

Abstract
During mammalian neocortical development, neural precursor cells generate neurons first and astrocytes later. The cell fate switch from neurons to astrocytes is a key process generating proper numbers of neurons and astrocytes. Although the intracellular mechanisms regulating this cell fate switch have been well characterized, extracellular regulators are still largely unknown. Here, we uncovered that fibroblast growth factor (FGF) regulates the cell fate switch from neurons to astrocytes in the developing cerebral cortex using mice of both sexes. We found that the FGF signaling pathway is activated in radial glial cells of the ventricular zone at time points corresponding to the switch in cell fate. Our loss- and gain-of-function studies using in utero electroporation indicate that activation of FGF signaling is necessary and sufficient to change cell fates from neurons to astrocytes. We further found that the FGF-induced neuron–astrocyte cell fate switch is mediated by the MAPK pathway. These results indicate that FGF is a critical extracellular regulator of the cell fate switch from neurons to astrocytes in the mammalian cerebral cortex.
SIGNIFICANCE STATEMENT Although the intracellular mechanisms regulating the neuron–astrocyte cell fate switch in the mammalian cerebral cortex during development have been well studied, their upstream extracellular regulators remain unknown. By using in utero electroporation, our study provides in vivo data showing that activation of FGF signaling is necessary and sufficient for changing cell fates from neurons to astrocytes. Manipulation of FGF signaling activity led to drastic changes in the numbers of neurons and astrocytes. These results indicate that FGF is a key extracellular regulator determining the numbers of neurons and astrocytes in the mammalian cerebral cortex, and is indispensable for the establishment of appropriate neural circuitry.
Keywords: astrocyte, cell fate switch, cerebral cortex, FGF, neuron
Introduction
Neural precursor cells (NPCs) undergo a characteristic temporal pattern of differentiation in the developing cerebral cortex wherein neurons are generated first, followed by astrocytes and finally oligodendrocytes (Qian et al., 2000; Hirabayashi et al., 2009; Wang et al., 2016). The cell fate switch from neurons to astrocytes is critical not only for determining how many neurons and astrocytes are ultimately made, but also for the establishment of neural circuitry (Miller and Gauthier, 2007; Freeman, 2010; Bronstein et al., 2017). This cell fate switch consists of two distinct molecular processes: the cessation of neurogenesis and the initiation of astrocytogenesis. It has been proposed that the neuron–astrocyte cell fate switch is governed by both extracellular cues and intracellular mechanisms (Rowitch and Kriegstein, 2010; Kang et al., 2012).
One of the most relevant controls of the neuron–astrocyte cell fate switch would be the intrinsic epigenetic changes in the NPCs. A previous study showed that Polycomb group (PcG) proteins, which are well known epigenetic regulators, play key roles in chromatin dynamics and the neuron–astrocyte cell fate switch in the developing cerebral cortex (Hirabayashi et al., 2009). High mobility group B2, which serves as an enhancer of Polycomb (Déjardin et al., 2005), was reported to regulate PcG proteins and consequently the neuron–astrocyte cell fate switch (Bronstein et al., 2017). Furthermore, recent pioneering studies investigated the intracellular signaling mechanisms of the neuron–astrocyte cell fate switch in the developing mouse cerebral cortex and demonstrated the involvement of the gp130/JAK/STAT pathway and the MEK/MAPK pathway. Loss-of-function of gp130 and STAT3 resulted in an excess of neurons along with impaired astrocytogenesis (Nakashima et al., 1999; He et al., 2005; Cao et al., 2010). Similarly, MEK1/2 deletion led to extended neurogenesis and a persistent failure of astrocytogenesis (Li et al., 2012). In contrast to the intracellular mechanisms, the extracellular mechanisms regulating the neuron–astrocyte cell fate switch remain largely unknown. Although ciliary neurotrophic factor, which is an extracellular regulator of the gp130/JAK/STAT pathway, was reported to suppress neurogenesis and promote astrocytogenesis in culture, it did not show obvious effects in vivo (Masu et al., 1993; DeChiara et al., 1995).
Because the MEK/MAPK pathway is activated by growth factors, and fibroblast growth factor receptors (Fgfrs) are expressed in the developing cerebral cortex, we focused on fibroblast growth factor (FGF). Here, we uncovered that FGF regulates the cell fate switch from neurons to astrocytes in the developing mouse cerebral cortex. We found that the FGF signaling pathway was activated in radial glial cells (RGCs) of the ventricular zone (VZ) at time points corresponding to the cell fate switch. Activation of FGF signaling suppressed neurogenesis and promoted astrocytogenesis. Furthermore, inhibition of FGF signaling by dominant-negative FGFR3 promoted neurogenesis but inhibited astrocytogenesis. These results indicate that FGF is a critical extracellular regulator of the cell fate switch from neurons to astrocytes in the mammalian cerebral cortex.
Materials and Methods
Animals.
ICR mice were purchased from SLC (Hamamatsu) and reared on a normal 12 h light/dark schedule. The day of conception and that of birth were counted as embryonic day (E)0 and postnatal day (P)0, respectively. Mouse pups of both sexes were used for the experiments. All procedures were performed in accordance with protocols approved by the Animal Care Committee of Kanazawa University.
In utero electroporation (IUE) procedure.
In utero electroporation using mice was performed as described previously with slight modifications (Fukuchi-Shimogori and Grove, 2001; Tabata and Nakajima, 2001; Saito, 2006; Wakimoto et al., 2015; Hoshiba et al., 2016). Briefly, pregnant ICR mice were anesthetized, and the uterine horns were exposed. Approximately 1–2 μl of DNA solution was injected into the lateral ventricle of embryos using a pulled glass micropipette. Each embryo within its uterus was placed between tweezer-type electrodes (CUY650 P0.5-3, NEPA Gene). Square electric pulses (40 V, 50 ms) were passed five times at 1 s intervals using the electroporator. Care was taken to quickly place embryos back into the abdominal cavity to avoid excessive temperature loss. The wall and skin of the abdominal cavity were sutured, and embryos were allowed to develop normally.
Plasmids.
pCAG-EGFP, pCAG-FGF8 and pCAG-sFGFR3c were described previously (Sehara et al., 2010; Masuda et al., 2015; Matsumoto et al., 2017). We combined the piggyBac (PB) transposase system and in utero electroporation to induce stable expression of transgenes. PB-CAG-EiP and pCAG-PBase were described previously (Matsui et al., 2014; Kim et al., 2016). Plasmids were purified using the EndoFree Plasmid Maxi kit (Qiagen). For gain-of-function experiments, a mixture of pCAG-EGFP (0.5 mg/ml) plus either pCAG-FGF8 or pCAG control plasmid (1 mg/ml) in PBS was used. For loss-of-function experiments, a mixture of PB-CAG-EiP (1.6 mg/ml), pCAG-PBase (0.4 mg/ml) plus either pCAG-sFGFR3c or empty vector plasmid (3 mg/ml) in PBS was used. SASA-MEK, a dominant-negative form of MEK (DN-MEK), was kindly provided by Dr. Eisuke Nishida (RIKEN Center for Biosystems Dynamics Research). Two serines, which are important for MEK activation, were replaced with alanines in SASA-MEK (Zheng and Guan, 1994; Gotoh et al., 1999). SASA-MEK was inserted into pCAG plasmid. Before in utero electroporation procedures, Fast Green solution was added to a final concentration of 0.3% to monitor the injection.
FGFR inhibitor treatment.
We used the FGFR inhibitor NVP-BGJ398, which is a potent and selective inhibitor of FGF receptors 1, 2, and 3 (Guagnano et al., 2011). To label both astrocytes and neurons with GFP, in utero electroporation was performed at E15.5 using a mixture of PB-CAG-EiP (1.6 mg/ml) and pCAG-PBase (0.4 mg/ml). Pregnant mothers were then treated with either NVP-BGJ398 (10 mg/kg body weight; ChemieTek) or vehicle solution (2:1 mix of PEG-300/5% glucose) by oral gavage twice per day, four times in total.
Tissue preparation.
Tissue preparation was performed as described previously (Hayakawa and Kawasaki, 2010; Iwai et al., 2013). After mice were deeply anesthetized, embryonic brains were fixed and incubated overnight in 4% paraformaldehyde (PFA)/PBS at 4°C. Postnatal mice were transcardially perfused with 4% PFA/PBS. Brains were dissected and postfixed with overnight immersion in 4% PFA/PBS, cryoprotected with 2-d immersion in 30% sucrose/PBS, and embedded in OCT compound. Coronal sections of 14 μm or 50 μm thickness were made using a cryostat.
Immunohistochemistry.
Immunohistochemistry was performed as described previously with slight modifications (Kawasaki et al., 2000; Toda et al., 2013). Briefly, free-floating sections were permeabilized with 0.3% Triton X-100/PBS and incubated overnight with primary antibodies in 2% bovine serum albumin (BSA)/PBS, then washed with PBS three times for 5 min each. The sections were incubated with secondary antibodies and Hoechst 33342 in 2% BSA/PBS for 2 h, and then washed with PBS three times for 5 min each. The sections were mounted on slides with Mowiol (Sigma-Aldrich). Antibodies used for immunostaining were as follows: rat anti-GFP antibody (Nacalai Tesque; RRID:AB_10013361), mouse anti-NeuN antibody (Millipore; RRID:AB_2314889), rabbit anti-NeuN antibody (Cell Signaling Technology; RRID:AB_2630395), rabbit anti-Cux1 antibody (Santa Cruz Biotechnology; RRID:AB_2261231), rabbit anti-S100β antibody (Synaptic Systems; RRID:AB_2620024), goat anti-NDRG2 antibody (Santa Cruz Biotechnology; RRID:AB_2150312), mouse anti-GFAP antibody (Sigma-Aldrich; RRID:AB_477010), mouse anti-phospho-histone H3 (pHH3) antibody (Millipore; RRID:AB_310016), rabbit anti-phospho-p44/42 MAPK (ERK1/2) antibody (Cell Signaling Technology; RRID:AB_2315112), mouse anti-Pax6 antibody (Abcam; RRID:AB_1566562), rabbit anti-Tbr2 antibody (Abcam; RRID:AB_778267), donkey secondary antibodies conjugated to AlexaFluor 488 (Molecular Probe), donkey secondary antibodies conjugated to Cy3 (Jackson ImmunoResearch), and goat secondary antibodies conjugated to AlexaFluor 647 (Molecular Probe).
In situ hybridization.
In situ hybridization was performed as described previously with slight modifications (Kawasaki et al., 2004; Iwai et al., 2013; Ebisu et al., 2017). Briefly, sections of 14 μm thickness were incubated overnight with digoxigenin-labeled RNA probes in hybridization buffer (50% formamide, 5× saline-sodium citrate buffer, 5× Denhardt's solution, 0.3 mg/ml yeast RNA, 0.1 mg/ml herring sperm DNA, and 1 mm dithiothreitol). The sections were then incubated with an alkaline phosphatase-conjugated anti-digoxigenin antibody (Roche) and were visualized using NBT/BCIP as substrates. In some experiments, the sections were then subjected to Hoechst 33342 staining.
Combination of in situ hybridization and immunostaining was described previously (Matsumoto et al., 2017). Briefly, after hybridization, sections were incubated with mouse anti-Pax6 antibody (Abcam; RRID:AB_1566562) and rabbit anti-Tbr2 antibody (Abcam; RRID:AB_778267). After being incubated with secondary antibodies, the sections were visualized with NBT/BCIP.
RNA probes for mouse Fgfr1, Fgfr2, and Fgfr3 were described previously (Matsumoto et al., 2017). To make RNA probes for mouse Etv5 and Spry2, cDNA fragments were amplified with RT-PCR using the following primers. Etv5, 5′-ATACCTTGGCTCGGGAGG-3′ and 5′-AAGCTGTGTCCAGGCTGC-3′; Spry2, 5′-GCAATTAACCCTCACTAAAGG-3′, and 5′-TAATACGACTCACTATAGGG-3′. After PCR products were confirmed by DNA sequencing, they were inserted into the pCRII-TOPO vector by using a TOPO TA Cloning Kit (Invitrogen), and digoxigenin-labeled RNA probes were made. To make RNA probes for mouse proteolipid protein (PLP), cDNA fragments were amplified with RT-PCR using the following primers. PLP, 5′-ccGAATTCagtcagagtgccaaagacatg-3′, and 5′-ggGTCGACtcagaacttggtgcctcg-3′. After PCR products were confirmed by DNA sequencing, they were inserted into the pBluescript vector, and digoxigenin-labeled RNA probes were made.
To measure the average intensities of Etv5 and Spry2 mRNA signals, the regions corresponding to the VZ were selected using the “Freehand selections” function of ImageJ. Average signal intensities in the regions were measured using the “ROI manager” tool of ImageJ.
Microscopy.
Epifluorescence microscopy was performed with a BZ-X710 microscope (KEYENCE). Confocal microscopy was performed with a FLUOVIEW FV10i (Olympus) and an LSM 5 PASCAL (Carl Zeiss).
Experimental design and statistical analysis.
For immunohistochemistry, three coronal sections containing abundant GFP-positive signals at three distinct rostrocaudal levels from each animal were used. Coronal sections were stained with Hoechst 33342 and anti-GFP antibody plus either anti-NeuN antibody, anti-S100β antibody or anti-NDRG2 antibody. GFP-positive regions in the mediolateral cerebral cortex corresponding to the primary somatosensory cortex were analyzed. Images captured using an BZ-X710 microscope were used for quantification. The background signal was removed by subtracting the average signal intensity of negative cells. Then, the numbers of cells positive for NeuN, S100β, NDRG2, or GFP in the gray matter of the electroporated areas were counted using the “cell counter” tool of ImageJ software.
For quantification of the percentages of GFP-positive cells coexpressing NeuN, NDRG2, or S100β, three sections per brain were used. At least 400 GFP-positive cells per brain were examined. For quantification of the numbers of NeuN-positive cells and Cux1-positive cells in layer 2/3, the numbers of immunopositive cells in 200 × 200 μm areas of layer 2/3 were counted. To minimize the variation in the numbers of positive cells depending on the positions of coronal sections along the anterior–posterior axis, the numbers of positive cells on the electroporated side were divided by those on the contralateral side. For quantification of astrocytes, the numbers of immunopositive cells were counted in 200 × 200 μm areas of GFP-positive regions of the cerebral cortex, and were then divided by those in areas on the contralateral side for normalization. For quantification of the ratio of the number of GFP-positive neurons to that of GFP-positive astrocytes, the numbers of immunopositive cells in columns spanning the entire thickness of the gray matter were counted. Five columns per section in the cortical regions containing most abundant GFP-positive cells were analyzed.
For quantification of the percentages of pHH3-positive/S100β-positive cells, the percentages of S100β-positive astrocytes coexpressing pHH3 were calculated in cortical areas of 1.5 mm width. For normalization, the percentages of S100β-positive cells coexpressing pHH3 on the electroporated side were divided by those on the contralateral side.
For quantification of the numbers of PLP-positive oligodendrocytes, the numbers of PLP-positive cells were counted in cortical areas of 750 μm width. For normalization, the numbers of PLP-positive cells on the electroporated side were divided by those on the contralateral side.
Values in graphs are mean ± SEM. An unpaired two-tailed Student's t test was used to determine statistical significance. P values <0.05 were considered as statistically significant.
Results
FGF signaling is activated during the neuron–astrocyte cell fate switch in the developing mouse cerebral cortex
Previous observations showed that astrocytogenesis in the mouse cerebral cortex began ∼E17 (Wasylyk et al., 1998; Fan et al., 2005). We therefore analyzed the expression of Fgfr1, 2, and 3 at E15 and E18. In situ hybridization showed that Fgfr1, 2, and 3 were preferentially expressed in the germinal zones including the VZ and the subventricular zone at E15, and these Fgfrs continued to be expressed in the VZ at E18 (Fig. 1A).
Figure 1.

FGF signaling is activated during the neuron–astrocyte cell fate switch in the developing mouse cerebral cortex. Immunohistochemistry and in situ hybridization were performed using 14 μm coronal sections of the mouse cerebral cortex at E15 and E18. A, In situ hybridization for Fgfr1, 2, and 3 in the germinal zones of the mouse cerebral cortex. Scale bar, 50 μm. B, Immunohistochemistry for pMAPK and in situ hybridization for Etv5 and Spry2. Scale bar, 50 μm. C, D, Quantification of Etv5 and Spry2 mRNA signal intensities in the VZ. The signal intensities were increased between E15 and E18 [unpaired Student's t test, *p = 0.0082 (C) and 0.005 (D)]. E, F, In situ hybridization for Etv5 or Spry2 followed by immunostaining for Pax6 and Tbr2 at E18. The areas within the boxes at the top were magnified and shown in the bottom. Note that Etv5 and Spry2 are expressed in Pax6-positive cells but not in Tbr2-positive cells. Scale bars: top, 100 μm; bottom, 25 μm. IZ, Intermediate zone; SVZ, subventricular zone.
Because MAPK is phosphorylated and activated in response to Fgfrs stimulation, we examined activation of FGF signaling by using anti-phospho-p44/42 MAPK (pMAPK) antibody. Consistent with the expression of Fgfrs (Fig. 1A), pMAPK was observed mainly in the germinal zones (Fig. 1B, top). pMAPK was mainly located in the cytoplasm, although it was also weakly observed in the nucleus (Fig. 2). Because activation of the MEK/MAPK pathway increases the expressions of Ets variant gene 5 (Etv5) and Sprouty2 (Spry2) (Yordy and Muise-Helmericks, 2000; Tsang and Dawid, 2004), we performed in situ hybridization for Etv5 and Spry2. Etv5 and Spry2 were abundantly expressed in the germinal zones at E15 and E18 (Fig. 1B, middle and bottom). Our quantification revealed that the expression levels of Etv5 mRNA in the VZ were significantly higher at E18 compared with those at E15 (E15: 13.28 ± 1.75; E18: 23.85 ± 1.28; p = 0.0082; unpaired Student's t test, n = 3 animals for each condition; Fig. 1C). In line with the Etv5 signals, Spry2 signals in the VZ were also significantly higher at E18 compared with those at E15 (E15: 11.11 ± 2.13; E18: 31.49 ± 2.95; p = 0.005; unpaired Student's t test, n = 3 animals for each condition; Fig. 1D). These data suggest that the MEK/MAPK pathway is activated during the neuron–astrocyte cell fate switch.
Figure 2.
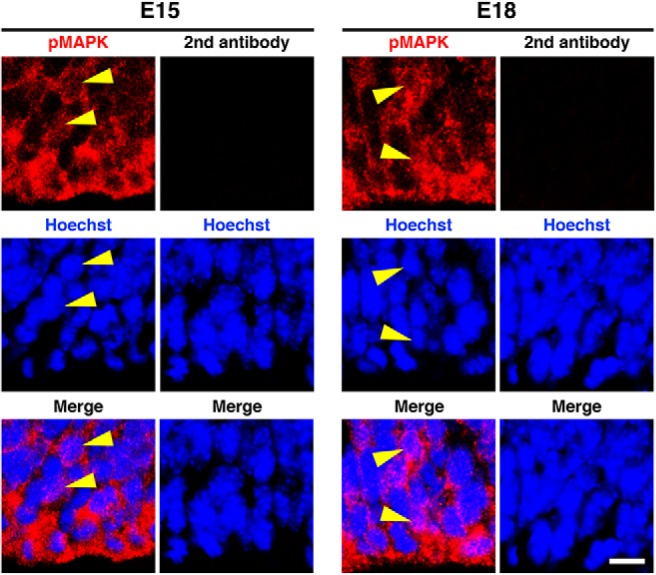
Subcellular distribution patterns of pMAPK signals. Immunohistochemistry for pMAPK was performed using 14 μm coronal sections of the mouse cerebral cortex at E15 and E18. Images in the VZ were taken using a confocal microscope. Arrowheads indicate the positions of the nuclei. Note that no signal was observed without pMAPK antibody (second antibody). Scale bar, 20 μm.
The germinal zones of the developing cerebral cortex consist of Pax6-positive RGCs and Tbr2-positive intermediate progenitor cells. To identify which cell type expresses Etv5 mRNA and Spry2 mRNA, we costained for Pax6 and Tbr2. We found that Etv5-positive and Spry2-positive signals were mostly distributed in Pax6-positive/Tbr2-negative cells, suggesting that they are mainly RGCs (Fig. 1E,F). These results suggest that FGF signaling is highly activated in RGCs when the neuron–astrocyte cell fate switch is occurring in the developing cerebral cortex.
Activation of FGF signaling strongly inhibits neurogenesis and induces astrocytogenesis
To investigate the role of FGF signaling in the neuron–astrocyte cell fate switch, pCAG-EGFP plus either pCAG-FGF8 or control plasmid was introduced in the mouse cerebral cortex at E15.5 by in utero electroporation (Fig. 3A). The pups were fixed at P15, and sections of the cerebral cortex were subjected to immunohistochemistry. As reported previously (Hoshiba et al., 2016), in the control brains electroporated with GFP, all GFP-positive cells were positive for NeuN and located in layer 2/3 (Fig. 3B–D, top). Moreover, these cells were negative for S100β (Fig. 3E–G, top) and NDRG2 (Fig. 4A–C, top). These data suggest that all GFP-positive cells acquired a neuronal cell fate and migrated into upper layers of the cerebral cortex.
Figure 3.
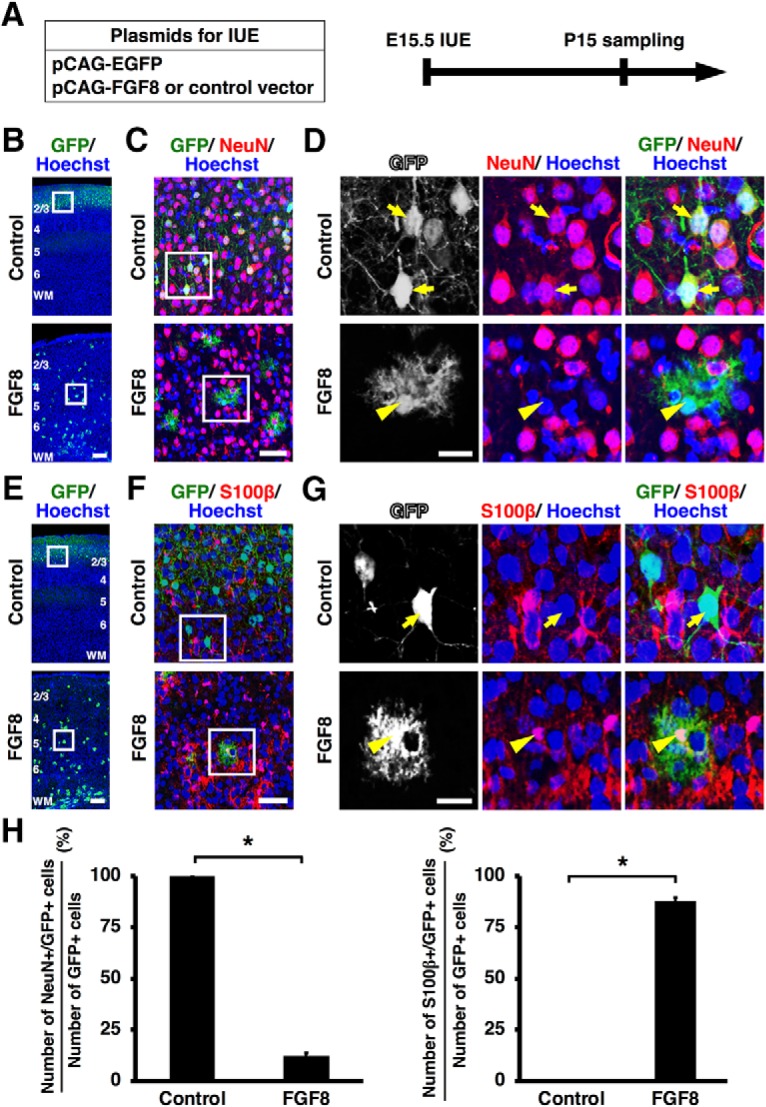
Activation of FGF signaling induces the cell fate switch of GFP-positive cells from neurons to astrocytes in vivo. A, Experimental schematic. pCAG-EGFP plus either pCAG-FGF8 or pCAG control vector was introduced into the mouse cerebral cortex at E15.5 by IUE, and coronal sections were prepared at P15. B–D, Sections were stained with anti-GFP and anti-NeuN antibodies and Hoechst 33342. B, Lower-magnification images. The areas within the boxes in B were magnified and shown in C, and the areas within the boxes in C were magnified and shown in D. All GFP-positive cells migrated into layer 2/3 in the control brain, whereas they were ubiquitously located throughout the cortex of the FGF8-electroporated brain. GFP-positive cells in the control brain showed neuronal morphologies (arrows), whereas they exhibited astrocytic star-shaped morphologies with small nuclei and many fine branches in the FGF8-electroporated brain (arrowhead). GFP-positive cells in the control brain coexpressed NeuN (arrows), whereas GFP-positive cells in FGF8-electroporated brain were negative for NeuN (arrowhead). E–G, Sections were stained with anti-GFP and anti-S100β antibodies plus Hoechst 33342. E, Lower-magnification images. The areas within the boxes in E were magnified and shown in F, and the areas within the boxes in F were magnified and shown in G. Note that GFP-positive cells in the control brain were S100β-negative (arrow), whereas those in FGF8-transfected brains coexpressed S100β (arrowhead). Numbers indicate layers in the cortex. WM, White matter. Scale bars: B, E, 200 μm; C, F, 50 μm; D, G, 20 μm. H, The percentages of GFP-positive cells coexpressing NeuN (left) or S100β (right). Note that GFP-positive neurons were drastically decreased, and GFP-positive astrocytes were markedly increased by FGF8 (unpaired Student's t test, *p < 0.0001). Error bars represent mean ± SEM.
Figure 4.

Activation of FGF signaling induces the cell fate switch of GFP-positive cells from neurons to astrocytes in vivo. pCAG-EGFP plus either pCAG-FGF8 or pCAG control vector was introduced into the mouse cerebral cortex at E15.5 by IUE, and coronal sections were prepared at P15. A–C, Sections were stained with anti-GFP and anti-NDRG2 antibodies plus Hoechst 33342. A, Lower-magnification images. The areas within the boxes in A were magnified and shown in B, and the areas within the boxes in B were magnified and shown in C. Note that GFP-positive cells in the control brain were NDRG2-negative (arrow), whereas those in FGF8-transfected brains coexpressed NDRG2 (arrowhead). Numbers indicate layers in the cortex. WM, White matter. Scale bars: A, 200 μm; B, 50 μm; C, 20 μm. D, The percentages of GFP-positive cells coexpressing NDRG2. Note that NDRG2-positive astrocytes were markedly increased by FGF8 (unpaired Student's t test, *p < 0.0001). Error bars represent mean ± SEM.
Surprisingly, when FGF signaling was activated by introducing FGF8, cell fate was drastically changed. We found that most GFP-positive cells exhibited astrocytic star-shaped morphologies with small nuclei and many fine branches (Fig. 3B–G, bottom). Furthermore, high-magnification images revealed that these GFP-positive cells did not express NeuN and instead expressed the astrocyte markers S100β and NDRG2 (Figs. 3D,G, bottom, 4C, bottom) (Grosche et al., 2013; Flügge et al., 2014). We then performed triple immunostaining for GFP and NeuN plus either S100β or NDRG2, and quantified the percentages of GFP-positive cells coexpressing NeuN, S100β, or NDRG2. The percentages of GFP-positive cells coexpressing NeuN were significantly reduced by FGF8 (control: 100%; FGF8: 12.11 ± 1.65%; p < 0.0001; unpaired Student's t test, n = 3 animals for each condition), whereas those of GFP-positive cells coexpressing S100β or NDRG2 were markedly increased (S100β, control: 0%; FGF8: 87.91 ± 1.65%; p < 0.0001; unpaired Student's t test, n = 3 animals for each condition; NDRG2, control: 0%; FGF8: 86.94%± 3.24%; p < 0.0001; unpaired Student's t test, n = 3 animals for each condition; Figs. 3H, 4D). Thus, we concluded that GFP-positive cells changed their cell fates from neurons to astrocytes in response to FGF activation. Interestingly, we found that GFP-positive cells, which were located in layer 2/3 of the control cerebral cortex, were widely distributed throughout the FGF8-transfected cortex (Fig. 3B,E). This is consistent with the fact that astrocytes are distributed throughout the cerebral cortex.
We also performed immunostaining for NeuN and found that NeuN-positive layer 2/3 neurons were markedly decreased by FGF8 (Fig. 5A,B). The number of NeuN-positive layer 2/3 neurons in the electroporated side of the brain was counted and divided by that of NeuN-positive layer 2/3 neurons in the contralateral side. As we expected, NeuN-positive layer 2/3 neurons were significantly reduced in response to FGF8 (control: 1.01 ± 0.01; FGF8: 0.71 ± 0.06; p = 0.0069; unpaired Student's t test, n = 3 animals for each condition; Fig. 5C). We further measured the percentage of NeuN-positive cells in layer 2/3. The percentage of NeuN-positive cells in the transfected side was divided by that of NeuN-positive cells in the contralateral side. The percentage of NeuN-positive cells in layer 2/3 was also significantly reduced by FGF8 (control: 1.02 ± 0.01; FGF8: 0.8 ± 0.03; p = 0.0023; unpaired Student's t test, n = 3 animals for each condition; Fig. 5D). Consistent results were obtained using immunostaining for Cux1, which is a marker for upper layer neurons of the cerebral cortex (Nieto et al., 2004). Cux1-positive layer 2/3 neurons were significantly reduced in response to FGF8 (control: 1.1 ± 0.04; FGF8: 0.53 ± 0.8; p = 0.0027; unpaired Student's t test, n = 3 animals for each condition; Fig. 6A–C). The ratio of the percentage of Cux1-positive cells in layer 2/3 of the transfected side to that of the contralateral side was also significantly reduced by FGF8 (control: 1.05 ± 0.08; FGF8: 0.57 ± 0.05; p = 0.0064; unpaired Student's t test, n = 3 animals for each condition; Fig. 6D).
Figure 5.

Activation of FGF signaling decreased neurons and increased astrocytes in vivo. pCAG-EGFP plus either pCAG-FGF8 or pCAG control vector was introduced into the mouse cerebral cortex at E15.5 by IUE, and coronal sections were prepared at P15. Sections were stained for GFP plus either NeuN (A–D) or NDRG2 (E–G), and Hoechst 33342. A–D, FGF8 overexpression reduced layer 2/3 neurons. The areas within the boxes in A were magnified and shown in B. Fewer NeuN-positive neurons were observed in layer 2/3 of the FGF8-transfected cortex. C, Quantification of the number of NeuN-positive cells in layer 2/3. The number of layer 2/3 neurons was significantly suppressed by FGF8 (unpaired Student's t test, *p = 0.0069). D, The percentage of cells in layer 2/3 coexpressing NeuN. The percentage was significantly reduced by FGF8 (unpaired Student's t test, *p = 0.0023). E–G, FGF8 overexpression increased astrocytes. The areas within the boxes in E were magnified and shown in F. NDRG2-positive astrocytes were markedly increased by FGF8 (arrowheads). G, Quantification of the number of NDRG2-positive cells in the cerebral cortex. Astrocytes were significantly increased by FGF8 (unpaired Student's t test, *p < 0.0001). Error bars represent mean ± SEM. Scale bars: A, 200 μm; B, 50 μm; E, 100 μm; F, 20 μm. Numbers indicate layers in the cortex. WM, White matter.
Figure 6.

Activation of FGF signaling decreased neurons and increased astrocytes in vivo. pCAG-EGFP plus either pCAG-FGF8 or pCAG control vector was introduced into the mouse cerebral cortex at E15.5 by IUE, and coronal sections were prepared at P15. Sections were stained for GFP plus either Cux1 (A–D) or S100β (E–G), and Hoechst 33342. A–D, FGF8 overexpression reduced layer 2/3 neurons. The areas within the boxes in A were magnified and shown in B. Fewer Cux1-positive neurons were observed in layer 2/3 of the FGF8-transfected cortex. C, Quantification of the number of Cux1-positive cells in layer 2/3. The number of layer 2/3 neurons was significantly suppressed by FGF8 (unpaired Student's t test, *p = 0.0027). D, The percentage of cells in layer 2/3 coexpressing Cux1. The percentage was significantly reduced by FGF8 (unpaired Student's t test, *p = 0.0064). E–G, FGF8 overexpression increased astrocytes. The areas within the boxes in E were magnified and shown in F. S100β-positive astrocytes were markedly increased by FGF8 (arrowheads). G, Quantification of the number of S100β-positive cells in the cerebral cortex. Astrocytes were significantly increased by FGF8 (unpaired Student's t test, *p = 0.0006). Error bars represent mean ± SEM. Scale bars: A, 200 μm; B, 50 μm; E, 100 μm; F, 20 μm. Numbers indicate layers in the cortex.
Next, we performed NDRG2 and S100β immunohistochemistry and found that NDRG2-positive and S100β-positive astrocytes were markedly increased by FGF8 at P15 (Figs. 5E,F, 6E,F). To quantify the number of astrocytes, we counted the numbers of S100β-positive cells and NDRG2-positive cells in the cerebral cortex of the electroporated side and divided them by those in the contralateral side. The numbers of astrocytes were markedly increased by FGF8 (NDRG2, control: 0.99 ± 0.02; FGF8: 1.9 ± 0.02; p < 0.0001; unpaired Student's t test, n = 3 animals for each condition; S100β, control: 0.91 ± 0.02; FGF8: 1.96 ± 0.11; p = 0.0006; unpaired Student's t test, n = 3 animals for each condition; Figs. 5G, 6G). These results suggest that activation of FGF signaling induces the cell fate switch from neurons to astrocytes. Consistent with this idea, when we overexpressed FGF8 by IUE at E15.5 and performed GFAP immunostaining at P0, we found that GFAP-positive cells appeared earlier in the FGF8-electroporated cortex than in the control cortex (Fig. 7). Together, these results indicate that activation of FGF signaling is sufficient to change cell fates from neurons to astrocytes.
Figure 7.
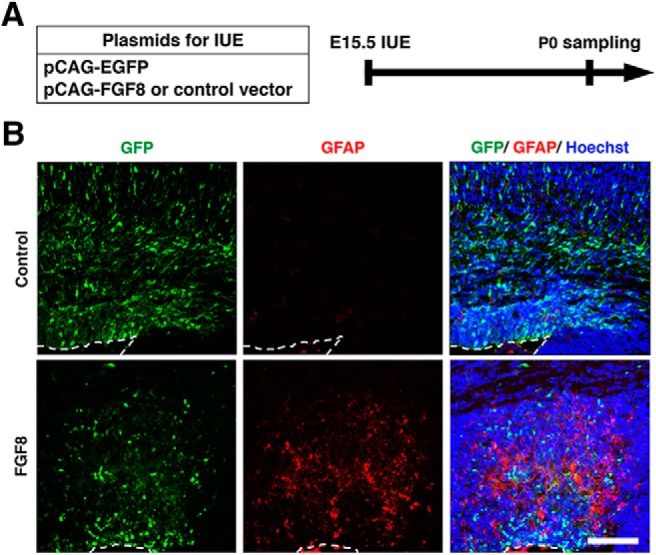
Activation of FGF signaling induces an earlier appearance of astrocytes in the cortex. A, Experimental schematic. pCAG-EGFP plus either pCAG-FGF8 or pCAG control vector was introduced into the mouse cerebral cortex at E15.5 by IUE, and coronal sections were prepared at P0. B, Sections were stained for GFP plus GFAP and Hoechst 33342. Images of the germinal zones are shown. The broken lines indicate the surface of the lateral ventricle. Scale bar, 200 μm.
Inhibition of FGF signaling promotes neurogenesis at the expense of astrocytogenesis
Because activation of FGF signaling promoted the neuron–astrocyte cell fate switch, we next examined whether inhibition of FGF signaling extends neurogenesis at the expense of astrocytogenesis. To examine the effect of inhibition of FGF signaling on the neuron–astrocyte cell fate switch, we used a soluble form of FGFR3 (sFGFR3c), which is a dominant-negative form of FGFR (Fukuchi-Shimogori and Grove, 2001; Matsumoto et al., 2017). Because IUE at E15.5 introduces pCAG-EGFP plasmid only into layer 2/3 neurons but not into astrocytes, we combined IUE with the piggyBac transposon system, which integrates transgenes into the genome. The piggyBac transposon system is a binary system with a helper plasmid (pCAG-PBase) providing piggyBac transposase (PBase), and the donor plasmid (PB-CAG-EiP) providing the CAG-GFP transgene between the 5′ and 3′ terminal repeats (Chen and LoTurco, 2012). We co-electroporated PB-CAG-EiP and pCAG-PBase plus either pCAG-sFGFR3c or control plasmid into the cerebral cortex at E15.5, and the samples were collected at P10 (Figs. 8A, 9A). As expected, not only GFP-positive layer 2/3 neurons (Fig. 8B, control, arrows) but also numerous GFP-positive astrocytes were observed in the control cortex using this method (Fig. 8B, control, arrowheads).
Figure 8.

Inhibition of FGF signaling promotes neurogenesis at the expense of astrocytogenesis. A, Experimental schematic. The piggyBac transposon system (pCAG-PBase and PB-CAG-EiP) plus either pCAG-sFGFR3c or pCAG control vector was co-electroporated at E15.5, and the coronal sections were prepared at P10. B, Immunohistochemistry for GFP, NeuN, and S100β was performed. The areas within the boxes on the left were magnified and shown on the right. Note that IUE with the piggyBac system induced GFP expression not only in neurons (arrows), but also astrocytes (arrowheads) in the control brain. Inhibition of FGF signaling by sFGFR3c increased the number of GFP-positive/NeuN-positive neurons (arrows) and decreased the number of GFP-positive/S100β-positive astrocytes (arrowheads). Numbers indicate layers in the cortex. Scale bars: left, 100 μm; right, 50 μm. C, The percentage of GFP-positive cells coexpressing S100β. Inhibition of FGF signaling significantly decreased astrocytes (unpaired Student's t test, *p = 0.0023). D, The percentage of GFP-positive cells coexpressing NeuN. Inhibition of FGF signaling increased neurons (unpaired Student's t test, *p = 0.0023). E, The ratio of the number of GFP-positive neurons to that of GFP-positive astrocytes (unpaired Student's t test, *p = 0.012). Error bars represent mean ± SEM.
Figure 9.

Inhibition of FGF signaling promotes neurogenesis at the expense of astrocytogenesis. A, Schematic of experiment. The piggyBac transposon system (pCAG-PBase and PB-CAG-EiP) plus either pCAG-sFGFR3c or pCAG control vector was co-electroporated at E15.5, and the coronal sections were prepared at P10. B, Immunohistochemistry for GFP, NeuN, and NDRG2 was performed. The areas within the boxes on the left were magnified and shown on the right. Inhibition of FGF signaling by sFGFR3c increased the number of GFP-positive/NeuN-positive neurons (arrows) and decreased the number of GFP-positive/NDRG2-positive astrocytes (arrowheads). Numbers indicate layers in the cortex. Scale bars: left, 100 μm; right, 50 μm. C, The percentage of GFP-positive cells coexpressing NDRG2. Inhibition of FGF signaling significantly decreased astrocytes (unpaired Student's t test, *p = 0.0017). D, The percentage of GFP-positive cells coexpressing NeuN. Inhibition of FGF signaling increased neurons (unpaired Student's t test, *p = 0.0017). E, The ratio of the number of GFP-positive neurons to that of GFP-positive astrocytes (unpaired Student's t test, *p = 0.0069). Error bars represent mean ± SEM.
We then performed triple immunostaining for GFP, NeuN and S100β to assess the numbers of GFP-positive/NeuN-positive neurons and GFP-positive/S100β-positive astrocytes. Strikingly, overexpression of sFGFR3c significantly decreased GFP-positive astrocytes (control: 62.29 ± 1.17%; sFGFR3c: 33.47 ± 4.02%; p = 0.0023; unpaired Student's t test, n = 3 animals for each condition; Fig. 8B,C), whereas it increased GFP-positive neurons (control: 37.71 ± 1.17%; sFGFR3c: 66.53 ± 4.02%; p = 0.0023; unpaired Student's t test, n = 3 animals for each condition; Fig. 8B,D). Consistently, the ratio of the number of GFP-positive neurons to that of GFP-positive astrocytes was significantly increased by sFGFR3c (control: 0.61 ± 0.03; sFGFR3c: 2.07 ± 0.33; p = 0.012; unpaired Student's t test, n = 3 animals for each condition; Fig. 8E). We also performed triple immunostaining for GFP, NeuN and NDRG2 and obtained consistent results. sFGFR3c significantly decreased GFP-positive astrocytes (control: 58.81 ± 0.79%; sFGFR3c: 36.78 ± 2.82%; p = 0.0017; unpaired Student's t test, n = 3 animals for each condition; Fig. 9B,C), whereas it increased GFP-positive neurons (control: 41.19 ± 0.79%; sFGFR3c: 63.22 ± 2.82%; p = 0.0017; unpaired Student's t test, n = 3 animals for each condition; Fig. 9B,D). The ratio of the number of GFP-positive neurons to that of GFP-positive astrocytes was significantly increased by sFGFR3c (control: 0.73 ± 0.02; sFGFR3c: 1.82 ± 0.21; p = 0.0069; unpaired Student's t test, n = 3 animals for each condition; Fig. 9E). Altogether, these results indicate that inhibition of FGF signaling promotes neurogenesis at the expense of astrocytogenesis in the developing mouse cerebral cortex.
To further confirm the role of FGF signaling in the neuron–astrocyte cell fate switch, we performed an alternative method to inhibit FGF signaling. IUE was performed at E15.5 with a mixture of pCAG-PBase and PB-CAG-EiP to label both astrocytes and neurons with GFP. Pregnant mothers were then treated with either NVP-BGJ398 (10 mg/kg body weight) or vehicle solution (PEG-300/5% glucose) by oral gavage twice per day, four times in total. Sections were prepared at P10, and triple immunostaining for GFP, NeuN and S100β was performed to assess the numbers of GFP-positive/NeuN-positive neurons and GFP-positive/S100β-positive astrocytes (Fig. 10A). Consistent with the results obtained using sFGFR3c, NVP-BGJ398 significantly decreased GFP-positive astrocytes (control: 63.31 ± 0.86%; NVP-BGJ398: 38.11 ± 1.99%; p = 0.0003; unpaired Student's t test, n = 3 animals for each condition; Fig. 10B,C) and increased GFP-positive neurons (control: 36.69 ± 0.86%; NVP-BGJ398: 61.89 ± 1.99%; p = 0.0003; unpaired Student's t test, n = 3 animals for each condition; Fig. 10B,D). Furthermore, the ratio of the number of GFP-positive neurons to that of GFP-positive astrocytes was significantly increased by NVP-BGJ398 (control: 0.59 ± 0.02; NVP-BGJ398: 1.65 ± 0.13; p = 0.0011; unpaired Student's t test, n = 3 animals for each condition; Fig. 10E). Together, our findings indicate that activation of FGF signaling is necessary and sufficient to change cell fates from neurons to astrocytes.
Figure 10.

The effect of an FGFR inhibitor on the neuron–astrocyte cell fate switch. A, Experimental schematic. pCAG-PBase and PB-CAG-EiP were electroporated at E15.5, and the pregnant mothers were treated with the FGFR inhibitor NVP-BGJ398. Coronal sections were prepared at P10. B, Immunohistochemistry for GFP, NeuN, and S100β. The areas within the boxes on the left were magnified and shown on the right. Inhibition of FGF signaling by NVP-BGJ398 increased the number of GFP-positive/NeuN-positive neurons (arrows) and decreased the number of GFP-positive/S100β-positive astrocytes (arrowheads). Numbers indicate layers in the cortex. Scale bars: left, 100 μm; right, 50 μm. C, The percentage of GFP-positive cells coexpressing S100β. Inhibition of FGF signaling significantly decreased astrocytes (unpaired Student's t test, *p = 0.0003). D, The percentage of GFP-positive cells coexpressing NeuN. Inhibition of FGF signaling increased neurons (unpaired Student's t test, *p = 0.0003). E, The ratio of the number of GFP-positive neurons to that of GFP-positive astrocytes (unpaired Student's t test, *p = 0.0011). Error bars represent mean ± SEM.
The MAPK pathway mediates the FGF-induced neuron-to-astrocyte cell fate switch
Because it has been demonstrated that the MAPK pathway is strongly activated by FGF, we examined whether the MAPK pathway is activated in the developing mouse cerebral cortex after introduction of FGF8. We performed IUE to express FGF8 at E15.5, and sampling was performed at P0. Immunohistochemistry demonstrated that pMAPK signal in the VZ was strongly enhanced by FGF8 (Fig. 11, top). Consistently, in situ hybridization showed that Etv5 expression in the VZ was greatly increased by FGF8 (Fig. 11, bottom). These results suggest that FGF overexpression strongly activates the MAPK pathway in the VZ of the mouse cerebral cortex.
Figure 11.

The MAPK pathway was activated in the germinal zones of the mouse cerebral cortex by the introduction of FGF8 by IUE. FGF8 was electroporated at E15.5, and sampling was performed at P0. Coronal sections were stained with anti-pMAPK antibody or subjected to in situ hybridization for Etv5. Overexpression of FGF8 drastically increased pMAPK and Etv5 signals in the germinal zones of the cerebral cortex. Scale bar, 200 μm.
Because the MAPK pathway was reported to mediate the neuron-glia cell fate switch (Li et al., 2012), it seemed possible that FGF is the extracellular upstream regulator of the neuron–astrocyte cell fate switch through the MAPK pathway. To test this possibility, we expressed GFP and FGF8 plus either control plasmid or a dominant negative form of MEK (DN-MEK), in which two serines were replaced with alanines (Zheng and Guan, 1994; Gotoh et al., 1999), in the cerebral cortex at E15.5. Sections were prepared at P10 and stained with anti-GFP, anti-NeuN and anti-S100β antibodies (Fig. 12A). We found that DN-MEK significantly increased the percentage of GFP-positive cells which acquired NeuN-positive neuronal fate (FGF8: 2.51 ± 0.91%; FGF8+DN-MEK: 18.27 ± 2.34%; p = 0.0009; unpaired Student's t test; n = 4 animals for FGF8 and n = 3 animals for FGF8+DN-MEK; Fig. 12B,C). These results indicate that the MAPK pathway is involved in the neuron–astrocyte cell fate switch induced by FGF signaling.
Figure 12.
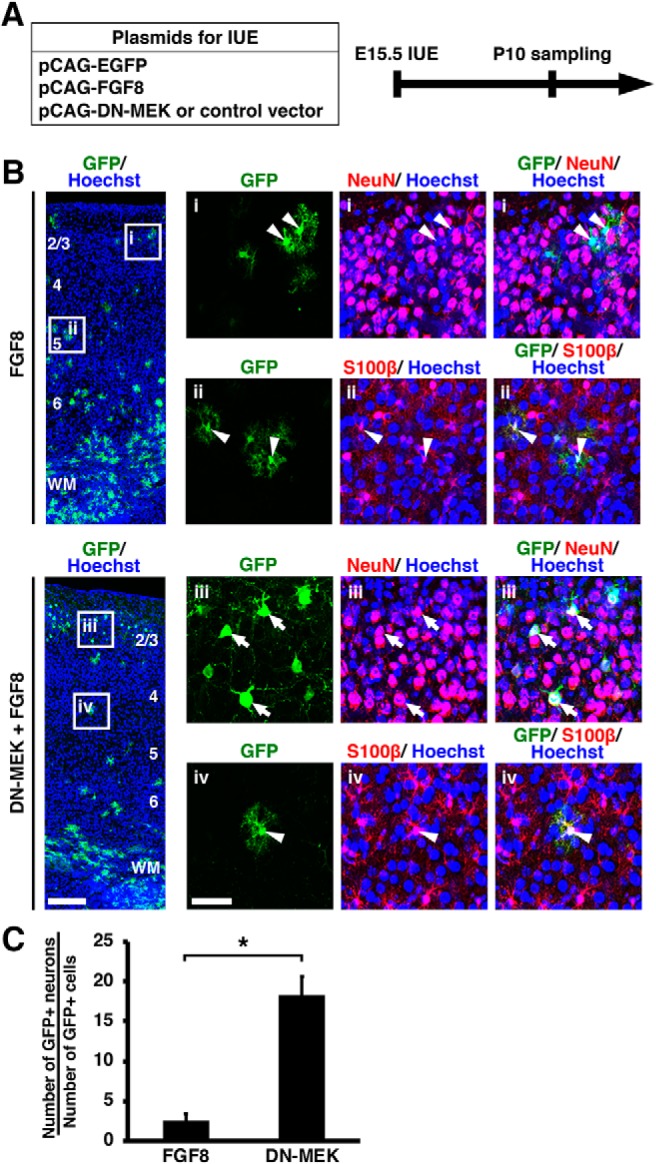
The MAPK pathway mediates the FGF-induced neuron-to-astrocyte cell fate switch. A, Experimental schematic. pCAG-EGFP and pCAG-FGF8 plus either pCAG-DN-MEK or pCAG control vector were co-electroporated at E15.5, and the coronal sections were prepared at P10. B, Immunohistochemistry for GFP, NeuN, and S100β. The areas within the boxes on the left were magnified and shown on the right. As shown in Figures 3 and 4, IUE with pCAG-FGF8 produced many GFP-positive astrocytes (FGF8, arrowheads). Inhibition of MAPK signaling by DN-MEK increased the number of GFP-positive/NeuN-positive neurons (DN-MEK+FGF8, arrows) and decreased the number of GFP-positive/S100β-positive astrocytes (DN-MEK+FGF8, arrowheads). Numbers indicate layers in the cortex. WM, White matter. Scale bars: left, 200 μm; right, 50 μm. C, The percentage of GFP-positive cells coexpressing NeuN. The numbers of immunopositive cells in the gray matter and the white matter were counted. Inhibition of MAPK signaling significantly increased neurons (unpaired Student's t test, *p = 0.0009). Error bars represent mean ± SEM.
Activation of FGF signaling increases astrocyte proliferation and oligodendrocyte development
It was reported that astrocytes increased postnatally in the rodent brain (Bandeira et al., 2009), and this increase involved local proliferation of differentiated astrocytes (Ge et al., 2012). Therefore, it remained possible that FGF activation also enhanced cell proliferation of astrocytes. To test this possibility, we introduced FGF8 by electroporation at E15.5 and accessed astrocyte proliferation at P3 using double immunostaining for pHH3 and S100β (Fig. 13A). The percentage of S100β-positive cells coexpressing pHH3 was increased by FGF8 (control: 1.01 ± 0.05; FGF8: 1.33 ± 0.08, p = 0.0282; unpaired Student's t test, n = 3 animals for each condition; Fig. 13B,D). This result suggests that the increase in astrocytes by FGF activation involves not only the neuron–astrocyte cell fate switch but also astrocyte proliferation.
Figure 13.
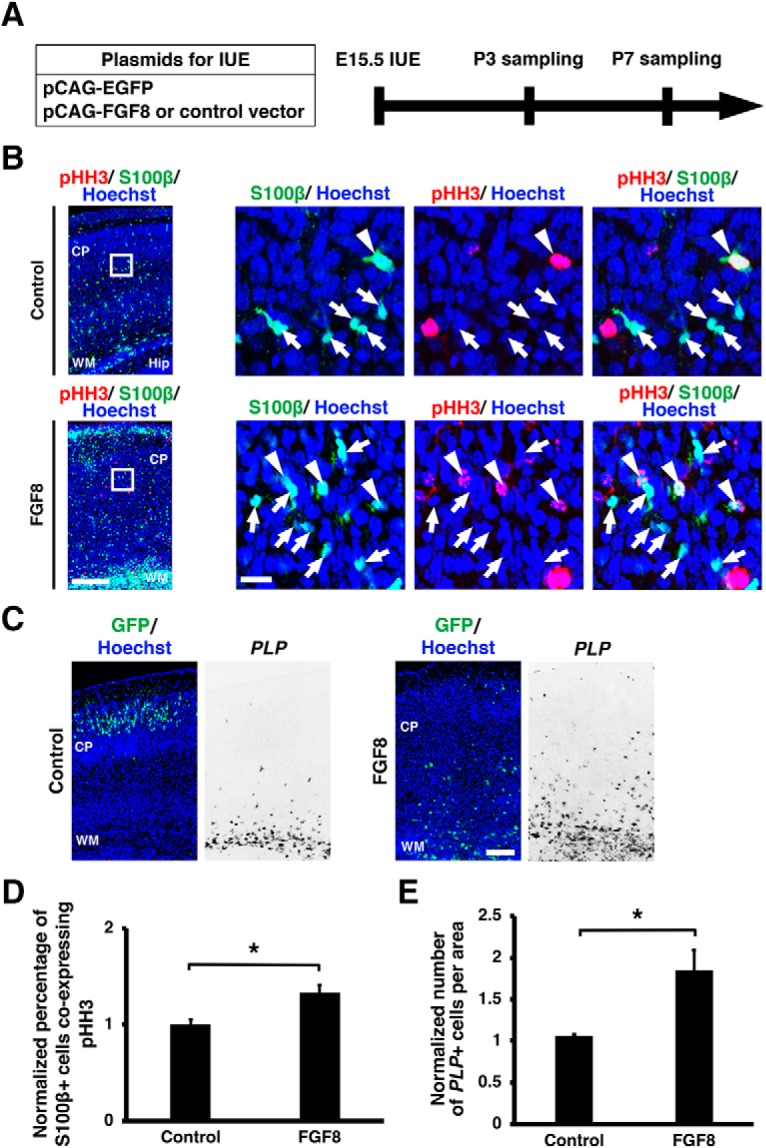
Activation of FGF signaling promotes astrocyte proliferation and oligodendrogenesis. A, Schematic of experiment. pCAG-EGFP plus either pCAG-FGF8 or pCAG control vector was introduced into the mouse cerebral cortex by IUE at E15.5, and coronal sections were prepared at P3 for pHH3 and S100β immunostaining and at P7 for PLP in situ hybridization. B, Immunohistochemistry for pHH3 and S100β. Arrows indicate S100β-positive/pHH3-negative cells, whereas arrowheads indicate S100β-positive cells coexpressing pHH3. Scale bars: left, 200 μm; right, 20 μm. C, In situ hybridization for PLP. PLP-positive cells were increased by FGF8. Scale bar, 200 μm. D, Quantification of the percentage of S100β-positive cells coexpressing pHH3 in the cortex. The percentage of S100β-positive cells coexpressing pHH3 in the electroporated side of the cortex were divided by those in the contralateral non-electroporated cortex (unpaired Student's t test, *p = 0.0282). Error bars represent mean ± SEM. E, Quantification of the number of PLP-positive cells in the cortex. The numbers of PLP-positive cells in the electroporated cortex were divided by those in the other non-electroporated cortex (unpaired Student's t test, *p = 0.0289). Error bars represent mean ± SEM. CP, cortical plate; Hip, Hippocampus; WM, white matter.
In addition, we examined whether FGF signaling also affects oligodendrocyte development. FGF8 was electroporated at E15.5, and sections prepared at P7 were subjected to in situ hybridization for PLP, which is a marker for oligodendrocytes (Fig. 13A,C). PLP-positive cells were increased by FGF8 electroporation (control: 1.07 ± 0.01; FGF8: 1.85 ± 0.24; p = 0.0289; unpaired Student's t test, n = 3 animals for each condition; Fig. 13C,E). This result is consistent with previous studies showing the involvement of FGF signals in oligodendrocyte development (McKinnon et al., 1990; Bansal et al., 1996; Oh et al., 2003; Furusho et al., 2011). It seems likely that FGF signaling also regulates oligodendrocyte development.
Discussion
Here we have shown, through both gain- and loss-of-function experiments, that FGF signaling is a crucial extracellular regulator of the neuron–astrocyte cell fate switch. Our experiments involving in situ hybridization and immunohistochemistry demonstrated that FGF signaling is activated in the VZ when the neuron–astrocyte cell fate switch occurs. Activation of FGF signaling suppressed neurogenesis and enhanced astrocytogenesis. Furthermore, inhibition of FGF signaling extended neurogenesis at the expense of astrocytogenesis. Together, our in vivo research revealed FGF signaling as the first discovered extracellular regulator of the neuron–astrocyte cell fate switch in the developing mouse cerebral cortex.
In vivo and in vitro experiments of the neuron–astrocyte cell fate switch
Generation of precise numbers of both neurons and astrocytes during development is an important step for brain functions and homeostatic maintenance (Miller and Gauthier, 2007). In the embryonic cerebral cortex, NPCs generate neurons followed by astrocytogenesis (Qian et al., 2000; Guillemot and Zimmer, 2011; Sloan and Barres, 2014). Cell fate switch from neurons to astrocytes is crucial for matching numbers in each cell lineage (Freeman, 2010; Wang et al., 2016).
Recent pioneering research has uncovered the intracellular mechanisms of the neuron–astrocyte cell fate switch in the developing mouse cerebral cortex. Recently, the MEK/MAPK pathway has received much attention as a regulator of the neuron-glia cell fate switch. This pathway has been shown in vitro and in vivo to promote astrocytogenesis and inhibit neurogenesis (Rajan and McKay, 1998; Li et al., 2012; Wang et al., 2012). The next important question was determining the extracellular mechanisms regulating the activation of the MEK/MAPK pathway, leading to the transition from neurogenesis to astrocytogenesis in the mammalian developing neocortex. Here we uncovered that FGF signaling is necessary and sufficient for the neuron–astrocyte cell fate switch and that the MEK/MAPK pathway is involved in the effect of FGF on the neuron–astrocyte cell fate switch in the developing cerebral cortex. FGF is the first extracellular signaling molecule shown to be responsible for the neuron–astrocyte cell fate switch in vivo.
Beside the MEK/MAPK pathway, the JAK/STAT pathway was also reported to regulate the cell fate switch of NPCs through both promotion of astrocytogenesis and inhibition of neurogenesis (Bonni et al., 1997; Nakashima et al., 1999; Gu et al., 2005; Cao et al., 2010; Namihira and Nakashima, 2013), but the extracellular regulators of the JAK/STAT pathway regulating the cell fate switch are still unclear. Cardiotrophin-1 (CT-1) was proposed to be an upstream regulator of this pathway (Martynoga et al., 2012) because CT-1 promoted astrocytogenesis (Ochiai et al., 2001). CT-1, however, failed to affect neurogenesis (Barnabé-Heider et al., 2005), suggesting that CT-1 is involved solely in astrocytogenesis rather than in the neuron–astrocyte cell fate switch. Similarly, BMP4 was reported to promote astrocytogenesis, whereas it failed to affect neurogenesis (Mabie et al., 1999; Gomes et al., 2003).
FGF signaling regulates cell fate switch from neurons to astrocytes
Here we have shown that FGF signaling plays a crucial role in the neuron–astrocyte cell fate switch in the developing cerebral cortex. Currently, it is unclear which member in the FGF family is responsible for the neuron–astrocyte cell fate switch. FGF2 was reported to suppress neuronal differentiation and facilitate astrocyte differentiation in vitro (Song and Ghosh, 2004). However, in vivo studies using FGF2 mutant mice did not show clear evidence of its involvement in the neuron–astrocyte cell fate switch (Ortega et al., 1998; Vaccarino et al., 1999). Another study reported that FGF8 acts as a cell fate regulator by promoting astrocyte differentiation and inhibiting neurogenesis of a primary culture model of E15 rat cerebral cortex (Hajihosseini and Dickson, 1999). However, FGF8 was reported to be expressed in midline zipper glia along the interhemispheric fissure, rather than in the cerebral cortex, of the developing mouse brain (Gobius et al., 2016), raising the possibility that FGFs other than FGF8 are responsible for the neuron–astrocyte cell fate switch. Unfortunately, FGF8 mutant embryos cannot survive beyond E9.5, which was a limitation of studying FGF8 involvement in the neuron–astrocyte cell fate switch in vivo (Sun et al., 1999). Future investigation is necessary to uncover the entire picture of the molecular mechanisms underlying the neuron–astrocyte cell fate switch.
Mechanisms of the neuron–astrocyte cell fate switch downstream of FGF signaling
Here we showed that the MEK/MAPK pathway was activated by FGF in the developing mouse cerebral cortex. pMAPK signals were markedly enhanced by FGF. Furthermore, we demonstrated that DN-MEK suppressed the FGF-induced cell fate switch from neurons to astrocytes, suggesting that the FGF-induced cell fate switch from neurons to astrocytes is mediated by the MAPK pathway. We also found that the expression of Etv5, which is a downstream molecule of the MAPK pathway, was increased by FGF. Furthermore, a previous study reported that overexpression of Etv5 by IUE induced the neuron–astrocyte cell fate switch in the mouse cerebral cortex (Li et al., 2012). A plausible scenario would be that the cell fate switch triggered by FGF signaling is mediated by the MEK/MAPK pathway through Etv5.
It should be noted that when Etv5 was transfected, only 20% of transfected cells became astrocytes (Li et al., 2012). In contrast, our results showed that almost all GFP-positive cells became astrocytes in response to FGF. The stronger effect on the neuron–astrocyte cell fate switch by overexpression of FGF8 compared with that of Etv5 can be partially explained by the presence of intracellular pathways other than Etv5. Because the JAK/STAT pathway is also involved in the neuron–astrocyte cell fate switch, and because stimulation of FGFR activates the JAK/STAT pathway (Ebong et al., 2004; Yang et al., 2009), the JAK/STAT pathway could also mediate the neuron–astrocyte cell fate switch downstream of FGF signaling. Another possible mechanism involved in the FGF-induced neuron–astrocyte cell fate switch would be Notch/Sox9/NFIA signaling (Sloan and Barres, 2014). Previously, NFIA was shown to act downstream of Notch signaling to initiate the expression of astrocyte-specific genes during cortical development (Deneen et al., 2006; Namihira et al., 2009; Tchieu et al., 2019). Sox9 is considered to be a glial fate determinant in the spinal cord (Stolt et al., 2003) and is capable of inducing NFIA expression and subsequently associating with NFIA to promote glial lineage progression (Kang et al., 2012). Interestingly, it was previously reported that FGF promoted Sox9 expression in the hindbrain of developing zebrafish and in the developing mouse pancreas (Esain et al., 2010; Seymour et al., 2012). It seems possible that the FGF-induced neuron–astrocyte cell fate switch is partially mediated by the interaction between FGF signaling and Sox9/NFIA signaling.
Footnotes
This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology; Japan Agency for Medical Research and Development, Daiichi Sankyo Foundation of Life Science; Kawano Masanori Memorial Public Interest Incorporated Foundation for Promotion of Pediatrics; Mitsubishi Foundation; Takeda Science Foundation; the Kanazawa University SAKIGAKE project 2018; and the Kanazawa University CHOZEN project. We thank Dr. Eisuke Nishida (RIKEN Center for Biosystems Dynamics Research), Dr. Knut Woltjen and Dr. Akitsu Hotta (Kyoto University) for providing plasmids, and Dr. Tomohisa Toda, Dr. Haruka Ebisu, Kazumi Masuta, Shotaro Inoue, Zachary Blalock, and Kawasaki laboratory members for their helpful support.
The authors declare no competing financial interests.
References
- Bandeira F, Lent R, Herculano-Houzel S (2009) Changing numbers of neuronal and non-neuronal cells underlie postnatal brain growth in the rat. Proc Natl Acad Sci U S A 106:14108–14113. 10.1073/pnas.0804650106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal R, Kumar M, Murray K, Morrison RS, Pfeiffer SE (1996) Regulation of FGF receptors in the oligodendrocyte lineage. Mol Cell Neurosci 7:263–275. 10.1006/mcne.1996.0020 [DOI] [PubMed] [Google Scholar]
- Barnabé-Heider F, Wasylnka JA, Fernandes KJ, Porsche C, Sendtner M, Kaplan DR, Miller FD (2005) Evidence that embryonic neurons regulate the onset of cortical gliogenesis via cardiotrophin-1. Neuron 48:253–265. 10.1016/j.neuron.2005.08.037 [DOI] [PubMed] [Google Scholar]
- Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME (1997) Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science 278:477–483. 10.1126/science.278.5337.477 [DOI] [PubMed] [Google Scholar]
- Bronstein R, Kyle J, Abraham AB, Tsirka SE (2017) Neurogenic to gliogenic fate transition perturbed by loss of HMGB2. Front Mol Neurosci 10:153. 10.3389/fnmol.2017.00153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao F, Hata R, Zhu P, Nakashiro K, Sakanaka M (2010) Conditional deletion of Stat3 promotes neurogenesis and inhibits astrogliogenesis in neural stem cells. Biochem Biophys Res Commun 394:843–847. 10.1016/j.bbrc.2010.03.092 [DOI] [PubMed] [Google Scholar]
- Chen F, LoTurco J (2012) A method for stable transgenesis of radial glia lineage in rat neocortex by piggyBac mediated transposition. J Neurosci Methods 207:172–180. 10.1016/j.jneumeth.2012.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChiara TM, Vejsada R, Poueymirou WT, Acheson A, Suri C, Conover JC, Friedman B, McClain J, Pan L, Stahl N, Ip NY, Yancopoulos GD (1995) Mice lacking the CNTF receptor, unlike mice lacking CNTF, exhibit profound motor neuron deficits at birth. Cell 83:313–322. 10.1016/0092-8674(95)90172-8 [DOI] [PubMed] [Google Scholar]
- Déjardin J, Rappailles A, Cuvier O, Grimaud C, Decoville M, Locker D, Cavalli G (2005) Recruitment of Drosophila polycomb group proteins to chromatin by DSP1. Nature 434:533–538. 10.1038/nature03386 [DOI] [PubMed] [Google Scholar]
- Deneen B, Ho R, Lukaszewicz A, Hochstim CJ, Gronostajski RM, Anderson DJ (2006) The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron 52:953–968. 10.1016/j.neuron.2006.11.019 [DOI] [PubMed] [Google Scholar]
- Ebisu H, Iwai-Takekoshi L, Fujita-Jimbo E, Momoi T, Kawasaki H (2017) Foxp2 regulates identities and projection patterns of thalamic nuclei during development. Cereb Cortex 27:3648–3659. 10.1093/cercor/bhw187 [DOI] [PubMed] [Google Scholar]
- Ebong S, Yu CR, Carper DA, Chepelinsky AB, Egwuagu CE (2004) Activation of STAT signaling pathways and induction of suppressors of cytokine signaling (SOCS) proteins in mammalian lens by growth factors. Invest Ophthalmol Vis Sci 45:872–878. 10.1167/iovs.03-0311 [DOI] [PubMed] [Google Scholar]
- Esain V, Postlethwait JH, Charnay P, Ghislain J (2010) FGF-receptor signalling controls neural cell diversity in the zebrafish hindbrain by regulating olig2 and sox9. Development 137:33–42. 10.1242/dev.038026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, Martinowich K, Chin MH, He F, Fouse SD, Hutnick L, Hattori D, Ge W, Shen Y, Wu H, ten Hoeve J, Shuai K, Sun YE (2005) DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development 132:3345–3356. 10.1242/dev.01912 [DOI] [PubMed] [Google Scholar]
- Flügge G, Araya-Callis C, Garea-Rodriguez E, Stadelmann-Nessler C, Fuchs E (2014) NDRG2 as a marker protein for brain astrocytes. Cell Tissue Res 357:31–41. 10.1007/s00441-014-1837-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MR. (2010) Specification and morphogenesis of astrocytes. Science 330:774–778. 10.1126/science.1190928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi-Shimogori T, Grove EA (2001) Neocortex patterning by the secreted signaling molecule FGF8. Science 294:1071–1074. 10.1126/science.1064252 [DOI] [PubMed] [Google Scholar]
- Furusho M, Kaga Y, Ishii A, Hébert JM, Bansal R (2011) Fibroblast growth factor signaling is required for the generation of oligodendrocyte progenitors from the embryonic forebrain. J Neurosci 31:5055–5066. 10.1523/JNEUROSCI.4800-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge WP, Miyawaki A, Gage FH, Jan YN, Jan LY (2012) Local generation of glia is a major astrocyte source in postnatal cortex. Nature 484:376–380. 10.1038/nature10959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobius I, Morcom L, Suárez R, Bunt J, Bukshpun P, Reardon W, Dobyns WB, Rubenstein JL, Barkovich AJ, Sherr EH, Richards LJ (2016) Astroglial-mediated remodeling of the interhemispheric midline is required for the formation of the corpus callosum. Cell Rep 17:735–747. 10.1016/j.celrep.2016.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes WA, Mehler MF, Kessler JA (2003) Transgenic overexpression of BMP4 increases astroglial and decreases oligodendroglial lineage commitment. Dev Biol 255:164–177. 10.1016/S0012-1606(02)00037-4 [DOI] [PubMed] [Google Scholar]
- Gotoh I, Fukuda M, Adachi M, Nishida E (1999) Control of the cell morphology and the S phase entry by mitogen-activated protein kinase kinase. A regulatory role of its N-terminal region. J Biol Chem 274:11874–11880. 10.1074/jbc.274.17.11874 [DOI] [PubMed] [Google Scholar]
- Grosche A, Grosche J, Tackenberg M, Scheller D, Gerstner G, Gumprecht A, Pannicke T, Hirrlinger PG, Wilhelmsson U, Hüttmann K, Härtig W, Steinhäuser C, Pekny M, Reichenbach A (2013) Versatile and simple approach to determine astrocyte territories in mouse neocortex and hippocampus. PLoS One 8:e69143. 10.1371/journal.pone.0069143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu F, Hata R, Ma YJ, Tanaka J, Mitsuda N, Kumon Y, Hanakawa Y, Hashimoto K, Nakajima K, Sakanaka M (2005) Suppression of Stat3 promotes neurogenesis in cultured neural stem cells. J Neurosci Res 81:163–171. 10.1002/jnr.20561 [DOI] [PubMed] [Google Scholar]
- Guagnano V, Furet P, Spanka C, Bordas V, Le Douget M, Stamm C, Brueggen J, Jensen MR, Schnell C, Schmid H, Wartmann M, Berghausen J, Drueckes P, Zimmerlin A, Bussiere D, Murray J, Graus Porta D (2011) Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamin o]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J Med Chem 54:7066–7083. 10.1021/jm2006222 [DOI] [PubMed] [Google Scholar]
- Guillemot F, Zimmer C (2011) From cradle to grave: the multiple roles of fibroblast growth factors in neural development. Neuron 71:574–588. 10.1016/j.neuron.2011.08.002 [DOI] [PubMed] [Google Scholar]
- Hajihosseini MK, Dickson C (1999) A subset of fibroblast growth factors (Fgfs) promote survival, but fgf-8b specifically promotes astroglial differentiation of rat cortical precursor cells. Mol Cell Neurosci 14:468–485. 10.1006/mcne.1999.0800 [DOI] [PubMed] [Google Scholar]
- Hayakawa I, Kawasaki H (2010) Rearrangement of retinogeniculate projection patterns after eye-specific segregation in mice. PLoS One 5:e11001. 10.1371/journal.pone.0011001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Ge W, Martinowich K, Becker-Catania S, Coskun V, Zhu W, Wu H, Castro D, Guillemot F, Fan G, de Vellis J, Sun YE (2005) A positive autoregulatory loop of jak-STAT signaling controls the onset of astrogliogenesis. Nat Neurosci 8:616–625. 10.1038/nn1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi Y, Suzki N, Tsuboi M, Endo TA, Toyoda T, Shinga J, Koseki H, Vidal M, Gotoh Y (2009) Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 63:600–613. 10.1016/j.neuron.2009.08.021 [DOI] [PubMed] [Google Scholar]
- Hoshiba Y, Toda T, Ebisu H, Wakimoto M, Yanagi S, Kawasaki H (2016) Sox11 balances dendritic morphogenesis with neuronal migration in the developing cerebral cortex. J Neurosci 36:5775–5784. 10.1523/JNEUROSCI.3250-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai L, Ohashi Y, van der List D, Usrey WM, Miyashita Y, Kawasaki H (2013) FoxP2 is a parvocellular-specific transcription factor in the visual thalamus of monkeys and ferrets. Cereb Cortex 23:2204–2212. 10.1093/cercor/bhs207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang P, Lee HK, Glasgow SM, Finley M, Donti T, Gaber ZB, Graham BH, Foster AE, Novitch BG, Gronostajski RM, Deneen B (2012) Sox9 and NFIA coordinate a transcriptional regulatory cascade during the initiation of gliogenesis. Neuron 74:79–94. 10.1016/j.neuron.2012.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Mizuseki K, Nishikawa S, Kaneko S, Kuwana Y, Nakanishi S, Nishikawa SI, Sasai Y (2000) Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron 28:31–40. 10.1016/S0896-6273(00)00083-0 [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Crowley JC, Livesey FJ, Katz LC (2004) Molecular organization of the ferret visual thalamus. J Neurosci 24:9962–9970. 10.1523/JNEUROSCI.2165-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SI, Oceguera-Yanez F, Sakurai C, Nakagawa M, Yamanaka S, Woltjen K (2016) Inducible transgene expression in human iPS cells using versatile all-in-one piggyBac transposons. Methods Mol Biol 1357:111–131. 10.1007/7651_2015_251 [DOI] [PubMed] [Google Scholar]
- Li X, Newbern JM, Wu Y, Morgan-Smith M, Zhong J, Charron J, Snider WD (2012) MEK is a key regulator of gliogenesis in the developing brain. Neuron 75:1035–1050. 10.1016/j.neuron.2012.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabie PC, Mehler MF, Kessler JA (1999) Multiple roles of bone morphogenetic protein signaling in the regulation of cortical cell number and phenotype. J Neurosci 19:7077–7088. 10.1523/JNEUROSCI.19-16-07077.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynoga B, Drechsel D, Guillemot F (2012) Molecular control of neurogenesis: a view from the mammalian cerebral cortex. Cold Spring Harb Perspect Biol 4:a008359. 10.1101/cshperspect.a008359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masu Y, Wolf E, Holtmann B, Sendtner M, Brem G, Thoenen H (1993) Disruption of the CNTF gene results in motor neuron degeneration. Nature 365:27–32. 10.1038/365027a0 [DOI] [PubMed] [Google Scholar]
- Masuda K, Toda T, Shinmyo Y, Ebisu H, Hoshiba Y, Wakimoto M, Ichikawa Y, Kawasaki H (2015) Pathophysiological analyses of cortical malformation using gyrencephalic mammals. Sci Rep 5:15370. 10.1038/srep15370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui H, Fujimoto N, Sasakawa N, Ohinata Y, Shima M, Yamanaka S, Sugimoto M, Hotta A (2014) Delivery of full-length factor VIII using a piggyBac transposon vector to correct a mouse model of hemophilia A. PLoS One 9:e104957. 10.1371/journal.pone.0104957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto N, Shinmyo Y, Ichikawa Y, Kawasaki H (2017) Gyrification of the cerebral cortex requires FGF signaling in the mammalian brain. eLife 6:e29285. 10.7554/eLife.29285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon RD, Matsui T, Dubois-Dalcq M, Aaronson SA (1990) FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron 5:603–614. 10.1016/0896-6273(90)90215-2 [DOI] [PubMed] [Google Scholar]
- Miller FD, Gauthier AS (2007) Timing is everything: making neurons versus glia in the developing cortex. Neuron 54:357–369. 10.1016/j.neuron.2007.04.019 [DOI] [PubMed] [Google Scholar]
- Nakashima K, Wiese S, Yanagisawa M, Arakawa H, Kimura N, Hisatsune T, Yoshida K, Kishimoto T, Sendtner M, Taga T (1999) Developmental requirement of gp130 signaling in neuronal survival and astrocyte differentiation. J Neurosci 19:5429–5434. 10.1523/JNEUROSCI.19-13-05429.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namihira M, Nakashima K (2013) Mechanisms of astrocytogenesis in the mammalian brain. Curr Opin Neurobiol 23:921–927. 10.1016/j.conb.2013.06.002 [DOI] [PubMed] [Google Scholar]
- Namihira M, Kohyama J, Semi K, Sanosaka T, Deneen B, Taga T, Nakashima K (2009) Committed neuronal precursors confer astrocytic potential on residual neural precursor cells. Dev Cell 16:245–255. 10.1016/j.devcel.2008.12.014 [DOI] [PubMed] [Google Scholar]
- Nieto M, Monuki ES, Tang H, Imitola J, Haubst N, Khoury SJ, Cunningham J, Gotz M, Walsh CA (2004) Expression of cux-1 and cux-2 in the subventricular zone and upper layers II-IV of the cerebral cortex. J Comp Neurol 479:168–180. 10.1002/cne.20322 [DOI] [PubMed] [Google Scholar]
- Ochiai W, Yanagisawa M, Takizawa T, Nakashima K, Taga T (2001) Astrocyte differentiation of fetal neuroepithelial cells involving cardiotrophin-1-induced activation of STAT3. Cytokine 14:264–271. 10.1006/cyto.2001.0883 [DOI] [PubMed] [Google Scholar]
- Oh LY, Denninger A, Colvin JS, Vyas A, Tole S, Ornitz DM, Bansal R (2003) Fibroblast growth factor receptor 3 signaling regulates the onset of oligodendrocyte terminal differentiation. J Neurosci 23:883–894. 10.1523/JNEUROSCI.23-03-00883.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega S, Ittmann M, Tsang SH, Ehrlich M, Basilico C (1998) Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc Natl Acad Sci U S A 95:5672–5677. 10.1073/pnas.95.10.5672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Shen Q, Goderie SK, He W, Capela A, Davis AA, Temple S (2000) Timing of CNS cell generation: a programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron 28:69–80. 10.1016/S0896-6273(00)00086-6 [DOI] [PubMed] [Google Scholar]
- Rajan P, McKay RD (1998) Multiple routes to astrocytic differentiation in the CNS. J Neurosci 18:3620–3629. 10.1523/JNEUROSCI.18-10-03620.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowitch DH, Kriegstein AR (2010) Developmental genetics of vertebrate glial-cell specification. Nature 468:214–222. 10.1038/nature09611 [DOI] [PubMed] [Google Scholar]
- Saito T. (2006) In vivo electroporation in the embryonic mouse central nervous system. Nat Protoc 1:1552–1558. 10.1038/nprot.2006.276 [DOI] [PubMed] [Google Scholar]
- Sehara K, Toda T, Iwai L, Wakimoto M, Tanno K, Matsubayashi Y, Kawasaki H (2010) Whisker-related axonal patterns and plasticity of layer 2/3 neurons in the mouse barrel cortex. J Neurosci 30:3082–3092. 10.1523/JNEUROSCI.6096-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour PA, Shih HP, Patel NA, Freude KK, Xie R, Lim CJ, Sander M (2012) A Sox9/Fgf feed-forward loop maintains pancreatic organ identity. Development 139:3363–3372. 10.1242/dev.078733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan SA, Barres BA (2014) Mechanisms of astrocyte development and their contributions to neurodevelopmental disorders. Curr Opin Neurobiol 27:75–81. 10.1016/j.conb.2014.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MR, Ghosh A (2004) FGF2-induced chromatin remodeling regulates CNTF-mediated gene expression and astrocyte differentiation. Nat Neurosci 7:229–235. 10.1038/nn1192 [DOI] [PubMed] [Google Scholar]
- Stolt CC, Lommes P, Sock E, Chaboissier MC, Schedl A, Wegner M (2003) The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes Dev 17:1677–1689. 10.1101/gad.259003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Meyers EN, Lewandoski M, Martin GR (1999) Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes Dev 13:1834–1846. 10.1101/gad.13.14.1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata H, Nakajima K (2001) Efficient in utero gene transfer system to the developing mouse brain using electroporation: visualization of neuronal migration in the developing cortex. Neuroscience 103:865–872. 10.1016/S0306-4522(01)00016-1 [DOI] [PubMed] [Google Scholar]
- Tchieu J, Calder EL, Guttikonda SR, Gutzwiller EM, Aromolaran KA, Steinbeck JA, Goldstein PA, Studer L (2019) NFIA is a gliogenic switch enabling rapid derivation of functional human astrocytes from pluripotent stem cells. Nat Biotechnol 37:267–275. 10.1038/s41587-019-0035-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda T, Homma D, Tokuoka H, Hayakawa I, Sugimoto Y, Ichinose H, Kawasaki H (2013) Birth regulates the initiation of sensory map formation through serotonin signaling. Dev Cell 27:32–46. 10.1016/j.devcel.2013.09.002 [DOI] [PubMed] [Google Scholar]
- Tsang M, Dawid IB (2004) Promotion and attenuation of FGF signaling through the Ras-MAPK pathway. Sci STKE 2004:pe17. 10.1126/stke.2282004pe17 [DOI] [PubMed] [Google Scholar]
- Vaccarino FM, Schwartz ML, Raballo R, Nilsen J, Rhee J, Zhou M, Doetschman T, Coffin JD, Wyland JJ, Hung YT (1999) Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat Neurosci 2:246–253. 10.1038/6350 [DOI] [PubMed] [Google Scholar]
- Wakimoto M, Sehara K, Ebisu H, Hoshiba Y, Tsunoda S, Ichikawa Y, Kawasaki H (2015) Classic cadherins mediate selective intracortical circuit formation in the mouse neocortex. Cereb Cortex 25:3535–3546. 10.1093/cercor/bhu197 [DOI] [PubMed] [Google Scholar]
- Wang S, Liang Q, Qiao H, Li H, Shen T, Ji F, Jiao J (2016) DISC1 regulates astrogenesis in the embryonic brain via modulation of RAS/MEK/ERK signaling through RASSF7. Development 143:2732–2740. 10.1242/dev.133066 [DOI] [PubMed] [Google Scholar]
- Wang Y, Kim E, Wang X, Novitch BG, Yoshikawa K, Chang LS, Zhu Y (2012) ERK inhibition rescues defects in fate specification of Nf1-deficient neural progenitors and brain abnormalities. Cell 150:816–830. 10.1016/j.cell.2012.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasylyk B, Hagman J, Gutierrez-Hartmann A (1998) Ets transcription factors: nuclear effectors of the ras-MAP-kinase signaling pathway. Trends Biochem Sci 23:213–216. 10.1016/S0968-0004(98)01211-0 [DOI] [PubMed] [Google Scholar]
- Yang X, Qiao D, Meyer K, Friedl A (2009) Signal transducers and activators of transcription mediate fibroblast growth factor-induced vascular endothelial morphogenesis. Cancer Res 69:1668–1677. 10.1158/0008-5472.CAN-07-6385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yordy JS, Muise-Helmericks RC (2000) Signal transduction and the ets family of transcription factors. Oncogene 19:6503–6513. 10.1038/sj.onc.1204036 [DOI] [PubMed] [Google Scholar]
- Zheng CF, Guan KL (1994) Activation of MEK family kinases requires phosphorylation of two conserved Ser/Thr residues. EMBO J 13:1123–1131. 10.1002/j.1460-2075.1994.tb06361.x [DOI] [PMC free article] [PubMed] [Google Scholar]