

Abstract
Camptothecin (CPT) has been shown to block disassembly of the topoisomerase-I (Topo I)/DNA cleavable complex. However, the poor aqueous solubility, intrinsic instability and severe toxicity of CPTs have limited their clinical applications. Herein, we report the design and synthesis of H2O-soluble and orally bioavailable hexacyclic CPT derivatives. By analysis of a virtual chemical library and cytotoxicity screening in vitro, 9 and 11 were identified as potential pro-drugs and chosen for further characterization in vivo. Both compounds exhibited remarkable anticancer and anti-inflammation efficacies in animals, and improved drug-like profiles.
Keywords: DNA Topoisomerase I, Camptothecin, Sepsis
Graphical Abstract
INTRODUCTION
Half a century has passed since the structure of Camptothecin (CPT), a plant alkaloid originally extracted from the Chinese tree, Camptotheca acuminata, was identified. CPT was first isolated in 1958 by Wall and Wani and has been demonstrated to be an effective agent for cancer chemotherapy.1 However, the early clinical trials of CPT were all terminated due to its poor solubility and stability as well as unpredictable severe toxicity.2-4 In addition, the highly electrophilic lactone E ring of CPT derivatives could rapidly undergo hydrolysis to an inactive, H2O-soluble carboxylic acid which is ionized at physiological pH (shown in Scheme 1).5-8
Scheme 1.

Equilibrium of between the lactone form and the open carboxylate form of camptothecin.
In the late 1980s, CPT was found to be capable of inhibiting the DNA topoisomerase-I (Topo I) protein, a finding which resurrected this field.9-15 Results indicated that CPT can bind to the transient Topo I/DNA cleavage complex resulting in Topo I-mediated DNA breakage, which subsequently disturbs cellular processes such as replication, transcription and DNA repair, and finally causing cell death. Mutation experiments and X-ray crystallography both confirmed that CPT interacts with three key amino residues of Topo I (Asn722, Arg364 and Asp533) as well as with DNA base pairs.16, 17 Non-covalent interactions between CPT and Topo I/DNA binary complex are formed while base stacking interactions and hydrogen bond interactions also play vital roles in the stabilized ternary complex (Figure 1).
Figure 1.

Binding patterns of CPT in DNA-Topo I-CPT ternary complex (PDB entry: 1T8I). TGP is a sulfhydryl modified guanosine. DA, DT and DG represent DNA adenosine, thymidine and guanosine, respectively.
Due to challenges in production and derivatization of natural products, a large number of investigations on semi-synthesis and total synthesis of CPT analogs have been reported. These have stimulated exhaustive SAR studies aiming at improving both solubility and stability. Most successes were achieved by introducing multiple substituents on the A and B rings, specifically at carbons 7, 9, 10 and 11 (Scheme 2). This exercise led to two molecules, topotecan and irinotecan, which were ultimately approved by FDA for the treatment of various cancers, including colon cancer and ovarian cancer.18-23 Nevertheless, the toxicity associated with both drugs is severe, and includes, but is not limited to gastrointestinal toxicity, myelosuppression and other side effects,24 which constrains the doses that can be safely administered. As a result, there has been long, but unsuccessful search for a better analogue with improved oral bioavailability, attenuated toxicity profile and a superior therapeutic window.
Scheme 2.

Multi-substitution at 7, 9, 10 and 11 positions.
CPTs have long been used as anticancer agents, but a recent study indicates that blockage of Topo I by CPT suppresses the host immune response and protects mice against death from lethal inflammation in vivo.25 Sepsis, for example, is a leading cause of death in hospitalized patients due to excessive host response to infection, and there is still a need for better treatments.26 This finding expands the possibility of CPTs as a therapy against life-threatening host response caused by bacteria and/or virus infections.
In the present work, by constructing a virtual chemical library, we rationally designed and synthesized several novel hexacyclic CPT analogs. The cellular antitumor activities of these analogs were then assessed against 15 different cancer cell lines. Two potent candidates were finally identified, exhibiting improved in vivo efficacy in a xenograft model of human liver cancer as well as a bacterial lipopolysaccharide (LPS)-induced sepsis model.
RESULTS AND DISCUSSION
From a drug-like property perspective, the solubility of CPT is poor and its in vivo stability is also low. To overcome these problems, structure-activity relationship (SAR) studies have been performed in the past decades, and led to the discovery of several semisynthetic and synthetic derivatives that are currently in clinical assessment. According to the SAR information from previous studies, the aromatic rings A and B are essential for biological activity. Saturating either ring destroys the activity even at high concentrations.27, 28 The activity is retained in hexacyclic systems, but tetracyclic or tricyclic structures show complete loss of biological activity, indicating that at least a pentacyclic ring structure is required to preserve the activity. No activity is observed when the D ring is replaced by a benzene ring.29 Hydrolysis of the lactone E ring leads to a notable decrease in anticancer activity.30 Optimizations of ring A in many cases however, improve the activity of CPT, while modifications of the remainder of CPT are less acceptable.31
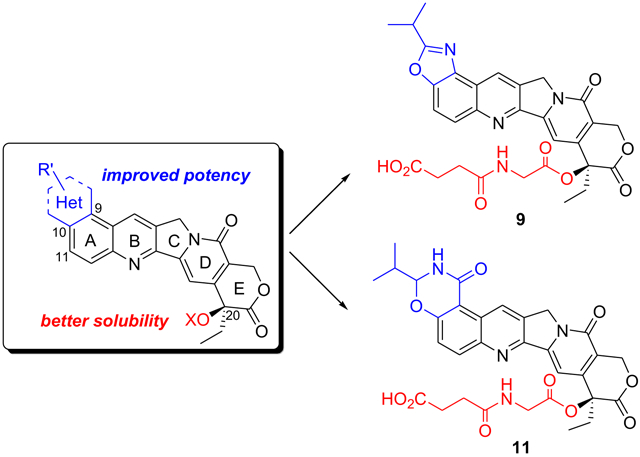
The potential of hexacyclic CPT derivatives, in which position 9 and position 10 of ring A are linked via a five- or six-membered heterocycle, was further explored in the light of the above information. In our effort to design novel hexacyclic CPTs, we first constructed a virtual chemical library in which 5- or 6-membered ring fragments were fused onto the 9,10 positions of CPT, and 682 hexacyclic CPTs were enumerated. Then all the generated compounds were docked and ranked by the Glide module in Schrodinger 9.0.32 Compounds 1, 2 and 3 (see Chart 1) were chosen from 30 top-ranked molecules based on the ease of synthesizing these compounds and potential drug-likeness properties, and were synthesized for subsequent biological assessment. In addition, further substitutions on the 1,3-oxazole ring of compound 2, yielding compounds 4 - 7 were explored to determine the preferred sidechains. As mentioned above, the in vivo hydrolysis of the lactone E ring results in a marked decrease in activity. Many studies have reported esterification of CPT as an effective pro-drug strategy. Giovanella and colleagues validated that esterification of CPT at 20-OH could stabilize the lactone ring.33, 34 The enhanced stability and solubility by ester-CPT derivatives help to prolong circulatory retention and improve tumor accumulation, which undoubtedly lead to improved activity in vivo.35-37 To enhance the stability of the E ring and at the same time improve compound solubility and bioavailability, modifications on the lactone ring were carried out by introducing a similar and well-known hydrophilic chain structure to the 20-OH group of compounds 1 to 4, producing analogues 8 to 11.
Chart 1.

Structures and synthesis of hexacyclic CPT derivatives.
In Vitro Anticancer Activity of Hexacyclic CPTs.
The cytotoxicity of all the synthesized hexacyclic CPT derivatives was evaluated against eleven human cancer cell lines (A549, MCF-7, MDA-MB-231, HepG-2, KB, BEL-7402, SMMC-7721, MGC80-3, SK-N-SH, QGY-7703 and SGC-7901) by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. These cell lines cover different types of human cancers, including lung cancer, breast cancer, hepatoma, oral cutaneous carcinoma, gastric cancer and neuroblastoma. The FDA-approved drug, topotecan, was used as a reference inhibitor. The results of the in vitro cytotoxicity of all the compounds are summarized in Table 1. Most of the designed hexacyclic CPTs exhibit anti-proliferative activity comparable or superior to that of topotecan. The cytotoxicity against two cancer cell types, HepG-2 and MGC80-3, is obviously higher compared with that of other cell lines. In particular, compound 4 can potently inhibit both HepG-2 and MGC80-3 at very low concentrations, and the inhibitory IC50 values of compound 4 (0.016 and 0.018 μM, respectively) are ~10-fold lower than those of topotecan (0.237 μM and 0.190 μM, respectively). The inhibitory IC50 values of compound 2 against HepG-2 and MGC80-3 are 0.180 and 0.231 μM, respectively, which are >10-fold higher than those of compound 4, indicating that substitution in the 1,3-oxazole ring by an isopropyl group significantly increases the activity. The purpose of modifying the 20-position hydroxyl of the E ring is to improve H2O solubility and in vivo bioavailability, so even though the in vitro activities of compounds 9 to 11 against HepG-2 are slightly decreased compared with those of the unmodified compounds, and may be caused by changes in cellular permeability and/or different sensitivity to transporter proteins (the expressions are distinct in various cells) that pump foreign substances out of cells, the in vivo activities of these compounds were expected to be improved.
Table 1.
Anticancer activity of hexacyclic CPTs against eleven human cancer cell lines.
| Cell line | Compound IC50 (μM) a | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Topotecan | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
| A549 | 1.424 | 1.028 | 2.057 | 2.169 | 1.394 | 1.124 | 3.371 | 1.502 | 0.092 | 0.170 | 1.832 | 6.472 |
| MCF-7 | 2.135 | 1.157 | 2.314 | 10.846 | 4.640 | 2.135 | 7.865 | 6.438 | > 10 | > 10 | > 10 | > 10 |
| MDA-MB-231 | 1.898 | 0.488 | 1.542 | 1.323 | 1.439 | 0.562 | 2.135 | 1.996 | 0.348 | 0.425 | 0.641 | 5.016 |
| HepG-2 | 0.237 | 0.064 | 0.180 | 1.106 | 0.016 | 0.063 | 0.079 | 0.071 | 0.055 | 0.060 | 0.348 | 0.971 |
| KB | 2.610 | 3.342 | 2.571 | 9.761 | 2.204 | 2.247 | 2.202 | 2.039 | 0.916 | 0.935 | 7.143 | 7.282 |
| BEL-7402 | 7.118 | 10.54 | 2.442 | > 10 | 3.480 | 2.022 | 2.135 | 1.931 | 0.275 | 0.425 | 2.015 | > 10 |
| SMMC-7721 | 11.627 | > 10 | 3.085 | > 10 | 10.441 | > 10 | 11.011 | > 10 | > 10 | 8.503 | > 10 | > 10 |
| MGC80-3 | 0.190 | 0.514 | 0.231 | 0.390 | 0.018 | 0.085 | 0.090 | 0.054 | 0.064 | 0.062 | 0.275 | 0.971 |
| SK-N-SH | 0.093 | 0.463 | NT b | 0.325 | NT b | NT b | NT b | NT b | 0.348 | 0.51 | 0.549 | 0.194 |
| QGY-7703 | 11.864 | > 10 | 11.568 | > 10 | 11.601 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 |
| SGC-7901 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 |
Values are the mean of at least three data points.
Not Tested.
In Vivo Antitumor Activity of Compounds 9 and 11.
Preliminary in vitro cell-based evaluation demonstrated that hexacyclic CPTs showed potent activity against various cancer cell lines, especially HepG-2 and MGC80-3. Before starting in vivo studies, we first tested the aqueous solubility of compounds 9 and 11. Topotecan is a soluble analog of camptothecin, which is insoluble in H2O, but its solubility is still lower than 1 mg/mL. As shown in Figure 2a, the solubility of both compounds 9 and 11 (as sodium salts) is 5.07 and 3.40 mg/mL, respectively, much higher than that of topotecan and parental compounds 4 and 3 (< 2 μg/mL), suggesting that compounds 9 and 11 might have better therapeutic efficacy over topotecan in vivo, especially following oral administration. To test this hypothesis, the in vivo antitumor activities were assessed in Balb/C nude mice bearing an HepG-2 hepatoma xenograft. In order to determine the best doses, the maximum tolerated dose (MTD) was measured first. As can be observed in Figure 2a, both compounds 9 and 11 are tolerated better than repeated administration of topotecan. Topotecan is tolerated only up to 2 mg/kg with daily intravenous dosing for one week, while compounds 9 and 11 can be tolerated at up to 30 and 10 mg/kg dose, suggesting that the short-term toxicity of compounds 9 and 11 is lower than that of topotecan. In single intravenous administration, compounds 9 and 11 are even tolerated up to 60 and 16 mg/kg, respectively.
Figure 2.

(a) Aqueous solubility and maximum tolerated doses (MTD) of compounds 9, 11 and topotecan. The acute and short-term toxicity was determined after i.v. administration of drugs (single administration or repeated daily administration for seven days) with increasing doses to mice; (b) Balb/C nude mice bearing HepG-2 hepatoma xenografts were treated with compound 9 at i.v. doses of 15 and 30 mg/kg or topotecan at i.v. dose of 2.0 mg/kg once a week for 30 days followed by 2 weeks’ observation; (c) Nude mice bearing HepG-2 hepatoma xenografts were treated with compound 11 at oral dose of 1.3 mg/kg once a day and i.v. doses of 4.0 and 8.0 mg/kg once a week for 30 days followed by 2 weeks’ observations. Tumor volume was recorded and plotted against time, in days.
Based on the above results, the in vivo antitumor activities of topotecan, and compounds 9 and 11 in a HepG-2 xenograft model were examined using indicated dosage (Figures 2b and 2c), and the toxicities of these compounds were assessed by the observation of the body weight, general appearance and histopathologic examination. Topotecan was intravenously administrated in the highest dose (the same dose as MTD, 2 mg/kg) as a reference compound. All three compounds significantly suppress tumor growth, and both compounds 9 and 11 display dose-responsive antitumor efficacy. The final tumor growth inhibition (TGI) rates for topotecan (i.v., 2.0 mg/kg, q7d), compound 9 (i.v., 15 mg/kg, q7d) and 11 (i.v., 30 mg/kg, q7d) are 74%, 86% and 90%, respectively, suggesting that compound 9 shows superior antitumor efficacy than topotecan even at half the MTD dose (15 mg/kg). Similarly, the TGI rates of compound 11 from different doses are all higher than that of topotecan, reaching 83% (i.v., 4 mg/kg, q7d), 93% (i.v., 8 mg/kg, q7d) and 93% (p.o., 1.3 mg/kg, qd). Notably, the oral administration of compound 11 of 1.3 mg/kg gave rise to antitumor efficacy similar to that from an i.v. dose of 8 mg/kg of 11. Though given orally daily, the total oral dose is almost identical to q7d i.v. dose of 8 mg/kg, which implies that compound 11 can be absorbed well through oral administration. The data for oral ingestion of compound 9 was not collected since most mice failed to reach the end point of this experiment. The daily oral dose of compound 9 (5 mg/kg) was lower than the MTD, but the MTD was determined via i.v. injection for up to seven days. One possible explanation is that compound 9 is metabolized into (a) more toxic compound(s) via oral administration.
In order to determine the in vitro activity against Topo I, both Topo I inhibitory activity assay and Topo I-mediated DNA cleavage assay were performed. Topo I and supercoiled DNA were incubated with or without the test compounds. The inhibitory activity was detected on 1% EB-free gel which could separate super-coiled DNA from relaxed and nicked DNA. As shown in the Figures 3a and 3b, CPT can inhibit Topo I activity at concentrations of both 50 μM and 100 μM. Such inhibition is represented by the increased amount of super-coiled DNA that is not relaxed or cleaved. Compound 9 exhibits weak inhibition and compound 11 is almost inactive against Topo I at the concentrations of both 50 μM and 100 μM. Similar results are observed in the Topo I-mediated DNA cleavage assay which was carried out on EB-containing gel. CPT, topotecan and compound 9 can increase the amount of nicked DNA at concentrations of 100 μM or 50 μM, but compound 11 failed to stabilize Topo I-DNA cleavage complex at a concentration of 50 μM. Both compound 9 and 11 were hypothesized to release their parent compound 4 and 3 in vivo. As we can see in Figure 4, the activity of compound 3 against Topo I is high, but compound 4 is a weak inhibitor of Topo I which is similar to compound 9. Compound 3 is much more active than compound 11 and is even slightly more active than CPT.
Figure 3.

Topo I inhibitory activity of CPT, compound 9 and 11 at 50 μM (a), and at 100 μM (b). Samples were analyzed on 1% EB-free agarose gel. Lane 1, super-coiled DNA plasmid (DNA); lane 2, DNA + Topo I; lane 3, DNA + Topo I + CPT; lane 4, DNA + Topo I + compound 9; lane 5, DNA + Topo I + compound 11. (c) Topo I-mediated DNA cleavage gels. Samples were analyzed on 1% EB-containing agarose gel. Lane 1, DNA; lane 2, DNA + Topo I; lane 3, DNA + Topo I + 50 μM Topotecan; lane 4, DNA + Topo I + 50 μM CPT; lane 5, DNA + Topo I + 100 μM compound 9; lane 6, DNA + Topo I + 50 μM compound 9; lane 7, DNA + Topo I + 100 μM compound 11; lane 8, DNA + Topo I + 50 μM compound 11.
Figure 4.

(a) Topo I inhibitory activity of CPT, compound 4 and 3 at 50 μM. Samples were analyzed on 1% EB-free agarose gel. Lane 1, super-coiled DNA plasmid (DNA); lane 2, DNA + Topo I; lane 3, DNA + Topo I + CPT; lane 4, DNA + Topo I + compound 4; lane 5, DNA + Topo I + compound 3. (b) Topo I-mediated DNA cleavage gels. Samples were analyzed on 1% EB-containing agarose gel. Lane 1, DNA; lane 2, DNA + Topo I; lane 3, DNA + Topo I + CPT; lane 4, DNA + Topo I + compound 4; lane 5, DNA + Topo I + compound 3
Pathological anatomy of viscera of all xenograft mice was also carried out and the data collected are summarized in the Supporting Information (Table S2). The dominating side effects observed are gastrointestinal flatulence and splenomegaly. Notably, oral administration of compound 11 increases the occurrence of gastrointestinal flatulence and might affect hepatic status compared with the i.v. dosage, but these side effects have little influence on mouse survival. The body weight change of mice during treatment was recorded and plotted against time, in days in Figures 5a and 5b. It was found that the body weight of the untreated mice steadily increased over 35 days, but the body weight of mice treated with topotecan actually decreased slightly. However, treatment with compounds 9 or 11 at different doses had no major influence on body weight in comparison to treatment by topotecan, which on the other hand indicates that compounds 9 and 11 are probably less toxic than much higher doses of topotecan. Compared with topotecan, compounds 9 and 11 showed improved antitumor activity without acute and short-term in vivo toxicity.
Figure 5.

Change in body weight of the treated mice. (a) Balb/C nude mice bearing HepG-2 hepatoma xenografts were treated with compound 9 at i.v. doses of 15 or 30 mg/kg or topotecan at an i.v. dose of 2.0 mg/kg once a week for 30 days; (b) Nude mice bearing HepG-2 hepatoma xenografts were treated with compound 11 at an oral dose of 1.3 mg/kg once a day and i.v. doses of 4.0 and 8.0 mg/kg once a week for 30 days. Body weight percentage change was plotted against time, in days.
Compounds 9 and 11 Protect from Lethal Inflammation and Death In Vivo
Over the past decades, CPTs have become some of the most widely used anticancer drugs.38 In 2016, CPT was reported to suppress the expression of inflammatory genes and rescue mortality caused by excessive inflammation.25 This prompted us to characterize the effectiveness of compounds 9 and 11 in in vivo sepsis models. As shown in Figure 6a, only 20% of animals survived LPS-induced septic shock, and oral administration of 10 mg/kg compound 9 rescued 80% of animals. The protective effect of compound 11 is even better. Although it is administered in relatively lower doses, 100% of the animals orally treated with 4 mg/kg 11 were rescued (Figure 6b). Furthermore, the survival rate of the animals receiving oral administration was higher than by intravenous injection, which is in accordance with the results from in vivo antitumor models. This phenomenon possibly suggests that the compound may act as a pro-drug that undergoes hydrolysis and/or metabolic processes in vivo and the activity of the metabolites is higher, or there are other protein targets that are inhibited. In order to assess this hypothesis, the preliminary pharmacokinetic properties of compound 9 and 11 were studied. As shown in Figure 7, The concentration of both compound 9 (data not detectable) and 11 (Figure 7c) in plasma through oral administration is extremely low, and the corresponding parent compounds 4 and 3 are also not released. For i.v. administration, compound 4 is also not detected in plasma after giving 2 mg/kg compound 9, and very low concentration of compound 3 is identified after giving 2 mg/kg compound 11 intravenously. These results indicate that both compound 9 and 11 are not hydrolyzed into their parent compound 4 and 3. The exact metabolite(s) of compound 9 and 11 are unclear. In addition, the expressions of two inflammatory cytokines, IL-6 and TNF-α in mouse serum were suppressed when treated with compound 9 or 11 (Figure 6c and 5d). These data suggest that compounds 9 and 11 provide effective protection in LPS-induced models of lethal inflammation at the organism level.
Figure 6.

Compounds 9 and 11 protect against LPS-induced sepsis in vivo. (a) Survival curves of C57BL/6C mice treated with compound 9 in response to LPS injection; (b) Survival curves of mice treated with compound 11 in response to LPS injection; (c) Expression of IL-6 in mice serum; (d) Expression of TNF-α in mice serum.
Figure 7.

Pharmacokinetic properties of compound 9 and 11. Pharmokinetics were studied in male C57BL/6 mice after (a) single intravenous administration of 2 mg/kg Compound 9; (b) single intravenous administration of 2 mg/kg Compound 11; (c) single oral administration of 10 mg/kg Compound 11; (d) single intravenous administration of 10 mg/kg Compound 11. Plasma levels of the compounds were determined by LC-MS/MS analysis at the indicated time points.
CONCLUSIONS
CPT compounds are among the first classes of anticancer agents, which have been demonstrated to stabilize the Topo I/DNA complex.9 However, the poor solubility, inherent instability and unpredictable severe toxicity of CPTs are unsatisfactory24, 39 for therapeutic use. In the present work, a novel kind of hexacyclic CPT derivatives was first designed and synthesized. These compounds are very promising and exhibit favorable biological properties, including improved solubility, reduced toxicity and enhanced efficacy. An in vitro cytotoxicity assay suggests that these compounds can efficiently suppress the growth of multiple cancer cell lines, especially HepG-2 and MGC80-3. Further in vivo antitumor activities of pro-drugs 9 and 11 were determined using HepG-2 hepatoma xenograft models, and the results indicate that compounds 9 and 11 exhibit improved in vivo antitumor activity but decreased toxicity compared with topotecan. Compounds 9 and 11 can also suppress the expression of several inflammatory cytokines and rescue mortality caused by excessive inflammation in in vivo sepsis models. Compound 9 and 11 were designed based on the pro-drug concept, but the PK results indicate that both compounds are not hydrolyzed into their parent compound 4 and 3, which implies that compound 9 and 11 might convert into new active compounds to work in vivo. These data qualify compounds 9 and 11 as not only promising anticancer agents but also potential drug candidates for treatment of lethal inflammation.
EXPERIMENTAL SECTION
General Information.
All solvents were distilled according to standard methods prior to use. All reagents and chemicals were purchased and used without further purification unless specified otherwise. Solvents for flash column chromatography were technical grade and distilled prior to use. Thin-layer chromatography (TLC) was carried out on Huanghai silica gel plates with HSGF 254. Chromatograms were visualized based on UV absorbance (254 nm) with proper stains. Flash column chromatography was conducted using Qingdao Haiyang Chemical HG/T2354-92 silica gel (200-300 mesh) with the specified solvent system in the corresponding experiment. Proton (1H) NMR and carbon (13C) NMR data were recorded on Bruker nuclear resonance spectrometers (400 MHz for 1H and 100 MHz for 13C). Chemical shifts (δ) in ppm are reported relative to the residual signals of chloroform (1H 7.26 ppm and 13C 77.16 ppm). 13C NMR spectra were recorded with total proton decoupling. Multiplicities are described as: s (singlet), bs (broad singlet), d (doublet), t (triplet), q (quartet), m (multiplet); and coupling constants (J) are reported in Hertz (Hz). The purity was determined by liquid chromatography-mass spectrometry (LC-MS). The purity of all final compounds was 95% or higher.
(S)-10-Amino-4-ethyl-4,9-dihydroxy-1H-pyrano[3',4':6,7]indolizino[1,2-b]qui noline-3,14(4H,12H)-dione (S1).
65% HNO3 was added to a solution of 10-hydroxycamptothecin (180 mg, 0.5 mmol) in AcOH (1 mL). The reaction mixture was stirred at r.t. overnight. The reaction was diluted with H2O (2.5 mL) to give a brown suspension, which was filtered and washed with H2O. The resulting brown solid was suspended in MeOH (5 mL) and hydrogenated using at 50° C for 2 h using Pd/C (10% w/w, 53 mg, 0.05 mmol) and a hydrogen balloon. When TLC showed complete consumption of the starting material, the reaction mixture was filtered and washed thoroughly with MeOH. The resulting solution was concentrated to give 133 mg (70% crude yield) of compound S1 as a pale grey solid, which was used without further purification. 1H NMR (400 MHz, DMSO) δ 9.80 (bs, 1H), 8.73 (s, 1H), 7.73 – 7.61 (m, 1H), 7.37 (q, J = 9.0 Hz, 2H), 7.23 (s, 1H), 6.47 (s, 1H), 5.40 (s, 2H), 5.23 (s, 2H), 4.27 – 4.04 (m, 1H), 3.15 (d, J = 4.9 Hz, 1H), 1.90 – 1.78 (m, 2H), 0.87 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 173.00 (s), 157.35 (s), 150.52 (s), 149.43 (s), 146.60 (s), 143.94 (s), 140.70 (s), 130.02 (s), 127.44 (s), 125.60 (s), 121.74 (s), 118.70 (s), 118.43 (s), 117.58 (s), 96.19 (s), 72.86 (s), 65.69 (s), 50.59 (s), 30.68 (s), 8.22 (s).
(S)-4-Ethyl-4,9-dihydroxy-3,14-dioxo-3,4,12,14-tetrahydro-1H-pyrano[3',4':6, 7]indolizino[1,2-b]quinoline-10-carbaldehyde (S2).
A solution of 10-hydroxycamptothecin (100 mg, 0.27 mmol) and HMTA (80 mg, 0.55 mmol) in TFA (10 mL) was heated to reflux for 20 h under argon. The reaction mixture was concentrated. H2O (20 mL) was added and the mixture was stirred for 1 h. Additional H2O (20 mL) was added and the pH was adjusted to 8-9 using saturated aqueous NaHCO3 solution. The aqueous layer was washed with EtOAc (10 mL), acidified to pH ~1.5 using 2N HCl and extracted using EtOAc (20 mL*5). The combined organic phase was washed with 1N HCl, H2O and brine, dried over Na2SO4, filtered and concentrated. The residue was passed through a short plug of silica gel, thoroughly washed using a mixture of MeOH and DCM (1:50), and concentrated to give S2 (79 mg, 73% crude yield), which was used without further purification. 1H NMR (400 MHz, DMSO) δ 11.97 (bs, 1H), 10.61 (s, 1H), 9.50 (s, 1H), 8.13 (d, J = 9.3 Hz, 1H), 7.45 (d, J = 9.3 Hz, 1H), 7.15 (s, 1H), 6.46 (s, 1H), 5.36 (s, 2H), 5.08 (s, 2H), 1.94 – 1.69 (m, 2H), 0.87 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 191.65 (s), 172.90 (s), 164.91 (s), 157.07 (s), 150.29 (s), 150.15 (s), 145.61 (s), 143.13 (s), 138.74 (s), 132.72 (s), 127.21 (s), 126.73 (s), 123.57 (s), 118.93 (s), 112.88 (s), 96.48 (s), 72.77 (s), 65.68 (s), 50.80 (s), 30.79 (s), 8.21 (s).
(S)-4-Ethyl-4,9-dihydroxy-3,14-dioxo-3,4,12,14-tetrahydro-1H-pyrano[3',4':6, 7]indolizino[1,2-b]quinoline-10-carboxamide (S3).
S2 (880 mg, 2.2 mmol), hydroxylamine hydrochloride salt (400 mg, 5.8 mmol) and sodium formate (2 g, 29.4 mmol) were mixed in formic acid (200 mL). The reaction was refluxed overnight. The solvent was removed. The residue was passed through a short plug of silica gel, thoroughly washed using a mixture of MeOH and DCM (1:50) and concentrated. The residue was dissolved in TFA (4.8 mL). Concentrated H2SO4 (1.2 mL) was added slowly. The mixture was heated at 95 °C for 16 h, then cooled to r.t. and concentrated. The residue was passed through a short plug of silica gel, thoroughly washed using a mixture of MeOH and DCM (1:10), and concentrated to give S3 (69 mg, 30% yield), which was used without further purification. 1H NMR (400 MHz, DMSO) δ 11.66 (bs, 1H), 11.52 (bs, 1H), 9.30 (s, 1H), 8.85 (s, 1H), 8.01 (d, J = 9.2 Hz, 1H), 7.61 (d, J = 9.2 Hz, 1H), 7.23 (s, 1H), 6.51 (s, 1H), 5.39 (s, 2H), 5.20 (s, 2H), 1.86 (dd, J = 14.0, 6.9 Hz, 2H), 0.85 (d, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 172.92 (s), 157.33 (s), 157.25 (s), 150.48 (s), 149.83 (s), 146.48 (s), 146.01 (s), 143.79 (s), 132.61 (s), 131.07 (s), 127.64 (s), 127.11 (s), 123.15 (s), 118.79 (s), 110.08 (s), 96.50 (s), 72.82 (s), 65.69 (s), 50.75 (s), 30.72 (s), 8.23 (s).
(S)-8-Ethyl-8-hydroxy-8H-isoxazolo[4,5-f]pyrano[3',4':6,7]indolizino[1,2-b]quinoline-9,12(11H,14H)-dione (1).
Triethylamine (15 μL, 0.15 mmol) was added to a solution of S2 (50 mg, 0.13 mmol) and hydroxylamine hydrochloride (11 mg, 0.15 mmol) in EtOH (10 mL). The reaction mixture was stirred at 95 °C for 20 h. The reaction was cooled to room temperature and concentrated. The residue was dissolved in EtOAc (10 mL) and washed with H2O (15 mL). The aqueous layer was extracted with EtOAc (10 mL*2). The combined organic layer was washed with H2O and brine, dried over Na2SO4, filtered and concentrated. The residue was dissolved in THF (10 mL), and DIAD (25 mg, 0.12 mmol) was added, followed by PPh3 (32 mg, 0.12 mmol). The mixture was stirred at r.t. for 5 h. The solvent was removed and the residue was purified by silica gel column chromatography eluting with MeOH:DCM = 1:50 to give compound 1 as a pale yellow solid (26 mg, 53% yield from S2). Melting point: 228.1-229.8°C. 1H NMR (500 MHz, DMSO-d6) δ 9.89 (s, 1H), 9.12 (s, 1H), 8.33 (d, J = 9.3 Hz, 1H), 8.25 (d, J = 9.3 Hz, 1H), 7.33 (s, 1H), 6.53 (s, 1H), 5.41 (s, 2H), 5.34 (s, 2H), 1.87 (qd, J = 14.1, 7.3 Hz, 2H), 0.88 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 172.93, 161.84, 157.20, 152.07, 150.48, 147.12, 146.48, 145.74, 133.19, 132.20, 128.07, 122.44, 119.45, 116.94, 115.01, 97.04, 72.84, 65.70, 50.87, 30.77, 8.24.
(S)-8-Ethyl-8-hydroxy-8H-oxazolo[4,5-f]pyrano[3',4':6,7]indolizino[1,2-b]quinoline-9,12(11H,14H)-dione (2).
S1 (133 mg, 0.35 mmol) was dissolved in EtOH (2 mL) and triethyl orthoformate (96 mg, 0.65 mmol) was added. The reaction mixture was heated to reflux overnight under argon. The solvent was removed and the residue was purified by silica gel chromatography eluting with MeOH:DCM = 1:50 to give compound 2 as a pale yellow solid (84 mg, 62% from S1). Melting point: 162.4-163.7°C. 1H NMR (500 MHz, DMSO-d6) δ 9.10 (s, 1H), 9.03 (s, 1H), 8.31 (d, J = 9.2 Hz, 1H), 8.21 (d, J = 9.2 Hz, 1H), 7.35 (s, 1H), 6.52 (s, 1H), 5.43 (s, 2H), 5.33 (s, 2H), 1.87 (m, 2H), 0.88 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 172.90, 156.94, 155.17, 151.74, 150.27, 147.50, 146.23, 145.42, 135.07, 130.70, 127.65, 125.75, 121.10, 119.27, 115.64, 96.84, 72.79, 65.67, 50.67, 30.81, 8.25.
(9S)-9-Ethyl-9-hydroxy-3-isopropyl-2,3-dihydro-[1,3]oxazino[5,6-f]pyrano[3',4':6,7]indolizino[1,2-b]quinoline-1,10,13(9H,12H,15H)-trione (3).
pTSA monohydrate (1.4 mg, 0.0075 mmol) and isobutyraldehyde (41 uL, 0.45 mmol) were added to the solution of S3 (60 mg, 0.15 mmol) in a mixture of toluene (0.5 mL) and DMSO (0.25). The reaction mixture was stirred at 110 °C for 16 h. Toluene was removed and the DMSO solution was diluted with DCM (5 mL) and washed with H2O and brine. The organic layer was dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with MeOH:DCM = 1:50 to give compound 3 as a pale yellow solid (49 mg, 71% yield from S3). Melting point: 256.8-258.2°C. 1H NMR (400 MHz, DMSO-d6) δ 9.78 (s, 1H), 8.96 (s, 1H), 8.24 (dd, J = 9.2, 1.9 Hz, 1H), 7.56 (dd, J = 9.2, 4.3 Hz, 1H), 7.33 – 7.23 (m, 1H), 6.50 (d, J = 2.4 Hz, 1H), 5.41 (s, 2H), 5.24 (m, 3H), 2.14 (m, 1H), 1.94 – 1.78 (m, 2H), 1.09 (t, J = 6.1 Hz, 6H), 0.88 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 172.89, 163.65, 159.21, 157.17, 150.94, 150.44, 150.39, 145.72, 144.87, 136.41, 131.82, 129.33, 127.40, 122.31, 119.22, 110.53, 96.79, 88.41, 72.82, 65.69, 50.93, 30.75, 17.32, 16.35, 8.21.
(S)-8-Ethyl-8-hydroxy-2-isopropyl-8H-oxazolo[4,5-f]pyrano[3',4':6,7]indolizino[1,2-b]quinoline-9,12(11H,14H)-dione (4).
S1 (61 mg, 0.16 mmol) was dissolved in THF (1 mL) and isobutyraldehyde (17 mg, 0.24 mmol) was added. The reaction mixture was heated at 50 °C for 6 h and DDQ (55 mg, 0.24 mmol) was added in one portion. The resulting mixture was stirred at 50 °C for another 10 h. The solvent was removed and the residue was purified by silica gel chromatography eluting with MeOH:DCM = 1:50 to give compound 4 as a pale yellow solid (34 mg, 50% yield from S1). Melting point: 154.3-155.2°C. 1H NMR (300 MHz, DMSO-d6) δ 9.08 (s, 1H), 8.25 (d, J = 9.2 Hz, 1H), 8.14 (d, J = 9.2 Hz, 1H), 7.35 (s, 1H), 6.55 (s, 1H), 5.43 (s, 2H), 5.32 (s, 2H), 3.43 (d, J = 6.9 Hz, 1H), 1.87 (dd, J = 11.5, 4.5 Hz, 2H), 1.46 (d, J = 6.9 Hz, 6H), 0.88 (m, 3H). 13C NMR (75 MHz, DMSO-d6) δ 172.93, 171.85, 156.95, 151.43, 150.32, 147.93, 145.94, 145.50, 135.89, 130.32, 126.62, 125.67, 120.71, 119.13, 115.17, 96.85, 72.82, 65.63, 50.64, 30.76, 28.75, 20.52, 8.23.
(S)-2-Butyl-8-ethyl-8-hydroxy-8H-oxazolo[4,5-f]pyrano[3',4':6,7]indolizno[1,2-b]quinoline-9,12(11H,14H)-dione (5).
This compound was synthesized according to the same procedure used for compound 4 using S1 and valeraldehyde (pale yellow solid, 58 mg, 82% yield). Melting point: 151.1-153.2°C. 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 8.13 (d, J = 9.1 Hz, 1H), 8.03 (d, J = 9.1 Hz, 1H), 7.27 (s, 1H), 6.52 (s, 1H), 5.40 (s, 2H), 5.20 (s, 2H), 3.04 (t, J = 7.4 Hz, 2H), 1.85 (dd, J = 15.3, 7.7 Hz, 4H), 1.44 (dd, J = 14.7, 7.3 Hz, 2H), 0.95 (t, J = 7.3 Hz, 3H), 0.89 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 172.95, 168.26, 157.18, 151.91, 150.43, 148.20, 146.24, 145.82, 136.31, 130.86, 126.74, 126.01, 120.99, 119.31, 115.42, 96.94, 72.85, 65.71, 50.90, 30.78, 28.78, 28.06, 22.12, 14.06, 8.24.
(S)-8-Ethyl-8-hydroxy-2-isobutyl-8H-oxazolo[4,5-f]pyrano[3',4':6,7]indolizno[1,2-b]quinoline-9,12(11H,14H)-dione (6).
This compound was synthesized according to the same procedure for 4 using S1 and isovaleraldehyde (pale yellow solid, 53 mg, 75% yield). Melting point: 150.8-152.5°C. 1H NMR (400 MHz, DMSO-d6) δ 8.98 (s, 1H), 8.16 (d, J = 9.1 Hz, 1H), 8.06 (d, J = 9.2 Hz, 1H), 7.29 (s, 1H), 6.52 (s, 1H), 5.41 (s, 2H), 5.24 (s, 2H), 2.94 (d, J = 7.1 Hz, 2H), 2.27 (dt, J = 13.5, 6.7 Hz, 1H), 1.92 – 1.82 (m, 2H), 1.03 (d, J = 6.6 Hz, 6H), 0.89 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 172.95, 167.51, 157.21, 152.00, 150.45, 148.25, 146.31, 145.87, 136.36, 130.97, 126.80, 126.10, 121.07, 119.35, 115.48, 96.97, 72.86, 65.71, 50.95, 37.19, 30.78, 27.51, 22.63, 8.25.
(S)-8-Ethyl-8-hydroxy-2-(pyridin-2-yl)-8H-oxazolo[4,5-f]pyrano[3',4':6,7]indolizino[1,2-b]quinoline-9,12(11H,14H)-dione (7).
This compound was synthesized according to the same procedure for 4 using S1 and picolinaldehyde (dark red solid, 16 mg, 22% yield). Melting point > 300°C. 1H NMR (300 MHz, DMSO-d6) δ 9.16 (s, 1H), 8.83 (s, 1H), 8.44 – 8.32 (m, 2H), 8.23 (d, J = 9.3 Hz, 1H), 8.10 (s, 1H), 7.66 (s, 1H), 7.35 (s, 1H), 6.54 (s, 1H), 5.42 (s, 2H), 5.35 (s, 2H), 1.86 (dd, J = 12.0, 6.6 Hz, 2H), 0.89 (t, J = 7.0 Hz, 3H). Due to the poor solubility of this compound, 13C NMR could not be obtained using common NMR solvents.
(S)-4-((2-((8-Ethyl-9,12-dioxo-9,11,12,14-tetrahydro-8H-oxazolo[4,5-f]pyrano[3',4':6,7]indolizino[1,2-b]quinolin-8-yl)oxy)-2-oxoethyl)amino)-4-oxobutanoic acid (8).
A solution of compound 2 (101 mg, 0.26 mmol), Sc(OTf)3 (79 mg, 0.16 mmol) and DMAP (94 mg, 0.78 mmol) in dry DCM (2 mL) was stirred at room temperature for 30 min then Boc-Gly-OH (136 mg, 0.78 mmol) was added in one portion. The resulting mixture was stirred at room temperature for 30 min before DCC (272 mg, 1.3 mmol) was added. The reaction was stirred at room temperature overnight, filtered through celite and concentrated. The residue was passed through a short plug of silica gel, washed using a mixture of MeOH and DCM (1:100), and concentrated. The residue was dissolved in DCM (2 mL) and TFA (1 mL) was added dropwise. The reaction mixture was stirred at rt for 30 min and concentrated. The residue was dissolved in DMF (2 mL). Succinic anhydride (97 mg, 0.97 mmol) and 4-methylpyridine (73 mg, 0.81 mmol) were added sequentially. The reaction was stirred at rt overnight. Removal of the solvent and purification using silica gel chromatography (elutent: MeOH:DCM = 1:50) afforded compound 8 as a pale yellow solid (97 mg, 68% yield). Melting point: 182.3-184.1°C. 1H NMR (400 MHz, DMSO-d6) δ 12.08 (s, 1H), 9.02 (s, 1H), 8.98 (s, 1H), 8.43 (t, J = 5.7 Hz, 1H), 8.23 (d, J = 9.2 Hz, 1H), 8.15 (d, J = 9.2 Hz, 1H), 7.13 (s, 1H), 5.50 (s, 2H), 5.35 – 5.21 (m, 2H), 4.19 (dd, J = 17.0, 5.8 Hz, 1H), 4.02 (dd, J = 17.0, 5.8 Hz, 1H), 2.49-2.37 (m, 4H), 2.22 – 2.13 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 174.14, 174.05, 172.06, 169.58, 167.51, 156.92, 155.34, 152.02, 147.77, 146.52, 146.32, 145.58, 135.33, 131.22, 127.86, 126.11, 121.53, 119.36, 115.90, 95.59, 76.66, 66.80, 50.95, 30.92, 30.16, 29.39, 29.26, 7.98.
(S)-4-((2-((8-Ethyl-2-isopropyl-9,12-dioxo-9,11,12,14-tetrahydro-8H-oxazolo[4,5-f]pyrano[3',4':6,7]indolizno[1,2-b]quinolin-8-yl)oxy)-2-oxoethyl)amino)-4-oxobutanoic acid (9).
This compound was synthesized according to the same procedure for 8 from 4 and was obtained as a pale yellow solid (66 mg, 43% yield). Melting point: 194.8.8-196.4°C. 1H NMR (400 MHz, DMSO) δ 12.07 (s, 1H), 9.06 (s, 1H), 8.44 (s, 1H), 8.21 (d, J = 9.0 Hz, 1H), 8.13 (d, J = 8.9 Hz, 1H), 7.15 (s, 1H), 5.50 (s, 2H), 5.30 (s, 2H), 4.22 – 4.14 (m, 1H), 4.03 −3.97 (m, 1H), 3.43 – 3.38 (m, 1H), 2.43 (d, J = 5.5 Hz, 2H), 2.38 (s, 2H), 2.16 (d, J = 6.5 Hz, 2H), 1.46 (d, J = 6.8 Hz, 6H), 0.93 (d, J = 6.7 Hz, 3H). 13C NMR (101 MHz; DMSO-d6) δ 7.96, 20.66, 28.84, 29.38, 30.14, 30.86, 50.97, 66.80, 76.65, 95.57, 115.65, 119.29, 121.29, 126.20, 126.83, 131.05, 136.29, 145.53, 146.35, 146.43, 148.31, 151.92, 156.97, 167.56, 169.57, 172.03, 172.22, 174.16.
(S)-4-((2-((8-Ethyl-9,12-dioxo-9,11,12,14-tetrahydro-8H-isoxazolo[4,5-f]pyrano[3',4':6,7]indolizino[1,2-b]quinolin-8-yl)oxy)-2-oxoethyl)amino)-4-oxobutanoic acid (10).
This compound was synthesized according to the same procedure for 8 from 1 (pale yellow solid, 106 mg, 75% yield). Melting point: 201.5. −203.5°C. 1H NMR (400 MHz, DMSO-d6) δ 12.06 (s, 1H), 9.90 (s, 1H), 9.11 (s, 1H), 8.44 (t, J = 5.1 Hz, 1H), 8.32 (t, J = 10.3 Hz, 1H), 8.25 (d, J = 9.4 Hz, 1H), 7.15 (s, 1H), 5.48 (d, J = 8.6 Hz, 2H), 5.34 (s, 2H), 4.17 (dd, J = 17.8, 5.8 Hz, 1H), 4.00 (dd, J = 17.9, 5.6 Hz, 1H), 2.42 (t, J = 4.5 Hz, 2H), 2.40 – 2.35 (m, 2H), 2.16 (dd, J = 14.7, 7.4 Hz, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz; DMSO-d6) δ 7.96, 29.36, 30.12, 30.88, 50.81, 66.77, 76.63, 95.60, 115.00, 116.94, 119.35, 122.50, 128.07, 132.17, 133.08, 145.58, 146.23, 146.44, 147.11, 151.88, 156.89, 161.85, 167.50, 169.58, 172.07, 174.12.
4-((2-(((9S)-9-Ethyl-3-isopropyl-1,10,13-trioxo-1,2,3,9,10,12,13,15-octahydro-[1,3]oxazino[5,6-f]pyrano[3',4':6,7]indolizino[1,2-b]quinolin-9-yl)oxy)-2-oxoethyl)amino)-4-oxobutanoic acid (11).
This compound was synthesized according to the same procedure for 8 from 3 (pale yellow solid, 74 mg, 46% yield). Melting point: 222.4.−224.8°C. 1H NMR (400 MHz, DMSO-d6) δ 12.05 (s, 1H), 9.81 (d, J = 4.3 Hz, 1H), 8.99 (s, 1H), 8.42 (t, J = 6.0 Hz, 1H), 8.28 (d, J = 9.2 Hz, 1H), 7.59 (d, J = 9.3 Hz, 1H), 7.11 (s, 1H), 5.48 (s, 2H), 5.33 (t, J = 8.9 Hz, 2H), 5.21 (s, 1H), 4.15 (dd, J = 18.1 5.9 Hz, 1H), 3.99 (dd, J = 17.9, 5.4 Hz, 1H), 2.46 – 2.41 (m, 2H), 2.36 (dd, J = 12.1 5.7 Hz, 2H), 2.14 (d, J = 5.3 Hz, 3H), 1.09 (t, J = 6.0 Hz, 6H), 0.93 – 0.86 (m, 3H). 13C NMR (101 MHz; DMSO-d6) δ 7.97, 16.36, 17.34, 29.34, 30.09, 30.74, 30.83, 51.03, 66.78, 76.63, 88.44, 95.42, 110.66, 119.17, 122.50, 127.57, 129.46, 132.00, 136.43, 144.93, 145.50, 146.30, 150.94, 156.95, 159.30, 163.70, 167.55, 169.57, 172.01 174.16.
Molecular Modeling.
Construction of a virtual chemical library of CPT derivatives and all docking simulations were performed in Schrödinger 9.0.32 The brief workflow of construction and screening of virtual library is depicted in Figure S1. The first step is to generate five-membered and six-membered ring structures from small molecule drug database which was obtained from the BindingDB database.40 Enumeration of a virtual library was carried out based on the Interactive Enumeration and Docking module in Schrodinger 9.0, where CPT was set as a core-containing molecule and the ring fragment collections were incorporated into the 9- and 10-position of CPT. Preparations of the initial structure of Topo I/DNA complex (PDB codes: 1T8I) were performed using the Protein Preparation wizard module. All H2O molecules were deleted from the system and the missing hydrogen atoms were added. A restrained minimization was subsequently conducted, and the receptor grid for docking simulations was produced and centered on the original CPT based on the Receptor grid generation module. The scaling factor for van der Waals radii and partial atomic charge cutoff value were set to 0.8 and 0.15, respectively. Preparations of CPT ligands were all performed by the LigPrep module with protonated states generated at pH = 7.0 ± 2.0. Finally, all the compounds were docked into the binding pocket of Topo I/DNA complex using the Glide module with extra precision (XP).
Cell-based Cytotoxicity Assays.
Cells were collected and re-suspended in RPMI-1640 (or DMEM) growth media + 10% fetal bovine serum (FBS). The cells were seeded on 96-well plates at the concentration of 10,000 cells/well and incubated at 37 °C in 5% CO2 overnight. The following day, compounds with serial dilutions were added into the corresponding wells, and the plates were further incubated at 37 °C for 48 h. The number of living cells was characterized using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay by adding 5 mg/ml MTT into the wells, and the plates were then cultured for 4 h. The converted dye was then dissolved in MTT buffer overnight, and the absorbance of each well was finally measured at 570 nm with the control wavelength set to 650 nm. The half-inhibitory concentration (IC50) of the compound was calculated based on the fitted dose-response curve.
Topo I Inhibitory Activity and DNA Cleavage Assay.
Both the inhibitory activity and Topo I-mediated cleavage assays were carried out using the topoisomerase I drug screening kit (TopoGEN, Inc.) The reaction mixture (20 μl) contained 10 mM Tris-HCl (pH 7.9), 1 mM EDTA, pHOT1 plasmid DNA (250 ng), Topo I (7 units for inhibitory assay and 10 units for cleavage assay) and indicated drug concentrations (1% DMSO). Topo I reaction mixtures were incubated at 37°C for 30 min. Reactions were terminated by the addition of 2 μl 10% SDS, digested with 50 μg/ml proteinase K for 30 min and extracted with phenol/chloroform/isoamyl alcohol (25:24:1). After adding 2 μl 10×gel loading buffer (0.25% bromophenol blue, 50% glycerol), the samples were loaded onto the agarose gel. Two types of agarose gels (1%) were prepared. The Topo I inhibitory gel was prepared in the absence of EB (ethidium bromide), and the cleavage gel was prepared with 0.5 μg/mL EB. Gels were run at 2 V/cm for 2.5 h in 1×TAE running buffer. After electrophoresis, the EB-free gels were stained with 0.5 μg/mL EB for 25 min and de-stained in H2O for 15 min prior to photodocumentation. All the experiments were carried out independently more than three times and the representative gels were shown.
In Vivo Xenograft Studies.
Experiments were carried out by Suzhou Xishan Zhongke Drug Research & Development (China) Co., Ltd., and all in vivo experiments were approved and supervised by institutional animal care and use committee (Jiangsu Province, PR China) and carried out according to the guidance for the care and use of laboratory animals. Balb/C nude mice were obtained from ShanghaiSippr-BK laboratory animal Co. Ltd. HepG-2 cells in logarithmic phase were harvested and re-suspended in PBS (5×108 cells/mL). 0.2 mL cells were subcutaneously injected into the right flank of each mouse. When the tumor reached the designated size of 50 mm3, the mice were randomly assigned to treatment groups (8 mice per group). CTP compounds and topotecan were all dissolved in normal saline. Tumor volume (TV) was calculated as: TV = (L × W2)/2, where L is tumor length and W represents tumor width. Body weight change (BW) was measured using the formula below: BW = (W/W0) × 100%, where W refers to the body weight on a specific day and W0 is the body weight on the first day of treatment. Tumor growth inhibition (TGI) was determined as: TGI = [1 - (T - T0)/(C - C0)] × 100%, where T (on a specific day) and T0 (on the first day of treatment) stand for the mean tumor volumes of treatment groups, and C and C0 refer to the mean tumor volumes for control groups.
Determination of Maximum Tolerated Doses (MTD).
The acute and short-term toxicity was tested following administration of drugs with increasing doses to healthy animals. Drugs were intravenously injected (single i.v. dosage or daily i.v. dosage for one week) with each group composed of three mice. Behavioral changes after each administration were monitored hourly, and survival and side effects of the mice were recorded daily for up to 28 days. MTD was determined as the dose before at least one mouse was killed in the treatment.
In Vivo Pharmacokinetics (PK).
Pharmacokinetic properties of compound 11 were studied in male C57BL/6 mice (4 mice per group) after single oral (2 mg/kg) and intravenous administration (10 mg/kg). Mice were fasted overnight with free access to H2O and fed four h after dosing. The whole blood samples were collected via the retro-orbital sinus and plexus at indicated time points and centrifuged at 5500 rpm for 10 min to obtain plasma samples. Plasma levels of the compounds were determined by LC-MS/MS analysis.
LPS-Induced Lethal Inflammation Models.
These experiments were all carried out by CTI Biotechnology (Suzhou, China) Co., Ltd. All experiments were approved and supervised by institutional animal care and use committee (Jiangsu Province, PR China) and carried out according to the guidance for the care and use of laboratory animals. C57BL/6 mice were obtained from ShanghaiSippr-BK laboratory animal Co. Ltd. Mice were separated into eight groups (10 mice per group). For the sepsis model, mice were injected intraperitoneally with 10 mg/kg LPS (from Escherichia coli 0111, Sigma). Dexamethasone (Dex) was set as a positive control drug. For treatment groups, mice were intravenously or orally administrated assigned doses of compounds 0.5 h before LPS injection followed by the same doses of compounds 1 h, 4 h and 8 h after LPS treatment. During the experiments, mice were weighed daily and observed at least twice per day in case of death. To obtain the concentration of inflammatory cytokines, 0.5 ml blood per mouse was retro-orbitally adopted 72 h after LPS treatment, and centrifuged at 3,000 rpm for 10min to separate serum.
Supplementary Material
ACKNOWLEDGMENT
This study was supported by the National Key R&D Program of China (2016YFA0501701; 2016YFB0201700), the National Science Foundation of China (21575128; 81302679) and Shenzhen Science and Technology Innovation Commission (JCYJ20160226105602871).
ABBREVIATIONS
- MTD
maximum tolerated dose
- TGI
tumor growth inhibition.
Footnotes
Supporting Information.
Workflow of construction and screening of virtual chemical library
Solubility of analogues.
Stability of compound 9 and 11 in human liver microsomes.
Pathological anatomy of primary viscera.
NMR spectroscopic data for compounds 1 - 11.
Molecular formula strings of the synthesized compounds (CSV).
The authors declare no competing financial interest.
REFERENCES
- 1.Wall ME; Wani MC; Cook CE; Palmer KH; McPhail AT; Sim GA Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from camptotheca acuminata. J. Am. Chem. Soc. 1966, 88, 3888–3890. [Google Scholar]
- 2.Moertel CG; Reitemeier RJ; Schutt AJ; Hahn RG Phase ii study of strepozotocin (nsc-85998) in the treatment of advanced gastrointestinal cancer. Cancer Chemother. Rep. 1971, 55, 303–307. [PubMed] [Google Scholar]
- 3.Schaeppi U; Fleischman RW; Cooney DA Toxicity of camptothecin (nsc-100880). Cancer Chemother. Rep. 3 1974, 5, 25–36. [PubMed] [Google Scholar]
- 4.Gottlieb JA; Luce JK Treatment of malignant melanoma with camptothecin (nsc-100880). Cancer Chemother. Rep. 1972, 56, 103–105. [PubMed] [Google Scholar]
- 5.Burke TG; Mi Z The structural basis of camptothecin interactions with human serum albumin: Impact on drug stability. J. Med. Chem. 1994, 37, 40–46. [DOI] [PubMed] [Google Scholar]
- 6.Hertzberg RP; Caranfa MJ; Holden KG; Jakas DR; Gallagher G; Mattern MR; Mong SM; Bartus JOL; Johnson RK; Kingsbury WD Modification of the hydroxylactone ring of camptothecin: Inhibition of mammalian topoisomerase i and biological activity. J. Med. Chem. 1989, 32, 715–720. [DOI] [PubMed] [Google Scholar]
- 7.Jaxel C; Kohn KW; Wani MC; Wall ME; Pommier Y Structure-activity study of the actions of camptothecin derivatives on mammalian topoisomerase i: Evidence for a specific receptor site and a relation to antitumor activity. Cancer Res. 1989, 49, 1465–1469. [PubMed] [Google Scholar]
- 8.Burke TG Chemistry of the camptothecins in the bloodstream. Drug stabilization and optimization of activity. Ann. N. Y Acad. Sci. 1996, 803, 29–31. [DOI] [PubMed] [Google Scholar]
- 9.Hsiang YH; Hertzberg R; Hecht S; Liu LF Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase i. J. Biol. Chem. 1985, 260, 14873–14878. [PubMed] [Google Scholar]
- 10.Yang C; Song Z; Goto M; Liu Y i.; Hsieh KY; Morris Natschke SL; Zhao Y; Zhang J; Lee K Design, synthesis, and cytotoxic activity of novel 7-substituted camptothecin derivatives incorporating piperazinyl-sulfonylamidine moieties. Bioorg. Med. Chem. Lett. 2017, 27, 3959–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X; Tang K; Wang H; Liu Y; Bao B; Fang Y; Zhang X; Lu W Design, synthesis, and biological evaluation of new cathepsin b-sensitive camptothecin nanoparticles equipped with a novel multifuctional linker. Bioconj. Chem. 2016, 27, 1267–1275. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y; Chen X; Ding J; Yu L; Ma D; Ding J Improved solubility and bioactivity of camptothecin family antitumor drugs with supramolecular encapsulation by water-soluble pillar[6]arene. ACS Omega 2017, 2, 5283–5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang F; Zhu G; Jacobson O; Liu Y; Chen K; Yu G; Ni Q; Fan J; Yang Z; Xu F; Fu X; Wang Z; Ma Y; Niu G; Zhao X; Chen X Transformative nanomedicine of an amphiphilic camptothecin prodrug for long circulation and high tumor uptake in cancer therapy. ACS Nano 2017, 11, 8838–8848. [DOI] [PubMed] [Google Scholar]
- 14.Westover D; Ling X; Liu X; Lam H; Gongora C; Rio MD; Li F Abstract 829: The novel camptothecin derivative and iap inhibitor fl118 is an effective treatment for irinotecan-refractory colorectal cancer. Cancer Res. 2014, 74, 829.24335958 [Google Scholar]
- 15.Capranico G; Marinello J; Chillemi G Type i DNA topoisomerases. J. Med. Chem. 2017, 60, 2169–2192. [DOI] [PubMed] [Google Scholar]
- 16.Staker BL; Feese MD; Cushman M; Pommier Y; Zembower D; Stewart L; Burgin AB Structures of three classes of anticancer agents bound to the human topoisomerase i-DNA covalent complex. J. Med. Chem. 2005, 48, 2336–2345. [DOI] [PubMed] [Google Scholar]
- 17.Staker BL; Hjerrild K; Feese MD; Behnke CA; Burgin AB; Stewart L The mechanism of topoisomerase i poisoning by a camptothecin analog. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 15387–15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ten BHW; Gore M; Carmichael J; Gordon A; Malfetano J; Hudson I; Broom C; Scarabelli C; Davidson N; Spanczynski M; Bolis G; Malmström H; Coleman R; Fields SC; Heron JF Topotecan versus paclitaxel for the treatment of recurrent epithelial ovarian cancer. J. Clin. Oncol. 1997, 15, 2183–2193. [DOI] [PubMed] [Google Scholar]
- 19.Long HJ; Bundy BN; Grendys EC; Benda JA; McMeekin DS; Sorosky J; Miller DS; Eaton LA; Fiorica JV Randomized phase iii trial of cisplatin with or without topotecan in carcinoma of the uterine cervix: A gynecologic oncology group study. J. Clin. Oncol. 2005, 23, 4626–4633. [DOI] [PubMed] [Google Scholar]
- 20.O'Brien MER; Ciuleanu TE; Tsekov H; Shparyk Y; Čučeviá B; Juhasz G; Thatcher N; Ross GA; Dane GC; Crofts T Phase iii trial comparing supportive care alone with supportive care with oral topotecan in patients with relapsed small-cell lung cancer. J. Clin. Oncol. 2006, 24, 5441–5447. [DOI] [PubMed] [Google Scholar]
- 21.Cunningham D; Pyrhönen S; James RD; Punt CJA; Hickish TF; Heikkila R; Johannesen TB; Starkhammar H; Topham CA; Awad L; Jacques C; Herait P Randomised trial of irinotecan plus supportive care versus supportive care alone after fluorouracil failure for patients with metastatic colorectal cancer. The Lancet 1998, 352, 1413–1418. [DOI] [PubMed] [Google Scholar]
- 22.Hartmann JT; Issels RD; Nicolo KS; Grünwald V; Hertenstein B; Papesch E; Krause S; Sturm I Topotecan plus cyclophosphamide in adults with relapsed or refractory pediatric-type sarcoma: A retrospective analysis from the german sarcoma medical oncology group (aio). Invest. New Drugs 2015, 33, 1115–1122. [DOI] [PubMed] [Google Scholar]
- 23.Kitai Y; Kawasaki T; Sueyoshi T; Kobiyama K; Ishii KJ; Zou J; Akira S; Matsuda T; Kawai T DNA-containing exosomes derived from cancer cells treated with topotecan activate a sting-dependent pathway and reinforce antitumor immunity. The Journal of Immunology 2017, 198, 1649–1659. [DOI] [PubMed] [Google Scholar]
- 24.Creemers G; Lund B; Verweij J Topoisomerase i inhibitors: Topotecan and irenotecan. Cancer Treat. Rev. 1994, 20, 73–96. [DOI] [PubMed] [Google Scholar]
- 25.Rialdi A; Campisi L; Zhao N; Lagda AC; Pietzsch C; Ho JSY; Martinez-Gil L; Fenouil R; Chen X; Edwards M; Metreveli G; Jordan S; Peralta Z; Munoz-Fontela C; Bouvier N; Merad M; Jin J; Weirauch M; Heinz S; Benner C; van Bakel H; Basler C; García-Sastre A; Bukreyev A; Marazzi I Topoisomerase 1 inhibition suppresses inflammatory genes and protects from death by inflammation. Science 2016, 352, aad7993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singer M; Deutschman CS; Seymour CW; Shankarhari M; Annane D; Bauer M; Bellomo R; Bernard GR; Chiche JD; Coopersmith CM The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 2016, 315, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crow RT; Crothers DM Structural modifications of camptothecin and effects on topoisomerase i inhibition. J. Med. Chem. 1992, 35, 4160–4164. [DOI] [PubMed] [Google Scholar]
- 28.Wani MC; Ronman PE; Lindley JT; Wall ME Plant antitumor agents. 18. Synthesis and biological activity of camptothecin analogs. J. Med. Chem. 1980, 23, 554–560. [DOI] [PubMed] [Google Scholar]
- 29.Nicholas AW; Wani MC; Manikumar G; Wall ME; Kohn KW; Pommier Y Plant antitumor agents. 29. Synthesis and biological activity of ring d and ring e modified analogues of camptothecin. J. Med. Chem. 1990, 33, 972–978. [DOI] [PubMed] [Google Scholar]
- 30.Wani MC; Nicholas AW; Wall ME Plant antitumor agents. 28. Resolution of a key tricyclic synthon, 5'(rs)-1,5-dioxo-5'-hydroxy-2'h,5'h,6'h-6'-oxopyrano[3',4'-f].Delta.6,8-tetrahydroindolizine: Total synthesis and antitumor activity of 20(s)- and 20(r)-camptothecin. J. Med. Chem. 1987, 30, 2317–2319. [DOI] [PubMed] [Google Scholar]
- 31.Martino E; Della Volpe S; Terribile E; Benetti E; Sakaj M; Centamore A; Sala A; Collina S The long story of camptothecin: From traditional medicine to drugs. Bioorg. Med. Chem. Lett. 2017, 27, 701–707. [DOI] [PubMed] [Google Scholar]
- 32.Schrödinger, M. Version 9.0. Schrödinger LLC, New York, NY: 2009. [Google Scholar]
- 33.Cao Z; Harris N; Kozielski A; Vardeman D; Stehlin JS; Giovanella B Alkyl esters of camptothecin and 9-nitrocamptothecin: Synthesis, in vitro pharmacokinetics, toxicity, and antitumor activity. J. Med. Chem. 1998, 41, 31–37. [DOI] [PubMed] [Google Scholar]
- 34.Zhao H; Lee C; Sai P; Choe YH; Boro M; Pendri A; Guan S; Greenwald RB 20-o-acylcamptothecin derivatives: Evidence for lactone stabilization. J. Org. Chem. 2000, 65, 4601–4606. [DOI] [PubMed] [Google Scholar]
- 35.Greenwald RB; Pendri A; Conover C; Gilbert C; Yang R; Xia J Drug delivery systems. 2. Camptothecin 20-o-poly(ethylene glycol) ester transport forms. J. Med. Chem. 1996, 39, 1938–1940. [DOI] [PubMed] [Google Scholar]
- 36.Deshmukh M; Chao P; Kutscher HL; Gao D; Sinko PJ A series of α-amino acid ester prodrugs of camptothecin: In vitro hydrolysis and a549 human lung carcinoma cell cytotoxicity. J. Med. Chem. 2010, 53, 1038–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wadkins RM; Potter PM; Vladu B; Marty J; Mangold G; Weitman S; Manikumar G; Wani MC; Wall ME; Von Hoff DD Water soluble 20 (s)-glycinate esters of 10, 11-methylenedioxycamptothecins are highly active against human breast cancer xenografts. Cancer Res. 1999, 59, 3424–3428. [PubMed] [Google Scholar]
- 38.Takimoto CH; Wright J; Arbuck SG Clinical applications of the camptothecins. Biochim. Biophys. Acta 1998, 1400, 107–119. [DOI] [PubMed] [Google Scholar]
- 39.Mathijssen RHJ; Loos WJ; Verweij J; Sparreboom A Pharmacology of topoisomerase i inhibitors irinotecan (cpt-11) and topotecan. Curr. Cancer Drug Targets 2002, 2, 103–123. [DOI] [PubMed] [Google Scholar]
- 40.Gilson MK; Liu T; Baitaluk M; Nicola G; Hwang L; Chong J Bindingdb in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.