into the vastus lateralis was well tolerated at doses up to and including 24 mg/kg, up to 6 injections per dose and up to 5 mL per injection. The percentage of subjects with at least 1 AE was lower in the pooled obiltoxaximab group (33.3%) than in the pooled placebo group (55.6%). The most common adverse events reported by subjects in the obiltoxaximab group were injection‐site pain, dizziness, and vomiting, and most adverse events were mild in severity. No hypersensitivity reactions were observed, and no subject developed ATAs during the study period of 71 days. Following intramuscular administration of obiltoxaximab, local tolerability was acceptable, and no injection‐site abscesses were observed. No relationship was seen for either the overall incidence of AEs or any individual AE with increasing obiltoxaximab dose, number of intramuscular injections, or injection volume.
Although the literature is sparse regarding guidelines for maximum injection volumes for various intramuscular injection sites in humans, maximum injection volumes of 5 mL have been previously proposed for gluteal or thigh muscles.19 From among the potential intramuscular injection sites, we chose the vastus lateralis as the preferred site in the current study because it is easily accessible in a sitting or lying position, the muscle is relatively large and can accommodate larger fluid volumes, regional blood flow is higher than in the gluteus maximus, and it contains no major blood vessels or nerve structures, thus minimizing potential pain and discomfort. In addition, the uptake of drugs from this site has been shown to result in higher serum concentrations than from the gluteus maximus.20, 21, 22, 23
Intravenous obiltoxaximab is approved in the United States for the treatment of inhalational anthrax from B. anthracis in combination with appropriate antibacterial drugs in adult and pediatric patients. The most significant adverse reactions following intravenous administration were hypersensitivity and anaphylaxis.11 Intravenous obiltoxaximab is also approved for prophylaxis when alternative therapies are not available or not appropriate but should only be used for prophylaxis when its benefit for prevention of inhalational anthrax outweighs the risk of hypersensitivity and anaphylaxis. Obiltoxaximab was previously shown to be efficacious when administered intravenously or intramuscularly for pre‐ and postexposure prophylaxis in animal models of inhalational anthrax.13 In the current study hypersensitivity reactions, including anaphylaxis, were not observed following intramuscular administration of obiltoxaximab to healthy subjects at doses up to 24 mg/kg. Absorption of obiltoxaximab following intramuscular injection in humans is much slower (median Tmax, 6 to 9 days) than following intravenous administration,11 and it is unknown whether this contributed to the absence of hypersensitivity reactions seen in this study.
Declaration of Conflicting Interests
C.F.N., T.S.L., and R.G. are employees of or were consultants for Elusys Therapeutics, Inc., during the conduct of this study. A.K. is an employee of Covance Clinical Research Unit, Inc.
Funding
This study was supported by federal funds from ASPR/BARDA under contract HHSO0100201000034C from the Department of Health and Human Services.
Prior Presentations
Nagy C, Mondick J, King A, et al. Safety, pharmacokinetics and immunogenicity of intramuscular (IM) administration of obiltoxaximab (ETI‐204) to healthy humans. Presented at the 2016 ASM Biodefense and Emerging Diseases Research Meeting; February 8–10, 2016; Arlington, Virginia.
Acknowledgments
Study monitoring was performed by contract personnel with sponsor oversight. Data collection and statistical analyses were performed by Covance Clinical Data, Analysis, and Reporting Organization, Covance, Madison, Wisconsin. The authors designed the study and reviewed and analyzed the data. All the authors vouch for the completeness and accuracy of the data presented and approved the final article.
References
- 1. Dixon TC, Meselson M, Guillemin J, Hanna PC. Anthrax. N Engl J Med. 1999;341:815–826. [DOI] [PubMed] [Google Scholar]
- 2. Mock M, Fouet A. Anthrax. Annu Rev Microbiol. 2001;55:647–671. [DOI] [PubMed] [Google Scholar]
- 3. Jernigan JA, Stephens DS, Ashford DA, et al. Bioterrorism‐related inhalational anthrax: the first 10 cases reported in the United States. Emerg Infect Dis. 2001;7:933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guarner J, Jernigan JA, Shieh WJ, et al. Pathology and pathogenesis of bioterrorism‐related inhalational anthrax. Am J Pathol. 2003;163:701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Inglesby TV, O'Toole T, Henderson DA, et al. Anthrax as a biological weapon, 2002: updated recommendations for management. JAMA. 2002;287:2236–2252. [DOI] [PubMed] [Google Scholar]
- 6. Holty JEC, Bravata DM, Liu H, Olshen RA, McDonald KM, Owens DK. Systematic review: a century of inhalational anthrax cases from 1900 to 2005. Ann Intern Med. 2006;144:270–280. [DOI] [PubMed] [Google Scholar]
- 7. Hendricks KA, Wright ME, Shadomy SV, et al. Centers for disease control and prevention expert panel meetings on prevention and treatment of anthrax in adults. Emerg Infect Dis. 2014;20:e130687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abramova FA, Grinberg LM, Yampolskaya OV, Walker DH. Pathology of inhalational anthrax in 42 cases from the Sverdlovsk outbreak of 1979. Proc Natl Acad Sci U S A. 1993;90:2291–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. FDA Product Development Under the Animal Rule : Guidance for Industry. Silver Spring, MD: US Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). 2015.
- 10. Yamamoto BJ, Shadiack AM, Carpenter S, et al. Efficacy projection of obiltoxaximab for treatment of inhalational anthrax across a range of disease severity. Antimicrob Agents Chemother. 2016;60:5787–5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nagy CF, Leach TS, Hoffman, JH , Czech, A , Carpenter SE, Guttendorf, R . Pharmacokinetics and tolerability of obiltoxaximab: A report of 5 healthy volunteer studies. Clin Ther. 2016;38:2083–2097. [DOI] [PubMed] [Google Scholar]
- 12. Nagy CF, Mondick J, Serbina N, et al. Animal‐to‐human dose translation of obiltoxaximab for treatment of inhalational anthrax under the US FDA Animal Rule. Clin Transl Sci. 2017;10:12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yamamoto BJ, Shadiack AM, Carpenter S, et al. Obiltoxaximab prevents disseminated Bacillus anthracis infection and improves survival during pre‐and postexposure prophylaxis in animal models of inhalational anthrax. Antimicrob Agents Chemother. 2016;60:5796–5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventative Vaccine Trials: Guidance for Industry. Silver Spring, MD: US Department of Health and Human Services Food and Drug Administration, Center for Biologics Evaluation and Research (CBER); 2007. [Google Scholar]
- 15. Greenblatt DJ. Intramuscular injection of drugs. N Engl J Med. 1976;295:542–546. [DOI] [PubMed] [Google Scholar]
- 16. Lobo ED, Hansen RJ, Balthasar, JP . Antibody pharmacokinetics and pharmacodynamics. J. Pharm Sci. 2004;93:2645–2668. [DOI] [PubMed] [Google Scholar]
- 17. Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84:548–558. [DOI] [PubMed] [Google Scholar]
- 18. Zhao L, Ji P, Li Z, Roy P, Sahajwalla CG. The antibody drug absorption following subcutaneous or intramuscular administration and its mathematical description by coupling physiologically based absorption process with the conventional compartment pharmacokinetic model. J Clin Pharmacol. 2013;53:314–325. [DOI] [PubMed] [Google Scholar]
- 19. Roger MA, King L. Drawing up and administering intramuscular injections: a review of the literature. J Adv Nurs. 2000;31:574–582. [DOI] [PubMed] [Google Scholar]
- 20. Hopkins U, Arias CY. Large‐volume IM injections: a review of best practices. Oncol Nurse Advis. Jan/Feb 2013. http://www.onclologyNurseAdvisor.com. [Google Scholar]
- 21. Evans, EF , Proctor, JD , Fratkin, MJ , Velandia J, Wasserman, AJ . Blood flow in muscle groups and drug absorption. Clin Pharmacol Ther. 1975;17:44–47. [DOI] [PubMed] [Google Scholar]
- 22. Newton M, Newton DW, Fudin J. Reviewing the big three injection routes. Nursing. 1992;22:34–42. [DOI] [PubMed] [Google Scholar]
- 23. Subramanian GM, Cronin PW, Poley G, et al. A phase 1 study of PAmAb, a fully human monoclonal antibody against Bacillus anthracis protective antigen, in healthy volunteers. Clin Infect Dis. 2005;41:12–20. [DOI] [PubMed] [Google Scholar]