

Abstract
Synaptic transmission occurs when an action potential triggers neurotransmitter release via the fusion of synaptic vesicles with the presynaptic membrane, driven by the formation of SNARE complexes composed of the vesicular (v)-SNARE synaptobrevin and the target (t)-SNAREs Snap-25 and syntaxin-1. Neurotransmitters are also released spontaneously, independent of an action potential, through the fusion of synaptic vesicles with the presynaptic membrane. The major neuronal vSNAREs, synaptobrevin-1 and synaptobrevin-2, are expressed at the developing neuromuscular junction (NMJ) in mice, but their specific roles in NMJ formation and function remain unclear. Here, we examine the NMJs in mutant mouse embryos lacking either synaptobrevin 1 (Syb1lew/lew) or synaptobrevin 2 (Syb2−/−), and those lacking both (Syb1lew/lewSyb2−/−). We found that, compared with controls: (1) the number and size of NMJs was markedly increased in Syb2−/− and Syb1lew/lewSyb2−/− mice, but not in Syb1lew/lew mice; (2) synaptic vesicle density was markedly reduced in Syb1lew/lewSyb2−/− NMJs; and (3) evoked neurotransmission was markedly reduced in Syb2−/− NMJs and completely abolished in Syb1lew/lewSyb2−/− NMJs. Surprisingly, however, spontaneous neurotransmission persists in the absence of both Syb1 and Syb2. Furthermore, spontaneous neurotransmission remains constant in Syb1lew/lewSyb2−/− NMJs despite changing Ca2+ levels. These findings reveal an overlapping role for Syb1 and Syb2 (with Syb2 being dominant) in developing NMJs in mice. Moreover, because spontaneous release becomes Ca2+-insensitive in Syb1lew/lewSyb2−/− NMJs, our findings suggest that synaptobrevin-based SNARE complexes play a critical role in conferring Ca2+ sensitivity during spontaneous release.
SIGNIFICANCE STATEMENT Neurotransmitters can be released at synapses with (evoked) or without (spontaneous) the influence of action potentials. Whereas evoked neurotransmission requires Ca2+ influx, those underlying the spontaneous neurotransmission may occur with or without Ca2+. Our findings show that, in the absence neuronal vSNARE synaptobrevin-1 and synaptobrevin-2, evoked neurotransmission is completely abolished; however, spontaneous synaptic transmission not only persists but even increased. Furthermore, spontaneous synaptic transmission that is normally highly Ca2+-sensitive became Ca2+-independent upon deletion of vSNARE synaptobrevin-1 and synaptobrevin-2. These findings reveal distinct mechanisms for evoked and spontaneous neurotransmitter release. Moreover, these findings suggest that synaptobrevin-based SNARE complexes play critical roles in conferring Ca2+ sensitivity during spontaneous neurotransmission at developing neuromuscular synapses in mice.
Keywords: evoked release, KO mice, neuromuscular junction, spontaneous release, synapse formation, synaptic transmission
Introduction
Neurotransmitters mediate neuronal communication when they are released from presynaptic vesicles at the chemical synapse. Their release may be evoked (i.e., triggered by the propagation of action potentials [APs] invading the presynaptic nerve terminal) or spontaneous (i.e., occurring independent of APs) (Barrett and Stevens, 1972; Goda and Stevens, 1994; Kaeser and Regehr, 2014; Schneggenburger and Rosenmund, 2015). Emerging evidence suggests that the mechanisms underlying the evoked and spontaneous release of neurotransmitters are differentially regulated. For example: (1) the evoked and spontaneous release of neurotransmitters uses separate pools of vesicles (Sara et al., 2005; Fredj and Burrone, 2009; Kavalali et al., 2011) and is independently regulated at active zones (Melom et al., 2013); (2) neurotransmitters are released spontaneously from the growth cones of embryonic neurons (Hume et al., 1983; Young and Poo, 1983), implicating a role in nerve growth and axon guidance as membrane is added to the tip of the growth cone (Futerman and Banker, 1996); (3) spontaneous release is selectively affected by certain pharmacological regents, such as mefloquine, a drug that is widely used for the treatment of malaria (McArdle et al., 2006); (4) spontaneous release has been implicated in the regulation of dendritic protein synthesis (Sutton et al., 2004), synapse maturation and function (Sutton et al., 2006; Choi et al., 2014), and homeostatic synaptic plasticity (Turrigiano, 2012); and (5) certain domains in the syntaxin-1, a neuronal component of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex, participate in evoked neurotransmission but not in spontaneous neurotransmission (Zhou et al., 2013).
The mechanisms underlying evoked neurotransmission have been fairly well defined (Südhof, 2013a), but those regulating spontaneous neurotransmission remain less clear. Both require the fusion of synaptic vesicles (SVs) with the presynaptic membrane, which leads to exocytosis and consequent release of neurotransmitters into the synaptic cleft. This fusion is driven by the formation of SNARE complexes composed of the vesicular (v)-SNARE synaptobrevin/vesicle-associated membrane protein (VAMP) in the vesicular compartment, the target (t)-SNARE synaptosomal-associated protein-25 (Snap-25) and syntaxin-1 in the presynaptic membrane, and their interactions with Sec1/Munc-18-like (SM) protein Munc18–1 and with Munc-13 (for review, see Jahn and Scheller, 2006; Südhof and Rothman, 2009; Jahn and Fasshauer, 2012; Rizo and Südhof, 2012; Südhof, 2013b; Brunger et al., 2018). Evoked release also requires an influx of Ca2+ (Dodge and Rahamimoff, 1967; Llinás et al., 1981; Augustine et al., 1987), which binds to synaptotagmins (Syt) to trigger fusion (Südhof, 2013a; Rizo and Xu, 2015; Brunger et al., 2018). By contrast, spontaneous neurotransmission may occur with or without Ca2+ (for review, see Schneggenburger and Rosenmund, 2015). This raises a number of questions: (1) How do the mechanisms that drive Ca2+-independent versus Ca2+-dependent spontaneous neurotransmission differ from each other? (2) Does Ca2+-independent spontaneous release occur in the same SV pools as those in which Ca2+-sensitive spontaneous release occurs? (3) How is Ca2+ sensitivity conferred during spontaneous release?
The major neuronal synaptobrevin proteins, synaptobrevin-1 (Syb1) and synaptobrevin-2 (Syb2), are expressed abundantly during the development of the nervous system (Hepp and Langley, 2001). We have previously demonstrated that Syb1 plays a crucial role in Ca2+-triggered neurotransmission at the neuromuscular synapse (Liu et al., 2011): this study was performed using Syb1 mutant mice resulted from a spontaneous mutation that causes a premature stop codon and Syb1 null (Syb1lew/lew) (Nystuen et al., 2007). However, the other major neuronal synaptobrevin isoform Syb2 was also expressed at the neuromuscular junction (NMJ) in Syb1lew/lew mice (Liu et al., 2011). This finding raises the possibility that a deficiency in Syb1 may have been compensated by the presence of Syb2. In the present study, we examined mutant mice deficient in Syb2 and in those deficient in both Syb1 and Syb2. We found that a deficiency in either Syb2 (Syb2−/−) or both Syb1 and Syb2 (Syb1lew/lewSyb2−/−) can lead to a marked increase in intramuscular nerve branching and precocious maturation of the NMJ during development. More importantly, in Syb1lew/lewSyb2−/− NMJs, evoked synaptic transmission was completely abolished, whereas spontaneous synaptic transmission was increased significantly in terms of both frequency and amplitude. Moreover, spontaneous release in Syb1lew/lewSyb2−/− NMJs was insensitive to Ca2+. These findings suggest that synaptobrevin plays an important role in conferring the Ca2+ sensitivity during spontaneous release.
Materials and Methods
Mice.
Mutant mice deficient in VAMP1/Syb1 were obtained from the The Jackson Laboratory. These mutant mice [RRID:IMSR_JAX:004626, C3H/HeSnJ-Vamp1lew/GrsrJ, lethal wasting (lew), MGI: 2449158; J:81354] (Harris et al., 2003) arose spontaneously as a result of a G190T transversion that resulted in a premature stop codon. The resultant homozygous lew mutant mice (Vamp1/Syb1lew/lew, thereafter as Syb1lew/lew) are Syb1 nulls (Nystuen et al., 2007; Liu et al., 2011). Mice deficient in Syb2 (Syb2−/−) (MGI: 2179745) were generated through targeted homologous recombination, as described previously (Schoch et al., 2001). Mice deficient in both Syb1 and Syb2 (Syb1lew/lewSyb2−/−) had to be generated using a two-stage breeding scheme because neither Syb1lew/lew nor Syb2−/− mice are viable: Syb1lew/lew mice usually die within 2 weeks after birth (Nystuen et al., 2007; Liu et al., 2011), and Syb2−/− mice die at birth (Schoch et al., 2001). We generated Syb1+/lewSyb2+/− double-heterozygous mice by crossing Syb1+/lew mice with Syb2+/− mice. We then bred Syb1+/lewSyb2+/− mice together to generate Syb1lew/lewSyb2−/− double homozygotes. These double homozygotes (Syb1lew/lewSyb2−/−) died at birth, however. This led us to focus on the development of the NMJ during embryonic stages. For NMJ studies, we analyzed mouse embryos at two embryonic stages: E14.5 and E18.5, of either sex. At E14.5, we analyzed 6 embryos (3 pairs of Syb2−/− and WT littermate control). At E18.5, a total of 304 embryos from 44 litters were obtained, including 20 with the genotype of Syb1lew/lewSyb2−/− (Table 1). Morphologic and electrophysiologic findings in these NMJs were indistinguishable among the Syb1+/+Syb2+/+, Syb1+/lewSyb2+/−, Syb1+/lewSyb2+/+, and Syb1+/+Syb2+/− embryos; for this reason, they were pooled together to serve as controls. All experimental protocols followed National Institutes of Health guidelines for the ethical treatment of animals and were approved by the University of Texas Southwestern Institutional Animal Care and Use Committee.
Table 1.
A summary of mouse embryos resulted from interbreeding of Syb1+/lewSyb2+/− micea
| Genotype | Observed number | Expected number | p (χ2 test) |
|---|---|---|---|
| Control | 166 | 171 | 0.436 |
| Syb1lew/lew | 55 | 57 | |
| Syb2−/− | 63 | 57 | |
| Syb1lew/lewSyb2−/− | 20 | 19 |
aA total of 304 embryos (E18.5) from 44 litters were genotyped. Control embryos included Syb1+/+Syb2+/± and Syb1+/lewSyb2+/±. Syb1lew/lew embryos included Syb1lew/lewSyb2+/+ and Syb1lew/lewSyb2+/−. Syb2−/− embryos included Syb1+/+Syb2−/− and Syb1+/lewSyb2−/−. The expected numbers were calculated from Mendelian ratios of 9:3:3:1.
Immunofluorescence staining.
The NMJs underwent immunofluorescence staining using procedures described previously (Liu et al., 2008; Chen et al., 2011). Briefly, embryonic diaphragm muscles (E18.5) were dissected and fixed in 2% PFA in a 0.1 M phosphate buffer, pH 7.3. After several washes, the muscles were incubated with Texas red-conjugated α-bungarotoxin (α-bgt) (2 nM, Invitrogen) for 30 min to label the AChRs. To determine the expression of SNARE proteins at the NMJ, frozen sections were made from diaphragm muscles (E18.5) and labeled with anti-Syb1 (rabbit polyclonal, P938, 1:500) or anti-Syb2 (rabbit polyclonal, P939, 1:1000) (Liu et al., 2011), anti-VAMP4 (Synaptic Systems, catalog #136002, RRID:AB_887816, 1:500), anti-VAMP7 (Abcam, catalog #ab36195, RRID:AB_2212928, 1:3000), and anti-Vti1a (BD Bioscience, catalog #611220, RRID:AB_398752, 1:500) antibodies. To determine whether Syb1 and Syb2 were coexpressed at the same NMJ, diaphragm muscle sections were labeled with anti-Syb1 (rabbit polyclonal, P938, 1:500) and anti-Syb2 (mouse monoclonal, CL69.1, 1:3000) (Schoch et al., 2001). To visualize developing nerves and nerve terminals, we labeled whole-mount diaphragm muscles with a mixture of antineurofilament (Millipore Bioscience Research Reagents, catalog #AB1981, RRID:AB_10013484, 1:1000) and anti-synaptotagmin2 (Syt2, 1:1000) antibodies (Pang et al., 2006b), then applied FITC-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, catalog #111-095-144, RRID:AB_2337978). Images were acquired using a Laser Scanning Microscope 510 or Laser Scanning Microscope 880 confocal microscope (Carl Zeiss). Endplate sizes, including area, perimeter, and Ferret's diameter (the length of the greatest axis), were measured using the National Institutes of Health ImageJ program (ImageJ, RRID:SCR_003070).
Quantification of nerve branching.
To quantify nerve branching, we used the method as described previously (Kaplan et al., 2018). First, confocal images were obtained from the same regions of diaphragm muscles (dorsal right quadrant). Seven vertical lines were drawn perpendicular to the long axis of muscle fibers, starting from the first at the center of the endplate band, then extending 300 μm to the left and 300 μm to the right, at 100 μm intervals. Then, the numbers of intersections between nerve branches and vertical lines were manually counted, and the values were normalized to the intersection numbers at the middle line.
Quantification of endplate on teased single muscle fibers.
Whole-mount diaphragm muscles (E18.5) were dissected in PBS and incubated in 0.2% collagenase II (Worthington Biochemical) in DMEM for 45 min. After collagenase incubation, the diaphragm muscles were fixed in 1% PFA (30 min) and then labeled with FITC-conjugated α-bgt (FITC-α-bgt, 2 nm, 30 min). After a brief wash in PBS, the diaphragm muscles were labeled with Texas red-conjugated phalloidin (1:100 in 0.1 M phosphate buffer, 0.01% saponin) overnight at 4°C, followed by a brief wash in PBS. Next, the diaphragm muscles were transferred onto a glass slide in a drop of antifade mountain medium. Under a dissecting microscope, the diaphragm muscles were manually teased using fine needles. The teased muscle fibers were then covered with coverslip and examined under microscope. Images with single muscle fibers were acquired using Laser Scanning Microscope 510 confocal microscope (Carl Zeiss), and the number of AChR patches on each muscle fibers were counted and quantified.
Western blot.
Spinal cord tissues (E18.5 and P14, either sex) were homogenized, separated by SDS-PAGE, and transferred to a nitrocellulose membrane. The membrane was then probed with polyclonal antibodies against Syb1 (P938, 1:500) or Syb2 (P441, 1:1000), and monoclonal antibodies against α-tubulin (Sigma-Aldrich, catalog #T9026, RRID:AB_477593, 1:1000), which serve as a loading control. Then the membrane was incubated with peroxidase-conjugated goat anti-rabbit or anti-mouse secondary antibodies (Jackson ImmunoResearch Laboratories, catalog #111-035-003, RRID:AB_2313567, catalog #115-035-003, RRID:AB_10015289, 1:10,000), followed by enhanced chemiluminescence and imaged with Bio-Rad ChemiDoc MP Imaging System.
Electrophysiology.
Electrophysiological recordings were performed in embryonic diaphragm muscles (E18.5) with the phrenic nerves attached to the muscle, using the procedures described previously (Liu et al., 2008, 2011). Briefly, glass microelectrodes (resistance 20–40 mΩ) filled with 2 m potassium citrate and 10 mm potassium chloride were used to make intracellular recordings in postsynaptic muscle cells. Changes in membrane potentials were measured using an intracellular amplifier (AxoClamp-2B), digitized with Digidata 1332 (Molecular Devices), and analyzed using pClamp software (pClamp, RRID:SCR_011323). Evoked endplate potentials (EPPs) were triggered by applying suprathreshold electrical stimulation to the phrenic nerve (2–5 V, 0.1 ms) through a suction electrode. Recordings were performed at room temperature in normal Ringer's solution according to Liley (1956a,b): 136.8 mM NaCl, 5 mM KCl, 12 mM NaHCO3, 1 mM NaH2PO4, 1 mM MgCl2, 2 mM CaCl2, and 11 mM d-glucose, with an osmolality at 300 mOsm and a pH of 7.3.
Ringer's solutions containing one of three concentrations of Ca2+ (1, 2, and 4 mM) were prepared by adjusting the concentration of CaCl2 to 1, 2, and 4 mM, respectively; a Ca2+-free Ringer's solution was prepared by omitting CaCl2 and adding 1 mM EGTA to remove any residual Ca2+. The nerve-muscle preparations were equilibrated in the Ca2+-free Ringer's solution for 60 min before recording.
Electron microscopy.
Embryonic diaphragm muscles were processed for electron microscopy using procedures described previously (Chen et al., 2011; Liu et al., 2011). Briefly, rib cages were fixed overnight at 4°C in a mixture of fixatives containing 1% glutaraldehyde and 4% PFA in 0.1 M phosphate buffer, pH 7.4. The samples were then rinsed with 0.1 M phosphate buffer, pH 7.4. The diaphragm muscles with both phrenic nerves attached were then dissected, trimmed to small pieces, and postfixed with 1% osmium tetroxide for 3 h, on ice. The tissues were then dehydrated in a graded series of ethanol, infiltrated, and embedded in Epon 812 (Poly/Bed 812 Embedding Kit, with DMP-30, Ted Pella). Ultrathin sections (70 nm) were prepared, mounted on Formvar-coated grids, and then stained with uranyl acetate and lead citrate. Electron microscopy images were acquired using a Tecnai electron microscope operated at 120 kV.
A total of 129 nerve terminals from Syb1lew/lewSyb2−/− mutant mouse embryos (E18.5, N = 4 embryos of either sex) and 131 nerve terminals from control embryos (E18.5, N = 5 embryos of either sex) underwent morphometric analysis. The control embryos included 2 Syb1+/+Syb2+/+ and 3 Syb1+/lewSyb2+/+ that were littermates of the Syb1lew/lewSyb2−/− mutants (collected from 3 litters). The following measurements were made from each presynaptic nerve terminal profile, according to previously defined criteria (Kelly and Zacks, 1969): nerve terminal area, nerve terminal perimeter length, and synaptic contact length. Synaptic contact length was defined as the length of presynaptic membrane opposed to the postsynaptic membrane. The synaptic contact proportion was the synaptic contact length expressed as a percentage of terminal perimeters. SVs were defined as a clear vesicle with a diameter of 40–50 nm, and dense core vesicle (DCV) was defined as a larger vesicle (diameter: 60–80 nm) containing a dense core. Individual SVs were counted manually in each nerve terminal to obtain a total SV number. SV density was calculated as total SV number per nerve terminal area. An active zone was defined as the specific site on the presynaptic plasma membrane in which SVs were clustered and included at least one docked SV (abutting the plasma membrane). When SV clustering was observed along the terminal plasma membrane that was not facing the postsynaptic side, it was considered as an ectopic SV cluster. Other membranous subcellular structures were counted per nerve terminal. These included multilamellar bodies, which contained a membrane lamellar structure; multivesicular bodies, which contained multiple small internal vesicles; vacuole-like structures, defined as clear, membrane-limited organelles larger than a DCV (typically >100 nm in diameter) that included objects of various (round to irregular) shapes; and tubular structures-thin, membrane-limited organelles ∼30 nm in diameter and >100 nm in length. A two-tailed t test was applied to evaluate statistical differences between mutants and controls.
Statistical analysis.
Data were presented as mean ± SEM. Paired Student's t test was performed to determine statistical difference in nerve branching between control and mutant diaphragm muscles. χ2 test was used for mouse embryo survival analyses (Table 1). Kolmogorov–Smirnov test was used to analyze cumulative probabilities of miniature EPP (mEPP) frequency, amplitude, and rise time. Two-tailed Student's t test was used to determine statistical differences between mutant and control groups. A p value of <0.05 is considered to be statistically significant.
In addition, a χ2 Goodness of Fit test was used to determine whether the distribution of mEPP frequency fits Poisson distribution. Specifically, the expected probability was calculated using Poisson function. Then χ2 test was used to determine the difference between observed probability and expected probability. The distribution of mEPP is considered to fit a Poisson distribution if p value by χ2 test is >0.05.
Results
Synaptobrevin deficiency leads to an increase in nerve branching and synaptogenesis
To determine the role of Syb1/Syb2 in the formation and function of the NMJ, we generated mutant mice deficient in both Syb1 and Syb2 (Syb1lew/lewSyb2−/−) by interbreeding of Syb1+/lewSyb2+/− double-heterozygous mice. Consistent with our previous observation that both Syb1 and Syb2 are expressed at the NMJs in juvenile mice (Liu et al., 2011), immunofluorescence staining using Syb1 or Syb2 specific antibodies revealed that both Syb1 and Syb2 were abundantly expressed at motor nerve terminals in WT embryos (Fig. 1A) but were absent in Syb1lew/lewSyb2−/− embryos (Fig. 1B). In contrast, other SNARE proteins, including VAMP4, Vti1a, and VAMP7, were either not present or barely detected at the NMJs in either WT (Fig. 1A) or Syb1lew/lewSyb2−/− embryos (Fig. 1B). Triple-labeling with AlexaFluor-647-conjugated α-bgt, anti-Syb1(rabbit polyclonal), and anti-Syb2 (mouse monoclonal) antibodies showed that Syb1 and Syb2 were coexpressed at the NMJ in the WT muscle (E18.5; Fig. 1C). Western blot analyses of spinal cord homogenates showed that Syb1 expression is markedly increased at P14, compared with that at E18.5 (Fig. 1D).
Figure 1.

Synaptobrevin vSNAREs (Syb1 and Syb2) are coexpressed at developing motor nerve terminals and spinal cords. A, B, Frozen section of embryonic diaphragm muscles (E18.5) from WT (A) and Syb1lew/lewSyb2−/− mice (B), doubly labeled with Texas red-conjugated α-bgt for postsynaptic AChRs (red, arrowhead) and specific antibodies against a SNARE protein (green): Syb1 (VAMP1), Syb2 (VAMP2), VAMP4, VAMP7, or Vti1a. Both Syb1 and Syb2 are detected at the NMJ in the WT (A) but not in Syb1lew/lewSyb2−/− mice (B). No detectable level of VAMP4, VAMP7, or Vti1a was found at the NMJ in WT or Syb1lew/lewSyb2−/− mice. C, Diaphragm muscle sections (E18.5) were triply labeled with AlexaFluor-647-conjugated α-bgt, anti-Syb1(rabbit polyclonal, green), and anti-Syb2 (mouse monoclonal, red) antibodies. Merged image shows that Syb1 and Syb2 are coexpressed at the NMJ in the WT (C, left panels) but absent in Syb1lew/lewSyb2−/− mice (C, right panels). D, Western blot analyses of spinal cord homogenates at E18.5 (WT and Syb1lew/lewSyb2−/−) and P14 (WT). Syb1 expression is markedly increased at P14, compared with that at E18.5. Scale bars: A, B, 20 μm; C, 10 μm.
To assess the role of Syb1 and Syb2 in the NMJ, we stained whole-mount embryonic diaphragm muscles using a mixture of antibodies against neurofilament protein NF150 (anti-NF150) and antibodies against Syt2 (anti-Syt2) to label presynaptic nerves, and used Texas red-conjugated α-bgt to label postsynaptic AChRs. Strikingly, Syb2 deficiency led to excessive nerve branching in developing diaphragm muscles as early as E14.5 (Fig. 2A,B). In WT embryos, intramuscular nerves were confined to a narrow band along the central region of the diaphragm muscle (Fig. 2A, left). In contrast, in Syb2−/− embryos, intramuscular nerve branches extended to a broad region of the diaphragm muscle (Fig. 2A, right). This expansion of innervation territory and increase in nerve branching were also evident at E18.5 (Fig. 2C,D), suggesting that the increases in innervation persisted throughout the stages during NMJ formation.
Figure 2.

Synaptobrevin deficiency leads to excessive intramuscular nerve branching. Innervation pattern of embryonic diaphragm muscles, revealed by antineurofilament (anti-NF150) antibodies (A) or by a mixture of antibodies (anti-NF150 and anti-Syt2, C). Intramuscular nerves extend orderly branches largely confined to the central region of the muscle in control muscles. In contrast, excessive nerve branches are present over a broad region of the muscle in both Syb2−/− and Syb1lew/lewSyb2−/− mice. B, D, Quantification of nerve branching. The number of nerve branches at E14.5 is significantly (p = 0.0013, paired Student's t test) increased in Syb2−/− mice compared with control (B). Similarly, the number of nerve branches at E18.5 is significantly increased in both Syb2−/− (p = 0.0029) and Syb1lew/lewSyb2−/− mice (p = 0.0027), compared with control (D). Scale bars: A, 400 μm; C, 200 μm.
The labeling of diaphragm muscles with α-bgt revealed that AChR clusters were confined to a narrow band (endplate band, Fig. 3A, dashed lines) along the central regions of the muscle in both control and Syb1lew/lew mice (Fig. 3A). The endplate bands were markedly broader in Syb2−/− (Fig. 3A,C) and Syb1lew/lewSyb2−/− mice (Fig. 3A,C), compared with control. Associated with an expansion of the endplate band, the number of individual endplates was markedly increased in both Syb2−/− and Syb1lew/lewSyb2−/− mice, compared with control (Fig. 3D).
Figure 3.

Synaptobrevin deficiency leads to precocious maturation of neuromuscular synapses. Whole mounts of embryonic diaphragm muscles (E18.5) were doubly labeled with Texas red-conjugated α-bgt for AChRs (red) and a mixture of antibodies (anti-NF150 and anti-Syt2) for the nerve (green). A, Low-power images illustrating the distribution pattern of AChR clusters (endplates). AChR clusters are confined to a narrow band (endplate band, outlined by dashed lines) along the central regions of the muscle in control and Syb1lew/lew muscles. By contrast, AChR clusters are broadly distributed in Syb2−/− and Syb1lew/lewSyb2−/− muscles. Quantification of endplate bandwidth and endplate number is illustrated in C and D, respectively. B, High-magnification views of individual NMJs. Nerve terminals (B, top) are juxtaposed with postsynaptic AChR clusters (B, middle panels), as shown in the merged images (B, bottom panels). C–F, Quantification of endplate bandwidth, endplate number, perforated endplates, and individual endplate size. The average width of the endplate band is markedly increased in Syb1lew/lewSyb2−/− (396 ± 44 μm, N = 3 mice) and Syb2−/− NMJs (367 ± 55 μm, N = 3 mice), compared with controls (166 ± 15 μm, N = 3 mice) or with Syb1lew/lew NMJs (135 ± 13 μm, N = 3 mice) (C). The number of endplates (per mm2) is also markedly increased in Syb1lew/lewSyb2−/− (383 ± 4, N = 3 mice) and Syb2−/− NMJs (326 ± 15, N = 3 mice), compared with controls (228 ± 36, N = 3 mice) or with Syb1lew/lew NMJs (206 ± 32, N = 3 mice) (D). E, Quantification shows that the percentage of perforated endplates is markedly increased in Syb1lew/lewSyb2−/− (32.6 ± 9.0%, n = 356 endplates, N = 3 mice), compared with the control (6.6 ± 1.2%, n = 279 endplates, N = 3 mice), Syb1lew/lew (8.2 ± 0.2%, n = 355 endplates, N = 3 mice), or Syb2−/− mice (7.9 ± 0.7%, n = 274 endplates, N = 3 mice). Furthermore, the size of individual endplates is significantly (p = 0.007, Student's t test) increased in Syb1lew/lewSyb2−/− NMJs (107.6 ± 5.7 μm2, n = 260 endplates, N = 3 mice), compared with controls (78.0 ± 6.3 μm2, n = 168 endplates, N = 3 mice) (F). Scale bars: A, 200 μm; B, 20 μm.
Furthermore, the number of AChR clusters per muscle fiber was markedly increased in Syb1lew/lewSyb2−/− mice compared with control. This was measured by counting AChR cluster numbers from teased single muscle fibers in E18.5 diaphragm muscles in both control and Syb1lew/lewSyb2−/− mice (Fig. 4). We isolated a total of 1372 muscle fibers from control (N = 3 mice) and 1498 muscle fibers from Syb1lew/lewSyb2−/− mice (N = 3 mice). The majority of these muscle fibers contained a single AChR cluster (AChR patch), and the rest of the muscle fibers carried 2 clusters (Fig. 4A,B). However, the proportion of muscle fibers that carry 2 clusters was significantly increased in Syb1lew/lewSyb2−/− mice compared with control (Fig. 4C).
Figure 4.

Analyses of endplate number in teased single muscle fibers (E18.5). A, B, Confocal images of teased single muscle fibers were double-labeled with FITC-conjugated α-bgt for AChR clusters (AChR patches) and Texas red-conjugated phalloidin for muscle fibers. C, Quantification of muscle fibers that contain either 1 or 2 patches. A total of 1372 muscle fibers from control (N = 3 mice) and 1498 muscle fibers from Syb1lew/lewSyb2−/− mice were analyzed. In control mice, 99 ± 0.3% bear a single patch, and 1.0 ± 0.3% muscle fibers bear 2 patches. By contrast, in Syb1lew/lewSyb2−/− mice, 95.5 ± 0.3% bear a single patch, and 4.5 ± 0.3% muscle fibers bear 2 patches. The proportion of muscle fibers that carries 2 patches is significantly increased in Syb1lew/lewSyb2−/− mice compared with control.
High-power views of individual NMJs obtained from controls and Syb1lew/lew, Syb2−/−, and Syb1lew/lewSyb2−/− mice showed that all of the AChR clusters lay adjacent to nerve terminals, indicating that the NMJs were established in both control and mutant mice (Fig. 3B). Notable differences between control and mutant NMJs were nonetheless observed. The majority of the endplates in control mice appeared ovoid in shape (Fig. 3B), and the majority of endplates in mice lacking both Syb1 and Syb2 were larger than in the controls (Fig. 3F). The latter were also perforated (Fig. 3B,E, arrowheads), thus taking on characteristics that are normally not seen until after birth (Slater, 1982; Marques et al., 2000). The appearance of these embryonic endplates (E18.5) thus suggests precocious NMJ maturation in the mutant mice.
Using electron microscopy, we found that the overall appearance of the NMJ was similar between controls and mutant (Syb1lew/lewSyb2−/−) mice (Fig. 5). Given that synapse elimination had not yet occurred, multiple (5–6) nerve terminals were seen lying adjacent to each muscle fiber (Fig. 5A,C), Syb1lew/lewSyb2−/− mice (5.59 ± 3.1, n = 162 nerve terminals, N = 4 mice), similar to control (6.35 ± 2.98, n = 165 nerve terminals, N = 5 mice). Thus, for any given plane, a similar number of nerve terminals would be expected in controls versus mutant (Syb1lew/lewSyb2−/−) mice. Both control and mutant nerve terminals contained an abundance of small, clear SVs (each with a diameter of 40–50 nm) and well-defined basal laminae (Fig. 5E,F). They differed markedly, however, in terms of SV density and synaptic contact length (Fig. 5). SVs were abundantly present in both control and mutant (Syb1lew/lewSyb2−/−) NMJs (Fig. 5B,D), but the number and density of SVs were markedly lower in Syb1lew/lewSyb2−/− NMJs compared with controls (Table 2). The synaptic contact length along presynaptic nerve terminals was also significantly reduced in mutant (Syb1lew/lewSyb2−/−) NMJs compared with controls, but the number of docked SVs was similar in both groups of mice (Table 2).
Figure 5.

Ultrastructure of the NMJ in Syb1lew/lewSyb2−/− mice. A, C, Low-power electron micrographs of the NMJs in diaphragm muscle (E18.5) obtained from a control (A) and a Syb1lew/lewSyb2−/− mouse (C). In both control and Syb1lew/lewSyb2−/− mice, clusters of 5–6 presynaptic nerve terminal profiles (Nt, pseudo-colored in gold) are juxtaposed against postsynaptic muscles to form the NMJ. B, D, High-power views of the NMJ, indicated by the red box for control (A) and Syb1lew/lewSyb2−/− mice (C). E, F, High-power views of the NMJ in control (E) and Syb1lew/lewSyb2−/− mice (F). An abundance of small, clear SVs and well-defined basal lamina (BL) is present in both genotypes. Arrow indicates docked SV. The number of DCVs is notably increased in the presynaptic nerve terminals of Syb1lew/lewSyb2−/− NMJs (F) compared with controls (E). Scale bars: A, B, 1000 nm; C–F, 500 nm.
Table 2.
Morphometric analysis of NMJs in control and Syb1lew/lewSyb2−/− embryosa
| Variable | Control | Syb1lew/lewSyb2−/− |
|---|---|---|
| Nerve terminal area (μm2) | 0.54 ± 0.06 | 0.46 ± 0.04 |
| Nerve terminal perimeter (μm) | 3.45 ± 0.22 | 2.98 ± 0.16 |
| Synaptic contact length (μm) | 1.14 ± 0.08 | 0.78 ± 0.05** |
| Synaptic contact proportion | 33.5 ± 1.0 | 27.3 ± 1.0*** |
| Synaptic vesicle number (per terminal) | 43.4 ± 4.5 | 31.9 ± 2.9* |
| Synaptic vesicle density (per μm2) | 91.7 ± 4.3 | 78.2 ± 3.8* |
| Docked synaptic vesicle number (per active zone) | 1.47 ± 0.09 | 1.59 ± 0.12 |
| DCV number (per terminal) | 0.56 ± 0.08 | 1.49 ± 0.17*** |
| Ectopic synaptic vesicle cluster (per terminal) | 0.023 ± 0.013 | 0.031 ± 0.015 |
| Vacuole-like structures (per terminal) | 1.77 ± 0.18 | 1.23 ± 0.13 |
| Tubular structures (per terminal) | 0.63 ± 0.13 | 0.38 ± 0.60 |
aMorphometric analysis was performed on EM images of the NMJs from control (n = 131 nerve terminal profiles, N = 5 mice) and Syb1lew/lewSyb2−/− mice (n = 129 nerve terminal profiles, N = 4 mice) at E18.5. The definition of parameters is described in Material and Methods. Docked synaptic vesicle numbers were counted from 68 nerve terminals in control and 37 nerve terminals in Syb1lew/lewSyb2−/− embryos. Data are mean ± SEM.
*p < 0.05;
**p < 0.001;
***p < 0.0001; two-tailed Student's t test.
By contrast, the mutant nerve terminals contained a considerably greater number of DCVs (large vesicles 60–80 nm in diameter that contain a dense core) compared with controls (Table 2). Among the 131 nerve terminals examined in control animals (N = 5 mice), 87% contained no more than one DCV, 11% contained 2–3 DCVs, and ∼2% contained ≥4 DCVs. By contrast, 24% of the nerve terminals in the Syb1lew/lewSyb2−/− NMJs (n = 129, N = 4 mice) contained 2–3 DCVs and 12% contained ≥4 DCVs. Despite these differences, the overall fine structure of the NMJ was fairly well preserved in the mutant animals.
Synaptobrevin is required for evoked neurotransmission
We next performed electrophysiological studies of EPPs in postsynaptic diaphragm muscle cells in response to phrenic nerve activity. EPPs reflect the AP- and calcium-dependent release of neurotransmitters from presynaptic nerve terminals (Katz, 1969). We found that suprathreshold stimulation of the nerve generated EPPs in controls and in mice lacking Syb1 (Syb1lew/lew) or Syb2 (Syb2−/−), but not in mice lacking both Syb1 and Syb2 (Syb1lew/lewSyb2−/−) (Fig. 6A). The average EPP amplitude in animals lacking Syb1 was similar to that in controls (13.37 ± 1.33 mV vs 13.33 ± 0.73 mV, respectively), but it was significantly reduced in mice lacking Syb2 compared with controls (9.43 ± 0.97 mV, n = 22 cells from 7 mice) (p < 0.05), and completely blocked in animals lacking both Syb1 and Syb2 (n = 65 cells from 11 mice) (Fig. 6B). This finding suggests that either Syb1 or Syb2 must be present for evoked neurotransmission to occur in embryonic NMJs.
Figure 6.

Deficiency in both Syb1 and Syb2 leads to a total blockade of evoked synaptic transmission at the NMJ. A, Representative traces of EPPs recorded from an acutely isolated phrenic nerve/diaphragm preparation (E18.5) in response to suprathreshold electrical stimulation to the phrenic nerve. Electrical stimulation of the nerve evoked an EPP in control, Syb1lew/lew, and Syb2−/− mice but failed to evoke an EPP response in Syb1lew/lewSyb2−/− mice. Arrowheads point to stimulus artifacts. B, Quantification of average EPP amplitudes in control, Syb1lew/lew, Syb2−/−, and Syb1lew/lewSyb2−/− mice. EPPs are similar between controls (13.33 ± 0.73 mV, n = 28 cells, N = 7 mice) and Syb1lew/lew NMJs (13.37 ± 1.33 mV, n = 18 cells, N = 4 mice), but are significantly (p < 0.05, t test) reduced in Syb2−/− NMJs (9.43 ± 0.97 mV, n = 22 cells, N = 7 mice) and completely blocked in Syb1lew/lewSyb2−/− NMJs (n = 65 cells, N = 11 mice).
Synaptobrevin deficiency leads to an increase in spontaneous neurotransmitter release
Despite a total absence of EPP, spontaneous release of neurotransmitter, which appeared as mEPPs, was readily detected in Syb1lew/lewSyb2−/− mice (Figs. 7, 8). The mEPPs in both WT and Syb1lew/lewSyb2−/− NMJs occurred stochastically; their frequencies (Fig. 8A) fitted to a Poisson distribution (p > 0.05, χ2 Goodness of Fit test), and their amplitudes (Fig. 8B) and rise time (Fig. 8C) varied over a broad range. These features were similar to those previously reported in WT NMJs (Boyd and Martin, 1956; Liley, 1957; Hubbard and Schmidt, 1963; Gage and Hubbard, 1965; Rotshenker and Rahamimoff, 1970; Bennett and Pettigrew, 1974; Dennis et al., 1981).
Figure 7.

Synaptobrevin deficiency leads to an increase in spontaneous synaptic transmission. A, Sample traces of spontaneous mEPPs recorded in embryonic diaphragm muscles (E18.5) in normal Ringer's solution. Each horizontal line indicates 30 superimposed traces (1 s each). mEPP frequency is markedly increased in Syb1lew/lewSyb2−/− mice compared with control, Syb1lew/lew, or Syb2−/− mice. B–D, Cumulative probability plots of mEPP frequency (B), amplitude (C), and rise time (D). There was no statistical difference in cumulative probability of mEPP amplitude and rise time (10%–90% rise time) among control, Syb1lew/lew, Syb2−/−, and Syb1lew/lewSyb2−/− mice. However, cumulative probability of mEPP frequency in both Syb2−/− and Syb1lew/lewSyb2−/− mice was significantly increased (right-shifted) compared with control or Syb1lew/lew mice (p < 0.001 by Kolmogorov–Smirnov test). E–G, Quantification of mEPP frequency, amplitude, and rise time. Average value of mEPP frequency is increased >5-fold in Syb1lew/lewSyb2−/− (5.94 ± 0.54 min−1) and >2-fold in Syb2−/− mice (2.28 ± 0.36 min−1), compared with that in control (0.89 ± 0.12 min−1) or Syb1lew/lew mice (0.77 ± 0.09 min−1) (E). The average mEPP amplitude is also significantly increased in Syb2−/− (3.48 ± 0.18 mV) and Syb1lew/lewSyb2−/− mice (3.37 ± 0.14 mV), compared with that in control (2.89 ± 0.14 mV) or Syb1lew/lew mice (3.04 ± 0.19 mV) (F). Furthermore, mEPP rise time (10%–90%) is significantly increased in Syb2−/− (6.62 ± 0.38 ms) and Syb1lew/lewSyb2−/− (7.64 ± 0.37 ms) mice, compared with that in control (5.21 ± 0.35 ms) mice (G). The number of cells (n) and mice (N) analyzed: controls, n = 55, N = 10; Syb1lew/lew, n = 49, N = 9; Syb2−/−, n = 63, N = 12; and Syb1lew/lewSyb2−/−, n = 65, N = 11.
Figure 8.

Histogram distribution of mEPP frequency, amplitude, and rise time (10%–90%) in control, Syb1lew/lew, Syb2−/−, and Syb1lew/lewSyb2−/− mice (E18.5). A, Frequency distribution histograms are generated from mEPPs recorded from control (190 mEPPs), Syb1lew/lew (166 mEPPs), Syb2−/− (335 mEPPs), and Syb1lew/lewSyb2−/− (996 mEPPs) mice. By χ2 Goodness of Fit test, the distribution of mEPP fits a Poisson distribution (p > 0.05), indicating that individual spontaneous release was independent and the probability of the release occurred in a random manner. B, C, Histogram distribution of mEPP amplitude and rise time (10%–90%). The number of cells (n) and mice (N) analyzed: controls, n = 55, N = 10; Syb1lew/lew, n = 49, N = 9; Syb2−/−, n = 63, N = 12; and Syb1lew/lewSyb2−/−, n = 65, N = 11.
The mEPPs between control and synaptobrevin-deficient mice differed considerably, however, in terms of their frequency, which was markedly increased in both Syb2−/− and Syb1lew/lewSyb2−/− NMJs, increasing by >50% to 2.28 ± 0.36 min−1 (n = 63 cells from 12 mice) and by >500% to 5.94 ± 0.54 min−1 (n = 65 cells from 11 mice), respectively, compared with controls (0.89 ± 0.12 min−1; n = 55 cells from 10 mice) (Fig. 7B,E).
Furthermore, the mEPPs in Syb1lew/lewSyb2−/− NMJs exhibited a marked increase in amplitude and rise time (10%–90%) compared with controls. We analyzed a total of 190 mEPPs from control NMJs (n = 55 cells, N = 10 mice) and 996 mEPPs from Syb1lew/lewSyb2−/− NMJs (n = 63 cells, N = 11 mice) (Fig. 9). Scatter plots of mEPP amplitude against rise time show that the majority of mEPPs in control NMJs appeared within the bottom left corner of the graph (Fig. 9C), indicating a uniformity in amplitude and rise time. Occasionally, a small number of mEPPs in control NMJs exhibited larger amplitude or slower rise time. Hence, we designated twice the average of mEPP amplitude (≈6 mV) or rise time (≈10 ms) as the criterion to distinguish large mEPPs, or slow mEPPs, respectively. By this criterion, there were 3.2% (6 of 190) large mEPPs and 4.2% (8 of 190) slow mEPPs in control NMJs. In contrast, in Syb1lew/lewSyb2−/− NMJs, there were 10.4% (104 of 996) large mEPPs and 19.8% (197 of 996) slow mEPPs (Fig. 9D). The increased population of large or slow mEPPs in Syb1lew/lewSyb2−/− NMJs was also demonstrated as the “tail” to the right in histogram distribution of mEPP amplitude (>6 mV) (Fig. 8B) or rise time (>10 ms) (Fig. 8C).
Figure 9.

Syb1lew/lewSyb2−/− NMJs exhibit an increase in mEPP amplitude and rise time. A, B, Sample mEPP traces recorded in control (A) and Syb1lew/lewSyb2−/− mice (B). In both control and Syb1lew/lewSyb2−/− mice, recordings from 2 muscle fibers are shown (1 on the left panel and 1 on the right panel). Each panel includes all mEPP events recorded in the same muscle fiber during a 3 min period. Within each panel, each trace represents a 1 s recording in which mEPP events occurred. Traces that did not contain any mEPPs are not shown. mEPPs appear in a broad range of amplitude and time course. Examples of fast (arrows) and slow (arrowheads) mEPPs are shown in B. C, D, Scatter plots displaying mEPP amplitude (x axis) versus 10%–90% rise time (y axis). Each dot represents a single mEPP recorded in control or Syb1lew/lewSyb2−/− mice. The data displayed in the graphs are pooled from 190 events in control mice (n = 55 cells, N = 10 mice) and 996 events in Syb1lew/lewSyb2−/− mice (n = 63 cells, N = 11 mice).
Synaptobrevin confers Ca2+ sensitivity for spontaneous neurotransmitter release
It has previously been shown that spontaneous release at NMJs increases with an increase in extracellular [Ca2+]o, either during a resting state (Boyd and Martin, 1956; Liley, 1956b; Hubbard, 1961; Hubbard et al., 1968; Losavio and Muchnik, 1997) or during membrane depolarization (i.e., in the presence of high potassium levels) (Liley, 1956a; Angleson and Betz, 2001). To further characterize spontaneous release in Syb1lew/lewSyb2−/− NMJs, we recorded mEPPs at various calcium concentrations ([Ca2+]o: 0, 1, 2, and 4 mM) in Ringer's solution. We found that mEPP frequency increased with the calcium concentration (Fig. 10A,B) in controls but remained virtually unchanged in Syb1lew/lewSyb2−/− NMJs (Fig. 10A,B).
Figure 10.
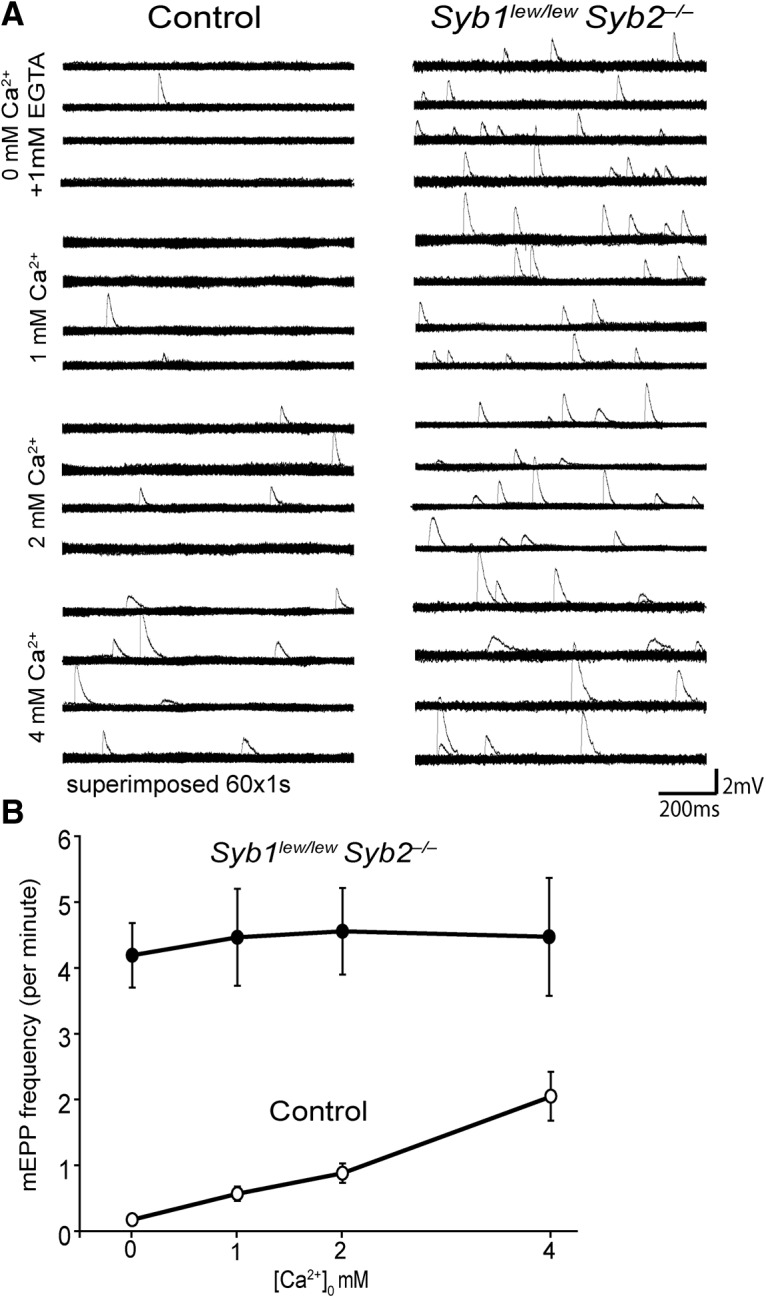
Spontaneous neurotransmitter release in Syb1lew/lewSyb2−/− NMJs is insensitive to extracellular Ca2+. A, Sample traces of mEPPs recorded in Ringer's solution at increasing extracellular Ca2+ concentration ([Ca2+]0, 0, 1, 2, and 4 mM). A continuous recording of 4 min is shown for each cell. Each horizontal line indicates a 1 min continuous recording containing 60 superimposed 1 s traces; 1 mM EGTA was added to 0 mM Ca2+ Ringer's solution, and the muscle samples were incubated in this solution for 60 min before recording. B, Quantification of mEPP frequencies. mEPP frequencies correlated with increases in extracellular calcium in control mice but remained insensitive to extracellular calcium in Syb1lew/lewSyb2−/− mice. mEPP frequencies (events/min) in controls were as follows: 0 mm Ca2+, 0.18 ± 0.06; 1 mM Ca2+, 0.57 ± 0.11; 2 mM Ca2+, 0.88 ± 0.15; and 4 mM Ca2+, 2.05 ± 0.37. mEPP frequencies in Syb1lew/lewSyb2−/− mice were as follows: 0 mM Ca2+, 4.19 ± 0.49; 1 mM Ca2+, 4.46 ± 0.74; 2 mM Ca2+, 4.55 ± 0.66; and 4 mM Ca2+, 4.47 ± 0.89. Sample sizes: 3 pairs of control and Syb1lew/lewSyb2−/− mice were analyzed. Controls, n = 20 cells; Syb1lew/lewSyb2−/−, n = 18 cells.
Similarly, the mEPP frequency in Syb1lew/lewSyb2−/− NMJs was insensitive to changes in calcium levels when the membrane was depolarized by a high concentration of potassium (40 mM) (Fig. 11A–C). The same condition induced a massive increase in mEPP frequency in control and Syb1lew/lew NMJs (Fig. 11B). High potassium also led to a marked increase in mEPP frequency in Syb2−/− NMJs, albeit not to the same extent as in controls or in Syb1lew/lew mice (Fig. 11B). Furthermore, mEPP frequency in control and Syb1lew/lew NMJs was increased dramatically as the concentration of extracellular calcium increased (Fig. 11C); in contrast, the same condition only led to a moderate increase in mEPP frequency in Syb2−/− NMJs (Fig. 11C) and no effect in in Syb1lew/lewSyb2−/− NMJs (Fig. 11C). Together, these data show that the spontaneous release in Syb1lew/lewSyb2−/− NMJs was insensitive to extracellular calcium.
Figure 11.

Spontaneous neurotransmitter release in Syb1lew/lewSyb2−/− NMJs is insensitive to extracellular calcium during membrane depolarization. A, Sample traces of mEPPs recorded in 40 mM K+ Ringer's solution containing three concentrations of calcium (1, 2, and 4 mM). Each horizontal line indicates a single trace recorded for 1 s. B, Comparison of mEPP frequency in normal Ringer's solution and in 40 mM K+ Ringer's solution at 2 mM Ca2+. mEPP frequency was increased 147-fold in controls (from 0.9 ± 0.2 to 131.7 ± 10.6 min−1), 188-fold in Syb1lew/lew (from 0.7 ± 0.1 to 132.5 ± 18.8 min−1), 18-fold in Syb2−/− (from 2.3 ± 0.3 to 42.4 ± 6.7 min−1), and 1.4-fold (from 6.5 ± 0.5 to 9.1 ± 0.8 min−1) in Syb1lew/lewSyb2−/− mutants. C, Potassium-induced mEPP frequency change at various concentrations of Ca2+ (1, 2, and 4 mM). mEPPs correlate with increases in Ca2+ in control or Syb1lew/lew mice but not in Syb1lew/lewSyb2−/− mice. In control and mutant mice, mEPP frequencies fit a Poisson distribution (p > 0.05, by χ2 Goodness of Fit test). Sample size: control, n = 24 muscle fibers, N = 5 mice; Syb1lew/lew, n = 15 muscle fibers, N = 4 mice; Syb2−/−, n = 29 muscle fibers, N = 6 mice; and Syb1lew/lewSyb2−/−, n = 20 muscle fibers, N = 4 mice. Data are mean ± SEM.
To determine whether spontaneous transmitter release at the NMJs in Syb1lew/lewSyb2−/− mice occurs in response to changes to intracellular Ca2+ levels, we equilibrated muscle samples (for 60 min) in calcium-free Ringer's solution containing both EGTA (1 mM) and cell-permeant calcium chelator BAPTA-AM (10 μm). When normal Ringer's solution was replaced with this calcium-free Ringer's solution, EPPs were completely abolished in control, Syb1lew/lew, Syb2−/−, and Syb1lew/lewSyb2−/− mice (Fig. 12D). Furthermore, mEPP frequencies (events/min) decreased in control mice (from 0.99 ± 0.13 to 0.32 ± 0.04, n = 28 cells, N = 4 mice), in Syb1lew/lew mice (from 1.20 ± 0.14 to 0.38 ± 0.05, n = 25 cells, N = 3 mice), and in Syb2−/− mice (from 2.57 ± 0.40 to 0.93 ± 0.12, n = 37 cells, N = 5 mice) (Fig. 12C). By contrast, mEPP frequencies in Syb1lew/lew Syb2−/− mice remained virtually unchanged (from 5.00 ± 0.61 to 4.84 ± 0.92, n = 34 cells, N = 4 mice) (Fig. 12C). These electrophysiological findings demonstrate that spontaneous neurotransmission is sensitive to calcium and that calcium sensitivity is abolished in the absence of both Syb1 and Syb2.
Figure 12.

Spontaneous transmitter release in Syb1lew/lewSyb2−/− NMJs persists despite the withdrawal of both extracellular and intracellular Ca2+. A, B, Sample traces of mEPPs recorded in normal Ringer's solution (2 mm Ca2+) (A), and in Ca2+-free Ringer's solution containing EGTA (1 mM) and BAPTA-AM (10 μm) (B). Each horizontal line indicates 30 superimposed 1 s traces. A 3 min continuous recording is shown for each cell. C, Bar graphs illustrating mEPP frequencies (events/min) recorded in normal Ringer's solution (open bar) and those in Ca2+-free Ringer's (gray bar) in control, Syb1lew/lew, Syb2−/−, and Syb1lew/lewSyb2−/− mice. mEPP frequencies were significantly (***) reduced in control (p = 8.45 × 10−7), Syb1lew/lew (p = 5.15 × 10−9), and Syb2−/− (p = 3.47 × 10−9) when normal Ringer's solution was replaced with Ca2+-free Ringer's solution. In contrast, no significant difference (p = 0.91) was found in mEPP frequencies in Syb1lew/lewSyb2−/− mice recorded in normal Ringer's solution versus Ca2+-free Ringer's solution. D, Sample EPP traces recorded in normal and calcium-free Ringer's solutions. EPPs were completely abolished in control, Syb1lew/lew, Syb2−/−, and Syb1lew/lewSyb2−/− muscles when recorded in calcium-free Ringer's solution (black arrowheads point to stimulus artifact). Controls, n = 28 cells, N = 4 mice; Syb1lew/lew, n = 25 cells, N = 3 mice; Syb2−/−, n = 37 cells, N = 5 mice; and Syb1lew/lewSyb2−/−, n = 34 cells, N = 4 mice. n.s., no significant difference (p = 0.91).
Discussion
In the present study, we have investigated the role of the major neuronal vSNAREs (Syb1 and Syb2) in the formation and function of the NMJ in mice. NMJs are fully formed in double-mutant mice deficient in both Syb1 and Syb2 (Syb1lew/lewSyb2−/−), indicating that neither Syb1 nor Syb2 is required for NMJ synaptogenesis. However, Syb1 and Syb2 are required for evoked neurotransmission because evoked synaptic transmission is completely blocked in Syb1lew/lewSyb2−/− NMJs. By contrast, spontaneous synaptic transmission not only persists but even increased in Syb1lew/lewSyb2−/− NMJs. Furthermore, spontaneous synaptic transmission that is normally highly Ca2+-sensitive became Ca2+-independent upon deletion of Syb1 and Syb2. These findings reveal distinct mechanisms for evoked and spontaneous neurotransmitter release, consistent with previous studies (Deitcher et al., 1998; Hua et al., 1998; Atasoy et al., 2008; Raingo et al., 2012; Ramirez et al., 2012). Moreover, because spontaneous release becomes Ca2+-insensitive in Syb1lew/lewSyb2−/− NMJs, these findings suggest that synaptobrevin-based SNARE complexes perform a critical role in conferring the Ca2+ sensitivity during spontaneous release.
The role of synaptobrevin in the development of the NMJ
Our findings indicate that neither Syb1 nor Syb2 is required for the formation of NMJs, but rather for the normal maturation of NMJs. In the absence of both Syb1 and Syb2, the innervation territories and endplate sizes were markedly increased, and synaptic contact length was significantly reduced. This characteristic phenotype is similar to the phenotype reported for mice lacking choline acetyltransferase (which is the enzyme required for biosynthesis of the neurotransmitter ACh) (Misgeld et al., 2002; Brandon et al., 2003), for mice lacking AChR α1 or AChR γ subunit (Liu et al., 2008; An et al., 2010), for mice lacking L-type Ca2+ channels (Cav1.1) (Powell et al., 1984; Rieger et al., 1984a, b; Chen et al., 2011), and for mice lacking Ca2+ influx through Cav1.1 and Ca2+ release from sarcoplasmic reticulum (Kaplan et al., 2018). The fact that mutations in these genes produce a similar phenotype suggests a common underlying pathway. The ultimate effect of these genetic mutations is the blockade of muscle activity, which suggests that a negative feedback mechanism mediated by muscle activity is involved in fine-tuning the development of the motor nerve terminals during normal NMJ synaptogenesis. In the absence of synaptic transmission at the NMJ, interruption of this negative feedback regulation causes hypertrophy of synaptic junctions, possibly via a mechanism akin to a homeostatic regulatory process.
The role of synaptobrevin in evoked neurotransmission
Our electrophysiological studies demonstrate that evoked neurotransmission at NMJs is completely blocked in the absence of synaptobrevin (i.e., in Syb1lew/lewSyb2−/− mutants, partially blocked in mice lacking Syb2 (Syb2−/−) only, and unaffected in mice lacking Syb1 (Syb1lew/lew) only. These results demonstrate that Syb2 is the major contributor for evoked neurotransmission at developing mouse NMJs, and that Syb1 and Syb2 perform an overlapping role in evoked neurotransmission. These findings are consistent with those of a previous report that the genetic deletion of Syb2 leads to a massive (>100-fold), but not complete, reduction in evoked neurotransmission at central (CNS) synapses (Schoch et al., 2001). The residual fusion activity in Syb2-deficient CNS synapses could reflect low levels of Syb1 (or other vesicular SNARE proteins). A recent report demonstrated that Syb1 is present in a subpopulation of hippocampal neurons and that there is a slight reduction in the frequency of mEPSCs when Syb1 is knocked down in Syb2-null neurons (Zimmermann et al., 2014). In vitro studies have shown that vesicle-axonal fusion requires only two synaptobrevin molecules (Sinha et al., 2011); therefore, a low level of Syb1 may still be sufficient for vesicle fusion to occur.
We have previously shown that evoked neurotransmitter release is significantly reduced in Syb1lew/lew postnatal mice (P14) (Liu et al., 2011). The apparent difference in physiological phenotype between embryonic and postnatal NMJs in Syb1lew/lew mice is likely due to changes in the relative level of Syb1 expression in embryonic versus postnatal motor nerve terminals. Our data show that Syb1 is expressed at a low level during embryonic stages and its expression increases markedly at postnatal stage (Fig. 1D). A similar switch of isoforms has been observed for synaptotagmins, where synaptotagmin-1 is the predominant isoform at birth but is completely replaced by synaptotagmin-2 during the first month of life in mice (Pang et al., 2006a,b). Similarly, the difference in levels of Syb1 and Syb2 expression might explain the differences in the effect of their deletions on neuromuscular synaptic transmission. Syb2 is probably expressed in considerably greater amounts than Syb1 in embryonic mouse NMJs. Thus, the deletion of Syb1 alone in embryonic NMJs has only a negligible effect. By contrast, the deletion of Syb2 could have a considerable effect on evoked synaptic transmission. The presence of Syb1 is essential, however, because the absence of both Syb1 and Syb2 completely blocks evoked neurotransmitter release. These data suggest that a functional redundancy of Syb1 and Syb2 exists in developing NMJs, and, importantly, demonstrate that an optimal level of evoked neurotransmitter release in developing NMJs requires both Syb1 and Syb2.
The role of synaptobrevin in spontaneous release
We have shown that spontaneous neurotransmitter release persists, whereas evoked neurotransmitter release is completely abolished, in Syb1lew/lewSyb2−/− NMJs. This indicates that AP-triggered, Ca2+-dependent vesicle fusion is completely blocked when both Syb1 and Syb2 are absent. Thus, the spontaneous release of neurotransmitters in Syb1lew/lewSyb2−/− NMJs must occur independently of Syb1 and Syb2. This conclusion is consistent with results from previous studies in which (1) spontaneous release persists at NMJs exposed to botulinum neurotoxin (Harris and Miledi, 1971; Spitzer, 1972; Boroff et al., 1974; Cull-Candy et al., 1976; Colméus et al., 1982; Hua et al., 1998); (2) genetic mutations in neuronal synaptobrevin (n-syb) in Drosophila resulted in persistent spontaneous synaptic transmission (Broadie et al., 1995; Sweeney et al., 1995; Deitcher et al., 1998); (3) genetic deletion of the t-SNARE protein Snap-25 blocked evoked, but not spontaneous, neurotransmitter release (Washbourne et al., 2002); (4) exocytosis in inner hair cell ribbon synapses was found to be insensitive to neurotoxins that cleave SNAREs and occurred in mutant mice deficient in SNAREs (Nouvian et al., 2011); (5) deletion of Syb2 from central synapses in mice largely abolished evoked release but did not block spontaneous release (Schoch et al., 2001); and (6) release could still be fully triggered in Syb2-deficient central synapses by a Ca2+ ionophore that led to massive Ca2+ influx in the terminals (Deák et al., 2009).
The observation that spontaneous release occurred in the absence of both Syb1 and Syb2 suggests that these spontaneous release events may involve other mechanisms than those using the major neuronal SNARE proteins. One such possibility is that spontaneous release may be mediated by other SNARE proteins that are expressed at a low level (i.e., at a level undetectable by immunofluorescence; Fig. 1) but are nonetheless sufficient for spontaneous synaptic activity. For example, These proteins include Vti1a, a ubiquitous protein that has been shown to mediate spontaneous fusion in hippocampal synapses (Antonin et al., 2000; Ramirez et al., 2012), VAMP4 (Raingo et al., 2012), or VAMP7 (Hua et al., 2011; Bal et al., 2013). Furthermore, it is also possible that spontaneous release of neurotransmitters may be mediated by VAMP3/cellubrevin, a SNARE protein that is broadly expressed in rodents (McMahon et al., 1993; Chilcote et al., 1995) and in humans (Bernstein and Whiteheart, 1999; Bock et al., 2001; Jahn and Scheller, 2006), and is capable of rescuing synaptic transmission defects in Syb2-deficient neurons (Deák et al., 2006).
In the absence of Syb1 and Syb2 (i.e., Syb1lew/lewSyb2−/−), the frequency of spontaneous neurotransmitter release at NMJs is markedly increased compared with controls. Our data suggest that at least two factors may contribute to the increase in mEPP frequency in Syb1lew/lewSyb2−/− NMJs: (1) increased size of NMJs (Fig. 3), and (2) increased number of NMJs per muscle fiber (Fig. 4). Increases in either NMJ size or NMJ numbers may lead to increase mEPP frequency (Bennett and Pettigrew, 1974; Dennis et al., 1981).
Furthermore, a considerable number of mEPPs in Syb1lew/lewSyb2−/− NMJs show unusual characteristics: they have a prolonged (slow) rise time with varied amplitudes. These mEPPs normally occur in WT NMJs at a low incidence (Liley, 1957). Furthermore, they are insensitive to nerve terminal depolarization or calcium, reminiscent of the “giant mEPPs” previously reported in studies of the NMJs poisoned with botulinum neurotoxin (Harris and Miledi, 1971; Spitzer, 1972; Boroff et al., 1974; Cull-Candy et al., 1976; Colméus et al., 1982; Thesleff et al., 1983; Kim et al., 1984; Hua et al., 1998), or the NMJs treated with 4-aminoquinoline (Molgó et al., 1982; Thesleff et al., 1983) or mefloquine (McArdle et al., 2006).
The underlying mechanism of these giant mEPPs remains to be further elucidated. It has been previously suggested that giant mEPPs are generated by multiquantal unit (Liley, 1957), or by a small pool of SV-like structures (Lupa et al., 1986), or constitutive secretion (Sellin et al., 1996). We consider the following possibilities that may lead to “giant mEPPs.” First, the rise time of mEPPs could be reduced if morphological abnormalities disrupt the alignment of active zones on the nerve terminal and endplate clusters of AChRs. We have examined numerous electron micrographs of the NMJs. Occasionally, we did observe ectopic vesicle clusters (e.g., vesicles docked at sites not directly opposite to postsynaptic membrane. However, such an incidence was rare and very hard to quantify (varied dependent upon the plane of electron microscopy sections). Second, for very large mEPPs, which rise slowly, it is also possible the large volume of transmitter release just acts over a wider than normal endplate area, and so, causes asynchronous AChR activation. Third, it is common that mEPPs recorded from the same muscle fiber (i.e., at the same synaptic sites) appeared with a broad range of rise times (some fast and some slow). This is because embryonic NMJs are typically innervated by multiple nerve terminals. Thus, the mEPPs recorded from each muscle fiber resulted from spontaneous releases originated from various nerve terminals, with various distance to the recording electrode, which may produce various rise times and amplitudes. Since incidence of giant mEPPs in Syb1lew/lewSyb2−/− NMJs was massively increased, this suggests that Syb1 and Syb2 are involved in regulating the generation of giant mEPPs. In other words, the presence of Syb1/Syb2 may regulate the occurrences of giant mEPPs in WT NMJs.
In mammalian NMJs, the frequency of mEPPs positively correlates with extracellular [Ca2+] (Boyd and Martin, 1956; Liley, 1956b; Hubbard et al., 1968); however, some mEPPs, although at markedly reduced frequency, could still be recorded in calcium-free solution, even in the presence of a chelating agent, such as EDTA and/or EGTA (Hubbard, 1961; Elmqvist and Feldman, 1965; Hubbard et al., 1968). A Ca2+ sensor(s) specific for spontaneous neurotransmission remains unknown. Emerging evidence suggests that multiple players, including synaptotagmins (Pang et al., 2006a; Maximov et al., 2007; Xu et al., 2009), Doc2 (Groffen et al., 2010; Pang et al., 2011), and complexins (Huntwork and Littleton, 2007; Maximov et al., 2009; Strenzke et al., 2009; Xue et al., 2009, 2010; Yang et al., 2010; Hobson et al., 2011; Martin et al., 2011; Lai et al., 2014), are involved in regulating spontaneous neurotransmission. In the absence of synaptotagmins, spontaneous release remains Ca2+-sensitive, albeit with a different Ca2+ dependence (Xu et al., 2009). We show that spontaneous release is normally increased as the Ca2+ concentration increases, and this Ca2+ sensitivity is lost in Syb1lew/lewSyb2−/− NMJs). These findings suggest that Syb1 and Syb2 are required to confer Ca2+ sensitivity during spontaneous neurotransmission, presumably because Syb1/Syb2-containing SNARE complexes interact with synaptotagmin Ca2+ sensors during spontaneous neurotransmission.
Footnotes
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant R01 NS055028 to W.L., Edward Mallinckrodt, Jr. Foundation to W.L., Cain Foundation in Medical Research to W.L., and Robert Packard Center for ALS Research at Johns Hopkins University to W.L. We thank Qiaohong Ye for excellent technical assistance; and Drs. Vanessa Routh, Joseph McArdle, Ben Szaro, and Jose Rizo-Rey for critical comments on manuscript drafts.
The authors declare no competing financial interests.
References
- An MC, Lin W, Yang J, Dominguez B, Padgett D, Sugiura Y, Aryal P, Gould TW, Oppenheim RW, Hester ME, Kaspar BK, Ko CP, Lee KF (2010) Acetylcholine negatively regulates development of the neuromuscular junction through distinct cellular mechanisms. Proc Natl Acad Sci U S A 107:10702–10707. 10.1073/pnas.1004956107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angleson JK, Betz WJ (2001) Intraterminal Ca(2+) and spontaneous transmitter release at the frog neuromuscular junction. J Neurophysiol 85:287–294. 10.1152/jn.2001.85.1.287 [DOI] [PubMed] [Google Scholar]
- Antonin W, Riedel D, von Mollard GF (2000) The SNARE Vti1a-beta is localized to small synaptic vesicles and participates in a novel SNARE complex. J Neurosci 20:5724–5732. 10.1523/JNEUROSCI.20-15-05724.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Ertunc M, Moulder KL, Blackwell J, Chung C, Su J, Kavalali ET (2008) Spontaneous and evoked glutamate release activates two populations of NMDA receptors with limited overlap. J Neurosci 28:10151–10166. 10.1523/JNEUROSCI.2432-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ, Charlton MP, Smith SJ (1987) Calcium action in synaptic transmitter release. Annu Rev Neurosci 10:633–693. 10.1146/annurev.ne.10.030187.003221 [DOI] [PubMed] [Google Scholar]
- Bal M, Leitz J, Reese AL, Ramirez DM, Durakoglugil M, Herz J, Monteggia LM, Kavalali ET (2013) Reelin mobilizes a VAMP7-dependent synaptic vesicle pool and selectively augments spontaneous neurotransmission. Neuron 80:934–946. 10.1016/j.neuron.2013.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EF, Stevens CF (1972) The kinetics of transmitter release at the frog neuromuscular junction. J Physiol 227:691–708. 10.1113/jphysiol.1972.sp010054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MR, Pettigrew AG (1974) The formation of synapses in striated muscle during development. J Physiol 241:515–545. 10.1113/jphysiol.1974.sp010670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein AM, Whiteheart SW (1999) Identification of a cellubrevin/vesicle associated membrane protein 3 homologue in human platelets. Blood 93:571–579. [PubMed] [Google Scholar]
- Bock JB, Matern HT, Peden AA, Scheller RH (2001) A genomic perspective on membrane compartment organization. Nature 409:839–841. 10.1038/35057024 [DOI] [PubMed] [Google Scholar]
- Boroff DA, del Castillo J, Evoy WH, Steinhardt RA (1974) Observations on the action of type A botulinum toxin on frog neuromuscular junctions. J Physiol 240:227–253. 10.1113/jphysiol.1974.sp010608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd IA, Martin AR (1956) Spontaneous subthreshold activity at mammalian neural muscular junctions. J Physiol 132:61–73. 10.1113/jphysiol.1956.sp005502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon EP, Lin W, D'Amour KA, Pizzo DP, Dominguez B, Sugiura Y, Thode S, Ko CP, Thal LJ, Gage FH, Lee KF (2003) Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J Neurosci 23:539–549. 10.1523/JNEUROSCI.23-02-00539.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadie K, Prokop A, Bellen HJ, O'Kane CJ, Schulze KL, Sweeney ST (1995) Syntaxin and synaptobrevin function downstream of vesicle docking in Drosophila. Neuron 15:663–673. 10.1016/0896-6273(95)90154-X [DOI] [PubMed] [Google Scholar]
- Brunger AT, Choi UB, Lai Y, Leitz J, Zhou Q (2018) Molecular mechanisms of fast neurotransmitter release. Annu Rev Biophys 47:469–497. 10.1146/annurev-biophys-070816-034117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Liu Y, Sugiura Y, Allen PD, Gregg RG, Lin W (2011) Neuromuscular synaptic patterning requires the function of skeletal muscle dihydropyridine receptors. Nat Neurosci 14:570–577. 10.1038/nn.2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilcote TJ, Galli T, Mundigl O, Edelmann L, McPherson PS, Takei K, De Camilli P (1995) Cellubrevin and synaptobrevins: similar subcellular localization and biochemical properties in PC12 cells. J Cell Biol 129:219–231. 10.1083/jcb.129.1.219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BJ, Imlach WL, Jiao W, Wolfram V, Wu Y, Grbic M, Cela C, Baines RA, Nitabach MN, McCabe BD (2014) Miniature neurotransmission regulates Drosophila synaptic structural maturation. Neuron 82:618–634. 10.1016/j.neuron.2014.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colméus C, Gomez S, Molgó J, Thesleff S (1982) Discrepancies between spontaneous and evoked synaptic potentials at normal, regenerating and botulinum toxin poisoned mammalian neuromuscular junctions. Proc R Soc Lond B Biol Sci 215:63–74. 10.1098/rspb.1982.0028 [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Lundh H, Thesleff S (1976) Effects of botulinum toxin on neuromuscular transmission in the rat. J Physiol 260:177–203. 10.1113/jphysiol.1976.sp011510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deák F, Shin OH, Kavalali ET, Südhof TC (2006) Structural determinants of synaptobrevin 2 function in synaptic vesicle fusion. J Neurosci 26:6668–6676. 10.1523/JNEUROSCI.5272-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deák F, Liu X, Khvotchev M, Li G, Kavalali ET, Sugita S, Südhof TC (2009) Alpha-latrotoxin stimulates a novel pathway of Ca2+-dependent synaptic exocytosis independent of the classical synaptic fusion machinery. J Neurosci 29:8639–8648. 10.1523/JNEUROSCI.0898-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitcher DL, Ueda A, Stewart BA, Burgess RW, Kidokoro Y, Schwarz TL (1998) Distinct requirements for evoked and spontaneous release of neurotransmitter are revealed by mutations in the Drosophila gene neuronal-synaptobrevin. J Neurosci 18:2028–2039. 10.1523/JNEUROSCI.18-06-02028.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MJ, Ziskind-Conhaim L, Harris AJ (1981) Development of neuromuscular junctions in rat embryos. Dev Biol 81:266–279. 10.1016/0012-1606(81)90290-6 [DOI] [PubMed] [Google Scholar]
- Dodge FA Jr, Rahamimoff R (1967) Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol 193:419–432. 10.1113/jphysiol.1967.sp008367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmqvist D, Feldman DS (1965) Calcium dependence of spontaneous acetylcholine release at mammalian motor nerve terminals. J Physiol 181:487–497. 10.1113/jphysiol.1965.sp007777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredj NB, Burrone J (2009) A resting pool of vesicles is responsible for spontaneous vesicle fusion at the synapse. Nat Neurosci 12:751–758. 10.1038/nn.2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman AH, Banker GA (1996) The economics of neurite outgrowth: the addition of new membrane to growing axons. Trends Neurosci 19:144–149. 10.1016/S0166-2236(96)80025-7 [DOI] [PubMed] [Google Scholar]
- Gage PW, Hubbard JI (1965) Evidence for a Poisson distribution of miniature end-plate potentials and some implications. Nature 208:395–396. 10.1038/208395a0 [DOI] [PubMed] [Google Scholar]
- Goda Y, Stevens CF (1994) Two components of transmitter release at a central synapse. Proc Natl Acad Sci U S A 91:12942–12946. 10.1073/pnas.91.26.12942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groffen AJ, Martens S, Díez Arazola R, Cornelisse LN, Lozovaya N, de Jong AP, Goriounova NA, Habets RL, Takai Y, Borst JG, Brose N, McMahon HT, Verhage M (2010) Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science 327:1614–1618. 10.1126/science.1183765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris AJ, Miledi R (1971) The effect of type D botulinum toxin on frog neuromuscular junctions. J Physiol 217:497–515. 10.1113/jphysiol.1971.sp009582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris BS, Johnson KR, Bronson RT, Davisson MT (2003) Lethal wasting (lew), a new mouse mutation causing prewean lethality. Available at http://www.informatics.jax.org/reference/J:81354.
- Hepp R, Langley K (2001) SNAREs during development. Cell Tissue Res 305:247–253. 10.1007/s004410100359 [DOI] [PubMed] [Google Scholar]
- Hobson RJ, Liu Q, Watanabe S, Jorgensen EM (2011) Complexin maintains vesicles in the primed state in C. elegans. Curr Biol 21:106–113. 10.1016/j.cub.2010.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua SY, Raciborska DA, Trimble WS, Charlton MP (1998) Different VAMP/synaptobrevin complexes for spontaneous and evoked transmitter release at the crayfish neuromuscular junction. J Neurophysiol 80:3233–3246. 10.1152/jn.1998.80.6.3233 [DOI] [PubMed] [Google Scholar]
- Hua Z, Leal-Ortiz S, Foss SM, Waites CL, Garner CC, Voglmaier SM, Edwards RH (2011) v-SNARE composition distinguishes synaptic vesicle pools. Neuron 71:474–487. 10.1016/j.neuron.2011.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard JI. (1961) The effect of calcium and magnesium on the spontaneous release of transmitter from mammalian motor nerve endings. J Physiol 159:507–517. 10.1113/jphysiol.1961.sp006824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard JI, Schmidt RF (1963) An electrophysiological investigation of mammalian motor nerve terminals. J Physiol 166:145–167. 10.1113/jphysiol.1963.sp007096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard JI, Jones SF, Landau EM (1968) On the mechanism by which calcium and magnesium affect the spontaneous release of transmitter from mammalian motor nerve terminals. J Physiol 194:355–380. 10.1113/jphysiol.1968.sp008413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume RI, Role LW, Fischbach GD (1983) Acetylcholine release from growth cones detected with patches of acetylcholine receptor-rich membranes. Nature 305:632–634. 10.1038/305632a0 [DOI] [PubMed] [Google Scholar]
- Huntwork S, Littleton JT (2007) A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat Neurosci 10:1235–1237. 10.1038/nn1980 [DOI] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D (2012) Molecular machines governing exocytosis of synaptic vesicles. Nature 490:201–207. 10.1038/nature11320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Scheller RH (2006) SNAREs: engines for membrane fusion. Nat Rev Mol Cell Biol 7:631–643. 10.1038/nrm2002 [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Regehr WG (2014) Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu Rev Physiol 76:333–363. 10.1146/annurev-physiol-021113-170338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MM, Sultana N, Benedetti A, Obermair GJ, Linde NF, Papadopoulos S, Dayal A, Grabner M, Flucher BE (2018) Calcium influx and release cooperatively regulate AChR patterning and motor axon outgrowth during neuromuscular junction formation. Cell Rep 23:3891–3904. 10.1016/j.celrep.2018.05.085 [DOI] [PubMed] [Google Scholar]
- Katz B. (1969) The release of neural transmitter substances. Liverpool: Liverpool UP. [Google Scholar]
- Kavalali ET, Chung C, Khvotchev M, Leitz J, Nosyreva E, Raingo J, Ramirez DM (2011) Spontaneous neurotransmission: an independent pathway for neuronal signaling? Physiology (Bethesda) 26:45–53. 10.1152/physiol.00040.2010 [DOI] [PubMed] [Google Scholar]
- Kelly AM, Zacks SI (1969) The fine structure of motor endplate morphogenesis. J Cell Biol 42:154–169. 10.1083/jcb.42.1.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YI, Lømo T, Lupa MT, Thesleff S (1984) Miniature endplate potentials in rat skeletal muscle poisoned with botulinum toxin. J Physiol 356:587–599. 10.1113/jphysiol.1984.sp015484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Diao J, Cipriano DJ, Zhang Y, Pfuetzner RA, Padolina MS, Brunger AT (2014) Complexin inhibits spontaneous release and synchronizes Ca2+-triggered synaptic vesicle fusion by distinct mechanisms. Elife 3:e03756. 10.7554/eLife.03756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liley AW. (1956a) The effects of presynaptic polarization on the spontaneous activity at the mammalian neuromuscular junction. J Physiol 134:427–443. 10.1113/jphysiol.1956.sp005655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liley AW. (1956b) An investigation of spontaneous activity at the neuromuscular junction of the rat. J Physiol 132:650–666. 10.1113/jphysiol.1956.sp005555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liley AW. (1957) Spontaneous release of transmitter substance in multiquantal units. J Physiol 136:595–605. 10.1113/jphysiol.1957.sp005784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Padgett D, Takahashi M, Li H, Sayeed A, Teichert RW, Olivera BM, McArdle JJ, Green WN, Lin W (2008) Essential roles of the acetylcholine receptor gamma-subunit in neuromuscular synaptic patterning. Development 135:1957–1967. 10.1242/dev.018119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sugiura Y, Lin W (2011) The role of synaptobrevin1/VAMP1 in Ca2+-triggered neurotransmitter release at the mouse neuromuscular junction. J Physiol 589:1603–1618. 10.1113/jphysiol.2010.201939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Steinberg IZ, Walton K (1981) Relationship between presynaptic calcium current and postsynaptic potential in squid giant synapse. Biophys J 33:323–351. 10.1016/S0006-3495(81)84899-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losavio A, Muchnik S (1997) Spontaneous acetylcholine release in mammalian neuromuscular junctions. Am J Physiol 273:C1835–C1841. 10.1152/ajpcell.1997.273.6.C1835 [DOI] [PubMed] [Google Scholar]
- Lupa MT, Tabti N, Thesleff S, Vyskocil F, Yu SP (1986) The nature and origin of calcium-insensitive miniature endplate potentials at rodent neuromuscular junctions. J Physiol 381:607–618. 10.1113/jphysiol.1986.sp016346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques MJ, Conchello JA, Lichtman JW (2000) From plaque to pretzel: fold formation and acetylcholine receptor loss at the developing neuromuscular junction. J Neurosci 20:3663–3675. 10.1523/JNEUROSCI.20-10-03663.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JA, Hu Z, Fenz KM, Fernandez J, Dittman JS (2011) Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr Biol 21:97–105. 10.1016/j.cub.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Shin OH, Liu X, Südhof TC (2007) Synaptotagmin-12, a synaptic vesicle phosphoprotein that modulates spontaneous neurotransmitter release. J Cell Biol 176:113–124. 10.1083/jcb.200607021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang ZP, Südhof TC (2009) Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science 323:516–521. 10.1126/science.1166505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArdle JJ, Sellin LC, Coakley KM, Potian JG, Hognason K (2006) Mefloquine selectively increases asynchronous acetylcholine release from motor nerve terminals. Neuropharmacology 50:345–353. 10.1016/j.neuropharm.2005.09.011 [DOI] [PubMed] [Google Scholar]
- McMahon HT, Ushkaryov YA, Edelmann L, Link E, Binz T, Niemann H, Jahn R, Südhof TC (1993) Cellubrevin is a ubiquitous tetanus-toxin substrate homologous to a putative synaptic vesicle fusion protein. Nature 364:346–349. 10.1038/364346a0 [DOI] [PubMed] [Google Scholar]
- Melom JE, Akbergenova Y, Gavornik JP, Littleton JT (2013) Spontaneous and evoked release are independently regulated at individual active zones. J Neurosci 33:17253–17263. 10.1523/JNEUROSCI.3334-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR (2002) Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron 36:635–648. 10.1016/S0896-6273(02)01020-6 [DOI] [PubMed] [Google Scholar]
- Molgó J, Gomez S, Polak RL, Thesleff S (1982) Giant miniature endplate potentials induced by 4-aminoquinoline. Acta Physiol Scand 115:201–207. 10.1111/j.1748-1716.1982.tb07066.x [DOI] [PubMed] [Google Scholar]
- Nouvian R, Neef J, Bulankina AV, Reisinger E, Pangrsic T, Frank T, Sikorra S, Brose N, Binz T, Moser T (2011) Exocytosis at the hair cell ribbon synapse apparently operates without neuronal SNARE proteins. Nat Neurosci 14:411–413. 10.1038/nn.2774 [DOI] [PubMed] [Google Scholar]
- Nystuen AM, Schwendinger JK, Sachs AJ, Yang AW, Haider NB (2007) A null mutation in VAMP1/synaptobrevin is associated with neurological defects and prewean mortality in the lethal-wasting mouse mutant. Neurogenetics 8:1–10. 10.1007/s10048-006-0068-7 [DOI] [PubMed] [Google Scholar]
- Pang ZP, Sun J, Rizo J, Maximov A, Südhof TC (2006a) Genetic analysis of synaptotagmin 2 in spontaneous and Ca(2+)-triggered neurotransmitter release. EMBO J 25:2039–2050. 10.1038/sj.emboj.7601103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Melicoff E, Padgett D, Liu Y, Teich AF, Dickey BF, Lin W, Adachi R, Südhof TC (2006b) Synaptotagmin-2 is essential for survival and contributes to Ca2+ triggering of neurotransmitter release in central and neuromuscular synapses. J Neurosci 26:13493–13504. 10.1523/JNEUROSCI.3519-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Bacaj T, Yang X, Zhou P, Xu W, Südhof TC (2011) Doc2 supports spontaneous synaptic transmission by a Ca(2+)-independent mechanism. Neuron 70:244–251. 10.1016/j.neuron.2011.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JA, Rieger F, Blondet B, Dreyfus P, Pinçon-Raymond M (1984) Distribution and quantification of ACh receptors and innervation in diaphragm muscle of normal and mdg mouse embryos. Dev Biol 101:168–180. 10.1016/0012-1606(84)90127-1 [DOI] [PubMed] [Google Scholar]
- Raingo J, Khvotchev M, Liu P, Darios F, Li YC, Ramirez DM, Adachi M, Lemieux P, Toth K, Davletov B, Kavalali ET (2012) VAMP4 directs synaptic vesicles to a pool that selectively maintains asynchronous neurotransmission. Nat Neurosci 15:738–745. 10.1038/nn.3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Khvotchev M, Trauterman B, Kavalali ET (2012) Vti1a identifies a vesicle pool that preferentially recycles at rest and maintains spontaneous neurotransmission. Neuron 73:121–134. 10.1016/j.neuron.2011.10.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger F, Powell JA, Pinçon-Raymond M (1984a) Extensive nerve overgrowth and paucity of the tailed asymmetric form (16 S) of acetylcholinesterase in the developing skeletal neuromuscular system of the dysgenic (mdg/mdg) mouse. Dev Biol 101:181–191. 10.1016/0012-1606(84)90128-3 [DOI] [PubMed] [Google Scholar]
- Rieger F, Cross D, Peterson A, Pinçon-Raymond M, Tretjakoff I (1984b) Disease expression in +-/+- —-mdg/mdg mouse chimeras: evidence for an extramuscular component in the pathogenesis of both dysgenic abnormal diaphragm innervation and skeletal muscle 16 S acetylcholinesterase deficiency. Dev Biol 106:296–306. 10.1016/0012-1606(84)90228-8 [DOI] [PubMed] [Google Scholar]
- Rizo J, Südhof TC (2012) The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices: guilty as charged? Annu Rev Cell Dev Biol 28:279–308. 10.1146/annurev-cellbio-101011-155818 [DOI] [PubMed] [Google Scholar]
- Rizo J, Xu J (2015) The synaptic vesicle release machinery. Annu Rev Biophys 44:339–367. 10.1146/annurev-biophys-060414-034057 [DOI] [PubMed] [Google Scholar]
- Rotshenker S, Rahamimoff R (1970) Neuromuscular synapse: stochastic properties of spontaneous release of transmitter. Science 170:648–649. 10.1126/science.170.3958.648 [DOI] [PubMed] [Google Scholar]
- Sara Y, Virmani T, Deák F, Liu X, Kavalali ET (2005) An isolated pool of vesicles recycles at rest and drives spontaneous neurotransmission. Neuron 45:563–573. 10.1016/j.neuron.2004.12.056 [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Rosenmund C (2015) Molecular mechanisms governing Ca(2+) regulation of evoked and spontaneous release. Nat Neurosci 18:935–941. 10.1038/nn.4044 [DOI] [PubMed] [Google Scholar]
- Schoch S, Deák F, Königstorfer A, Mozhayeva M, Sara Y, Südhof TC, Kavalali ET (2001) SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294:1117–1122. 10.1126/science.1064335 [DOI] [PubMed] [Google Scholar]
- Sellin LC, Molgó J, Törnquist K, Hansson B, Thesleff S (1996) On the possible origin of giant or slow-rising miniature endplate potentials at the neuromuscular junction. Pflugers Arch 431:325–334. 10.1007/BF02207269 [DOI] [PubMed] [Google Scholar]
- Sinha R, Ahmed S, Jahn R, Klingauf J (2011) Two synaptobrevin molecules are sufficient for vesicle fusion in central nervous system synapses. Proc Natl Acad Sci U S A 108:14318–14323. 10.1073/pnas.1101818108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater CR. (1982) Postnatal maturation of nerve-muscle junctions in hindlimb muscles of the mouse. Dev Biol 94:11–22. 10.1016/0012-1606(82)90063-X [DOI] [PubMed] [Google Scholar]
- Spitzer N. (1972) Miniature endplate potentials at mammalian neuromuscular junctions poisoned by botulinum toxin. Nat New Biol 237:26–27. 10.1038/newbio237026a0 [DOI] [PubMed] [Google Scholar]
- Strenzke N, Chanda S, Kopp-Scheinpflug C, Khimich D, Reim K, Bulankina AV, Neef A, Wolf F, Brose N, Xu-Friedman MA, Moser T (2009) Complexin-I is required for high-fidelity transmission at the endbulb of held auditory synapse. J Neurosci 29:7991–8004. 10.1523/JNEUROSCI.0632-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. (2013a) A molecular machine for neurotransmitter release: synaptotagmin and beyond. Nat Med 19:1227–1231. 10.1038/nm.3338 [DOI] [PubMed] [Google Scholar]
- Südhof TC. (2013b) Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80:675–690. 10.1016/j.neuron.2013.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Rothman JE (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323:474–477. 10.1126/science.1161748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Wall NR, Aakalu GN, Schuman EM (2004) Regulation of dendritic protein synthesis by miniature synaptic events. Science 304:1979–1983. 10.1126/science.1096202 [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM (2006) Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 125:785–799. 10.1016/j.cell.2006.03.040 [DOI] [PubMed] [Google Scholar]
- Sweeney ST, Broadie K, Keane J, Niemann H, O'Kane CJ (1995) Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron 14:341–351. 10.1016/0896-6273(95)90290-2 [DOI] [PubMed] [Google Scholar]
- Thesleff S, Molgó J, Lundh H (1983) Botulinum toxin and 4-aminoquinoline induce a similar abnormal type of spontaneous quantal transmitter release at the rat neuromuscular junction. Brain Res 264:89–97. 10.1016/0006-8993(83)91123-X [DOI] [PubMed] [Google Scholar]
- Turrigiano G. (2012) Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol 4:a005736. 10.1101/cshperspect.a005736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Benditó G, Molnár Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC (2002) Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5:19–26. 10.1038/nn783 [DOI] [PubMed] [Google Scholar]
- Xu J, Pang ZP, Shin OH, Südhof TC (2009) Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat Neurosci 12:759–766. 10.1038/nn.2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Lin YQ, Pan H, Reim K, Deng H, Bellen HJ, Rosenmund C (2009) Tilting the balance between facilitatory and inhibitory functions of mammalian and Drosophila complexins orchestrates synaptic vesicle exocytosis. Neuron 64:367–380. 10.1016/j.neuron.2009.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Craig TK, Xu J, Chao HT, Rizo J, Rosenmund C (2010) Binding of the complexin N terminus to the SNARE complex potentiates synaptic-vesicle fusogenicity. Nat Struct Mol Biol 17:568–575. 10.1038/nsmb.1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Kaeser-Woo YJ, Pang ZP, Xu W, Südhof TC (2010) Complexin clamps asynchronous release by blocking a secondary Ca(2+) sensor via its accessory alpha helix. Neuron 68:907–920. 10.1016/j.neuron.2010.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SH, Poo MM (1983) Spontaneous release of transmitter from growth cones of embryonic neurones. Nature 305:634–637. 10.1038/305634a0 [DOI] [PubMed] [Google Scholar]
- Zhou P, Pang ZP, Yang X, Zhang Y, Rosenmund C, Bacaj T, Südhof TC (2013) Syntaxin-1 N-peptide and Habc-domain perform distinct essential functions in synaptic vesicle fusion. EMBO J 32:159–171. 10.1038/emboj.2012.307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann J, Trimbuch T, Rosenmund C (2014) Synaptobrevin 1 mediates vesicle priming and evoked release in a subpopulation of hippocampal neurons. J Neurophysiol 112:1559–1565. 10.1152/jn.00340.2014 [DOI] [PubMed] [Google Scholar]