

Abstract
Bufalin is a major cardiotonic compound in the traditional Chinese medicine Chanshu preparations of toad skin secretions. Cell culture studies have suggested anti-cancer potential involving multiple cellular processes including differentiation, apoptosis, senescence and angiogenesis. In prostate cancer (PCa) cell models, P53-dependent and independent caspase-mediated apoptosis and androgen receptor (AR) antagonism have been described for bufalin at micromolar concentrations. Since a human pharmacokinetic study indicated single nanomolar bufalin was safely achievable in the peripheral circulation, we evaluated its cellular activity within range with the AR-positive and P53-wild type human LNCaP PCa cells in vitro. Our data show that bufalin induced caspase-mediated apoptosis at 20 nanomolar or higher concentration with concomitant suppression of AR protein and its best known target prostate specific antigen (PSA) and steroid receptor co-activator 1 and 3 (SRC-1, SRC-3). Bufalin exposure induced protein abundance of P53 (not mRNA) and P21CIP1 (CDKN1A), G2 arrest and increased senescence-like phenotype (SA-galactosidase). Small interference RNA knocking down of P53 attenuated bufalin-induced senescence, whereas knocking down of P21CIP1 exacerbated bufalin-induced caspase-mediated apoptosis. In vivo, daily i.p. injection of bufalin (1.5 mg/kg body weight) for 9 weeks delayed LNCaP subcutaneous xenograft tumor growth in NSG SCID mice with a 67% decrease of final weight without affecting body weight. Tumors from bufalin-treated mice exhibited increased phospho-P53 and SA-galactosidase without detectable caspase-mediated apoptosis or suppression of AR and PSA. Our data suggest potential applications of bufalin in therapy of PCa in patients or chemo-interception of prostate precancerous lesions, engaging a selective activation of P53-senescence.
Introduction
Prostate cancer (PCa) is the second leading cause of cancer death in American men. In spite of significant advances in hormonal- [1, 2], and taxane-based chemotherapeutic [3] [4] modalities that have improved patient survival measured by months, there is currently no cure for castration resistant prostate cancer (CRPC) following standard of care androgen deprivation therapy (ADT). Unfortunately the heavily treated CRPC does not respond to check point inhibitor immunotherapies that exert durable remission in some “high mutation load” malignancies such as melanoma and non-small cell lung cancer. New drugs with mechanisms distinct from current modalities will likely, therefore, help to improve patient survival to prevent or delay recurrence at different stages of a patient’s therapeutic course.
Chansu, a traditional Chinese medicine prepared from the skin and auricular gland secretions of giant toads, has been used to treat various cardiac diseases, infection and pain for millennium [5]. Chansu-containing prescriptions have been used for cancer treatment in China and are being evaluated clinically elsewhere [6]. Bufalin (Fig. 1A) is a major cardiotonic steroid-like compound isolated from Chansu [5, 7]. In cell culture models, bufalin exerts growth inhibitory and/or cytotoxic activity on leukemia [8–10] and solid cancers of many organ sites, including prostate [7, 11, 12] and breast [13–15]. Reported cellular responses include differentiation, apoptosis, cell cycle arrests, autophagy, anti-angiogenesis and anti-metastasis [5]. Some of these reports suggest a preferential growth inhibition of malignant cells over “normal” non-cancer cells. Most studies employed bufalin exposure concentrations in sub- to micromolar range, except the few early studies with leukemia differentiation induction [5] [9] [16]. For example, PCa cell culture studies reported concentrations up to 15 μM [7, 11, 12]. Within such ranges, bufalin was shown to induce caspase-mediated apoptosis in association with changes of BAX/BCL2 ratio, cytochrome release and cellular Ca++ level. It is noteworthy that one study reported the mediator role of P53 (TP53) tumor suppressor protein in micromolar bufalin-induced caspase activation and apoptosis of LNCaP cells [11].
Figure 1. Effects of bufalin on apoptosis in LNCaP, DU145 and PC-3 PCa cells and on AR, P53 and DDR signaling in relationship to apoptosis in LNCaP cells.
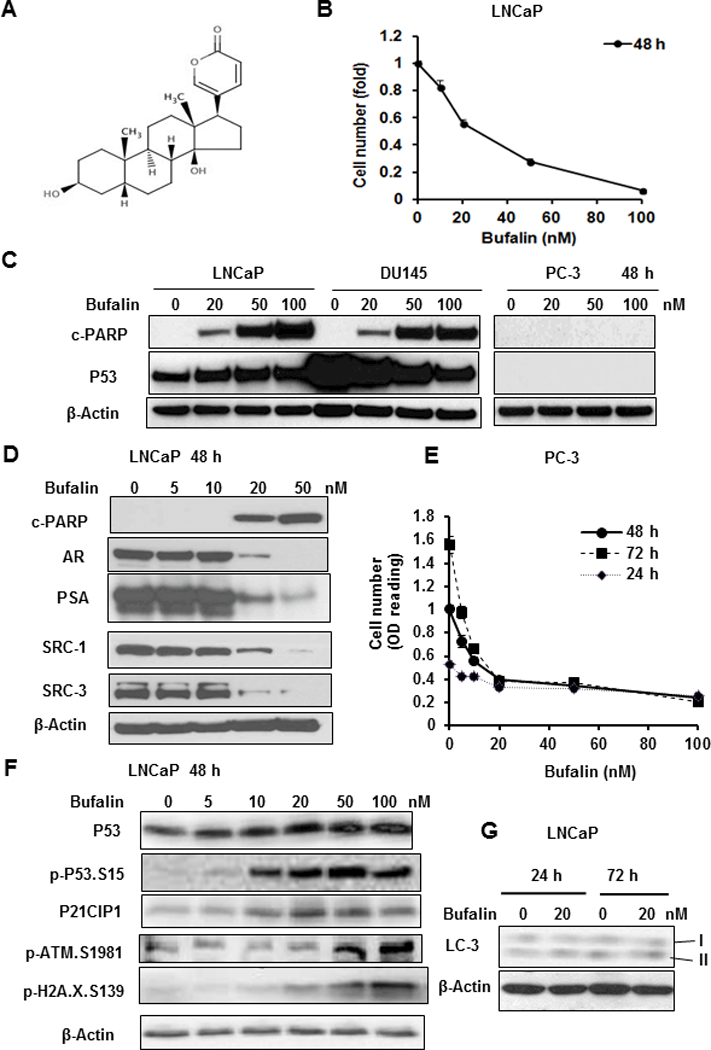
A, Chemical structure of bufalin. B. Concentration-dependent reduction of number of adherent LNCaP cells exposed to bufalin for 48 h. The data were shown as Mean ± SEM of crystal violet stain optical density, n=3 wells. C. Western blot detection of caspase-driven apoptotic marker (cleaved PARP) and P53 protein in LNCaP (wild type P53), DU145 (mutant P53) and PC-3 (P53 null) cells after bufalin treatment for 48 h. D, Western blot detection of c-PARP, AR, PSA, SRC-1, SRC-3 in LNCaP cells exposed to bufalin for 48 h. E. Growth suppression by bufalin on PC-3 cells estimated by crystal violet stain optical density, n=3 wells, Mean ± SEM. F, Western blot detection of P53, P21CIP1 and key DDR proteins in LNCaP cells exposed to bufalin for 48 h. G, Western blot detection of LC3 lipid modification as a marker of autophagy in LNCaP cells exposed to bufalin for 24 h and 72 h.
In light of the potent cardiotonic indication of bufalin, which also represents a major adverse effect of over-dosing, the therapeutic window is likely tight [5]. A human pharmacokinetic study with Huachansu (a commercial Chanshu preparation) at up to 8-fold higher doses than typical use showed that bufalin infusion could reach a plasma concentration of ~ 9 nM, without apparent side effects [6]. Therefore, the relevance of mechanisms studied in cell culture models should be validated with in vivo efficacy and safety data to facilitate meaningful translation into clinical benefits. Given the crucial role of androgen receptor (AR) signaling for PCa genesis and progression [17], the steroid-like backbone of bufalin and its reported AR antagonism [18] [19] and inhibition of select steroid receptor co-activator SRCs (SRC1 and SRC3 but not SRC2) in multiple cancer cell types [15], we evaluated the effects of bufalin in the AR-positive and P53-wild type human LNCaP PCa cells to explore the relationship of AR and P53 signaling pathways with cell cycle arrest and apoptosis induced by pharmacokinetically-relevant concentrations of bufalin. Owing to the multifaceted roles of P53 in regulating cell fates and survival such as apoptosis, autophagy and senescence [20] and the reported P53 activation by bufalin in the LNCaP model [11], we investigated the cross-talk of these molecular and cellular pathways with respect to bufalin action. Furthermore, we assessed the in vivo anti-cancer efficacy of a maximally-tolerated dose (MTD) of bufalin using LNCaP xenograft model in NSG SCID mice and examined the tumor tissues for key pathway molecules studied in vitro for concordance. Our data indicate P53-mediated cancer cell senescence as a primary cellular mechanism for the in vivo growth inhibitory efficacy of bufalin, with application implication for the adjuvant therapy of PCa in patients or for the chemoprevention of precancerous lesion progression.
Materials and Methods
Chemicals and cell culture
Bufalin (99.5% purity) was isolated as described previously [21]. siRNA for non-specific sequence (scramble) or targeting TP53 and CDKN1A (P21CIP1) were obtained from Santa Cruz Biotechnology (Paso Robles, CA). Human LNCaP PCa cells were purchased from American Type Culture Collection (ATCC, Manassas, VA) and used within 25 passages of thawing from cryopreservation. Authentication of the LNCaP cell line was accomplished by next-gen DNA whole genome sequencing (Macrogen Clinical Laboratory, Rockville, MD) and RNA-seq (Penn State Hershey Genome Sciences Facility, Hershey, PA). At the genotype level, the cells display XY gender trait and the characteristic T877A mutation in the AR gene [22]. At the mRNA level, these cells express AR, KLK3 (PSA), TP53 (P53) and CDKN1A (P21CIP1) mRNA. Cells were maintained in RPMI-1640 medium (ATCC, Manassas, VA) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Flowery Branch, GA) without antibiotics and incubated in standard 37°C and 5% CO2 humidified environment, except when indicated otherwise.
The DU145 and PC-3 PCa cells were also obtained from the American Type Culture Collection (ATCC, Manassas, VA) and used within 25 passages of thawing from cryopreservation. In terms of P53 functional status, DU145 cells contain non-functional mutant protein and PC3 cells are null [23] [24]. DU145 cells were cultured in Minimum Essential Eagle’s medium (ATCC) supplemented with 10% FBS without antibiotics. PC-3 cells were cultured in F-12K medium (ATCC) supplemented with 10% FBS without antibiotics. Mycoplasma testing was performed on LNCaP cells and was found negative.
Flow cytometry analyses
The LNCaP cells were harvested with trypsinization for flow cytometry as previously described [25]. Phospho-histone H3 (Ser10) was used as a marker of mitotic cells to distinguish the cell cycle phase between G2 and M. Cells were isolated and fixed (4% formaldehyde + 0.1% Triton X-100 in phosphate buffered saline (PBS) at room temperature for 10 minutes. The cells were washed with PBS and resuspened in 75 μl of 2% FBS in PBS. The cells were then incubated with anti-p-histone H3 (Ser10) (Cell Signaling Technology, Beverly, MA) antibody together with propidium iodide/RNAse A solution at room temperature in the dark for 60 minutes. The cells were rinsed two times and resuspended in 500 μl of 2% FBS in PBS and analyzed by flow cytometry with BD FACSverse (BD Biosciences, San Jose, CA).
Cell growth assays
The LNCaP and other PCa cells were exposed to the indicated treatment and duration, and then the adherent cells were quantized by crystal violet staining as described previously [26]. For long-term growth evaluation, we pretreated LNCaP cells with vehicle or 20 nM bufalin for 48 h and reseeded the same number of trypsin-recovered live cells in fresh complete growth medium for the indicated days in 6 cell-plates. The adherent cells were stained by crystal violet and photographed.
Immunoblotting
The whole cell lysate were prepared for immunoblotting as described previously [27]. Antibodies for P53, phospho-P53 (Ser15), P21CIP1, cleaved-PARP (c-PARP), cleaved caspase-3, SRC-1, SRC-3, HK2, phospho-ATM (Ser1981), and phospho-H2A.X (Ser139) were obtained from Cell Signaling Technology (Danvers, MA). Antibody for LC3 was purchased from MBL (Woburn, MA). Antibody for AR was obtained from Millipore (Billerica, MA) and PSA antibody was obtained from DAKO (Glostrup, Denmark). Antibody for β-actin was obtained from Sigma-Aldrich (St. Louis, MO). Positive and negative controls were included in immunoblot analyses whenever possible.
Assessment of senescence-associated-β-galactosidase (SA-β-gal) activity
Two methods were performed to assess the SA-β-gal activity at pH 6.0 in LNCaP cells or frozen tumor sections. For in situ detection, we used 5-bromo-4-chloro-3-indolyl-beta-D-galacto-pyranoside (x-gal) as a substrate to detect the SA-β-gal activity with a staining kit from Cell Signaling Technology (Danvers, MA). Cultured cells grown on 6-wells plates or frozen tumor tissues (10 μm thickness) mounted on slides were fixed with the fixative solution and stained with the staining working solution provided by the kit. The stained cells and tissues were viewed and photographed under a microscope. For the second method, we used 5-dodecanoylaminofluorescein di-β-D-galactopyranoside (C12FDG), obtained from Sigma-Aldrich (St. Louis, MO) as a substrate to detect SA-β-gal activity [28, 29]. Briefly, cells grown on 6-well plates were pretreated with 100 nM bafilomycin A1 (Sigma-Aldrich, St. Louis, MO) for 1 hour at 37°C and 5% CO2 humidified environment, and then were incubated with 33 μM C12FDG for another 2 hours. The SA-β-gal activity was either visualized under fluorescence microscopy or with a BD FACSVerse Flow Cytometer (BD Biosciences, San Jose, CA) following trypsinization. Nuclei were stained with 4’,6-diamidino-2-phenylindole, dihydrochloride (DAPI) (Thermo Fisher Scientific, Leesport, PA) for visualization under fluorescence microscopy. The flow cytometry data was analyzed with FlowJo software (FLOWJO, Ashland, OR).
Microarray Analysis of RNA transcriptome
The cells were treated with 0, 10 and 20 nM of bufalin for 24 h for concentration-dependent changes. Total RNA were extracted using RNeasy Mini kit (QIAGEN). The quality and quantity of the total RNA samples were determined using NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific, Leesport, PA). The RNA expression profiles of the indicated samples were performed on Illumina Human HT-12 Bead Chips (Illumina Inc., San Diego, CA). All RNA labeling and hybridization was performed at the BioMedical Genomics Center of the University of Minnesota according to the protocols specified by the manufacturers. Raw data were normalized by rank invariant normalization with Illumina BeadStudio 3.0 software. Data filter was performed on detection P-value (P ≤ 0.05) in at least 2/3 of the samples, signal intensity (≥100.0), and cut-off fold-change ≥1.1 and ≤0.9 between vehicle and bufalin-treated groups. For multiple probes of the same gene, the ratio of each probe signal intensity to vehicle was averaged. Thereafter, the normalized and filtered data consisted of 13758 genes. Differentially expressed gene sets were subjected to bioinformatic analysis using Ingenuity Pathway Analyses (IPA) software to corroborate affected signaling networks. Microarray data have been deposited with accession GSE118261.
Animal experiment
The animal study was approved by the Institutional Animal Care and Use Committee of Texas Tech University Health Sciences Center and carried out in the Amarillo campus facility. A total of 40 male NSG SCID mice at 8 weeks of age were obtained from The Jackson Laboratory (Bar Harbor, ME). One week after acclimation, the mice were randomly divided into 2 groups and subcutaneously inoculated with 1×106 LNCaP cells suspended in 100 μl of inoculum containing 50% RPMI-1640 medium and 50% Matrigel (BD Biosciences, San Jose, CA) in the lower right flank. One day post inoculation, the mice started to receive once daily solvent vehicle (5% alcohol in saline) or bufalin (1.5 mg/kg) treatments via intraperitoneal (i.p.) injection (7 days per week), respectively. The bufalin dosage was chosen based on published data [30]. Xenograft tumors were calipered once per week. The tumor volumes were calculated with a formula as L x W2 × 0.526. After 9 weeks of treatments, the mice were sacrificed 2 hours after the last dose; tumors, blood (plasma) and tissues were collected.
Immunohistochemistry (IHC) staining
IHC staining was performed as previously described [31]. After antigen retrieval, the sections were blocked with 6% horse serum and incubated with a primary antibody for cleaved caspase-3 (Cell Signaling Technology, Danvers, MA), phospho-P53 (Ser15) (R&D Systems, Minneapolis, MN), AR (Millipore, Billerica, MA), PSA (KLK3) (DAKO, Glostrup, Denmark). Biotinylated secondary antibody-HRP conjugate (Vector Labs, Burlingame, CA) was used at 1:200 dilutions. The sections were developed with 3.3’-diaminobenzidine and counterstained with hematoxylin (Sigma-Aldrich, St. Louis, MO). After dehydration and mounting in permount, images were captured under light microscopy.
Statistical analysis
When more than two groups are compared, ANOVA was used to analyze the statistical significance with appropriate post-hoc comparison among the specific groups. For comparison involving only 2 groups, Student’s t-test was used with α = 0.05 for statistical significance.
Results
Bufalin-induced suppression of AR and PSA protein abundance was not dissociable from apoptosis
Exposure of LNCaP cells to bufalin led to a strong concentration-dependent decrease of the number of adherent cells, estimated with crystal violet staining, with an IC50 value of ~25 nM when assessed at 48 h (Fig. 1B) and ~7 nM by 96 h (Supplement Fig. S1A). Under a phase contrast microscope, detached floaters typical of apoptotic response were easily observed in 50 nM bufalin exposed cells (Supplement Fig. S1B). To determine the contribution of apoptosis to the observed cell number suppression, we immunoblotted c-PARP as a molecular indicator of caspase-mediated death execution. Bufalin exposure for 48 h increased c-PARP readout in concentration-dependent manner with a threshold of 20 nM for apoptosis (Fig. 1C, D; Supplement Fig. S2A). Bufalin exposure for 24 h showed a similar pattern, albeit less intense (Supplement Fig. S2B).
The P53-mutant DU145 cells exhibited similar caspase-mediated c-PARP readouts as the P53-wild type LNCaP cells (Fig. 1C). On the other hand, the P53-null PC-3 cells did not show any detectable c-PARP with as high as 100 nM bufalin exposure (Fig. 1C). Nevertheless, bufalin induced cytostasis of PC-3 cells at 20 nM or higher as reflected by the time-independent steady cell number estimation (Fig. 1E). LNCaP and PC-3 cell lines express high level of constitutive AKT survival activity due to defective PTEN whereas DU145 cell line is low for this survival pathway due to wild type PTEN [32]. Therefore, the apoptosis sensitivity of DU145 cells to bufalin was more likely due to their low basal survival signaling than the non-functional mutant P53 protein [23] [24].
The protein level of AR and its best characterized transcriptional target PSA was decreased by bufalin at 20 nM or higher, mirroring the reciprocal increase of c-PARP (Fig. 1D; Supplement Fig. S2). Because bufalin in the range of 1–10 nM had been reported to down-regulate the abundance of two steroid receptor co-activator SRCs (SRC1 and SRC3 but not SRC2) in breast cancer and other cancer types [15], we immunoblotted SRC1 and SRC3 in bufalin-treated LNCaP cells to assess whether they were affected in our cellular model. As shown in Fig. 1D, these AR co-activators were suppressed by bufalin in a manner highly similar to AR and PSA suppression and not dissociable from apoptosis.
Microarray detection of mRNA of AR, KLK3 (PSA) and its family member KLK2 (Table 1) indicated little to modest effect of 24 h bufalin exposure on their steady state levels, but the mRNA level of another AR target gene NKX3–1 was decreased by as much as 4 folds. Whereas the lack of probes for SRC-1 and SRC-3 mRNA in our microarray precluded us from examining their responses to bufalin exposure, a related gene steroid receptor RNA activator 1 (SRA-1) was not affected by 24 h bufalin exposure (Table 1). Taken together, in LNCaP cells, the suppressing effects of bufalin on abundance of AR, PSA and SRC-1, −3 proteins were not separable from its apoptotic action and were not tightly linked to their steady state mRNA abundance, and therefore likely through post-transcriptional mechanism(s) of protein stability and/or protein synthesis.
Table 1.
Effect of bufalin on androgen receptor (AR) and select target genes and top-30 upregulated genes (24 h exposure) in LNCaP cells. Expressed as fold over control set as unity.
| SYMBOL | 10 nM bufalin | 20 nM bufalin | Major Functions | PMID |
|---|---|---|---|---|
| GAPDH | 0.94 | 0.86 | House keeping gene, glycolysis | |
| ACTB | 1.8 | 2.0 | Beta-actin, House keeping gene | |
| ACTG1 | 1.2 | 1.6 | Gamma- actin1 | |
| ACTA2 | 1.6 | 1.4 | Smooth muscle actin | |
| AR | 1.1 | 0.75 | Androgen receptor | |
| KLK3 (PSA) | 0.99 | 0.73 | AR target gene | |
| KLK2 | 0.68 | 0.58 | KLK family member | |
| KLK4 | 1.4 | 0.49 | KLK family member | |
| NKX3–1 | 0.57 | 0.21 | AR target gene | |
| SRA-1 | 0.91 | 0.79 | Steroid receptor RNA activator 1 | |
| TP53 | 1.2 | 1.0 | P53 tumor suppressor gene | |
| CYR61/CCNI | 10.6 | 25.2 | Senescence, DDR, P53, ROS, P16 activator | 22566095 |
| CTGF/CCN2 | 3.7 | 13.1 | Senescence, TGFβ target, cancer catabolism | 22684333 |
| PPP1R15A/GADD34 | 3.8 | 10.0 | Senescence, DDR, P53 activator | 14635196 |
| BCL6 | 6.0 | 7.4 | Senescence, P53 target, Apoptosis suppressor | 18524763 |
| CDKN1A (P21Cip1) | 7.2 | 7.2 | Senescence, P53 target, DDR, cycle arrest | 8242752 |
| HMOX1 | 2.7 | 14.3 | Cellular stress, ROS, P53 target | 17933770 |
| DUSP1 | 7.5 | 11.2 | Cellular stress, ROS, P53 target | 18403641 |
| DUSP5 | 3.1 | 5.3 | Cellular stress, ROS, P53 target | 12944906 |
| TXNIP | 5.7 | 5.5 | Cellular stress, ROS, P53 activator | 23823478 |
| IFRD1/PC4/Tis7 | 4.1 | 5.7 | Cellular stress, P53 activator | 17785449 |
| SDC4 | 3.9 | 7.5 | Cellular stress, hypoxia | 15141079 |
| TRIB1 | 3.8 | 5.9 | Cellular stress, ER cheperone, p53 inactivator | 25832642 |
| ATF3 | 5.8 | 12.7 | Cellular stress sensor, P53 target | 21927023 |
| TSC22D3 | 7.6 | 8.0 | Cellular stress sensor, osmotic | 17147695 |
| SIPA1L2 | 3.9 | 6.6 | Cellular stress response | 22178795 |
| IER5 | 3.2 | 7.3 | Cellular stress , chaperone, checkpoint | 23273978 |
| RNF103/Kf-1 | 3.2 | 5.4 | Cellular ER stress, ubiquitin ligase | 18675248 |
| NUPR1/Com1/p8 | 3.3 | 5.3 | Cellular stress, P53 interactor, P21 activator | 18690848 |
| GADD45B | 7.2 | 13.6 | Growth arrest, p53 target | 23948959 |
| GADD45A | 6.4 | 12.1 | DDR, p53 target | 10779360 |
| BTG2 | 4.3 | 5.4 | DDR, P53 target, cycle arrest | 8944033 |
| GDF15/NAG-1 | 9.0 | 7.6 | Tumor suppressor, P53 target | 23220538 |
| EFNA1 | 3.9 | 7.4 | Tumor suppressor, anti-proliferation | 17949899 |
| ERRFI1 | 3.9 | 7.2 | Tumor suppressor, anti-proliferation | 17351343 |
| TNFRSF12A | 5.1 | 9.1 | Androgen receptor repressed metastasis gene | 24970477 |
| ELF3 | 6.2 | 8.4 | Androgen receptor repressor | 23435425 |
| GABARAPL1 | 3.5 | 5.5 | Autophagy, maturation | 20404487 |
| ISG20L1 | 2.1 | 5.6 | Autophagy, P53 target | 20429933 |
| SLC38A2 | 5.2 | 6.6 | Solute transport | 21622135 |
| KLHL28 | 3.3 | 6.2 | Ubiquitination regulator | 21145461 |
Bufalin induced P53/P21CIP1 protein abundance, cellular stress and DNA damage response and senescence genes in preference over apoptosis and autophagy genes
Because P53 was found to mediate apoptosis induced by bufalin exposure in the micro-molar range [11] and P53 regulates not only apoptosis, but also senescence, autophagy, DNA damage response (DDR) and cellular oxidative stresses [20], we immunoblotted the extracts of LNCaP cells treated with increasing concentrations of bufalin for total P53 (Fig. 1C, 1F) and its best known transcriptional target P21CIP1(CDKN1A) (Fig. 1F). Bufalin treatment for 48 h led to an increased total P53 protein abundance at 10–20 nM as well as that of P21CIP1 in a concentration-dependent manner. We detected bufalin-induced increased phosphorylation of ATM kinase (p-ATM-Ser1981) and an increased DNA double strand break marker phospho-H2A.X (Ser139) that were concomitant with c-PARP (Fig. 1F). For autophagy marker LC-3 lipid modification, we failed to detect any bufalin-induced increase of the fast-moving form, LC-3-II at 24 or 72 h of exposure (Fig. 1G).
Microarray transcriptomic analyses of bufalin-induced changes at the mRNA level in LNCaP cells after 24 h exposure showed no change of TP53 mRNA (Table 1), consistent with well-known post-transcriptional (e.g., increased protein stability) and post-translational activation (e.g., phosphorylation) of P53 in cells undergoing DNA damage response (DDR). Bufalin exposure induced classical P53 target genes such as CDKN1A (P21CIP1), GADD45A involved in typical DDR, some classic and non-classical senescence genes (e.g., CCN1/CYR61, CCN2/CTGF, GADD34), a number of cellular, oxidative stress and endoplasmic reticulum (ER) stress genes (e.g., and GADD45B, HMOX1, DUSP1, DUSP5) and some unique tumor suppressors (e.g., GDF15/NAG-1) (Table 1, showing top 30 genes and Supplement Table S1). It is apparent that many of these top 30 upregulated genes were related to P53 in the capacity of direct transcriptional targets, activators or interactors.
Of pro-apoptotic P53 target genes, NOXA was moderately increased (4.2 – 4.5x) (Supplement Table S1) whereas BAX (1.5–1.8x) and PERP (1.5–1.8x) were within the fluctuation of the 3 actin genes (Table 1). Two non-classical autophagy genes, GABARAPL1 and ISG20L1, were within the top 30 bufalin-induced genes (Table 1), and P53-target autophagy gene DRAM1 was modestly increased (3.0–4.8X), but many other classical autophagy genes regardless of P53 dependence (e.g., ATG9A and ATG16L1) were within the fluctuation of the actin genes (Table 1; Supplement Table S1). A couple of AR-repressor/repressed genes such as TNFRSF12A, ELF3 were also among the top-30 upregulated genes, perhaps a reflection of the androgen sensitive nature of LNCaP cells. Bufalin exposure for 24 h decreased gene expression of many proliferative oncogene-like entities, mitochondrial function, DNA replication and chromatin, and cyclins (Supplement Tables S2, S3). Prompted by these changes and those indicating cellular senescence signaling (Table 1), we analyzed cell cycle arrest(s) and senescence next.
Bufalin exposure increased cellular senescence
Cell cycle distribution analyses showed increased sub G0/1 apoptotic fraction at bufalin exposure of 20 nM (Fig. 2A), consistent with the c-PARP pattern in Fig. 1C, D. However, the overall “apoptotic” fraction remained rather minor (~ 6% point above control). There was no enrichment of G1 or S phase cells upon bufalin exposure, but the “G2/M” fraction was moderately enriched (Fig. 2A). Staining for phospho-histone H3 (Ser10) as a marker of mitotic cells showed decreased % of mitotic cells in spite of increased bufalin-enriched “G2/M” % (Fig. 2A), revealing a G2 phase arrest rather than M phase arrest. Transcriptomic profiling of changes of cyclin genes (CCN) and minichromosome maintenance (MCM) genes showed greater effect on S-G2 CCNA2 as well as S phase helicase MCM 4, 6 core (Supplement Table S3), consistent with G2 arrest.
Figure 2. Effects of bufalin on cell cycle distribution and senescence-like phenotype in LNCaP cells.
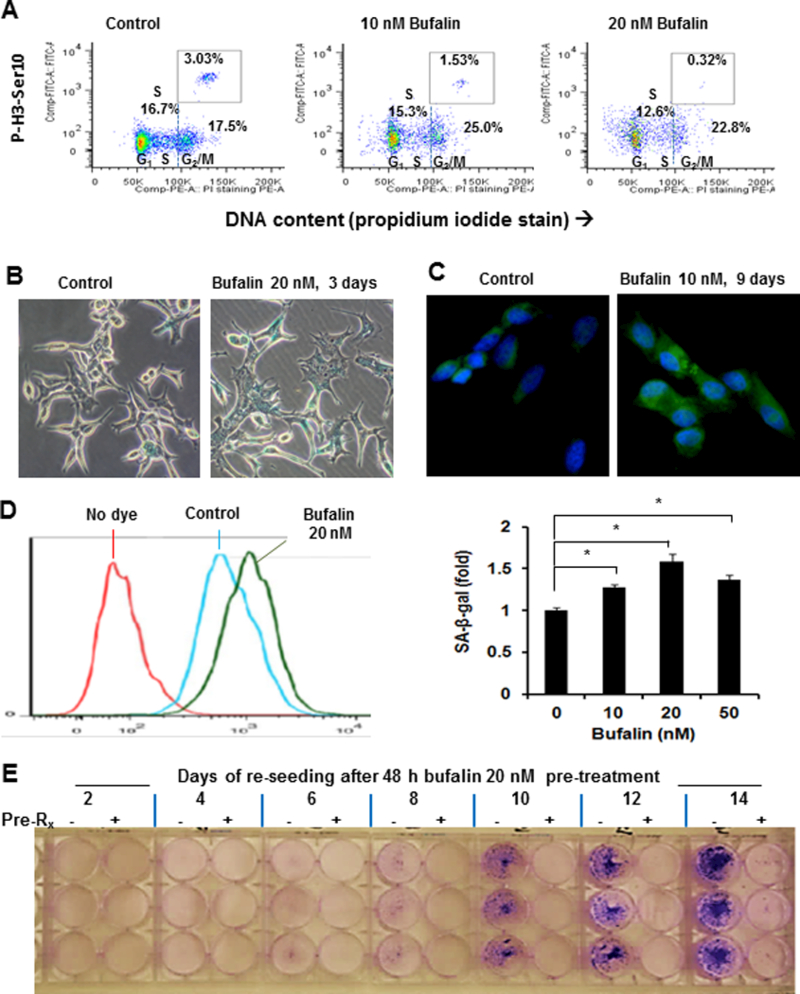
A, Flow cytometry analyses of cell cycle distribution in LNCaP cells exposed to bufalin for 48 h and of mitotic specific protein marker histone H3 Ser 10 phosphorylation. B, Representative light-microscopic image of SA-β-gal activity using x-gal as substrate in LNCaP cells exposed to 20 nM of bufalin treatment for 3 days. C, Representative fluorescent-microscopic image of SA-β-gal activity using C12FDG as substrate in LNCaP cells exposed to 10 nM of bufalin for 9 days. D, Flow cytometry detection of SA-β-gal activity using C12FDG as substrate in LNCaP cells exposed to 20 nM of bufalin treatments for 4 days. Bar graph shows the summary of SA-β-gal activity enhancement by bufalin. The experiments were done in triplicate. The data were shown as Mean ± SEM. *: p < 0.05. E. Durable growth inhibition of LNCaP cells by 48 h bufalin exposure in a re-seeding experiment. After 48 h of pretreatment with ethanol or 20 nM of bufalin, the cells were rinsed with PBS and harvested by trypsinization, then the same number of “live” cells were re-plated into new 6-well plates with fresh complete medium. Adherent cells/colonies were visualized by crystal violet staining over 2 to 14 days.
Cellular senescence is an irreversible terminal arrest, playing an especially important role in suppressing Pten-deficiency-driven prostate carcinogenesis [33]. We examined cellular senescence phenotype in bufalin-exposed LNCaP (PTEN-negative, high AKT activity) cells by evaluating SA-β-gal activity as a cytochemical marker (Fig. 2B) and observed increased proportion of cells with blue staining and flattened morphology. Using a fluorogenic substrate, we detected SA-β-gal activity with stronger senescence signal in the cytoplasm of bufalin-treated cells compared to the control cells under a fluorescence microscope (Fig. 2C). When quantified by flow cytometry (Fig. 2D), bufalin at 20 nM induced peak SA-β-gal signal. Higher concentrations of bufalin exposure compromised senescence detection due to apoptosis. At the mRNA expression level, bufalin exposure for 24 h induced a number of genes related to senescence, including the top upregulated gene CYR61/CCNI and its related family member CTGF/CCN2 and P53 target gene CDKN1A (P21CIP1) (Table 1).
As apoptosis eliminates cancer cells and senescence commits them to an irreversible terminal arrest fate, we tested whether bufalin pre-treatment at 20 nM for 48 h would lead to a persistent growth inhibitory action on LNCaP cells in a re-seeding experiment. With the same initial number of trypsin-harvested “live” bufalin-pretreated cells vs. control cells in fresh culture medium, the bufalin-pretreated cells almost completely lost their growth capability whereas the control cells grew exponentially, forming large visible colonies by 14 days (Fig. 2E).
P53-dependent upregulation of P21CIP1 by bufalin promoted senescence and attenuated apoptosis
To further probe the role of P53-P21CIP1 in bufalin-induced apoptosis and senescence in LNCaP cells, we used siRNA approach to knockdown (KD) expression of each. As shown in Fig. 3A, TP53 silencing by siRNA (lane 4) was effective at decreasing basal (lane 1) and scramble siRNA-transfection elicited (lane 2) P21CIP1 expression and blocked the induction of P21CIP1 after bufalin treatment (lane 8 vs. 5 & 6), indicating that both transfection- and bufalin-induced P21CIP1 expression were P53-dependent in LNCaP cells. However, knocking down CDKN1A (P21CIP1) did not decrease transfection-elicited (lane 3 vs. 2) and bufalin-induced P53 expression (lane 7 vs. 6), as expected of P53 role as a classic upstream regulator of P21CIP1. Notably, CDKN1A (P21CIP1)-silencing increased transfection-elicited apoptotic cleavage of PARP (lane 3 vs. 2) and bufalin-induced cleavage activation of caspase-3 and PARP (lane 7 vs. 6), supporting a pro-survival role of P21CIP1 in LNCaP cells. Due to the increased cell death in P21CIP1-KD cells, it was not possible to assess their senescence status. Knocking down P53 did not affect bufalin-induced caspase-driven c-PARP (lane 8 vs. 6). One interpretation was that siRNA-silencing of P53 disabled not only the P21CIP1-mediated protection against bufalin-induced apoptosis, but also decreased the other P53 transcriptional death targets, therefore the magnitude of overall death by bufalin was less than in P21CIP1-KD cells.
Figure 3. Impact of knocking down P53/P21CIP1 axis on bufalin induction of apoptosis and senescence in LNCaP cells.
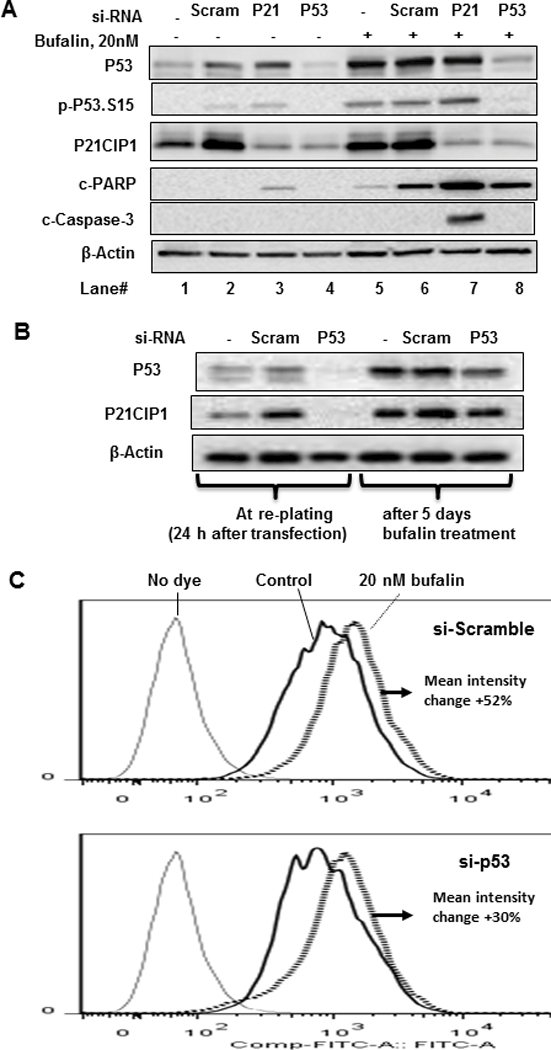
A, Western blot detection of P53 and P21CIP1 knocking down by siRNA and caspase-mediated PARP cleavage by bufalin after 24 h exposure. B, Western blot detection of P53 knocking down by siRNA 24 h after transfection and 5 days after initiation of bufalin treatment. C, Flow cytometry detection of SA-β-gal activity using C12FDG as substrate for scramble-RNA transfected vs. si-TP53-knockdown cells exposed to bufalin for 5 days.
To test the role of P53 induction by bufalin in senescence, we transfected LNCaP cells with TP53 siRNA for 24 h (Fig. 3B) and re-plated them to attach for 48 h, then treated with bufalin for 5 additional days before subjecting the cells to SA-β-gal flow cytometric detection. By the time SA-β-gal was assessed, transfected TP53 siRNA had lost considerable potency (Fig. 3B). In spite of the partial recovery of P53 and P21CIP1, we observed a substantial attenuation of bufalin-induced SA-β-gal (Fig 3C). Therefore, P53/P21CIP1 signaling induced by bufalin exposure likely promoted LNCaP cell fate switching from apoptosis to senescence.
Bufalin inhibited LNCaP xenograft tumor growth in NSG SCID mice
We randomized male NSG SCID mice inoculated with LNCaP cells into two groups (N = 20 per group) and administered solvent vehicle (5% ethanol in saline) or 1.5 mg/kg of bufalin, respectively, by once daily i.p. injection starting the day after inoculation. We chose this bufalin dosage based on a reported toxicity study [30]. After 9 weeks of treatments, the tumor take rate of the vehicle group was 80 % (16 out of 20), consistent with the reported tumor take rates of LNCaP xenograft in immuno-deficient mice. The take rate in bufalin group was 75% (15 out of 20). Compared to vehicle alone, bufalin significantly delayed LNCaP xenograft tumor growth (Fig. 4B and C) without affecting animal body weights (p > 0.05) (Fig. 4A). At necropsy, the mean tumor weight of bufalin-treated mice showed a decrease of approximately 67% (p < 0.05) compared to that of the vehicle-treated mice (Fig. 4D).
Figure 4. Effect of once daily i.p. injection of bufalin on the growth of subcutaneously inoculated LNCaP xenograft tumors in NSG SCID mice.
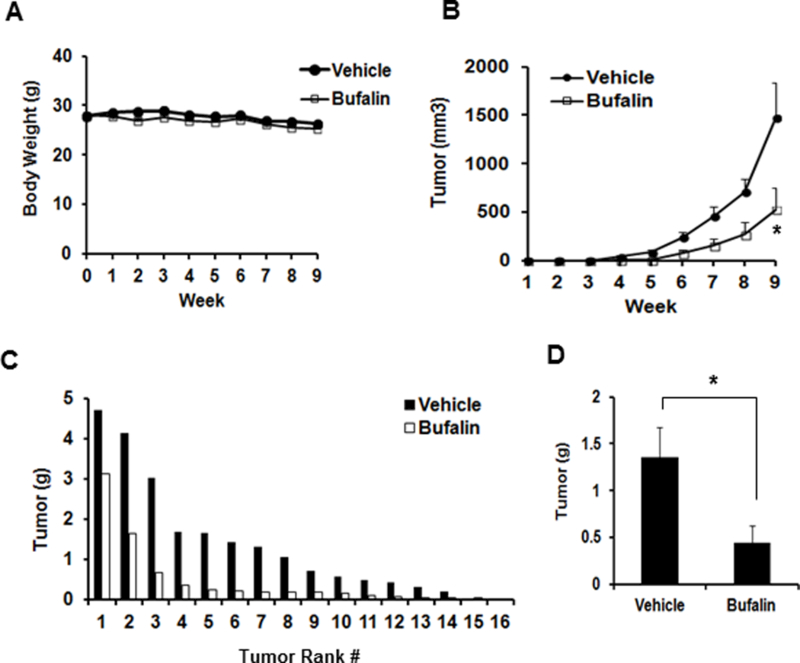
A, Mouse body weight. B. Growth curve of LNCaP xenograft tumors by tumor volumes, non-tumor bearing mice were excluded (effective N=15,16). C, Ranking order of tumor weight of vehicle- and bufalin-treated mice. D. Mean tumor weight at necropsy. The data were shown as Mean ± SEM. vehicle-treated (N=16); bufalin-treated. (N=15). *: p < 0.05.
Increased senescence in bufalin-treated LNCaP tumors without detectable increase of caspase-apoptosis
We selected the 3rd-8th ranked tumors from each group and analyzed them by H&E staining (Fig. 5A), immunohistochemistry (Fig. 5B) and immunoblot (Fig. 5C). In vehicle-treated tumors, H&E staining showed dark hematoxylin-stained regions that were densely packed with cells (Fig. 5A, left panel). There were variable and large eosinophilic necrotic areas (without nuclei, middle panel marked N) within these tumors (5 out of 5 vehicle control tumors >40% area). The control tumors displayed profound acute hemorrhage (profuse presence of erythrocytes on H&E section) such that their paraffin blocks appeared dark vs. the translucent appearance of the bufalin-treated tumor blocks (Supplement Fig. S3). The bufalin-treated tumors showed less necrotic proportion and their acute hemorrhage status ranged from scant to moderate. H&E staining revealed loosely packed cells in the bufalin-treated tumors (Fig. 5A, right panel). There was no evidence of increased in situ apoptotic cleavage of caspase-3 in the bufalin-treated tumors with IHC detection (Fig. 5B). Immunoblot analyses did not detect cleaved-caspase-3 or c-PARP in either group (Fig. 5C).
Figure 5. Biomarkers analyses of bufalin-treated LNCaP xenograft tumors.
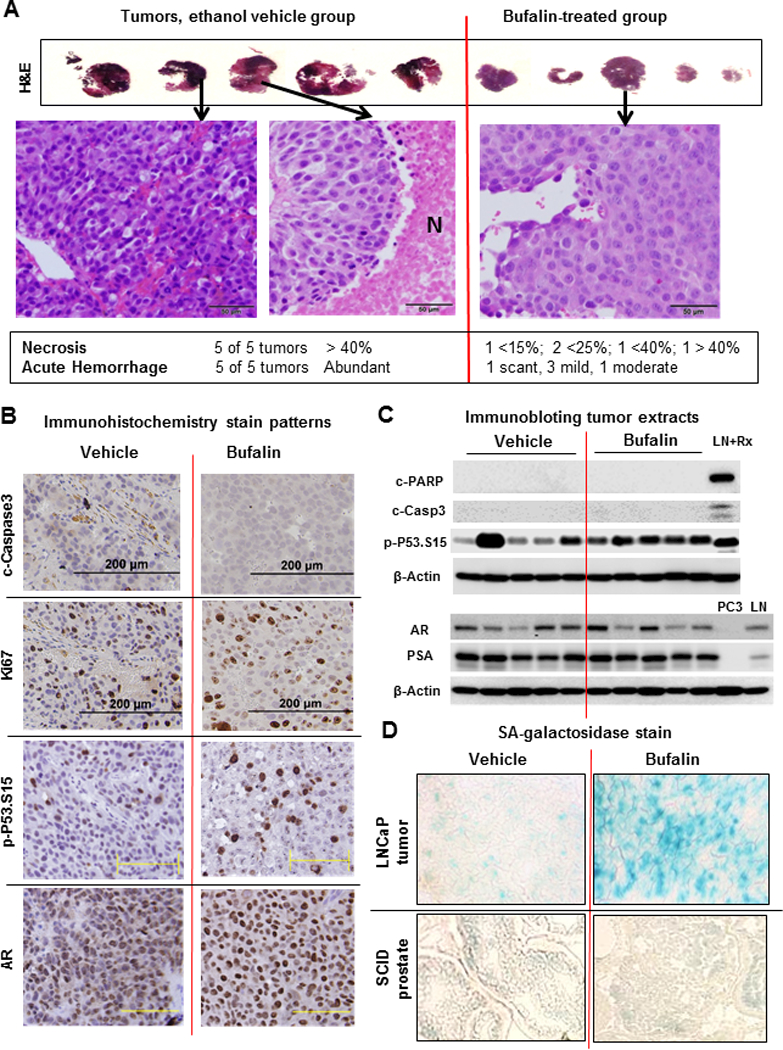
A, H&E-stained 3rd-8th ranked LNCaP xenograft tumors from ethanol vehicle- and bufalin-treated groups, respectively. Large eosinophilic areas without nuclei in vehicle group represent necrotic regions (marked N in middle panel). Scale bars, 50 micron. B, Representative IHC images of Ki-67 proliferation marker and apoptosis marker (c-caspase 3), p-P53.Ser15 and AR in tumors from vehicle- or bufalin-treated mice. Black scale bars, 200 micron; Yellow scale bars, 100 micron. C, Western blot detection of cleaved-PARP, cleaved-caspase-3, p-P53.Ser15, AR and PSA in tumors from vehicle- or bufalin-treated mice. LNCaP cells exposed to 1 μM doxorubicin (a DNA damage drug) treatment for 24 h were used as a positive control for apoptosis (LN+Rx). PC3 extract and LNCaP extract (LN) were used as negative and positive control for AR and PSA, respectively. D, Representative images of SA-β-gal staining of cryo-sections of LNCaP xenograft tumors and the normal prostate of NSG SCID mice treated with vehicle or bufalin.
The “proliferation biomarker” Ki67 IHC staining revealed large positive nuclei in bufalin-treated group and the proportion of positive cells were same as vehicle group, if not higher (Fig. 5B). The strong Ki67-staining large nuclei in the bufalin-treated tumors were therefore consistent with the in vitro observation that bufalin induced G2 arrest (Fig. 2A). The proportion of p-P53.Ser15 positive large nuclei was increased in bufalin-treated tumors (Fig. 5B), a finding consistent with immunoblot analyses (Fig. 5C). Yet, there was no detectable effect of bufalin on AR (Fig. 5B nuclear staining, 5C) and PSA (Fig. 5C). The abundance of p-AKT or HK2, two crucial kinases for PCa survival and growth [34], was not altered between the two tumor groups (Supplement Fig. S4).
Compared to the vehicle group, in situ SA-β-gal activity of frozen xenograft tumor sections of bufalin-treated mice was higher (blue staining in Fig. 5D, top right panel). In contrast, the prostate epithelium of the NSG SCID mice from vehicle group or bufalin group did not show a difference of the basal SA-β-gal readout (Fig. 5D, bottom panels). Taken together, the data suggest P53-mediated cellular senescence was a likely cancer-selective, primary cellular mechanism for bufalin to inhibit LNCaP PCa tumor growth in vivo.
Discussion
With a human pharmacokinetic study outcome in mind, i.e., i.v. infusion leading to a maximum level of ~ 9 nM at the end of the 2-h infusion [6], we investigated the cellular effects of relevant concentration range of bufalin in LNCaP cell culture model and discovered the induction of G2 arrest and increased senescence-like cellular phenotype as the primary cellular processes to inhibit LNCaP cancer cell proliferation (Fig. 2), involving P53-P21CIP1 as a crucial molecular signaling axis (Fig. 3). Whereas P21CIP1-knockdown significantly increased the apoptosis in response to bufalin (Fig. 3A), P53-knockdown attenuated the extent of senescence induction (Fig. 3C), supporting P53-P21CIP1 in our cell model as a cell fate determinant from apoptosis to senescence (See Supplement Fig. S5).
Whereas bufalin was found to suppress AR and PSA abundance in cell culture exposure model, these molecular readouts were not separable from the apoptotic action; nor was the reduction of SRC-1 and SRC-3 proteins (Fig. 1D). SRC1 and SRC3 are 2 universal coactivators of steroid receptors including AR [15]. Transcriptomic profiling of 24 h bufalin-exposed cells showed decreased AR-target NKX3–1 by as much as 4 folds at 20 nM bufalin whereas the PSA (KLK3) gene was not much affected within this time frame (Table 1). Therefore, bufalin might exert differential post-transcriptional as well as transcriptional controls over these AR targets. Given structure-activity relationship (SAR) investigation demonstrated bufalin as a poor ligand binding antagonist of AR [18, 19], the cell culture data suggest that bufalin-induced reduction of AR and its co-activators was caused by apoptosis and might contribute little to senescence signaling at the sub-apoptotic range of bufalin exposure.
We demonstrated that daily injection of a maximally-tolerated dose of bufalin significantly delayed LNCaP xenograft growth in vivo (Fig. 4), in association with a significant induction of SA-β-gal senescence (Fig. 5D) but without apparent changes of caspase-mediated apoptosis biomarkers or a suppression of AR and PSA (Fig. 5B and C). However, we could not rule out the possibility that apoptosis-susceptible cancer cells had already been eliminated from the xenograft tumors at the time of necropsy, thus enriching long-lived senescent cancer cells in the bufalin-treated mice. Given such caveats, it is imperative that we interpret the in vivo biomarker changes and mechanism of action with considerable reservation.
The mRNA profiling of 24 h bufalin-exposed LNCaP cells revealed gene signatures of P53-driven DNA damage response (DDR), oxidative cellular stress and endoplasmic reticulum stresses as well as senescence in top-30 upregulated genes (Table 1). Anchored by ATM/ATR-activated P53, DDR elicits multiple cellular responses ranging from transient responses (cell cycle arrest and DNA repair) to terminal fates (death or senescence) [35–39]. In vivo restoration of P53 function in P53-dysfuntional sarcomas or liver tumors led to significant tumor regression [40, 41] and surprisingly, through P53-mediated senescence and innate immune cells instead of apoptosis [40]. Recent studies further reveal P53-mediated mitosis skip in cellular senescence [42, 43]. Activation of P53 by DNA damage promotes G2 arrest, leading to cell cycle exit and senescence without entering mitosis [42, 43]. The mitosis skip is executed by P53 induction and upregulated P21CIP1 abundance to bind and inhibit the mitosis-promoting activity of nuclear cyclin B1 with its CDK [42]. Even transient activation of P53 without DNA damage was sufficient to lead to mitosis skip and senescence [40, 42]. Our in vivo bufalin-treated cancers showed large Ki67 positive nuclei (Fig. 5B), and increased SA-β-gal senescence staining (Fig. 5D). We speculate that the daily bufalin exposure in vivo (bufalin sharp spikes) promoted DDR and the P53-P21CIP1-mediated mitotic skip and senescence cascade, culminating to an enrichment of long-lived senescent cells (Supplement Fig. S5).
The wild type TP53 in most primary PCa [44] would be advantageous for bufalin-induction of senescence as a means to intercept PCa progression. Whereas our NSG SCID mice lacked key lymphocytes that had largely precluded a contribution of bufalin-induced immune responses, it is prudent to speculate possible ramifications of bufalin-activation of P53-senesence in immune competent hosts for PCa therapy or chemoprevention. As noted above, Tp53 gene restoration in liver tumors led to senescent cells producing various inflammatory cytokines and chemokines (known as senescence associated secretory phenotype, SASP) that activated innate immune cells including NK cells, macrophages and neutrophils [40]. The innate immune response was indispensable because blocking these immune cells significantly abolished preventive action of Tp53 gene reactivation [40]. A recent study shows that P53-expressing senescent cells secrete factors that promote macrophage activation towards tumor-inhibiting M1-state, whereas P53-deficient cells secrete those that skew macrophage activation towards tumor-promoting M2-state [45]. Our top-upregulated gene CYR61/CCN1 by bufalin (Table 1) encodes a matricellular protein that is secreted by tumor cells and stimulates macrophage activation towards tumor-inhibiting M1-state [46], suggesting a potential for macrophage activation towards M1-state by bufalin. Given that bufalin induces a P53-mediated senescence program, it is likely that bufalin may lead to even greater tumor suppression in immune competent hosts through coordination of cellular senescence (growth arrest) and innate immune responses (tumor clearance). Such hypothesis should be tested in immune-competent animal cancer models.
A recent paper addressed the poor aqueous solubility and cardiotoxicity of bufalin with chemical modification through phosphorylation of its 3-OH [14]. The authors showed that 3-phospho-bufalin was a water soluble prodrug suitable for in vivo administration and significantly improved the safety margin while effectively inhibited tumor growth in an orthotopic triple-negative breast cancer xenograft model. The development of 3-phospho-bufalin broadens the potential application of bufalin in cancer chemointerception and therapy for prostate and other organs.
In conclusion, our data suggest a probable primary in vivo mechanism of PCa suppression by bufalin through inducing P53-mediated cellular senescence. With the availability of safer 3-phospho-bufalin as pro-drug to deliver bufalin [14] or possible future targeted delivery through novel bufalin formulations, it may be practical to apply bufalin in adjuvant therapy of PCa recurrence in patients or the chemointerception of precancerous prostate lesions.
Supplementary Material
Acknowledgements
The work was supported by institutional start-up funds at Texas Tech University Health Sciences Center School of Pharmacy (J. Lu) and Penn State Hershey College of Medicine (J. Lu) and, in part, by National Institutes of Health National Cancer Institute grant R01 CA172169 (J. Lu). The authors thank Jinhui Zhang, PhD for guidance and assistance with microarray analyses and Deepkamal Karelia, PhD for assistance with immunoblot analyses of DU145 and PC-3 cells. They thank Yuka Imamura, PhD and the Penn State Cancer Institute Genomic Sciences Core facility for next-gen sequencing authentication of cell line and veterinarian pathologist Timothy Cooper, DVM, PhD of Department of Comparative Medicine at Penn State University Hershey College of Medicine for evaluating xenograft tumor histology sections.
Abbreviations
- AR
androgen receptor
- ADT
androgen deprivation therapy
- CRPC
castration resistant prostate cancer
- DDR
DNA damage response
- FBS
fetal bovine serum
- MTD
maximally-tolerated dose
- PBS
phosphate buffered saline
- PCa
Prostate cancer
- PSA
prostate specific antigen
- SA-β-gal
senescence-associated-β-galactosidase
- SASP
senescence associated secretory phenotype
Footnotes
Conflict of interest statement: The authors declare no potential conflicts of interest.
References
- 1.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Jr., Saad F et al. : Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 2011, 364(21):1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND et al. : Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012, 367(13):1187–1197. [DOI] [PubMed] [Google Scholar]
- 3.Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jones JA Jr, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M et al. : Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med 2004, 351(15):1513–1520. [DOI] [PubMed] [Google Scholar]
- 4.de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, Gravis G, Bodrogi I, Mackenzie MJ, Shen L et al. : Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 2010, 376(9747):1147–1154. [DOI] [PubMed] [Google Scholar]
- 5.Yin S, Jiang P, Ye M, Hu H, Lu J, Jiang C: A Critical Assessment of Anti-cancer Activities of Bufadienolides. Horizons in Cancer Research 2013, 52:63–88. [Google Scholar]
- 6.Meng Z, Yang P, Shen Y, Bei W, Zhang Y, Ge Y, Newman RA, Cohen L, Liu L, Thornton B et al. : Pilot study of huachansu in patients with hepatocellular carcinoma, nonsmall-cell lung cancer, or pancreatic cancer. Cancer 2009, 115(22):5309–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeh JY, Huang WJ, Kan SF, Wang PS: Effects of bufalin and cinobufagin on the proliferation of androgen dependent and independent prostate cancer cells. Prostate 2003, 54(2):112–124. [DOI] [PubMed] [Google Scholar]
- 8.Watabe M, Masuda Y, Nakajo S, Yoshida T, Kuroiwa Y, Nakaya K: The Cooperative Interaction of Two Different Signaling Pathways in Response to Bufalin Induces Apoptosis in Human Leukemia U937 Cells. Journal of Biological Chemistry 1996, 271(24):14067–14073. [DOI] [PubMed] [Google Scholar]
- 9.Jing Y, Ohizumi H, Kawazoe N, Hashimoto S, Masuda Y, Nakajo S, Yoshida T, Kuroiwa Y, Nakaya K: Selective inhibitory effect of bufalin on growth of human tumor cells in vitro: association with the induction of apoptosis in leukemia HL-60 cells. Japanese Journal of Cancer Research 1994, 85(6):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang L, Nakaya K, Yoshida T, Kuroiwa Y: Bufalin as a potent inducer of differentiation of human myeloid leukemia cells. Biochem Biophys Res Commun 1991, 178(2):686–693. [DOI] [PubMed] [Google Scholar]
- 11.Yu CH, Kan SF, Pu HF, Jea Chien E, Wang PS: Apoptotic signaling in bufalin- and cinobufagin-treated androgen-dependent and -independent human prostate cancer cells. Cancer Sci 2008, 99(12):2467–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhai XF, Fang FF, Liu Q, Meng YB, Guo YY, Chen Z: MiR-181a contributes to bufalin-induced apoptosis in PC-3 prostate cancer cells. BMC Complement Altern Med 2013, 13:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong Y, Yin S, Li J, Jiang C, Ye M, Hu H: Bufadienolide compounds sensitize human breast cancer cells to TRAIL-induced apoptosis via inhibition of STAT3/Mcl-1 pathway. Apoptosis 2011, 16(4):394–403. [DOI] [PubMed] [Google Scholar]
- 14.Song X, Zhang C, Zhao M, Chen H, Liu X, Chen J, Lonard DM, Qin L, Xu J, Wang X et al. : Steroid Receptor Coactivator-3 (SRC-3/AIB1) as a Novel Therapeutic Target in Triple Negative Breast Cancer and Its Inhibition with a Phospho-Bufalin Prodrug . PLoS One 2015, 10(10):e0140011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill TG, Wang J, Qi R, Matzuk AJ, Song X, Madoux F et al. : Bufalin is a potent small-molecule inhibitor of the steroid receptor coactivators SRC-3 and SRC-1. Cancer Res 2014, 74(5):1506–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen A, Yu J, Zhang L, Sun Y, Zhang Y, Guo H, Zhou Y, Mitchelson K, Cheng J: Microarray and biochemical analysis of bufalin-induced apoptosis of HL-60 Cells . Biotechnol Lett 2009, 31(4):487–494. [DOI] [PubMed] [Google Scholar]
- 17.Watson PA, Arora VK, Sawyers CL: Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 2015, 15(12):701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tian HY, Yuan XF, Jin L, Li J, Luo C, Ye WC, Jiang RW: A bufadienolide derived androgen receptor antagonist with inhibitory activities against prostate cancer cells. Chem Biol Interact 2014, 207:16–22. [DOI] [PubMed] [Google Scholar]
- 19.Yuan XF, Tian HY, Li J, Jin L, Jiang ST, Liu KW, Luo C, Middleton DA, Esmann M, Ye WC et al. : Synthesis of bufalin derivatives with inhibitory activity against prostate cancer cells. Nat Prod Res 2014, 28(11):843–847. [DOI] [PubMed] [Google Scholar]
- 20.Bieging KT, Mello SS, Attardi LD: Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 2014, 14(5):359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye M, Qu G, Guo H, Guo D: Novel cytotoxic bufadienolides derived from bufalin by microbial hydroxylation and their structure-activity relationships. J Steroid Biochem Mol Biol 2004, 91(1–2):87–98. [DOI] [PubMed] [Google Scholar]
- 22.Veldscholte J, Ris-Stalpers C, Kuiper GG, Jenster G, Berrevoets C, Claassen E, van Rooij HC, Trapman J, Brinkmann AO, Mulder E: A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun 1990, 173(2):534–540. [DOI] [PubMed] [Google Scholar]
- 23.Isaacs WB, Carter BS, Ewing CM: Wild-type p53 suppresses growth of human prostate cancer cells containing mutant p53 alleles. Cancer Res 1991, 51(17):4716–4720. [PubMed] [Google Scholar]
- 24.Carroll AG, Voeller HJ, Sugars L, Gelmann EP: p53 oncogene mutations in three human prostate cancer cell lines. Prostate 1993, 23(2):123–134. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Shaik AA, Xing C, Chai Y, Li L, Zhang J, Zhang W, Kim SH, Lu J, Jiang C: A synthetic decursin analog with increased in vivo stability suppresses androgen receptor signaling in vitro and in vivo. Invest New Drugs 2012, 30(5):1820–1829. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Kim KH, Zhang W, Guo Y, Kim SH, Lu J: Galbanic acid decreases androgen receptor abundance and signaling and induces G1 arrest in prostate cancer cells. Int J Cancer 2012, 130(1):200–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Won SH, Jiang C, Lee HJ, Jeong SJ, Lee EO, Zhang J, Ye M, Kim SH, Lu J: Tanshinones from Chinese medicinal herb Danshen (Salvia miltiorrhiza Bunge) suppress prostate cancer growth and androgen receptor signaling. Pharm Res 2012, 29(6):1595–1608. [DOI] [PubMed] [Google Scholar]
- 28.Noppe G, Dekker P, de Koning-Treurniet C, Blom J, van Heemst D, Dirks RW, Tanke HJ, Westendorp RG, Maier AB: Rapid flow cytometric method for measuring senescence associated beta-galactosidase activity in human fibroblasts. Cytometry A 2009, 75(11):910–916. [DOI] [PubMed] [Google Scholar]
- 29.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O: Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 2009, 4(12):1798–1806. [DOI] [PubMed] [Google Scholar]
- 30.Han KQ, Huang G, Gu W, Su YH, Huang XQ, Ling CQ: Anti-tumor activities and apoptosis-regulated mechanisms of bufalin on the orthotopic transplantation tumor model of human hepatocellular carcinoma in nude mice. World J Gastroenterol 2007, 13(24):3374–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Y, Srinivasan B, Xing C, Lu J: A new chalcone derivative (E)-3-(4-methoxyphenyl)-2-methyl-1-(3,4,5-trimethoxyphenyl)prop-2-en-1-one suppresses prostate cancer involving p53-mediated cell cycle arrests and apoptosis. Anticancer Res 2012, 32(9):3689–3698. [PubMed] [Google Scholar]
- 32.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R et al. : PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275(5308):1943–1947. [DOI] [PubMed] [Google Scholar]
- 33.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W et al. : Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436(7051):725–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, Guo X, Chang LJ, You MJ, Koochekpour S et al. : Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep 2014, 8(5):1461–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campisi J: Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2005, 120(4):513–522. [DOI] [PubMed] [Google Scholar]
- 36.Campisi J, d’Adda di Fagagna F: Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007, 8(9):729–740. [DOI] [PubMed] [Google Scholar]
- 37.Lossaint G, Besnard E, Fisher D, Piette J, Dulic V: Chk1 is dispensable for G2 arrest in response to sustained DNA damage when the ATM/p53/p21 pathway is functional. Oncogene 2011, 30(41):4261–4274. [DOI] [PubMed] [Google Scholar]
- 38.Rufini A, Tucci P, Celardo I, Melino G: Senescence and aging: the critical roles of p53. Oncogene 2013, 32(43):5129–5143. [DOI] [PubMed] [Google Scholar]
- 39.Wang X, Zeng L, Wang J, Chau JF, Lai KP, Jia D, Poonepalli A, Hande MP, Liu H, He G et al. : A positive role for c-Abl in Atm and Atr activation in DNA damage response. Cell Death Differ 2011, 18(1):5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW: Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445(7128):656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T: Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445(7128):661–665. [DOI] [PubMed] [Google Scholar]
- 42.Krenning L, Feringa FM, Shaltiel IA, van den Berg J, Medema RH: Transient activation of p53 in G2 phase is sufficient to induce senescence. Mol Cell 2014, 55(1):59–72. [DOI] [PubMed] [Google Scholar]
- 43.Johmura Y, Shimada M, Misaki T, Naiki-Ito A, Miyoshi H, Motoyama N, Ohtani N, Hara E, Nakamura M, Morita A et al. : Necessary and sufficient role for a mitosis skip in senescence induction. Mol Cell 2014, 55(1):73–84. [DOI] [PubMed] [Google Scholar]
- 44.Cancer Genome Atlas Research N: The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163(4):1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V et al. : Non-cell-autonomous tumor suppression by p53. Cell 2013, 153(2):449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bai T, Chen CC, Lau LF: Matricellular protein CCN1 activates a proinflammatory genetic program in murine macrophages. J Immunol 2010, 184(6):3223–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.